

Abstract
Ru-based olefin metathesis catalysts containing carbohydrate-derived NHCs from glucose and galactose were synthesized and characterized by NMR spectroscopy. 2D-NMR spectroscopy revealed the presence of Ru-C (benzylidene) rotamers at RT and the rate of rotation was measured using magnetization transfer and VT-NMR spectroscopy. The catalysts were found to be effective at ring-opening metathesis polymerization (ROMP), ring closing metathesis (RCM), cross metathesis (CM), and asymmetric ring opening cross metathesis (AROCM) and showed surprising selectivity in both CM and AROCM.
Introduction
The development of powerful air-stable catalysts has made olefin metathesis an indispensable tool in a variety of fields including organic synthesis, materials science, and biochemistry.1 Among the most versatile and robust catalysts are those based on ruthenium, the first of which was synthesized in 1992.2 The continued evolution of this catalyst via replacement of one phosphine by an N-heterocyclic carbine (NHC)3 and the other by a chelating ether moiety4 (Chart 1) has propelled advancements in a multitude of reactions including cross metathesis (CM), ring-closing metathesis (RCM), ring-opening cross metathesis (ROCM), and ring-opening metathesis polymerization (ROMP). Nevertheless, more stable and E/Z selective catalysts are still required for both laboratory and industrial applications.
Chart 1.

Previously Reported Ruthenium-Based Catalysts for Olefin Metathesis.
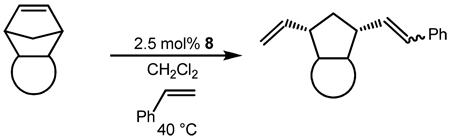
A common strategy for improving catalyst activity and selectivity involves modification of the NHC ligand. The majority of efforts thus far have focused on modification of the NHC backbone or aryl substituents.5 In general, N-aryl bulk was found to increase activity while increased backbone substitution decreased activity but increased catalyst lifetime.6 However, these structural studies were limited to catalysts with NHCs containing N-aryl substituents. NHC-based metathesis catalysts with Nakyl groups on the other hand have received relatively little attention due to their lower stability in solution.7,8 Recently, certain N-alkyl NHCs have demonstrated remarkable activity, including the traditionally difficult RCM of tetrasubstituted olefins.9
One class of N-alkyl substituents for NHCs which have not yet been explored for metathesis applications are carbohydrates. Carbohydrates are extremely abundant molecules and comprise some of the most important biological machinery in living organisms including glycolipids, glycoproteins, and nucleic acids. Thus, it is no surprise that their synthesis10 and their biological function continue to be studied extensively.11 As ligands, carbohydrates are advantageous because of their innate chirality and steric bulk in addition to their long history of synthetic manipulation and solubility in water. Indeed, carbohydrates have already shown promise as ligands for asymmetric catalysis12 and as chiral synthons.13 Additionally, carbohydrates have also been used as ligand scaffolding for platinum and other metals.14 Finally, carbohydrates possess multiple, modular stereocenters and a steric environment which can be tuned through the judicious choice of alcohol protecting groups. However, carbohydrate-based NHCs have only recently been synthesized, and, to the best of our knowledge, a rigorous study of their applications in transition metal catalysis or organocatalysis has not been undertaken.15 Therefore, with the goals of developing a new structural class of highly active, stable, stereoselective olefin metathesis catalysts, and determining the potential of carbohydrate-based NHCs in catalysis, we undertook the synthesis of catalysts containing carbohydrate-based NHCs.
