

Abstract
Nonalcoholic fatty liver disease, characterized by accumulation of triacylglycerols (TG) and other lipids in the liver, often accompanies obesity and is a risk factor for nonalcoholic steatohepatitis and fibrosis. To treat or prevent fatty liver, a thorough understanding of hepatic fatty acid and TG metabolism is crucial. To investigate the role of acyl CoA:diacylglycerol acyltransferase 1 (DGAT1), a key enzyme of TG synthesis, in fatty liver development, we studied mice with global and liver-specific knockout of Dgat1. DGAT1 was required for hepatic steatosis induced by high-fat diet and prolonged fasting, which are both characterized by delivery of exogenous fatty acids to the liver. Studies in primary hepatocytes showed that DGAT1 deficiency protected against hepatic steatosis by reducing synthesis and increasing the oxidation of fatty acids. In contrast, lipodystrophy (aP2-SREBP-1c436) and liver X receptor activation (T0901317), which increase de novo fatty acid synthesis in liver, caused steatosis independently of DGAT1. Pharmacologic inhibition of Dgat1 with antisense oligonucleotides protected against fatty liver induced by a high-fat diet. In conclusion, our findings identify a specific role for hepatic DGAT1 in esterification of exogenous fatty acids and indicate that DGAT1 contributes to hepatic steatosis induced by this mechanism.
Keywords: Triglyceride synthesis, obesity, diabetes, DGAT, fatty liver
Nonalcoholic fatty liver disease (NAFLD) is characterized by lipid accumulation in hepatocytes of people who consume little to no alcohol (1–3). Chronic lipid accumulation in the liver (hepatic steatosis) is a risk factor for nonalcoholic steatohepatitis, an inflammatory condition in some patients with fatty liver (4). NAFLD is the most common cause of abnormal liver enzyme tests (5), is associated with obesity and insulin resistance, and is increasing in prevalence, affecting ~30 million adults in the U.S., making it the most common liver disorder (2, 6). NAFLD portends epidemic problems for public health, and a better understanding of the pathways that regulate lipid accumulation in the liver is crucial for developing effective therapies for hepatic steatosis.
Lipids that accumulate in hepatic steatosis are mainly triacylglycerols (TGs). After adipose tissue, the liver has perhaps the largest capacity to synthesize and store TGs (7). TGs are products of the glycerol phosphate synthesis pathway, in which fatty acyl moieties are joined to glycerol via ester bonds (8, 9). FAs in the liver may come from exogenous sources (e.g., dietary fat or mobilization from white adipose tissue (WAT) during fasting) or from endogenous de novo synthesis promoted by leptin deficiency or high levels of circulating insulin and glucose (1, 10–12).
The final step in TG synthesis is catalyzed by DGAT enzymes. Mammals have two DGAT enzymes that are members of distinct gene families (13, 14). Both are expressed widely in tissues and in the livers of mice and humans (13, 15). Increased levels of DGAT1 mRNA, in particular, occur in human livers with NAFLD (16), underscoring the importance of defining the role of DGAT1 in this tissue. Mice lacking Dgat1 (Dgat1−/−) are viable, have reduced tissue TG levels, exhibit increased sensitivity to insulin and leptin, and are protected against diet-induced obesity through increased energy expenditure (17, 18). However, DGAT1’s function in hepatic steatosis has not been fully explored.
To investigate the role of DGAT1 in hepatic steatosis, we studied mice with global (17) and liver-specific knockout of Dgat1 under conditions that promote hepatic steatosis. These included a high-fat diet, fasting (in which lipids are mobilized from the WAT to the liver), and two conditions in which endogenous FA synthesis is greatly increased—genetically induced lipodystrophy (19) and treatment with the liver X receptor (LXR) agonist T0901317 (20). Relevant to clinical therapies, we also determined whether knockdown of Dgat1 expression with antisense oligonucleotides (ASO) protects against diet-induced hepatic steatosis.
Experimental Procedures
Mice
Dgat1−/− and wild-type mice (C57BL/6J background) were genotyped asdescribed (17). Mice were housed in a pathogen-free barrier facility (12-hlight/12-h dark cycle) and fed chow (5053 PicoLab Diet; Purina) or a high-fat diet (20% milk fat, 0.2% cholesterol by weight; TD 01064 Harlan-Teklad). Dgat1flox/flox mice were generated as described (Supplementary Methods). During fasting, mice had access to water. Dgat1−/− mice were crossed with aP2-SREBP-1c436 transgenic mice (Jackson Laboratory; 50% C57BL/6J, 50% SJL). Dgat1+/− aP2-SREBP-1c436 mice (75% C57BL/6J, 25% SJL) were crossed with Dgat1−/− (C57BL/6J) males to generate Dgat1−/− aP2-SREBP-1c436 mice (87.5% C57BL/6J, 12.5% SJL). All experiments were approved by the Committee on Animal Research, University of California, San Francisco.
Histological Analyses
Lipid Analyses
Livers were homogenized in buffer containing 50 mM Tris-HCl, pH 7.4, and 250 mM sucrose with complete protease inhibitor (Roche). Lipids were extracted with chloroform:methanol (2:1) and separated as described (21). TG bands were identified, scraped, and quantified spectrophotometrically (22).
Adenoviruses
Cre-, LacZ-, and GFP-expressing adenoviruses (Vector Development Laboratory, Baylor College; 2×1011 particles in 0.2 ml of PBS) were injected into a jugular vein (23) of Dgat1flox/flox mice (16–20 weeks old). After 4 weeks, mice were placed on a high-fat diet for 3 weeks or fasted for 20 h.
