

Abstract
Alzheimer disease (AD) is characterized by deposition of amyloid-β, tau, and other specific proteins that accumulate in the brain in detergent-insoluble complexes. AD also involves glutamatergic neurotransmitter system disturbances. Excitatory amino acid transporter 2 (EAAT2) is the dominant glutamate transporter in cerebral cortex and hippocampus. We investigated whether accumulation of detergent-insoluble EAAT2 is related to cognitive impairment and neuropathologic changes in AD by quantifying detergent-insoluble EAAT2 levels in hippocampus and frontal cortex of cognitively normal patients, patients with clinical dementia rating (CDR) = 0.5 (mildly impaired), and AD patients. Parkinson disease (PD) patients served as neurodegenerative disease controls. We found that Triton X-100-insoluble EAAT2 levels were significantly increased in patients with AD compared to controls, while Triton X-100-insoluble EAAT2 levels in CDR = 0.5 patients were intermediately elevated between control and AD subjects. Detergent-insolubility of Presenilin-1, a structurally similar protein, did not differ among the groups, thus arguing EAAT2 detergent-insolubility was not due to nonspecific cellular injury. These findings demonstrate that detergent-insoluble EAAT2 accumulation is a progressive biochemical lesion that correlates with cognitive impairment and neuropathologic changes in AD. These findings lend further support to the idea that dysregulation of the glutamatergic system may play a significant role in AD pathogenesis.
Keywords: Glutamate, Alzheimer disease, EAAT2, Excitotoxicity, Mild cognitive impairment, Protein aggregation, Oxidative stress, SLC1A2
Introduction
Aberrant glutamate stimulation has been proposed as a mechanism by which synapses and neurons are injured in Alzheimer disease (AD) (1). Excitatory amino acid transporter 2 (EAAT2; also called GLT-1) is the major glutamate transporter in forebrain that is responsible for a number of essential neuroprotective and regulatory functions that include preventing glutamate-mediated injury to neurons and synapses and regulating normal synaptic input specificity (2-5). Several reports indicate that EAAT2 levels are significantly reduced in AD (6-8), thus raising the possibility that glutamate dyshomeostasis plays a role in AD pathogenesis. In addition, EAAT2 is oxidatively damaged by exposure to amyloid-β (Aβ) (9-13). EAAT2 oxidation impairs glutamate uptake and promotes the formation of high molecular weight EAAT2 oligomers that are insoluble in detergents such as Triton X-100 (14-16).
These findings suggest that AD pathogenesis may disrupt EAAT2 via mechanisms that recapitulate those of other key AD-related molecules, most notably Aβ, which undergoes oxidation, misfolding and aggregation. Although the studies cited above establish that EAAT2 is biochemically and functionally damaged by Aβ-related processes, the potential disease relevance of these findings have not been examined in AD patients. There currently is little evidence at the protein level on the relationship between aberrant EAAT2 expression and the degree of cognitive loss and associated AD pathology. To address this important issue we measured Triton X-100-insoluble and Triton X-100-soluble EAAT2 in the hippocampus and frontal cortex of more than 100 clinically and pathologically well-characterized normal controls, patients with Clinical Dementia Rating (CDR) = 0.5 (17), and later-stage AD patients.
Materials and Methods
Patients
Subjects were from the Alzheimer's Disease Center (ADC), Oregon Health and Sciences University (OHSU), and Alzheimer's Disease Research Center (ADRC) at the University of Washington (Table). Control subjects and subjects with clinical dementia rating CDR = 0.5, (intended to approximate mild cognitive impairment), were participants in brain aging studies at the Oregon Aging/ADC. Subjects received annual neurologic and neuropsychologic evaluation, with CDR assigned by an experienced clinician. Controls had normal cognitive and functional examinations. CDR = 0.5 subjects were functionally intact on enrollment and progressed to a global CDR = 0.5 (no sub-scores greater than 0.5) at their last evaluation, within a year of autopsy. AD subjects were diagnosed by clinical team consensus conference, met NINDS-ADRDA diagnostic criteria for clinical AD, had CDR = >1.0, and neuropathologic confirmation at autopsy (after informed consent). Tissue use conformed to IRB-approved protocols. Neuropathologic assessment conformed to NIA-Reagan consensus criteria (18). The AD group included subjects with probable AD, moderate-to-frequent neuritic plaques, and Braak stage V-VI neurofibrillary tangles. Controls were clinically non-demented subjects with sparse or no neuritic plaques and neurofibrillary tangles ≤ Braak stage II. AD patients and controls with Lewy body disease involving the brainstem (including substantia nigra), amygdala, middle frontal gyrus, and patients with vascular brain disease manifested by grossly observed arterial territorial infarcts, grossly observed lacunar infarcts, or microvascular infarcts were excluded. PD patients had expected clinical signs, symptoms, and midbrain, but not cerebral cortex, Lewy body pathology.
Table. Patient Demographic and Pathologic Characteristics.
| Group | N | Male: Female | Age (y) | PMI (h) | CERAD Score | Braak Stage |
|---|---|---|---|---|---|---|
| Hippocampus | ||||||
| AD | 22 | 6:5 | 84.3 +/- 2.2 | 11.4 +/- 1.4 | Frequent (moderate-frequent) | VI (V-VI) |
| CDR=0.5 | 14 | 7:8 | 88.9 +/- 2.2 | 13.0 +/- 2.0 | Sparse (none-frequent) | IV (I-VI) |
| Norm | 13 | 8:5 | 87.0 +/- 1.7 | 9.8 +/- 1.3 | Sparse (none-sparse) | II (I-III) |
| PD | 4 | 1:1 | 81.0 +/- 5.7 | 17.8 +/- 2.2 | Sparse (none-sparse) | II (II-III) |
| Frontal Cortex | ||||||
| AD | 55 | 6:5 | 82.7 +/- 1.3 | 11.6 +/- 0.8 | Frequent (sparse-frequent) | VI (V-VI) |
| CDR=0.5 | 23 | 1:1 | 90.4 +/- 1.8 | 12.5 +/- 1.4 | Sparse (none-frequent) | III (I-VI) |
| Norm | 20 | 2:3 | 90.1 +/- 1.7 | 11.2 +/- 1.2 | None (non-sparse) | II (I-III) |
| PD | 4 | 3:1 | 84.9 +/- 3.7 | 10.5 +/- 4.3 | None (none-sparse) | II (I-III) |
Abbreviations: Alzheimer disease (AD), clinical dementia rating (CDR), normal controls (Norm), Parkinson disease (PD), number of subjects (N), Male:Female (M:F) ratio, age at death (Age), postmortem interval (PMI), consortium to establish a registry for Alzheimer disease (CERAD).