Results and Discussion
Synthesis and Characterization
Several groups have demonstrated that a carbohydrate containing imidazolium salt may be synthesized from the reaction of an alkyl or aryl imidazole with glucopyranosyl bromide.15 Along these lines, imidazolium salts 7a and 7b were synthesized in acceptable yields from the reaction of mesityl imidazole with 2,3,4,6-tetra-O-acetyl-α-D-glucopyranosyl bromide (6a) or 2,3,4,6-tetra-Oacetyl-α-D-galactopyranosyl bromide (6b), respectively, in the presence of silver triflate (Scheme 1) according to a previous report.15 Subsequent deprotonation with sodium tert-butoxide and reaction with catalyst 1 in THF afforded the desired complex (8a) following column chromatography on silica gel. Complex 8a was isolated as a single anomer (β) while 8b (along with 7b) was isolated as a ca. 1.2:1 mixture of β:α anomers.16 Other methods of NHC ligation including deprotonation with KHMDS or transmetalation from the silver complex17 failed to give significant yields of 8a/b.18 Both 8a and 8b were bench stable in the solid state and could be stored as a solution in C6H6 under a nitrogen or argon atmosphere for a period of at least 3 days as determined by 1H NMR spectroscopy.
Scheme 1.

Synthesis of catalyst 8a/b.
Characterization of complex 8a at 25 °C revealed the unusual presence of two benzylidene resonances at ca. 19.77 (s) and 20.78 (d) ppm in the 1H NMR spectrum (C6D6) both of which were correlated to the main ruthenium complex. Interestingly, the benzylidene resonances were found to exchange with one another using a 2D-NOESY experiment (Figure 1). Based on the spin multiplicities of these peaks, along with the 2D-NOESY spectrum, the observed exchange has been attributed to two rotameric species resulting from rotation about the benzylidene C–Ru bond (Scheme 2). At room temperature, such a process is more common among molybdenum and tungsten metathesis catalysts19 but has also been observed for Ru-based catalysts.20
Figure 1.

600 MHz 1H NMR NOESY for the benzylidene region of 8a in C6D6 at 25 °C. Positive peak intensity is colored red while negative intensity is colored blue. Mixing time = 0 ms (left) and 100 ms (right). Peak intensities are listed clockwise starting at the high field diagonal resonance. Left – 589.02, 49.36. Right – 1047.03, 71.91, 68.03, 30.99.
Scheme 2.

Equilibrium depicting rotation about Ru – C/benzylidene bond with anti/syn designation denoting relative position of benzylidene phenyl group to the NHC.
Alkylidene rotamers are not just structural curiosities, but also play an important role in the activity and selectivity of metathesis catalysts.19 Unfortunately, a crystal structure of either rotamer of 8a was unobtainable despite a variety of crystallization conditions. Therefore, in order to fully characterize the unique properties of 8a, a more in-depth structural study of the rotamers of 8a in solution was conducted using NMR spectroscopy.
Cooling a CD2Cl2 solution of 8a to −75 °C resulted in the freezing out of the benzylidene C–Ru bond rotation as well as the appearance of a new benzylidene resonance which can be attributed to slow rotation about the Ru–NHC bond (see Supporting Information).21 Moreoever, at this temperature, the benzylidene ortho protons also became well resolved, indicating that rotation about the C(carbene)–C(phenyl) bond is facile at RT. A graphical summary of observable dynamic processes in 8a at 25 °C is shown in Figure 3.22
Figure 3.

Eyring plot showing VT-NMR spectroscopy data for complex 8a. R2= 0.989, ΔH‡ = 16.1±0.8 kcal/mol, ΔS‡ = −4.4±2.5 cal/(mol·K).
From a magnetization transfer experiment23 conducted at 25 °C, ks/a and ka/s for the benzylidene rotamers were determined to be 1.01 s−1 and 5.28 s−1 respectively.24 These values correspond to a ΔG‡ of 17.42 kcal/mol for the forward reaction (syn to anti) which is consistent with previous reports of Ru–C/benzylidene rotation and also with the relative site population observed at 25 °C.20 Furthermore, a VT 1H NMR spectroscopy experiment with subsequent line shape analysis (see Supporting Information) yielded a value of 17.4 ± 0.2 kcal/mol for ΔG‡ at 25 °C, consistent with the value obtained from the magnetization transfer experiment. The 1H NMR spectrum of complex 8b looked qualitatively similar to that of 8a although no attempt was made to determine the kinetic parameters quantitatively. These results demonstrate the structural rigidity of 8a compared to catalysts such as 2 and 5 where bond rotation is more facile at 25 °C.20b
Following characterization, both 8a and 8b were subjected to a series of standard reactions for ROMP, RCM, and CM in order to evaluate their activity and selectivity compared with previously reported catalysts.30 Additionally, the effectiveness of 8a/b at asymmetric reactions was also of particular interest considering the chiral nature of the carbohydrate ligand. Therefore, asymmetric ring opening cross metathesis (AROCM) was chosen as a means of evaluating the performance of 8a/b in asymmetric reactions.