ASOs
Control and Dgat1 ASO (Isis Pharmaceuticals) with an 2′-O-(2-methoxy)-ethyl modification at the first and last five bases (24) were injected (50 mg/kg intraperitoneally) twice weekly for 5 weeks.
DGAT Activity Assays
Livers were homogenized in Buffer A (50 mM Tris-HCl, pH 7.4, 250 mM sucrose) with proteinase inhibitors (Roche Diagnostic). To prepare microsomes, homogenates were centrifuged three times at 4°C (600×g for 5 min, 10,000×g for 10 min, 100,000×g for 1 h); after each centrifugation, pellets were resuspended in Buffer A. DGAT assays were performed with microsome proteins (100 μg) in mix containing 100 mM MgCl2, 1.25 mg/ml bovine serum albumin, 200 μM 1,2-dioleoyl-sn-glycerol (Sigma-Aldrich) in acetone, and 25μM [14C]oleoyl-CoA (53.0 mCi/mmol)]. After 10 min at 37°C, lipids were extracted with chloroform:methanol (2:1, v:v) and separated on G-60 TLC plates with hexane:ethyl ether:acetic acid (80:20:1, v:v:v). TG bands were scraped, and radioactivity was measured by scintillation counting.
FA Oxidation Measurement
Hepatocytes were incubated with 3 ml of DMEM containing 200 μM [9,10-3H]oleic acid (10 μCi/ml) (Amersham) conjugated to 0.2% bovine serum albumin for 2 h. The medium was saved, cells were scraped with 1 ml of 0.1 N NaOH and 1 ml of water, and protein concentrations determined by Dc protein assay (Biorad). Medium (100 μl) was placed in a microcentrifuge tube, which was placed in a 20-ml scintillation vial containing 500 μl of water and sealed and heated at 50°C for ~18 h for [3H]H2O to equilibrate. Equilibrium efficiency was determined by similar analysis of 100 μl of 0.1 μCi/μl of [3H]H2O (~65%) or [9,10-3H]oleic acid (~0.5%). After cooling, microcentrifuge tubes removed, scintillation fluid (aqueous/nonaqueous) was added, and [3H]H2O was measured with a scintillation counter.
RNA Extraction and Real-Time PCR
RNA was extracted from livers with RNA STAT-60 (Tel-Test) and treated with DNase (Ambion). cDNA was synthesized from RNA (5μg) with Superscript II reverse transcriptase and random hexamers (Invitrogen). Real-time PCR primers (Supplementary Table 2) were selected withPrimer Express (version 1.5; Applied Biosystems). Two-step RT-PCR were performed with Sybrgreen (Applied Biosystems) and an ABI 9600.
Statistical Analyses
Values are mean ± SEM. Means were compared by t-test or ANOVA and Student-Newman-Keuls test.
Results
Hepatic DGAT1 Deficiency Protects against High-fat-diet-induced Fatty Liver
Mice were fed a High-fat diet for 3 weeks. Dgat1+/+ livers were pale and stained positively for lipids (Fig. 1A). Lipid staining was reduced in Dgat1−/− livers, consistent with prolonged (32 weeks) high-fat feeding (17). Sirius Red staining showed similar amounts of collagen in Dgat1+/+ and Dgat1−/− livers (not shown). Hepatic TG levels were similar in chow-fed Dgat1+/+ and Dgat1−/− mice but ~80% lower in Dgat1−/− mice after high-fat feeding (Fig. 1B). All classes of FAs in TG were reduced. FA synthesis was increased in hepatocytes from Dgat1+/+ mice fed a high-fat diet (Fig. 1C), consistent with reported effects (25), but reduced in hepatocytes from Dgat1−/− mice, as were mRNA levels of the lipogenic transcription factors Srebp1c and its targets FA synthase (Fasn) and stearoyl-CoA desaturase 1 (Scd1), and carbohydrate response element binding protein (Chrebp) and its target liver-pyruvate kinase (L-pk) (Fig. 1D). This may reflect the lower serum insulin levels found in Dgat1−/− than Dgat1+/+ mice (0.7 ± 0.1 vs. 1.6 ± 0.3 ng/ml, n=4–5/genotype, P<0.05). Protection was associated with increased FA oxidation in hepatocytes from fat-fed Dgat1−/− mice (Fig. 1E); no differences were found in chow-fed mice. Consistent with increased FA oxidation, AMP-activated kinase, which induces hepatic FA oxidation (26), was activated in livers of fat-fed Dgat1−/− mice fed (Supplementary Fig. 1). mRNA levels of several genes involved in FA oxidation genes were similar in Dgat1+/+ and Dgat1−/− mice (Fig. 1F), suggesting increased substrate flux.
Fig. 1.

Protection against hepatic steatosis in Dgat1−/− mice fed a high-fat diet. (A) Livers and Oil-Red-O (ORO)-stained liver sections in 12-week-old mice fed a high-fat diet for 3 weeks. (B) Fatty acid (FA) composition of hepatic TG in mice fed a chow or high-fat diet (age 8–12 weeks, n=5/genotype). SFA, saturated FAs; MUFA, monounsaturated FAs; PUFA, polyunsaturated FAs. P * <0.001 vs. control. (C) FA synthesis in hepatocytes from mice fed chow or high-fat diet. P * <0.05 vs. chow-fed mice. * P<0.001 vs. high-fat control. (D) mRNA levels of sterol regulatory element-binding protein 1c (Srebp1c), FA synthase (fasn), stearoyl-CoA desaturase 1 (scd1), carbohydrate response element binding protein (Chrebp), and liver-pyruvate kinase (L-pk) in livers of mice fed chow or high-fat diet. Genes were normalized to cyclophilin or 36B4 (age 8–12 weeks, n=5). *P<0.05 vs. control. (E) Rate of FA oxidation in hepatocytes from Dgat1+/+ or Dgat1−/− mice fed chow or high-fat diet (age 12–16 weeks, n=2–3/genotype). P * <0.001 vs. control. (F) mRNA levels of peroxisome proliferators-activated receptor α (Pparα), carnitine palmitoyl-transferase 1 (Cpt1), and acyl-CoA oxidase (Aox) in livers from fat-fed Dgat1+/+ or Dgat1−/− mice (age 8–12 weeks, n=5).