Enzyme-Linked Immunosorbent Assays
Brain samples were homogenized, sequentially extracted in 10 mM Tris, 1 mM ethylene glycol tetra-acetic acid, 1 mM dithiothreitol, 10% sucrose, and then extracted 3× with 1% Triton X-100, as previously described (19). Remaining detergent-insoluble material was extracted with 70% formic acid. Formic acid extracts of detergent-insoluble proteins were resolubilized and adsorbed onto 96-well plates as described elsewhere (19). EAAT2/GLT-1 was detected with antibodies AB12 and GLT-1A (20). Presenilin-1 (PS1) was detected with PS1 N-terminal fragment antisera (21). Aβ was detected with 4G8 (Covance/Signet Laboratories, Dedham, MA). Total tau was detected using anti-tau antibody (Dako, Carpinteria, CA). Enzyme-linked immunosorbent assay (ELISA) plates were developed using standard methods with horseradish peroxidase (HRP)-conjugated antibodies and tetramethylbenzidine substrate.
Immunohistochemistry and Biochemistry
Standard immunohistochemical methods were used to evaluate EAAT2 staining in paraffin-embedded postmortem brain sections. Slides were incubated with AB12 and developed using 3,3′-Diaminobenzidine (Vector Laboratories, Burlingame, CA). Double immunostaining labeled EAAT2 (DAB brown chromogen) and either total tau (Tau-2; Sigma, St. Louis, MO) or Ab (4G8) labeled with Vector Red (Vector Laboratories). Counterstains were omitted in double-label experiments. A Nikon Optiphot-2 microscope/Insight QE digital camera was used. Image acquisition was performed using Spot imaging software (Diagnostics Instruments, Sterling Heights, MI) and formatted with Photoshop. For each experiment, images were acquired and digitally processed under identical conditions. Digital image processing was limited to linear brightness and contrast adjustments performed identically on experimental and control images. Mouse brain extracts were solubilized in Laemmli sample buffer. Total Triton X-100 soluble protein concentrations were determined by the BCA method (Pierce, Rockford, IL) and Western blotted with AB12 or GLT-1A, and detected using HRP-conjugated antibodies and chemiluminescence.
Liquid Chromatography-Tandem Mass Spectrometry
Equal amounts of formic acid-extracted protein were prepared from 5 AD subjects (mean age: 80 years; gender: 3 female and 2 male; CERAD NP score: Moderate or Frequent; Braak Stage: VI), pooled, dried, dissolved in bicarbonate buffer, reduced, and treated with iodoacetamide before being subjected to trypsin digestion, as previously described (22). Eluted peptides were resuspended in 0.1% formic acid, separated by 2-dimensional microcapillary high-performance liquid chromatography, and amino acid sequences of separated peptides determined by tandem mass spectrometry (ThermoFinnigan, San Jose, CA). Proteins from the mixture were identified automatically using the SEQUEST program, which searched spectral data against the International Protein Database. Sensitivity and specificity of protein identifications were determined by PeptideProphet and ProteinProphet as previously described (22).
Statistics
Data were analyzed with analysis of variance (ANOVA), Pearson correlation tests, and Fisher exact tests using SPSS 15.0 (SPSS, Chicago, IL). We predicted that detergent-insoluble EAAT2 would be highest in later stage AD cases, lower in CDR = 0.5 cases, and lowest in normal controls and PD patients lacking concurrent AD pathology. To test specifically this a priori prediction, well-established ANOVA trend analyses methods (23, 24) were used with the trend weights -2, -2, 1, and 3 corresponding to the PD, normal controls, CDR = 0.5, and AD, groups, respectively. In keeping with accepted methods these trend weights were selected because they are orthogonal integers (sum to zero) and represent the originally predicted relationships among the study groups (23, 24). Specifically, we predicted that the levels of insoluble EAAT2 in the PD and normal control groups would be both comparable to each other and lower than the other 2 groups (−2, -2; PD and normal controls). The insoluble EAAT2 levels of the CDR = 0.5 group were predicted to be higher than PD and normal controls but lower than the AD group, while the AD group was expected to have the highest insoluble EAAT2 levels (+1, +3 for the CDR = 0.5 and AD groups, respectively). These trend analyses followed only statistically significant omnibus ANOVA results. Because this was the only trend predicted a priori, no other trend analyses were attempted.
Results
Case Material
Among the hippocampal sample AD, CDR = 0.5, normal control, and PD subject groups studied, differences in male:female ratios, postmortem intervals and ages at death were not statistically significant (Fisher exact tests: [9] = 0.993; F[3,49] = 1.770; and F[3,49] = 1.149, respectively). Group differences in Consortium to Establish a Registry for Alzheimer Disease (CERAD) scores and Braak stages were significant (Fisher exact tests: [9] = 46.385, p < 0.00001 and [18] = 71.067, p < 0.00001, respectively). For the frontal cortex subject sample group, differences in the male:female ratios and postmortem intervals were not significant (Fisher exact tests: [3] = 2.646, and F[3,98] = 0.245, respectively). Ages at death were statistically significant (F[3,98] = 5.708, p < 0.001), due primarily to the AD patients who tended to die earlier than other subjects. As expected, CERAD scores and Braak stages were statistically significant among the groups (Fisher exact test: [9] = 87.238, p < 0.00001 and [18] = 115.049, p < 0.00001, respectively) (Table).