ROMP Activity
The ROMP of strained olefinic ring systems is one of the earliest industrial applications of olefin metathesis and remains a popular tool for modern polymer synthesis.1 The effectiveness of catalysts 8a/b at ROMP was examined by measuring the rate of polymerization of cyclooctadiene (COD) (Figure 5). Despite a relatively slow initiation, both catalysts were able to reach >95% conversion within 2 h at 30 °C with an initial monomer to catalyst ratio of 100:1. As expected, both 8a and 8b showed similar kinetic behavior. Additionally, both 8a/b performed well compared with other metathesis catalysts, showing a much higher activity than phosphine-based catalyst 1 and similar activity to 5. On the other hand, 8a/b were less active than catalyst 2 which contains a completely saturated NHC ligand.25
Figure 5.

RCM conversion of DEDAM with catalysts 1 (○), 2 (△), 5 (+), 8a (□), and 8b (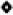 ). Conditions were 1 mol% catalyst, 0.1 M in substrate CD2Cl2 at 40 °C for 8a and 8b and at 30 °C for 1, 2, and 5.
). Conditions were 1 mol% catalyst, 0.1 M in substrate CD2Cl2 at 40 °C for 8a and 8b and at 30 °C for 1, 2, and 5.
Norbornene based substrates and cyclooctene (COE) could also be polymerized effectively using 8a/b with norbornene monomers showing an increase in rate due to the increase in ring strain. Characterization of the isolated polymers by GPC revealed high PDIs and molecular weights much larger than predicted which suggests a relatively slow catalyst initiation step compared to what is observed for fast initiating catalysts such as 2 or its bis-pyridine derivative.26
RCM Activity
Given the good activity of the catalysts in ROMP, we next focused on testing their activity in RCM, which is generally more demanding to the catalyst than ROMP.1 A standard reaction for testing the RCM activity of a particular catalyst is the ring closing of diethyl diallyl malonate (DEDAM) to the cyclopentene (Figure 6).30 Interestingly, 8a and 8b showed reproducibly different kinetic behavior when exposed to DEDAM even though they only differ at one stereocenter (C4).27 It is possible that the distinct behavior is due to one catalyst being more susceptible to a particular decomposition pathway. Another possibility is that the α anomer, which is observed in 8b but not 8a, is much more reactive than the β anomer under these specific reaction conditions.
Figure 6.


Conversion to desired cross product 12 and E/Z ratio using 1 (○), 2 (△), 5 (+), 8a (□), and 8b (). Data for 1, 2, and 5 obtained at 30 °C. E/Z ratio and conversion determined by GC relative to tridecane standard.