Because Dgat1 is widely expressed in murine tissues, protection of Dgat1−/− mice may reflect loss of DGAT1 in liver or other tissues affecting hepatic TG balance (27–29). In mice with adenovirus-mediated knockdown of Dgat1 in liver (LivD1KO mice) (Supplementary Fig. 1), hepatic Dgat1 mRNA levels were ~80% lower than in controls, and the knockdown was specific (Fig. 2A). FA synthesis and oxidation genes were similarly expressed in control and LivD1KO mice (Fig. 2A). DGAT activity was reduced by ~80% in livers of LivD1KO mice (Fig. 2B), or slightly less than in the global knockout (17), possibly because of residual activity in nonhepatocyte cells (30). After 3 weeks of high-fat feeding, LivD1KO livers were darker (Fig. 2C) and had ~50% lower hepatic TG levels (Fig. 2D), indicating some protection against steatosis.
Fig. 2.

Hepatic DGAT1 deficiency protects against hepatic steatosis induced by high-fat diet. (A) RT-PCR analysis of Dgat1 and Dgat2 mRNA in liver and epidydimal white adipose tissue (WAT). Male Dgat1flox/flox mice received adenoviruses expressing LacZ or Cre recombinase. After 4 weeks, mice were fed a high-fat diet for 3 weeks (age 16–20 weeks, n=5–6/genotype). * P<0.001 vs. control. (B) Microsomal hepatic DGAT activity as measured by the incorporation of [14C]-oleoyl-CoA into triglycerides (age 16–20 weeks, n=5–6/genotype). P * <0.001 vs. control. (C and D) Livers (C) and hepatic triglyceride content (D) in mice fed high-fat diet (age 16–20 weeks, n=5–6 per genotype). P * <0.05 vs. control.
Hepatic DGAT1 Deficiency Protects against Fasting-induced Hepatic Steatosis
During fasting, free fatty acids mobilized from the adipose tissue to liver can be esterified to form TG, which can accumulate and cause steatosis (31). To explore the role of DGAT1 in this process, we measured hepatic Dgat1 mRNA levels under different dietary conditions. Mice fasted for 16 h had almost 3-fold higher hepatic Dgat1 mRNA levels than mice fed ad libitum or after refeeding (Fig. 3A). Dgat2 mRNA levels were similar during these conditions.
Fig. 3.

Protection against fasting-induced hepatic steatosis in DGAT1-deficient and Liv-D1KO mice. (A) Real-time PCR analysis of Dgat1 and Dgat2 mRNA levels in livers of male mice fed ad libitum (Ad Lib), fasted for 16 h (Fast), or fasted for 24 h and refed for 12 h (Refed) (age 13–14 weeks, n=6–8/group). *P<0.001 vs. control. (B and C) Gross appearance of livers (B), and hepatic triglyceride content (C) after 20 hours of fasting (age 12 weeks, n=5/genotype). *P<0.001 vs. control. (D) Real-time PCR analysis of Dgat1, Dgat2, peroxisome proliferator-activated receptor alpha (Ppara), and target genes, carnitine-palmitoyl transferase 1(Cpt1), aldehyde dehydrogenase 3 family member A2 (Aldh3a2), enoyl-Coenzyme A, hydratase/3-hydroxyacyl Coenzyme A dehydrogenase (Ehhadh), and cytochrome P450, family 4, subfamily a, polypeptide 10 (Cypa10) mRNA levels in liver and gastrocnemius of 20 hour fasted mice. (age 16–20 weeks, n=4–5/genotype). *P<0.002 vs. control. (E) Microsomal hepatic DGAT activity measured by incorporation of [14C]-oleoyl-CoA into triglycerides (age 16–20 weeks, n=4/genotype). *P<0.001 vs. control. (F and G) Livers (F) and hepatic triglyceride content (G) in mice fasted for 20 h. (age 16–20 weeks, n=5/group). *P<0.01 vs. control. The experiment was repeated with similar results. (H) Triglyceride synthesis in hepatocytes challenged with palmitate (16:0) conjugated to bovine serum albumin as measured by incorporation of [14C]-Glycerol into triglycerides. The experiment was repeated with similar results.
We therefore examined hepatic steatosis in Dgat1+/+ and Dgat1−/− mice fasted for 20 h. Dgat1−/− livers were darker than Dgat1+/+ livers (Fig. 3B) and had ~70% lower TG content (Fig. 3C). Serum levels of the ketone β-hydroxybutyrate, a product of FA oxidation in the liver, were slightly elevated in fasted Dgat1−/− mice, although the differences did not reach significance (0.54 ± 0.13 versus 0.32 ± 0.07 mM, n = 5/genotype, P < 0.07; 8-h fast).