Detection of Detergent-Insoluble EAAT2 in AD by Mass Spectrometry
To examine whether detergent-insoluble EAAT2 accumulates in AD we performed mass spectrometry on tryptic digests of total detergent-insoluble proteins pooled from 5 autopsy-confirmed AD patients. A total of 348 statistically significant (p < 0.05) sequence-to-spectra matches corresponding to EAAT2 tryptic peptide fragments were identified (Fig. 1A). These fragments clustered in 6 primary domains located throughout EAAT2 (Fig. 1B), suggesting that the entire molecule was represented in the detergent-insoluble protein fractions.
Figure 1.

Detergent-insoluble excitatory amino acid transporter 2 (EAAT2) is present in Alzheimer disease frontal cortex. (A) A total of 348 EAAT2 tryptic fragments identified by liquid chromatography tandem mass spectrometry performed on Triton X-100-insoluble proteins from 5 AD patient cerebral cortex samples corresponded to the 24 unique EAAT2 sequences shown. (B) The EAAT2 tryptic fragments mapped to 6 primary structural regions indicated by gray highlighting located throughout the molecule. This EAAT2 model was modified from x-ray crystal structure results of Yernool (25). The primary transmembrane domains are indicated (I-VIII). Hairpin structures denoted as HP1 and HP2 form the glutamate-binding/gating domain.
These data confirm that Triton X-100-insoluble EAAT2 is present in the brains of AD patients. Despite the structural accuracy of liquid chromatography tandem mass spectrometry, this approach is not well suited for quantitative comparisons between affected and control subjects. To examine whether EAAT2 detergent-insolubility is elevated in AD we developed 2 independent EAAT2 ELISAs. This approach was adopted to facilitate quantification of the relatively large number of samples to be tested (102 cortex and 53 hippocampus samples measured in triplicate).
Specificity of EAAT2 Detection
AB12 is a polyclonal antibody that recognizes an amino-terminus epitope common to all EAAT2/GLT-1 isoforms; GLT-1A is a polyclonal antibody that recognizes the C-terminal domain of the dominant EAAT2 isoform expressed in brain (20). These antibodies were tested under a variety of conditions using brain tissue from GLT-1 wild-type (WT) and GLT-1 knockout (KO) mice. Figure 2 shows that AB12 and GLT-1A selectively recognized EAAT2/GLT-1 by ELISA (Fig. 2A). In addition, we verified that EAAT2/GLT-1 levels measured by these ELISAs increased as a linear function of protein assayed (linear regression for AB12 ELISA: r2 = 0.8799, F[1,23] = 168.6, p < 0.0001; and GLT-1A ELISA: r2 = 0.9567, F[1,23] = 507.8, p < 0.00001). AB12 and GLT-1A also detected GLT-1 by immunohistochemistry (Fig. 2B) and recognized Triton X-100-soluble GLT-1 via Western blots (Fig. 2C) from GLT-1 WT, but not GLT-1 knock-out mice. These data prove that AB12 and GLT-1A specifically recognize EAAT2.
Figure 2.
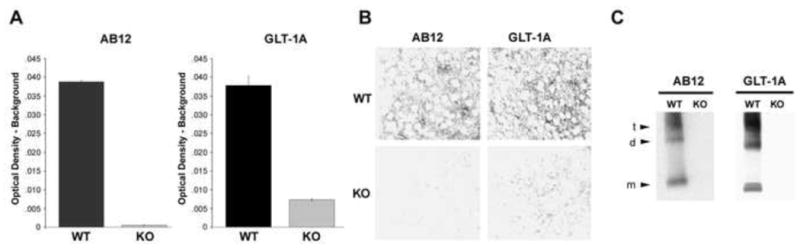
Specificity of excitatory amino acid transporter 2 (EAAT2) detection. Two different anti-EAAT2 antibodies (AB12 and GLT-1A that recognize the N- and C-terminus, respectively, were used. Pan-specific AB12 recognizes all EAAT2/GLT-1 isoforms; GLT-1A recognizes EAAT2a/GLT-1a, the most abundant EAAT2 isoform in cortex and hippocampus. Using brain tissue from GLT-1 wild-type (WT) and knock-out (KO) mice ELISAs (A), immunohistochemistry in cortex (B), and Western blots (C) prove the specificity of EAAT2/GLT-1 detection. EAAT2/GLT-1 is a homo-trimer that resolves as monomers (m), dimers (d), and apparent trimers (t) with approximate molecular weights of 70 kDa, 150 kDa, and >200 kDa, respectively. Error bars in A indicate standard error of the mean of WT and KO brain tissue samples measured in triplicate.
Detergent-Insoluble EAAT2 Aberrantly Accumulates in AD
Frontal cortex and hippocampus tissue samples were serially extracted X3 with Triton X-100 to remove detergent-soluble proteins and residual detergent-insoluble proteins were then solubilized with formic acid. Neutralized formic acid-extracted proteins and the first Triton X-100-soluble fraction were analyzed by ELISAs to determine the relative levels of detergent-insoluble and detergent-soluble EAAT2.
In hippocampus samples, detergent-insoluble EAAT2 levels were elevated in AD patients compared to normal controls and PD patients, whereas detergent-insoluble EAAT2 levels in CDR = 0.5 patients were intermediately elevated between the controls and later-stage AD patients (Fig. 3A, B). These differences in detergent-insoluble EAAT2 levels among groups detected either with AB12 or GLT-1A ELISAs were statistically significant (F[3,49] = 7.107, p < 0.0005 and F[3,49] = 6.733, p < 0.0007, respectively). These data suggest that EAAT2 becomes increasingly detergent-insoluble as AD-related pathology progresses from the less demented state to CDR = 0.5, and then becomes most pronounced by later stage AD. A contrast analysis tested the prediction that detergent-insoluble EAAT2 levels followed a statistically significant trend where: normal = PD < CDR = 0.5 < AD (AB12: t[49] = 3.685, p < 0.001; and GLT-1A: t[49] = 3.353, p < 0.002). The scatter plot in Figure 3C shows that detergent-insoluble EAAT2 levels in hippocampus measured by AB12 and GLT-1A ELISAs were positively correlated (r = 0.820, n = 53, p < 0.01), thereby confirming the close correspondence between 2 independent assays which show that EAAT2 detergent insolubility increases as AD pathology increases.