At a catalyst loading of 1 mol%, both 8a and 8b showed good performance during the RCM of DEDAM compared with catalysts 2 and 5, while 8b displayed similar activity to catalyst 1. Further reaction times or heating did not improve conversion significantly, but better results were achieved by increasing the catalyst loading to 5 mol% (not shown). Although we have not isolated any catalyst decomposition products, the early catalyst death of 8a/b during RCM indicates that the catalysts are particularly susceptible to decomposition pathways involving methylidene intermediates, similar to catalyst 1.28
CM Activity
Cross metathesis, in contrast to ROMP and RCM, does not possess a strong driving force that pushes the metathesis reaction to completion. Additionally, secondary metathesis events often change the stereochemistry of the desired product, eventually resulting in an excess of the thermodynamically more stable E product. Combined, these challenges often result in reactions with low yield and low selectivity. Controlling the stereochemistry of the olefin product in particular has been extraordinarily challenging although progress in this area is being made.29
In order to evaluate the activity and selectivity of catalysts 8a/b, the CM of allylbenzene and cis-diacetoxybutene was studied (Scheme 3).30 The formation of all reaction products including the desired cross product (12), trans-diacetoxybutene, and the E and Z isomers of the homocoupled allylbenzene were monitored over time via GC (Figure 7). Catalysts 8a/b reached similar levels of conversion compared with 1, 2, and 5 but maintained an exceptional E/Z ratio of around 3. Such a low E/Z ratio is unusual at high conversions where secondary metathesis events begin to favor the thermodynamic product. Furthermore, this result is also significant because the only difference between 5 and 8a/b is the replacement of a mesityl group with a carbohydrate, indicating that carbohydrates can have a substantial effect on catalyst selectivity. However, the low E/Z ratio appears to be more a result of catalyst decomposition as opposed to an inherent preference for one isomer over the other since adding a fresh batch of catalyst caused the E/Z ratio to increase to ca. 8 over a period of 5 h. No differences in either conversion or E/Z ratio were observed for catalysts 8a and 8b.
Scheme 3.

Standard CM using catalysts 8a/b.
AROCM Activity
A relatively recent application of ruthenium-based olefin metathesis is the AROCM of substituted norbornenes with terminal olefins.32 Given the relative selectivity observed during CM and the chiral nature of the sugar moiety attached to the NHC ligand, AROCM was attempted with the hope of observing enantiomeric selectivity. Exposing a variety of norbornene-based substrates to catalysts 8a/b in the presence of styrene for several hours at 40 °C resulted in complete conversion to the desired cis and trans products. As shown in Table 1, reactions ran in toluene generally outperformed those conducted in methylene chloride in terms of yield due to the greater long term stability of the catalysts in non-chlorinated solvents.31 Isolated yields were generally excellent while ee’s were poor compared to previously reported ruthenium-based catalysts.32 The extremely low yield and relatively high ee of entry 3 in Table 1 appears to be an anomaly that is specific to that substrate.32c Substrates from entries 3 and 4 were not tested with 8b due to their relatively low isolated yield. Despite the modest levels of enantioselectivity observed, these results demonstrate the potential of carbohydrate-based ligands as tools for asymmetric catalysis. Furthermore, the variety of commercially available carbohydrates and the ability to create a unique steric environment using different protecting groups should allow for the creation of carbohydrate–based catalysts which are more stereoselective.
Table 1.
AROCM with catalysts 8a/b.
 | ||||||
|---|---|---|---|---|---|---|
| Entry | Substrate | Catalyst | Solvent | Time (h) | ee % E(Z)a | Yield % (E:Z)b |
| 1 | 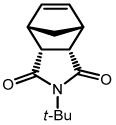 |
8a | CH2Cl2 | 8 | 11 (7) | 43 (1.3:1) |
| 8a | Toluene | 15 | 20 (7) | 88 (2.7:1) | ||
| 8b | Toluene | 15 | 11(3) | 86 (1.4:1) | ||
| 2 |  |
8a | CH2Cl2 | 15 | 22 (n.d.) | 36 (n.d.) |
| 8a | Toluene | 15 | 26 (n.d.) | 80 (n.d.) | ||
| 8b | Toluene | 15 | 19 (n.d.) | 73 (n.d.) | ||
| 3 | 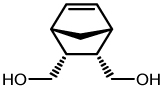 |
8a | Toluene | 10 | 75 (4) | 8 (2.7:1) |
| 4 | 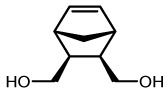 |
8a | Toluene | 5 | 20 (4) | 64 (0.7:1) |
ee% determined by chiral HPLC.
Isolated yield after column chromatography.