To determine if DGAT1 acts in a liver-specific manner during fasting, we examined steatosis in fasted mice. LivD1KO mice had ~75% lower hepatic Dgat1 mRNA levels than controls but similar hepatic Dgat2 mRNA levels and Dgat1 mRNA levels in gastrocnemius muscle (data not shown) (Fig. 3D). No apparent changes were found in the lipid oxidizing transcription factor, peroxisome proliferator-activated receptor alpha (Ppara), as well as target genes such as carnitine-palmitoyl transferase 1(Cpt1), aldehyde dehydrogenase 3 family member A2 (Aldh3a2), enoyl-Coenzyme A, hydratase/3-hydroxyacyl Coenzyme A dehydrogenase (Ehhadh), and cytochrome P450, family 4, subfamily a, polypeptide 10 (Cypa10). LivD1KO mice had ~80% lower hepatic DGAT activity (Fig. 3E), darker livers (Fig. 3F), and ~80% lower hepatic TG levels (Fig. 3G), a reduction similar to that in Dgat1−/− mice. Further, Dgat1−/− hepatocytes had reduced capacity to synthesize TG when FA concentrations were above 250 μM (Fig. 3H).
DGAT1 Deficiency Does Not Protect against Hepatic Steatosis Induced by Endogenous FA Synthesis
Congenital generalized lipodystrophy is characterized by paucity of adipose tissue, hyperinsulinemia, hyperglycemia, and hepatic steatosis. In aP2-SREBP-1c436 transgenic mice, a model of this disorder, leptin levels are reduced, leading to upregulation of FA synthesis genes and hepatic steatosis (11, 12, 19).
To determine if hepatic steatosis in this model requires DGAT1, we generated Dgat1−/− aP2-SREBP-1c436 mice. Hepatic Srebp1c expression was markedly elevated in aP2-SREBP-1c436 and Dgat1−/− aP2-SREBP-1c436 mice, as were mRNAs for SREBP1c targets and Dgat2 mRNA levels (Fig. 4A). In both genotypes, hepatic TG content was similarly increased to higher levels than in Dgat1+/+ and Dgat1−/− ontrols (Fig. 4B). Unlike livers of Dgat1+/+ and Dgat1−/− mice, livers of aP2-SREBP-1c436 and Dgat1−/− aP2-SREBP-1c436 mice were pale, enlarged, and stained positively for lipids (Fig. 4C).
Fig. 4.

DGAT1 deficiency does not protect against hepatic steatosis due to lipodystrophy or liver receptor X (LXR) activation. (A) Real-time PCR analysis of sterol regulatory element-binding protein (Srebp1c), fatty acid (FA) synthase (fasn), and stearoyl-CoA desaturase 1 (scd1) and Dgat2 mRNA levels in livers of male mice (age 24 weeks, n=5–7/genotype). P * <0.001, **P<0.05 vs. Dgat1+/+. (B) Hepatic triglyceride content in chow-fed male mice (age 24 weeks, n=7/genotype). P * <0.001 vs. Dgat1+/+ and Dgat1−/− mice. (C) Livers and Oil-Oed-O (ORO)-stained liver sections in 24-week-old male mice. (D) Real-time PCR analysis of FA synthase (Fasn) and stearoyl-CoA desaturase 1 (Scd1) mRNA levels in livers of mice treated with LXR agonist (T0901317, 50 mg/kg; n=5). P * <0.001 vs. Dgat1+/+ and Dgat1−/− mice. (E) Hepatic triglyceride content in male mice treate with 50-mg/kg T0901317 (n=5). P * <0.001 vs. Dgat1+/+ and Dgat1−/− mice.
To test the hypothesis that DGAT1 is not required for hepatic steatosis from increased de novo FA synthesis, we treated Dgat1+/+ and Dgat1−/− mice with an LXR agonist, T0901317, which activates FA synthesis in the liver and leads to hepatic steatosis (20). As expected, the mice had increased expression of Fasn and Scd1 (Fig. 4D). Consistent with our hypothesis, Dgat1+/+ and Dgat1−/− livers had similar degrees of steatosis after 2 weeks of treatment (Fig. 4E).
Inhibiting DGAT1 Protects against High-fat-diet-induced Fatty Liver
Next, we assessed ASO-mediated knockdown of Dgat1 as a treatment for fatty liver induced by a high-fat diet. Dgat1 ASO reduced hepatic Dgat1 mRNA levels without altering Dgat2 expression (Fig. 5A). ASO pretreatment reduced by ~40% the increase in hepatic TG content after 1 week of high-fat diet (Fig. 5B).
Fig. 5.
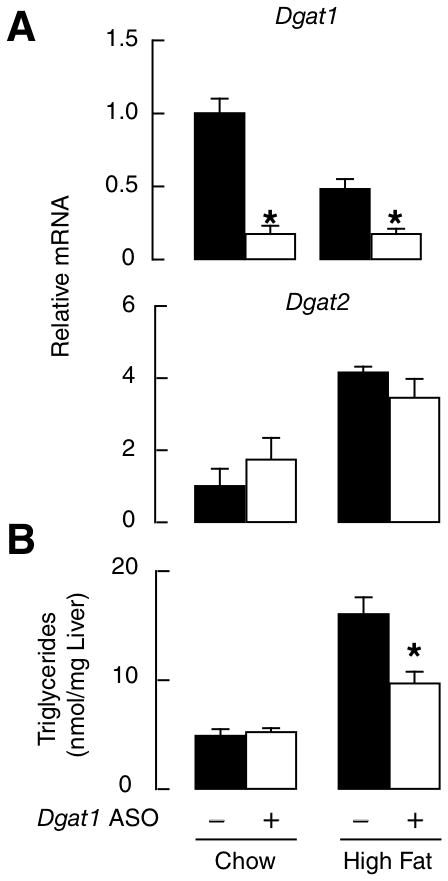
Pharmacologic inhibition of Dgat1 mRNA prevents hepatic steatosis in mice fed a high-fat diet. (A) Real-time PCR analysis of Dgat1 and Dgat2 mRNA after treatment with control or Dgat1 anisense oligonucleotides (ASO), administered twice weekly (50 mg/kg intraperitoneally). *P<0.01 vs. control. (B) Hepatic triglyceride content after 1 week of high-fat diet after pretreatment with control or Dgat1 ASO (n=5). *P<0.05 vs. control.