Figure 3.

Detergent-insoluble EAAT2 levels were increased in hippocampi of patients with Alzheimer disease (AD) pathology. (A) Total Triton X–100-insoluble proteins from Parkinson disease (PD, n = 4),normal control (Norm, n = 13), mild cognitive impairment (n = 14), and later stage AD (n = 22) patients were solubilized with formic acid and analyzed by AB12 ELISAs to quantify detergent-insoluble EAAT2 levels. (B) Total Triton X-100-insoluble EAAT2 levels were measured by GLT-1A ELISAs as in panel A. (C) Scatter plot shows correlation between insoluble EAAT2 levels measured by AB12 and GLT-1A ELISAs for all subjects (n = 53). P value indicates statistical significance determined by Pearson's correlation. (D) Total Triton X-100-insoluble levels of presenilin-1 (PS1) were measured in the same samples as shown in panels A and B. (E, F) Triton X-100-soluble EAAT2 levels from same samples as in panels A, B, and D were measured by AB12 and GLT-1A ELISAs, respectively. P values in panels A, B, D–F indicate results of overall single-factor ANOVAs.
In marked contrast to these findings, Triton X-100-insoluble PS1 levels (Fig. 3D) in the same samples did not differ among groups (F[3,49] = 2.141, n.s.). PS1 was chosen for comparison because it is localized prominently in astrocytes (26), which express approximately 80% of the EAAT2 in hippocampus and because overall PS1 expression levels are not affected by AD (27). These data argue that the detergent insolubility displayed by EAAT2 was not the result of generalized, non-specific cellular injury. Moreover, PS1 is structurally similar to EAAT2. Each molecule has 8 hydrophobic membrane-spanning domains with the N- and C- terminal domains localized intracellularly (27). Thus, the detergent insolubility profiles of EAAT2 obtained from the identical samples are unlikely to have arisen by non-specific protein-protein interactions that might have occurred during the extraction process.
We also examined detergent-soluble EAAT2 levels in the same hippocampal samples (Fig. 3E, F). In contrast to the significant trend of increasing EAAT2 detergent insolubility there was a statistically significant difference among groups in detergent-soluble EAAT2 levels detected by AB12 ELISAs (F[3,49] = 3.237, p < 0.030); detergent-soluble EAAT2 levels appeared to decrease with increasing AD pathology. Differences in detergent-soluble EAAT2 levels detected by GLT-1A ELISAs were not statistically significant (F[3,49] = 2.468, n.s.).
Because detergent-soluble EAAT2 levels were comparatively low in AD patients compared to normal controls while the detergent-insoluble EAAT2 levels were markedly elevated in the same samples, it is highly unlikely that the increased detergent-insoluble EAAT2 measured in AD patients could have been due to incomplete extraction of Triton X-100-soluble EAAT2 or that this reflects non-specific EAAT2 associations with detergent-insoluble amyloid plaques or neurofibrillary tangles (NFTs). This conclusion was further supported by the findings that detergent-soluble EAAT2 levels were not significantly correlated with detergent-insoluble Aβ levels measured by 4G8 ELISAs (AB12 vs. Aβ: r = -0.167, n = 53, n.s. and GLT-1A vs. Aβ: r = -0.162, n = 53, n.s.). Similarly, detergent-soluble EAAT2 levels were not correlated with insoluble tau levels measured by tau-2 ELISAs (AB12 vs. tau: r = -0.141, n = 53, n.s. and GLT-1A vs. tau: r = -0.131, n = 53, n.s.). These findings argue against the possibility of non-aggregating EAAT2 in the lysates associated non-specifically with the insoluble Aβ or insoluble tau.
As in the hippocampi, Triton X-100-insoluble EAAT2 levels were markedly elevated in AD frontal cortex compared to PD and normal control subjects (Fig. 4A, B). Detergent-insoluble EAAT2 levels measured by GLT-1A ELISAs in CDR = 0.5 subjects again fell between controls and AD levels (Fig. 4B), whereas increased detergent-insoluble EAAT2 levels measured by AB12 ELISAs in CDR = 0.5 cortex were less pronounced (Fig. 4A). These differences in detergent-insoluble EAAT2 levels among groups were significant (AB12 ELISA: F[3,98] = 8.743, p < 0.00003; GLT-1A ELISA: F[3,98] = 13.375, p < 0.00001). Detergent-insoluble EAAT2 levels in frontal cortex (Fig. 4A, B) followed the predicted trend: controls = PD < CDR = 0.5 < AD confirmed by statistically significant contrast test outcomes for both AB12 and GLT-1A ELISAs (t[98] = 3.100, p < 0.002 and t[98] = 4.087, p < 0.0001, respectively). Figure 4C shows a significant positive correlation between detergent-insoluble EAAT2 levels measured using AB12 vs. GLT-1A (r = 0.801, n = 102, p < 0.01). Triton X-100-insoluble PS1 levels in frontal cortex (Fig. 4D) did not differ significantly among groups (F[3,98] = 1.651, n.s.). In contrast to hippocampus (Fig. 3E, F), detergent-soluble EAAT2 expression levels in frontal cortex of normal controls, CDR = 0.5, and AD patients (Fig. 4E, F) were similar (AB12 ELISA: F[3,98] = 3.669, p < 0.015; GLT-1A ELISA F[3,98] = 1.330, n.s). Again, detergent-soluble EAAT2 levels in frontal cortex did not correlate with the levels of detergent-insoluble Aβ or tau measured in the same samples (AB12 vs. Aβ: r = -0.160, n = 102, n.s.; GLT-1A vs. Aβ: r = -0.008, n = 102, n.s.; AB12 vs. tau: r = -0.105, n = 102, n.s.; and GLT-1A vs. tau: r = -0.049, n = 102, n.s.), thus arguing against the possibility that incomplete extraction or non-specific EAAT2 protein-protein interactions occurred during tissue processing.
Figure 4.