Conclusions
Olefin metathesis catalysts incorporating carbohydrate-based NHCs have been synthesized and their structural characteristics and reactivity evaluated. These complexes are characterized by a relatively rigid structure due to the steric bulk of the carbohydrate, and in contrast to many N-alkyl NHCs, show excellent stability and good reactivity in a variety of olefin metathesis reactions including ROMP, RCM, CM, and AROCM. Furthermore, they also show surprising selectivity in CM compared to other catalysts, confirming that steric bulk plays a large role in influencing olefin geometry. Similarly, observable levels of enantioselectivity due to the chiral nature of the carbohydrate were also demonstrated. These results demonstrate the viability of using carbohydrate NHCs in olefin metathesis and establish them as a unique structural class of ligand. Finally, with the potential of carbohydrate–based NHCs in olefin metathesis proven, further improvements in catalyst activity and selectivity via modification of the sugar (steric) and NHC backbone (electronic) should be possible.
Supplementary Material
Figure 2.

Summary of observable rotational processes occurring in complex 8a.
Figure 4.

Conversion of COD with catalyst 1 (○), 2 (△), 5 (+), 8a (□,1 mol%), and 8b (, 1 mol%). Conditions were 1000:1 monomer to catalyst ratio in CD2Cl2 (0.1 M in monomer) at 30 °C.
Acknowledgement
We thank Dr. David VanderVelde and Prof. Dan O’ Leary for assisting with the 2D-NMR characterization and line shape analysis respectively and both for helpful discussions. We would also like to thank Prof. Brian Stoltz for the use of his groups’ Chiral HPLC. We gratefully acknowledge the financial support of the NSF and NIH (5R01 GM31332) and B.K.K thanks the NDSEG for a graduate fellowship. Finally, we thank Materia, Inc. for a generous donation of ruthenium complex 1.
Footnotes
Supporting Information Available:
1H, 13C, and 31P-NMR spectra for catalysts 8a and 8b along with 2D-NMR spectra. Detailed experimental procedures and data for benzylidene rotation rate determination. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Furstner A. Angew. Chem.-Int. Edit. 2000;39:3013. [Google Scholar]; (b) Trnka TM, Grubbs RH. Acc. Chem. Res. 2001;34:18. doi: 10.1021/ar000114f. [DOI] [PubMed] [Google Scholar]
- 2.(a) Nguyen ST, Johnson LK, Grubbs RH, Ziller JW. J. Am. Chem. Soc. 1992;114:3974. [Google Scholar]; (b) Schwab P, France MB, Ziller JW, Grubbs RH. Angew. Chem. Int. Ed. 1995;34:2039. [Google Scholar]; (c) Schwab P, Grubbs RH, Ziller JW. J. Am. Chem. Soc. 1996;118:100. [Google Scholar]
- 3.Scholl M, Ding S, Lee CW, Grubbs RH. Org. Lett. 1999;1:953. doi: 10.1021/ol990909q. [DOI] [PubMed] [Google Scholar]
- 4.(a) Kingsbury JS, Harrity JPA, Bonitatebus PJ, Hoveyda AH. J. Am. Chem. Soc. 1999;121:791. [Google Scholar]; (b) Garber SB, Kingsbury JS, Gray BL, Hoveyda AH. J. Am. Chem. Soc. 2000;122:8168. [Google Scholar]