Discussion
This study shows that hepatic steatosis induced in mice by a high-fat diet or fasting, which promote hepatic uptake of exogenous FAs, required hepatic DGAT1. DGAT1 was not required for hepatic steatosis induced by lipodystrophy or LXR activation, which upregulate endogenous de novo FA synthesis. Thus, DGAT1 has a specific role in esterifying exogenous FAs. Pretreatment with DGAT1-specific ASO reduced hepatic TG content, suggesting that pharmacologic inhibition of DGAT1 may prevent hepatic steatosis induced by a high-fat diet.
DGAT1 deficiency reduced high-fat-diet-induced hepatic steatosis, a condition involving uptake of dietary FAs and the activation of FA synthesis (25). On a 3-week, high-fat diet, global DGAT1 deficiency reduced hepatic TG by ~80%. Hepatic deletion of Dgat1 reduced hepatic TG by ~50%. The additional reduction with global Dgat1 deficiency may reflect loss of DGAT1 activity in tissues, such as WAT (through endocrine effects) (32) or small intestine (33). Reduced FA synthesis may also have contributed to protection against steatosis. Lipogenesis is regulated by Srebp1c, an insulin-responsive transcription factor that is typically upregulated in livers of obese mice (11), and Dgat1−/− mice had reduced mRNA levels of Srebp1c and FA synthesis enzymes. Reduced insulin levels in Dgat1−/− mice may explain their lower hepatic levels of Srebp1c, which may have resulted from the improved insulin sensitivity associated with Dgat1 deficiency (18). Expression of these genes was not changed in LivD1KO mice, suggesting that the changes in lipogenesis in Dgat1−/− mice are indirect and may result from loss of DGAT1 in nonhepatic tissues.
Our results also show a role for hepatic DGAT1 in fasting. During a fast, hepatic glycogen content is depleted, and the liver switches to lipids as fuel. Lipolysis is activated in WAT (34–36), and free FAs are mobilized to the liver, where they are oxidized to yield ketones or re-esterified to TG for storage or secretion in VLDL (37, 38). In a previous study of mice, a 16-h fast increased hepatic Dgat1 mRNA expression ~2.7-fold (37), suggesting a link between DGAT1 and steatosis in fasting. We confirmed this finding and, in studies of global and liver-specific Dgat1 gene inactivation, demonstrated a functional requirement for hepatic DGAT1 during fasting since these mice were protected against steatosis. In the absence of hepatic DGAT1 expression, FAs entering the liver from WAT during fasting are likely oxidized, as suggested by the increase in circulating ketones in Dgat1−/− mice during fasting. The reduced steatosis did not appear to reflect increased hepatic VLDL secretion, since serum TG levels were lower in fasted Dgat1−/− mice (Supplementary Fig. 2). Finally, in primary hepatocytes, DGAT1 was required for the esterification of exogenous FA (palmitate) at high concentrations of substrate, further supporting a role for DGAT1 in esterifying exogenous FAs.
In contrast, steatosis caused by increased endogenous de novo FA synthesis was independent of DGAT1. In lipodystrophies, leptin is deficient, and FA synthesis is activated and contributes to TG accumulation in the liver (11, 12, 19, 39). aP2-SREBP-1c436 and Dgat1−/− aP2-SREBP-1c436 mice had similar degrees of hepatic steatosis, indicating that DGAT1 was not required for steatosis in this lipodystrophy model. Similarly, in ob/ob mice, in which leptin deficiency leads to upregulated FA synthesis, DGAT1 deficiency did not protect against hepatic TG accumulation (18) (H. Chen, R. Farese: unpublished observations). Hepatic steatosis induced by LXR activation of de novo FA synthesis was also similar in wild-type and Dgat1−/− mice. Similar results were found with Dgat1 ASO treatment in mice administered LXR agonist T0901317 (Supplementary Fig. 3). In these situations of activated de novo lipogenesis, fatty acid oxidation is likely shutdown, and the effects of DGAT1 deficiency to activate this pathway may be obviated.
Our findings indicate that DGAT1 is not functionally linked to de novo FA synthesis and suggest that DGAT2 mediates TG synthesis in this situation. How the two enzymes couple to different sources of FAs is unknown. One possibility is functional compartmentalization, as DGAT2 physically associates with SCD1 (40), which desaturates newly synthesized FAs and functions in the de novo synthesis pathway (41). However, DGAT2 may not be exclusively linked to de novo synthesis, since ASO knockdown of Dgat2 reduced steatosis in fat-fed mice (24). Evidence for compartmentalization of DGAT enzymes exists. In tung tree cells, DGAT1 and DGAT2 localize to different subdomains of the ER, where they may synthesize different pools of TG within the cell (42). Mammalian DGAT1 and DGAT2 also localize to distinct regions in hepatoma cells (43).