Detergent-insoluble EAAT2 levels were increased in the frontal cortex of patients with Alzheimer disease (AD) pathology. (A) Total Triton X–100-insoluble proteins from Parkinson disease (PD, n = 4), normal control (Norm, n = 20), mild cognitive impairment (n = 23), and later stage AD (n = 55) patients were solubilized with formic acid and analyzed by AB12 ELISA to quantify detergent-insoluble EAAT2 levels. (B) Total Triton X-100-insoluble EAAT2 levels were measured by GLT-1A ELISAs as in panel A. (C) Scatter plot shows correlation between insoluble EAAT2 levels measured by AB12 and GLT-1A ELISAs for all subjects (n = 102). P value indicates statistical significance determined by Pearson's correlation. (D) Total Triton X-100-insoluble levels of presenilin-1 (PS1) were measured in the same samples as shown panels A and B. (E, F) Triton X-100-soluble EAAT2 levels from the same samples as in panels A, B, and D were measured by AB12 and GLT-1A ELISAs, respectively. P values in panels A, B, D–F indicate results of overall single factor ANOVAs.
EAAT2 Localization Appears Comparatively Normal in AD
In normal brain tissue EAAT2 is localized primarily, but not exclusively, in fine astrocytic processes that densely ramify throughout peri- and extra-synaptic domains (28. 29). We performed immunohistochemistry to determine whether aberrant EAAT2 expression was localized near amyloid plaques, in association with NFTs, or aberrantly accumulated in neuronal or astrocytic cell bodies.
Double-label immunostaining failed to reveal a consistent morphological association between EAAT2 expression and Aβ deposits in AD, which argues against the idea that EAAT2 accumulated in association with senile plaques in vivo (Fig. 5A, B). In keeping with the predominantly astrocytic expression pattern characteristic of EAAT2, we found a paucity of EAAT2 immunoreactivity in neuronal cell bodies and dendritic processes in both AD and normal control brains (Fig. 5C, D). The primary morphologically neuron-like EAAT2 expression pattern observed was that associated with apparent neurofibrillary ghost tangles in AD subjects that immunostained weakly for EAAT2 (not shown). Thus, we found limited evidence that EAAT2 was prominently associated with NFTs, a finding consistent with the fact that NFTs are localized primarily in neurons, but not in astrocytes in AD (30).
Figure 5.

Excitatory amino acid transporter 2 (EAAT2) immunohistochemistry (IHC) in Alzheimer disease (AD) and normal controls. (A) EAAT2 IHC using AB12 (brown) in AD frontal cortex reveals an irregular patchy astrocyte-like expression pattern that does not correspond with senile plaques immunostained red with the anti-amyloid-β (A-β) monoclonal antibody 4G8. (B) Higher magnification of the boxed region in panel A highlights the lack of correspondence between EAAT2 immunoreactivity and A-β deposits. Some plaques appeared to be surrounded by EAAT2 immunoreactivity, while other plaques were localized in EAAT2 immuno-negative domains. (C, D) EAAT2 was immunostained with AB12 (brown) in the hippocampal CA1 region of an AD (C) and a normal control (Norm, D). EAAT2 expression was primarily densely stained peri- and extra-synaptic EAAT2-positive puncta in the neuropil. (E) Apparent astrocyte immunostained with AB12 in AD frontal cortex where proximal and distal glial processes and the cell body display EAAT2 immunoreactivity. Prominent EAAT2 immunostaining in astrocyte-like cell bodies was more readily detected in AD than in controls. (F) AB12 immunostained apparent astrocytes in normal control revealed abundant EAAT2 immunoreactivity in proximal and distal processes, with less prominent cell body staining.
Close inspection of EAAT2 immunostaining revealed that EAAT2 was expressed in distal processes and to a lesser extent in cell bodies, both in AD and control brains. Nonetheless, astrocytes with prominent cell body EAAT2 immunostaining were more readily identified in AD frontal cortex than in normal control cortex (Fig. 5E, F). Despite this potentially interesting non-quantitative distinction between AD and control subjects, the overall immunohistochemical findings suggested that EAAT2 localization was not dramatically altered in AD patients compared to controls.
Discussion
Aberrant EAAT2 Detergent-Insolubility Is a Novel Biochemical Lesion in AD
Using 2 independent ELISA systems to measure detergent-insoluble EAAT2, along with corroborating mass spectrometry, we found that detergent-insoluble EAAT2 is aberrantly elevated in AD patient brains compared to controls and intermediately elevated in mildly impaired CDR = 0.5 patients with autopsy-confirmed early AD neuropathology. These data argue that EAAT2 detergent insolubility represents a progressive biochemical lesion of AD. The findings further suggest that EAAT2 belongs to class of specific proteins (Aβ and tau being the best-characterized examples) that display altered detergent solubility in AD, whereas other proteins (31) (including PS1) do not. Our findings are in keeping with previous work showing that glial fibrillary acidic protein becomes increasingly detergent-insoluble in AD (31). Taken together these findings support the idea that detergent insolubility is an aspect of the disease process not restricted to selective neuronal molecules, but also includes specific astrocytic proteins.
In contrast to detergent-insoluble EAAT2, detergent-soluble EAAT2 levels were reduced in AD patients compared to normal controls. These data are in keeping with previous human studies (6-8), and in findings from 2 AD mouse models (32, 33). Protein detergent insolubility does not appear to be recapitulated in AD mice models (data not shown). Thus, while it is currently not possible to examine detergent-insolubility in mice, the effects of AD-related pathology on soluble EAAT2/GLT-1 expression in AD patients and transgenic mice are nonetheless mutually supportive.
Our findings cannot address the mechanisms by which detergent-insoluble EAAT2 accumulates in AD. Potentially important insight comes from data showing that glutamate transporters are sensitive to biological conditions that promote reactive oxygen species (16, 34), and, significantly, oxidative stress is an early and persistent feature of AD (35). Post-translational oxidative EAAT2 modifications both inhibit glutamate uptake (16) and promote formation of detergent-insoluble high molecular weight multimers (34). This correspondence between detergent insolubility and reduced uptake suggests the possibility that increased levels of detergent-insoluble EAAT2 observed in AD brain tissue may reflect increasing EAAT2 dysfunction. This idea is consistent with data showing that Aβ generates oxidative radicals that impair glutamate uptake (36-37). In addition, Aβ impairs glutamate uptake in synaptosomes (9-13).