- 5.(a) Van Veldhuizen JJ, Garber SB, Kingsbury JS, Hoveyda AH. J. Am. Chem. Soc. 2002;124:4954. doi: 10.1021/ja020259c. [DOI] [PubMed] [Google Scholar]; (b) Despagnet-Ayoub E, Grubbs RH. Organometallics. 2005;24:338. [Google Scholar]; (c) Weigl K, Kohler K, Dechert S, Meyer F. Organometallics. 2005;24:4049. [Google Scholar]; (d) Funk TW, Berlin JM, Grubbs RH. J. Am. Chem. Soc. 2006;128:1840. doi: 10.1021/ja055994d. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Vehlow K, Maechling S, Blechert S. Organometallics. 2006;25:25. [Google Scholar]; (f) Anderson DR, Lavallo V, O'Leary DJ, Bertrand G, Grubbs RH. Angew. Chem. Int. Edit. 2007;46:7262. doi: 10.1002/anie.200702085. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Berlin JM, Campbell K, Ritter T, Funk TW, Chlenov A;, Grubbs RH. Org. Lett. 2007;9:1339. doi: 10.1021/ol070194o. [DOI] [PubMed] [Google Scholar]; (h) Stewart IC, Ung T, Pletnev AA, Berlin JM, Grubbs RH, Schrodi Y. Org. Lett. 2007;9:1589. doi: 10.1021/ol0705144. [DOI] [PubMed] [Google Scholar]; (i) Chung CK, Grubbs RH. Org. Lett. 2008;10:2693. doi: 10.1021/ol800824h. [DOI] [PubMed] [Google Scholar]; (j) Vougioukalakis GC, Grubbs RH. J. Am. Chem. Soc. 2008;130:2234. doi: 10.1021/ja075849v. [DOI] [PubMed] [Google Scholar]
- 6.Kuhn KM, Bourg JB, Chung CK, Virgil SC, Grubbs RH. J. Am. Chem. Soc. 2009;131:5313. doi: 10.1021/ja900067c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schuster O, Yang L, Raubenheimer HG, Albrecht M. Chem. Rev. 2009;109:3445. doi: 10.1021/cr8005087. [DOI] [PubMed] [Google Scholar]
- 8.(a) Weskamp T, Kohl FJ, Hieringer W, Gleich D, Herrmann WA. Angew. Chem. Int. Edit. 1999;38:2416. doi: 10.1002/(sici)1521-3773(19990816)38:16<2416::aid-anie2416>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]; (b) Ledoux N, Allaert B, Linden A, Van Der Voort P, Verpoort F. Organometallics. 2007;26:1052. [Google Scholar]; (c) Boydston AJ, Xia Y, Kornfield JA, Gorodetskaya IA, Grubbs RH. J. Am. Chem. Soc. 2008;130:12775. doi: 10.1021/ja8037849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Savoie J, Stenne B, Collins ShawnK. Adv. Syn. Catal. 2009;351:1826. [Google Scholar]
- 10.Hudlicky T, Entwistle DA, Pitzer KK, Thorpe AJ. Chem. Rev. 1996;96:1195. doi: 10.1021/cr9403300. [DOI] [PubMed] [Google Scholar]
- 11.(a) Gamblin DP, Scanlan EM, Davis BG. Chem. Rev. 2008;109:131. doi: 10.1021/cr078291i. [DOI] [PubMed] [Google Scholar]; (b) Murrey HE, Hsieh-Wilson LC. Chem. Rev. 2008;108:1708. doi: 10.1021/cr078215f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dieguez M, Pamies O, Claver C. Chem. Rev. 2004;104:3189. doi: 10.1021/cr0306889. [DOI] [PubMed] [Google Scholar]
- 13.Hollingsworth RI, Wang G. Chem. Rev. 2000;100:4267. doi: 10.1021/cr990374e. [DOI] [PubMed] [Google Scholar]
- 14.(a) Gyurcsik B, Nagy L. Coord. Chem. Rev. 2000;203:81. [Google Scholar]; (b) Steinborn D, Junicke H. Chem. Rev. 2000;100:4283. doi: 10.1021/cr9903050. [DOI] [PubMed] [Google Scholar]
- 15.(a) Nishioka T, Shibata T, Kinoshita I. Organometallics. 2007;26:1126–1128. [Google Scholar]; (b) Tewes F, Schlecker A, Harms K, Glorius F. J. Organomet. Chem. 2007;692:4593–4602. [Google Scholar]; (c) Shi JC, Lei N, Tong QS, Peng YR, Wei JF, Jia L. Eur. J. Inorg. Chem. 2007:2221. [Google Scholar]
- 16.Determined by 1H-NMR spectrum analysis of JHH coupling in 8b.
- 17.The silver complex was formed via deprotonation by silver (I) oxide. See ref. 13b for details.