In contrast to our findings, Choi et al. (44) showed that Dgat1 ASO did not block hepatic steatosis in rats fed a high-fat diet. However, the rats were treated with ASO after 3 days on a high-fat diet, whereas our mice were pretreated with ASO for 4 weeks before starting a 1-week high-fat diet. Moreover, our diet contained milk fat (57% saturated, 30% monounsaturated, and 3% polyunsaturated fat) and cholesterol, whereas theirs was rich in safflower oil (12% saturated, 13% monounsaturated, and 75% polyunsaturated fat). The composition of dietary fat can produce contrasting results in mice. For example, mice lacking liver FA-binding protein are protected against hepatic steatosis when challenged with a diet rich in saturated but not polyunsaturated fat (45). In db/db mice, Yamaguchi et al. (30) also showed that Dgat1 ASO reduced hepatic fibrosis, but did not protect against hepatic TG accumulation induced by a methionine choline–deficient diet, providing a role for DGAT1 in NASH. This finding is consistent with our finding that DGAT1 deficiency does not protect ob/ob (and db/db) mice against obesity, diabetes, and hepatic steatosis (18) (H. Chen, R. Farese: unpublished observations). This may be due in part to an induction of FA synthesis in ob/ob mice that drives TG synthesis mediated by DGAT2.
In conclusion, we showed that DGAT1 deficiency has a specific role in the development of steatosis due to exogenous FAs but not endogenous FA synthesis. Global and liver-specific inactivations of Dgat1 and knockdown of Dgat1 by ASO afforded protection from steatosis due to a high-fat diet. It remains to be determined how our findings in murine models translate to human disease. However, DGAT1 is expressed highly in human liver (15), and DGAT1 expression is increased in humans with NAFLD (16). Moreover, in a murine model of NASH, treatment with Dgat1 ASO reduced hepatic fibrosis (30). Since high-fat diets are common in individuals with obesity and hepatic steatosis, DGAT1 inhibition may be a useful strategy for treating hepatic steatosis.
Supplementary Material
Supplemental Table 1. Activation of fatty acid synthesis with LXR ligand T0901317 in control and Dgat1−/− mice
Supplemental Table 2. Primer sequence used for real–time PCR.
Acknowledgments
Financial Support
Supported by the NIH (R01-DK056084 to R.F., F31-DK065312 to C.V.), an American Physiological Society–Porter Physiology Fellowship (to C.V.), NIH/NCRR (CO6 RR018928 to Gladstone), and the J. David Gladstone Institutes.
We thank the UCSF Liver Center (NIH grant P30-DK26743) for assistance in isolating primary hepatocytes; Jay Horton and Joachim Herz for helpful advice; Jo Dee Fish for histological preparations; Sid Espineda for blastocyst microinjections; Gary Howard and Stephen Ordway for editorial assistance; Mijoung Chang and Daryl Jones for manuscript preparation; and Ryan Streeper, Charlie Harris, Jackie Maher, and Peter Tontonoz for comments on the manuscript.
Abbreviations
- ASO
antisense oligonucleotides
- DGAT1, acyl CoA
diacylglycerol acyltransferase 1
- FA
fatty acid
- LXR
liver X receptor
- NAFLD
nonalcoholic fatty liver disease
- TG
triacylglycerols
References
- 1.Browning JD, Horton JD. Molecular mediators of hepatic steatosis and liver injury. J Clin Invest. 2004;114:147–152. doi: 10.1172/JCI22422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Angulo P. Nonalcoholic fatty liver disease. N Engl J Med. 2002;346:1221–1231. doi: 10.1056/NEJMra011775. [DOI] [PubMed] [Google Scholar]
- 3.Clark J. The epidemiology of nonalcoholic fatty liver disease in adults. J Clin Gastroenterol. 2006;40:S5–S10. doi: 10.1097/01.mcg.0000168638.84840.ff. [DOI] [PubMed] [Google Scholar]
- 4.Neuschwander-Tetri B, Caldwell S. Nonalcoholic steatohepatitis: summary of an AASLD Single Topic Conference. Hepatology. 2003;37:1202–1219. doi: 10.1053/jhep.2003.50193. [DOI] [PubMed] [Google Scholar]
- 5.Daniel S, Ben-Menachem T, Vasudenvan G, Ma C, Blumenkehl M. Prospective evaluation of unexplained chronic liver transaminase abnormalities in asymptomatic and symptomatic patients. Am J Gastroenterol. 1999;94:3010–3014. doi: 10.1111/j.1572-0241.1999.01451.x. [DOI] [PubMed] [Google Scholar]
- 6.Ramesh S, Sanyal A. Evaluation and management of non-alcoholic steatohepatitis. J Hepatol. 2005;42:S2–12. doi: 10.1016/j.jhep.2004.11.022. [DOI] [PubMed] [Google Scholar]
- 7.Martin S, Parton RG. Lipid droplets: a unified view of a dynamic organelle. Nat Rev Mol Cell Biol. 2006;7:373–378. doi: 10.1038/nrm1912. [DOI] [PubMed] [Google Scholar]
- 8.Coleman RA, Lee DP. Enzymes of triacylglycerol synthesis and their regulation. Prog Lipid Res. 2004;43:134–176. doi: 10.1016/s0163-7827(03)00051-1. [DOI] [PubMed] [Google Scholar]