Until recently, EAAT2 had been widely accepted as an astrocyte-specific glutamate transporter but it is now clear that EAAT2 is also expressed by neurons (38). Such findings, in conjunction with a report that EAAT2 is sporadically expressed in tau-positive cortical and hippocampal neurons of some AD patients (39), raised the question as to whether increased AD-related detergent-insoluble EAAT2 reflects aberrant EAAT2 expression in neurons. We did observe EAAT2 immunoreactivity associated with apparent ghost tangles (neurofibrillary remnants of dead neurons), but absence of EAAT2 perikaryon immunoreactivity in either AD or normal control patients was much more striking. These findings suggest that EAAT2 does not markedly accumulate in somal or proximal dendritic regions of neurons in AD. It seems more likely that detergent-insoluble EAAT2 complexes accumulate in fine peri-synaptic distal astrocytic processes where the majority of EAAT2 is normally localized (28), but which are difficult to investigate by light microscopy. Our human postmortem specimens are not suitable for the electron microscopic approaches required to address this issue in a quantitative manner.
EAATs Regulates Multiple Critical Neuroprotective Functions in the Brain and Are Disturbed in AD
A family of 5 Na+-dependent high affinity glutamate transporters (referred to as EAAT1-5, also known as GLAST, GLT-1, EAAC1, EAAT4, and EAAT5, respectively) carry out the critical task of clearing glutamate, primarily into astrocytes, thereby maintaining glutamate at basal extracellular concentrations that recently have been estimated to be in the low nanomolar range (40). The importance of rapid clearing extracellular glutamate is illustrated by the consequences of injecting potent glutamate transport blockers in mice that die quickly of apparent acute glutamate toxicity (41). EAAT1, EAAT2, and neuron-specific EAAT3 are the primary glutamate transporters in the hippocampus and cortex; EAAT4 is expressed primarily in the cerebellum; EAAT5 is expressed mostly in the retina (42). Of these transporters, EAAT2 is responsible for the majority of glutamate clearance in forebrain and accounts for approximately 80% of the glutamate transporters in hippocampus (28, 29). The disproportionate expression of EAAT2 compared to other glutamate transporter subtypes is reflected by the dramatic phenotype of GLT-1 KO mice, which die shortly after birth due to seizures (2). The phenotypes of EAAT1/GLAST KO and EAAT3/EAAC1 KO mice are subtler. EAAT1/GLAST KO mice develop normally, but display defects in motor coordination related to cerebellar function (43). EAAT3/EAAC1 KO mice also breed normally but develop age-related neuronal loss (44). Nonetheless, both EAAT1 and EAAT3 expression are disturbed in AD (6, 44-46). Thus, in addition to EAAT2, multiple members of the Na+-dependent glutamate transporter family may contribute to AD-related pathogenic processes.
In addition to the critical role EAAT2 plays in preventing excitotoxicity (2), EAAT2 regulates stimulus-specific synaptic plasticity (5). EAAT2 loss has also been shown to impair activity-dependent glucose utilization (47). Impaired EAAT2 functions, even if initially latent or mild, may synergistically combine to promote increasingly pathogenic cycles of CNS dysfunction over time. In this regard, memantine, a drug hypothesized to temper excessive NMDA receptor activation has efficacy in treating AD (48). Such findings lend additional credence to the notion that disturbed glutamatergic signaling may have a significant role in AD pathogenesis. Our present findings offer further evidence that glutamate-related dysfunction may be an important feature of AD pathology and suggest the possibility that strategies aimed at enhancing the natural neuroprotective properties of astrocytes may open new therapeutic opportunities to treat AD.
Acknowledgments
Sources of support: Veteran's Affairs Office of Research and Development Medical Research Service (DGC), NIH institutional fellowships to K.D., J.M.F. (T32 AG000258), and to P.M. (T32 AG00057-31). University of Washington Alzheimer's Disease Research Center (AG5036) and the Oregon Alzheimer's Disease Center (5P30AG008017).