- 18.Unfortunately, in our hands, this methodology could not be extended to gluco- or galactopyransoyl bromides protected with benzyl (Bn), benzoyl (Bz), pivolate (Piv), or methyl (Me) groups. Attempts to form a phosphine-free catalyst via incorporation of a Hoveyda-type chelating benzylidene either by direct phosphine substitution of 3 or via cross metathesis of 8a/b with β-methyl isoprepoxy styrene were also unsuccessful.
- 19.(a) Oskam JH, Fox HH, Yap KB, McConville DH, Odell R, Lichtenstein BJ, Schrock RR. J. Organomet. Chem. 1993;459:185–198. [Google Scholar]; (b) Oskam JH, Schrock RR. J. Am. Chem. Soc. 1993;115:11831–11845. [Google Scholar]
- 20.(a) Grisi F, Costabile C, Gallo E, Mariconda A, Tedesco C, Longo P. Organometallics. 2008;27:4649–4656. [Google Scholar]; (b) Sanford M. Ph.D. Thesis. Pasadena: California Institute of Technology; 2001. [Google Scholar]
- 21.A 2D-NOESY experiment conducted at −75°C with a mixing time of 500 ms showed no exchange between the benzylidene rotamers (doublet and singlet) but small cross peaks between the two singlet resonances indicated that rotation about the Ru-NHC bond is not completely frozen out at this temperature. A low temperature VT-NMR experiment and subsequent line shape analysis yielded a ΔG† consistent with previous reports of NHC rotation (see ref. 20), although we cannot completely rule out another process such as oxygen coordination/decoordination at this time. See Supporting Information for relevant spectra and plots.
- 22.Although it is tempting to theorize that an oxygen from either an acetate or the sugar is coordinating to the metal center, we found no obvious indications of this during our NMR studies.
- 23.Sandström J. Dynamic NMR Spectroscopy. New York, New York: Academic Press Inc.; 1982. pp. 53–54. [Google Scholar]
- 24.See Supporting Information for experimental details.
- 25.At this time, we have been unable to synthesize a saturated variant of 8a or 8b.
- 26.Love JA, Sanford MS, Day MW, Grubbs RH. J. Am. Chem. Soc. 2003;125:10103. doi: 10.1021/ja027472t. [DOI] [PubMed] [Google Scholar]
- 27.The kinetic plots shown were reproducible for different kinetic runs and also for different batches of catalyst.
- 28.(a) Hong SH, Wenzel AG, Salguero TT, Day MW, Grubbs RH. J. Am. Chem. Soc. 2007;129:7961–7968. doi: 10.1021/ja0713577. [DOI] [PubMed] [Google Scholar]; (b) Hong SH, Chlenov A, Day MW, Grubbs RH. Angew. Chem. Int. Ed. 2007;46:5148–5151. doi: 10.1002/anie.200701234. [DOI] [PubMed] [Google Scholar]
- 29.Flook MM, Jiang AJ, Schrock RR, Müller P, Hoveyda AH. J. Am. Chem. Soc. 2009;131:7962–7963. doi: 10.1021/ja902738u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ritter T, Hejl A, Wenzel AG, Funk TW, Grubbs RH. Organometallics. 2006;25:5740–5745. [Google Scholar]
- 31.Kuhn KM, Bourg JB, Chung CK, Virgil SC, Grubbs RH. J. Am. Chem. Soc. 2009;131:5313. doi: 10.1021/ja900067c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.(a) Van Veldhuizen JJ, Garber SB, Kingsbury JS, Hoveyda AH. J. Am. Chem. Soc. 2002;124:4954–4955. doi: 10.1021/ja020259c. [DOI] [PubMed] [Google Scholar]; (b) Van Veldhuizen JJ, Gillingham DG, Garber SB, Kataoka O, Hoveyda AH. J. Am. Chem. Soc. 2003;125:12502–12508. doi: 10.1021/ja0302228. [DOI] [PubMed] [Google Scholar]; (c) Berlin JM, Goldberg SD, Grubbs RH. Angew. Chem. Int. Ed. 2006;45:7591–7595. doi: 10.1002/anie.200602469. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.