- 9.Dolinsky V, Gilham D, Alam M, Vance D, Lehner R. Triacylglycerol hydrolase: role in intracellular lipid metabolism. Cell Mol Life Sci. 2004;61:1633–1651. doi: 10.1007/s00018-004-3426-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yahagi N, Shimano H, Hasty A, Matsuzaka T, Ide T, Yoshikawa T, et al. Absence of sterol regulatory element-binding protein-1 (SREBP-1) ameliorates fatty livers but not obesity or insulin resistance in Lep(ob)/Lep(ob) mice. J Biol Chem. 2002;277:19353–19357. doi: 10.1074/jbc.M201584200. [DOI] [PubMed] [Google Scholar]
- 11.Shimomura I, Matsuda M, Hammer R, Bashmakov Y, Brown M, Goldstein J. Decreased IRS-2 and increased SREBP-1c lead to mixed insulin resistance and sensitivity in livers of lipodystrophic and ob/ob mice. Mol Cell. 2000;6:77–86. [PubMed] [Google Scholar]
- 12.Shimomura I, Hammer RE, Ikemoto S, Brown MS, Goldstein JL. Leptin reverses insulin resistance and diabetes mellitus in mice with congenital lipodystrophy. Nature. 1999;401:73–76. doi: 10.1038/43448. [DOI] [PubMed] [Google Scholar]
- 13.Cases S, Smith SJ, Zheng Y-W, Myers HM, Lear SR, Sande E, et al. Identification of a gene encoding an acyl CoA:diacylglycerol acyltransferase, a key enzyme in triacylglycerol synthesis. Proc Natl Acad Sci USA. 1998;95:13018–13023. doi: 10.1073/pnas.95.22.13018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cases S, Stone SJ, Zhou P, Yen E, Tow B, Lardizabal KD, et al. Cloning of DGAT2, a second mammalian diacylglycerol acyltransferase, and related family members. J Biol Chem. 2001;276:38870–38876. doi: 10.1074/jbc.M106219200. [DOI] [PubMed] [Google Scholar]
- 15.Farese RV, Jr, Cases S, Smith SJ. Triglyceride synthesis: Insights from the cloning of diacylglycerol acyltransferase. Curr Opin Lipidol. 2000;11:229–234. doi: 10.1097/00041433-200006000-00002. [DOI] [PubMed] [Google Scholar]
- 16.Kohjima M, Enjoji M, Higuchi N, Kato M, Kotoh K, Yoshimoto T, et al. Reevaluation of fatty acid metabolism-related gene expression in nonalcoholic fatty liver disease. Int J Mol Med. 2007;20:351–358. [PubMed] [Google Scholar]
- 17.Smith SJ, Cases S, Jensen DR, Chen HC, Sande E, Tow B, et al. Obesity resistance and multiple mechanisms of triglyceride synthesis in mice lacking DGAT. Nat Genet. 2000;25:87–90. doi: 10.1038/75651. [DOI] [PubMed] [Google Scholar]
- 18.Chen HC, Smith SJ, Ladha Z, Jensen DR, Ferreira LD, Pulawa LK, et al. Increased insulin and leptin sensitivity in mice lacking acyl CoA:diacylglycerol acyltransferase 1. J Clin Invest. 2002;109:1049–1055. doi: 10.1172/JCI14672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shimomura I, Hammer RE, Richardson JA, Ikemoto S, Bashmakov Y, Goldstein JL. Insulin resistance and diabetes mellitus in transgenic mice expressing nuclear SREBP-1c in adipose tissue: Model for congenital generalized lipodystrophy. Genes Dev. 1998;12:3182–3194. doi: 10.1101/gad.12.20.3182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schultz JR, Tu H, Luk A, Repa JJ, Medina JC, Li L, et al. Role of LXRs in control of lipogenesis. Genes Dev. 2000;14:2831–2838. doi: 10.1101/gad.850400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Folch J, Lees M, Stanley GHS. A simple method for the isolation and purification of total lipids from animal tissues. J Biol Chem. 1957;226:497–509. [PubMed] [Google Scholar]
- 22.Snyder F, Stephens N. A simplified spectrophotometric determination of ester groups in lipids. Biochim Biophys Acta. 1959;34:244–245. doi: 10.1016/0006-3002(59)90255-0. [DOI] [PubMed] [Google Scholar]
- 23.Rohlmann A, Gotthardt M, Willnow TE, Hammer RE, Herz J. Sustained somatic gene inactivation by viral transfer of Cre recombinase. Nat Biotechnol. 1996;14:1562–1565. doi: 10.1038/nbt1196-1562. [DOI] [PubMed] [Google Scholar]
- 24.Yu X, Murray S, Pandey S, Booten S, Bao D, Song X, et al. Antisense oligonucleotide reduction of DGAT2 expression improves hepatic steatosis and hyperlipidemia in obese mice. Hepatology. 2005;42:362–371. doi: 10.1002/hep.20783. [DOI] [PubMed] [Google Scholar]
- 25.Lin J, Yang R, Tarr P, Wu P, Handschin C, Li S, et al. Hyperlipidemic effects of dietary saturated fats mediated through PGC-1beta coactivation of SREBP. Cell. 2005;120:261–273. doi: 10.1016/j.cell.2004.11.043. [DOI] [PubMed] [Google Scholar]
- 26.Kahn B, Alquier T, Carling D, Hardie D. AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab. 2005;1:15–25. doi: 10.1016/j.cmet.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 27.Asilmaz E, Cohen P, Miyazaki M, Dobrzyn P, Ueki K, Fayzikhodjaeva G, et al. Site and mechanism of leptin action in a rodent form of congenital lipodystrophy. J Clin Invest. 2004;113:414–424. doi: 10.1172/JCI19511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xu A, Wang Y, Keshaw H, Xu L, Lam K, Cooper G. The fat-derived hormone adiponectin alleviates alcoholic and nonalcoholic fatty liver diseases in mice. J Clin Invest. 2003;112:91–100. doi: 10.1172/JCI17797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Donnelly K, Smith C, Schwarzenberg S, Jessurun J, Boldt M, Parks E. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest. 2005;115:1343–1351. doi: 10.1172/JCI23621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yamaguchi K, Yang L, McCall S, Huang J, Yu X, Pandey S, et al. Diacylglycerol acyltransferase 1 anti-sense oligonucleotides reduce hepatic fibrosis in mice with nonalcoholic steatohepatitis. Hepatology. 2008;47:625–35. doi: 10.1002/hep.21988. [DOI] [PubMed] [Google Scholar]