References
- 1.Lipton SA. Pathologically activated therapeutics for neuroprotection. Nat Rev Neurosci. 2007;8:803–8. doi: 10.1038/nrn2229. [DOI] [PubMed] [Google Scholar]
- 2.Tanaka K, Watase K, Manabe T, et al. Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science. 1997;276:1699–1702. doi: 10.1126/science.276.5319.1699. [DOI] [PubMed] [Google Scholar]
- 3.Namura S, Maeno H, Takami S, et al. Inhibition of glial glutamate transporter GLT-1 augments brain edema after transient focal cerebral ischemia in mice. Neurosci Lett. 2002;324:117–20. doi: 10.1016/s0304-3940(02)00193-3. [DOI] [PubMed] [Google Scholar]
- 4.Pardo AC, Wong V, Benson LM, et al. Loss of the astrocyte glutamate transporter GLT1 modifies disease in SOD1(G93A) mice. Exp Neurol. 2006;201:120–30. doi: 10.1016/j.expneurol.2006.03.028. [DOI] [PubMed] [Google Scholar]
- 5.Tsvetkov E, Shin RM, Bolshakov VY. Glutamate uptake determines pathway specificity of long-term potentiation in the neural circuitry of fear conditioning. Neuron. 2004;41:139–51. doi: 10.1016/s0896-6273(03)00800-6. [DOI] [PubMed] [Google Scholar]
- 6.Jacob CP, Koutsilieri E, Bartl J, et al. Alterations in expression of glutamatergic transporters and receptors in sporadic Alzheimer's disease. J Alzheimer's Dis. 2007;11:97–116. doi: 10.3233/jad-2007-11113. [DOI] [PubMed] [Google Scholar]
- 7.Masliah E, Alford M, DeTeresa R, et al. Deficient glutamate transport is associated with neurodegeneration in Alzheimer's disease. Ann Neurol. 1996;40:759–66. doi: 10.1002/ana.410400512. [DOI] [PubMed] [Google Scholar]
- 8.Abdul HM, Sama MA, Furman JL, et al. Cognitive decline in Alzheimer's disease is associated with selective changes in calcineurin/NFAT signaling. J Neurosci. 2009;29:12957–69. doi: 10.1523/JNEUROSCI.1064-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guo ZH, Mattson MP. Neurotrophic factors protect cortical synaptic terminals against amyloid and oxidative stress-induced impairment of glucose transport, glutamate transport and mitochondrial function. Cereb Cortex. 2000;10:50–57. doi: 10.1093/cercor/10.1.50. [DOI] [PubMed] [Google Scholar]
- 10.Guo Z, Ersoz A, Butterfield DA, et al. Beneficial effects of dietary restriction on cerebral cortical synaptic terminals: preservation of glucose and glutamate transport and mitochondrial function after exposure to amyloid beta-peptide, iron, and 3-nitropropionic acid. J Neurochem. 2000;75:314–20. doi: 10.1046/j.1471-4159.2000.0750314.x. [DOI] [PubMed] [Google Scholar]
- 11.Keller JN, Pang Z, Geddes JW, et al. Impairment of glucose and glutamate transport and induction of mitochondrial oxidative stress and dysfunction in synaptosomes by amyloid beta-peptide: Role of the lipid peroxidation product 4-hydroxynonenal. J Neurochem. 1997;69:273–84. doi: 10.1046/j.1471-4159.1997.69010273.x. [DOI] [PubMed] [Google Scholar]
- 12.Keller JN, Germeyer A, Begley JG, et al. 17Beta-estradiol attenuates oxidative impairment of synaptic Na+/K+-ATPase activity, glucose transport, and glutamate transport induced by amyloid beta-peptide and iron. J Neurosci Res. 1997;50:522–30. doi: 10.1002/(SICI)1097-4547(19971115)50:4<522::AID-JNR3>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 13.Lauderback CM, Hackett JM, Huang FF, et al. The glial glutamate transporter, GLT-1, is oxidatively modified by 4- hydroxy-2-nonenal in the Alzheimer's disease brain: The role of Abeta1- 42. J Neurochem. 2001;78:413–6. doi: 10.1046/j.1471-4159.2001.00451.x. [DOI] [PubMed] [Google Scholar]
- 14.Haugeto O, Ullensvang K, Levy LM, et al. Brain glutamate transporter proteins form homomultimers. J Biol Chem. 1996;271:27715–22. doi: 10.1074/jbc.271.44.27715. [DOI] [PubMed] [Google Scholar]
- 15.Trotti D, Rizzini BL, Rossi D, et al. Neuronal and glial glutamate transporters possess an SH-based redox regulatory mechanism. Eur J Neurosci. 1997;9:1236–43. doi: 10.1111/j.1460-9568.1997.tb01478.x. [DOI] [PubMed] [Google Scholar]
- 16.Trotti D, Rossi D, Gjesdal O, et al. Peroxynitrite inhibits glutamate transporter subtypes. J Biol Chem. 1996;271:5976–9. doi: 10.1074/jbc.271.11.5976. [DOI] [PubMed] [Google Scholar]
- 17.Hughes CP, Berg L, Danziger WL, et al. A new clinical scale for the staging of dementia. Br J Psychiatry. 1982;140:566–72. doi: 10.1192/bjp.140.6.566. [DOI] [PubMed] [Google Scholar]
- 18.Ball M, Braak H, Goethe J, et al. Consensus recommendations for the postmortem diagnosis of Alzheimer's disease. The National Institute on Aging, and Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer's Disease. Neurobiol Aging. 1997;18:S1–2. [PubMed] [Google Scholar]
- 19.Yang W, Woltjer RL, Sokal I, et al. Quantitative Proteomics Identifies Surfactant-Resistant {alpha}-Synuclein in cerebral cortex of parkinsonism-dementia complex of guam but not alzheimer's disease or progressive supranuclear palsy. Am J Pathol. 2007;171:993–1002. doi: 10.2353/ajpath.2007.070015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Williams SM, Sullivan RK, Scott HL, et al. Glial glutamate transporter expression patterns in brains from multiple mammalian species. Glia. 2005;49:520–41. doi: 10.1002/glia.20139. [DOI] [PubMed] [Google Scholar]
- 21.Yang Y, Kinney GA, Spain WJ, et al. Presenilin-1 and intracellular calcium stores regulate neuronal glutamate uptake. J Neurochem. 2004;88:1361–72. doi: 10.1046/j.1471-4159.2003.02279.x. [DOI] [PubMed] [Google Scholar]
- 22.Woltjer RL, Cimino PJ, Boutte AM, et al. Proteomic determination of widespread detergent-insolubility including A-beta but not tau early in the pathogenesis of Alzheimer's disease. FASEB J. 2005;19:1923–5. doi: 10.1096/fj.05-4263fje. [DOI] [PubMed] [Google Scholar]
- 23.Field A. Discovering Statistics Using SPSS. 2nd. London: Sage Publications; 2005. p. 777. [Google Scholar]
- 24.Winer BJ, Brown DR, Michels KM. Statistical Principles in Experimental Design. 3rd. McGraw-Hill Inc; 1991. p. 1057. [Google Scholar]
- 25.Yernool D, Boudker O, Jin Y, et al. Structure of a glutamate transporter homologue from Pyrococcus horikoshii. Nature. 2004;431:811–8. doi: 10.1038/nature03018. [DOI] [PubMed] [Google Scholar]
- 26.Cribbs DH, Chen LS, Bende SM, et al. Widespread neuronal expression of the presenilin-1 early-onset Alzheimer's disease gene in the murine brain. Am J Pathol. 1996;148:1797–1806. [PMC free article] [PubMed] [Google Scholar]
- 27.Johnston JA, Froelich S, Lannfelt L, Cowburn RF, et al. Quantification of presenilin-1 mRNA in Alzheimer's disease brains. FEBS Lett. 1996;394:279–84. doi: 10.1016/0014-5793(96)00969-6. [DOI] [PubMed] [Google Scholar]
- 28.Furness DN, Dehnes Y, Akhtar AQ, et al. A quantitative assessment of glutamate uptake into hippocampal synaptic terminals and astrocytes: New insights into a neuronal role for excitatory amino acid transporter 2 (EAAT2) Neuroscience. 2008;157:80–94. doi: 10.1016/j.neuroscience.2008.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lehre KP, Danbolt NC. The number of glutamate transporter subtype molecules at glutamatergic synapses: Chemical and stereological quantification in young adult rat brain. J Neurosci. 1998;18:8751–7. doi: 10.1523/JNEUROSCI.18-21-08751.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Feany MB, Dickson DW. Neurodegenerative disorders with extensive tau pathology: A comparative study and review. Ann Neurol. 1996;40:139–48. doi: 10.1002/ana.410400204. [DOI] [PubMed] [Google Scholar]
- 31.Woltjer RL, Sonnen JA, Sokal I, et al. Quantitation and mapping of cerebral detergent-insoluble proteins in the elderly. Brain Pathol. 2009;19:365–74. doi: 10.1111/j.1750-3639.2008.00190.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Malm TM, Iivonen H, Goldsteins G, et al. Pyrrolidine dithiocarbamate activates Akt and improves spatial learning in APP/PS1 mice without affecting beta-amyloid burden. J Neurosci. 2007;27:3712–21. doi: 10.1523/JNEUROSCI.0059-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Masliah E, Alford M, Mallory M, et al. Abnormal glutamate transport function in mutant amyloid precursor protein transgenic mice. Exp Neurol. 2000;163:381–7. doi: 10.1006/exnr.2000.7386. [DOI] [PubMed] [Google Scholar]
- 34.Trotti D, Danbolt NC, Volterra A. Glutamate transporters are oxidant-vulnerable: A molecular link between oxidative and excitotoxic neurodegeneration? Trends Pharmacol Sci. 1998;19:328–34. doi: 10.1016/s0165-6147(98)01230-9. [DOI] [PubMed] [Google Scholar]
- 35.Moreira PI, Santos MS, Oliveira CR, et al. Alzheimer disease and the role of free radicals in the pathogenesis of the disease. CNS Neurol Disord Drug Targets. 2008;7:3–10. doi: 10.2174/187152708783885156. [DOI] [PubMed] [Google Scholar]
- 36.Harris ME, Carney JM, Cole PS, et al. beta-Amyloid peptide-derived, oxygen-dependent free radicals inhibit glutamate uptake in cultured astrocytes: implications for Alzheimer's disease. Neuroreport. 1995;6:1875–9. doi: 10.1097/00001756-199510020-00013. [DOI] [PubMed] [Google Scholar]
- 37.Harris ME, Wang Y, Pedigo NW, Jr, et al. Amyloid beta peptide (25-35) inhibits Na+-dependent glutamate uptake in rat hippocampal astrocyte cultures. J Neurochem. 1996;67:277–86. doi: 10.1046/j.1471-4159.1996.67010277.x. [DOI] [PubMed] [Google Scholar]
- 38.Chen W, Mahadomrongkul V, Berger UV, et al. The glutamate transporter GLT1a is expressed in excitatory axon terminals of mature hippocampal neurons. J Neurosci. 2004;24:1136–48. doi: 10.1523/JNEUROSCI.1586-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thal DR. Excitatory amino acid transporter EAAT-2 in tangle-bearing neurons in Alzheimer's disease. Brain Pathol. 2002;12:405–11. doi: 10.1111/j.1750-3639.2002.tb00457.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Herman MA, Jahr CE. Extracellular glutamate concentration in hippocampal slice. J Neurosci. 2007;27:9736–41. doi: 10.1523/JNEUROSCI.3009-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shimamoto K, Sakai R, Takaoka K, et al. Characterization of novel L-threo-beta-benzyloxyaspartate derivatives, potent blockers of the glutamate transporters. Mol Pharmacol. 2004;65:1008–15. doi: 10.1124/mol.65.4.1008. [DOI] [PubMed] [Google Scholar]
- 42.Danbolt NC. Glutamate uptake. Prog Neurobiol. 2001;65:1–105. doi: 10.1016/s0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- 43.Watase K, Hashimoto K, Kano M, et al. Motor discoordination and increased susceptibility to cerebellar injury in GLAST mutant mice. Eur J Neurosci. 1998;10:976–88. doi: 10.1046/j.1460-9568.1998.00108.x. [DOI] [PubMed] [Google Scholar]
- 44.Aoyama K, Suh SW, Hamby AM, et al. Neuronal glutathione deficiency and age-dependent neurodegeneration in the EAAC1 deficient mouse. Nat Neurosci. 2006;9:119–26. doi: 10.1038/nn1609. [DOI] [PubMed] [Google Scholar]
- 45.Duerson K, Woltjer RL, Mookherjee P, et al. Detergent-Insoluble EAAC1/EAAT3 aberrantly accumulates in hippocampal neurons of alzheimer's disease patients. Brain Pathol. 2009;19:267–78. doi: 10.1111/j.1750-3639.2008.00186.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Scott HL, Pow DV, Tannenberg AE, et al. Aberrant expression of the glutamate transporter excitatory amino acid transporter 1 (EAAT1) in Alzheimer's disease. J Neurosci. 2002;22:RC206. doi: 10.1523/JNEUROSCI.22-03-j0004.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Voutsinos-Porche B, Bonvento G, Tanaka K, et al. Glial glutamate transporters mediate a functional metabolic crosstalk between neurons and astrocytes in the mouse developing cortex. Neuron. 2003;37:275–86. doi: 10.1016/s0896-6273(02)01170-4. [DOI] [PubMed] [Google Scholar]
- 48.Lipton SA. Paradigm shift in neuroprotection by NMDA receptor blockade: memantine and beyond. Nat Rev Drug Discov. 2006;5:160–70. doi: 10.1038/nrd1958. [DOI] [PubMed] [Google Scholar]