- 31.Hashimoto T, Cook W, Qi C, Yeldandi A, Reddy J, Raoa M. Defect in peroxisome proliferator-activated receptor alpha-inducible fatty acid oxidation determines the severity of hepatic steatosis in response to fasting. J Biol Chem. 2000;275:28918–28928. doi: 10.1074/jbc.M910350199. [DOI] [PubMed] [Google Scholar]
- 32.Chen HC, Jensen DR, Myers HM, Eckel RH, Farese RV., Jr Obesity resistance and enhanced glucose metabolismin mice transplanted with white adipose tissue lacking acyl CoA:diacylglycerol acyltransferase 1. J Clin Invest. 2003;111:1715–1722. doi: 10.1172/JCI15859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Buhman KK, Smith SJ, Stone SJ, Repa JJ, Wong JS, Knapp FF, Jr, et al. DGAT1 is not essential for intestinal triacylglycerol absorption or chylomicron synthesis. J Biol Chem. 2002;277:25474–25479. doi: 10.1074/jbc.M202013200. [DOI] [PubMed] [Google Scholar]
- 34.Zimmermann R, Strauss JG, Haemmerle G, Schoiswohl G, Birner-Gruenberger R, Riederer M, et al. Fat mobilization in adipose tissue is promoted by adipose triglyceride lipase. Science. 2004;306:1383–1386. doi: 10.1126/science.1100747. [DOI] [PubMed] [Google Scholar]
- 35.Finn P, Dice J. Proteolytic and lipolytic responses to starvation. Nutrition. 2006;22:830–844. doi: 10.1016/j.nut.2006.04.008. [DOI] [PubMed] [Google Scholar]
- 36.Owen O, Felig P, Morgan A, Wahren J, Cahill GF., Jr Liver and kidney metabolism during prolonged starvation. J Clin Invest. 1969;48:574–583. doi: 10.1172/JCI106016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Heijboer A, Donga E, Voshol P, Dang Z, Havekes L, Romijn J. Sixteen hours of fasting differentially affects hepatic and muscle insulin sensitivity in mice. J Lipid Res. 2005;46:582–588. doi: 10.1194/jlr.M400440-JLR200. [DOI] [PubMed] [Google Scholar]
- 38.Kersten S, Seydoux J, Peters J, Gonzalez F, Desvergne B, Wahli W. Peroxisome proliferator-activated receptor alpha mediates the adaptive response to fasting. J Clin Invest. 1999;103:1489–1498. doi: 10.1172/JCI6223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shimomura I, Bashmakov Y, Horton JD. Increased levels of nuclear SREBP-1c associated with fatty livers in two mouse models of diabetes mellitus. J Biol Chem. 1999;274:30028–30032. doi: 10.1074/jbc.274.42.30028. [DOI] [PubMed] [Google Scholar]
- 40.Man WC, Miyazaki M, Chu K, Ntambi J. Colocalization of SCD1 and DGAT2: implying preference for endogenous monounsaturated fatty acids in triglyceride synthesis. J Lipid Res. 2006;47:1928–1939. doi: 10.1194/jlr.M600172-JLR200. [DOI] [PubMed] [Google Scholar]
- 41.Miyazaki M, Kim Y-C, Gray-Keller MP, Attie AD, Ntambi JM. The biosynthesis of hepatic cholesterol esters and triglycerides is impaired in mice with a disruption of the gene for stearoyl-CoA desaturase 1. J Biol Chem. 2000;275:30132–30138. doi: 10.1074/jbc.M005488200. [DOI] [PubMed] [Google Scholar]
- 42.Shockey JM, Gidda SK, Chapital DC, Kuan JC, Dhanoa PK, Bland JM, et al. Tung Tree DGAT1 and DGAT2 Have Nonredundant Functions in Triacylglycerol Biosynthesis and Are Localized to Different Subdomains of the Endoplasmic Reticulum. Plant Cell. 2006;18:2294–2313. doi: 10.1105/tpc.106.043695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stone SS, Levin MC, Zhou P, Han J, Walther TC, Farese RV., Jr The endoplasmic reticulum enzyme, DGAT2, is found in mitochondria-associated membranes and has a mitochondrial targeting signal that promotes its association with mitochondria. J Biol Chem. 2008 doi: 10.1074/jbc.M805768200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Choi CS, Savage DB, Kulkarni A, Yu XX, Liu ZX, Morino K, Kim S, et al. Suppression of diacylglycerol acyltransferase-2 (DGAT2), but not DGAT1, with antisense oligonucleotides reverses diet-induced hepatic steatosis and insulin resistance. J Biol Chem. 2007;282:22678–22688. doi: 10.1074/jbc.M704213200. [DOI] [PubMed] [Google Scholar]
- 45.Newberry E, Kennedy S, Xie Y, Sternard B, Luo J, Davidson N. Diet-induced obesity and hepatic steatosis in L-Fabp−/− mice is abrogated with SF, but not PUFA, feeding and attenuated after cholesterol supplementation. Am J Physiol Gastrointest Liver Physiol. 2007;294:G307–314. doi: 10.1152/ajpgi.00377.2007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table 1. Activation of fatty acid synthesis with LXR ligand T0901317 in control and Dgat1−/− mice
Supplemental Table 2. Primer sequence used for real–time PCR.