

Abstract
The spectroscopic and chemical characterization of a new synthetic non-heme iron(IV)-oxo species [FeIV(O)(Me,HPytacn)(S)]2+ (2, Me,HPytacn = 1-(2′-pyridylmethyl)-4,7-dimethyl-1,4,7-triazacyclononane, S = CH3CN or H2O) is described. 2 has been prepared by reaction of [FeII(CF3SO3)2(Me,HPytacn)] (1) with peracetic acid. Complex 2 bears a tetradentate N4 ligand that leaves two cis- sites available for binding an oxo group and a second external ligand but, unlike related iron(IV)-oxo of tetradentate ligands, it is remarkably stable at room temperature (t1/2 > 2h at 288 K). Its ability to exchange the oxygen atom of the oxo ligand with water has been analyzed in detail by means of kinetic studies, and a mechanism has been proposed on the basis of DFT calculations. Hydrogen-atom abstraction from C-H bonds and oxygen atom transfer to sulfides by 2 have also been studied. Despite its thermal stability, 2 proves to be a very powerful oxidant that is capable of breaking the strong C-H bond of cyclohexane (BDE = 99.3 kcal·mol−1).
Keywords: Bioinorganic Chemistry, High Valent, Reaction Mechanisms, Iron, Nonheme Oxigenases
Introduction
Iron(IV)-oxo compounds are proposed as the active species in the catalytic cycles of several O2-activating non-heme iron enzymes such as isopenicillin N-synthase, pterin-dependent hydroxylases and 2-oxoglutarate-dependent oxygenases.[1–3] Quite recently, high spin (S = 2) iron(IV)-oxo intermediates have been trapped and spectroscopically characterized in five different non-heme iron enzymes: namely taurine:α-ketoglutarate dioxygenase (TauD),[4, 5] halogenase CytC3,[6] propyl-4-hydroxylase[7], tyrosine hydroxylase,[8] and aliphatic halogenase, SyrB2.[9] Parallel synthetic efforts have succeeded in the preparation of octahedral S = 2 [FeIV(O)(H2O)5]2+,[10] and octahedral S = 1 FeIV=O species using tetradentate N4 ligands with available trans-binding sites,[11, 12] tetradentate N4 ligands with available cis-binding sites,[13–17], pentadentate N5 ligands.[18–22] and a pentadentate N4S ligand.[23] Very recently, two examples of trigonal bipyramidal S = 2 iron(IV)-oxo complexes, which bears structural and electronic resemblance to the enzymatic non-heme intermediates, have been described.[24–26]
These metastable and highly oxidizing complexes have been characterized by various spectroscopic techniques including, in selected cases, X-ray crystallography.[11, 22, 25–27] The ability of synthetic non-heme FeIV(O) species to act as oxidants has been proved[28, 29] and several studies demonstrate that they are capable of performing several oxidation reactions including oxygen-atom transfer to sulfides and triphenylphosphine,[16, 30, 31] epoxidation[13, 19, 21] and possible dihydroxylation[21, 32] of alkenes, alcohol oxidation,[33] hydrogen atom abstraction,[18, 34, 35] aliphatic and aromatic hydroxylation,[36, 37] oxo-transfer to other FeII complexes,[38] and glutathione and peptide oxidation.[39, 40] On the other hand, by analogy to the better studied heme systems,[41] it is widely postulated that non-heme iron-oxo species can exchange their oxygen atom with water via a oxo-hydroxo tautomerism before attack to a substrate (Scheme 1). Thus indirect evidence for the participation of these compounds in the catalytic cycle of enzymes[42] and/or synthetic models[43] comes from the incorporation of oxygen from water into oxidized products. Recently, this has been experimentally proved in two synthetic non-heme ion(IV)-oxo species; well-defined [FeIV(O)(tmc)(CH3CN)]2+ and [FeIV(O)(N4Py)]2+ (Scheme 2) have been shown to undergo exchange with water (Scheme 1).[44]
Scheme 1.

Top: Proposed oxo-hydroxo tautomerism in heme systems. Bottom: Proposed oxo-hydroxo tautomerism in [FeIV(O)(tmc)(CH3CN)]2+ and [FeIV(O)(N4Py)]2+.
Scheme 2.

Schematic representation of synthetic FeIV(O) complexes relevant to this work.
Because the vast majority of non-heme iron oxygenases contain cis-labile binding sites, synthetic iron(IV)-oxo complexes with tripodal tetradentate ligands are especially relevant from a biological point of view. However, they have proved to be less stable than their pentadentate counterparts,[28] rendering their study more difficult. For this reason most of the studies have been performed using pentadentate N5 ligands (N4Py) or equatorial tetradentate N4 ligands with two labile sites in a trans configuration (tmc). In this work we address this issue and we describe the preparation and spectroscopic and chemical characterization of a new FeIV(O) species bearing the tripodal tetradentate Me,HPytacn ligand (Me,HPytacn = 1-(2′-pyridylmethyl)-4,7-dimethyl-1,4,7-triazacyclononane). This species shows an unprecedented high stability at room temperature allowing us to evaluate several biologically relevant aspects of its reactivity. These studies include the evaluation of its ability to perform oxygen-atom transfer to sulfides and hydrogen-atom abstraction reactions. Moreover, the exchange of its oxo group with the oxygen atom from H218O has been studied by means of kinetic and computational methods.
Results and Discussion
Preparation and characterization of the FeIV(O) species
Reaction of [FeII(CF3SO3)2(Me,HPytacn)] (1) (Scheme 3)[45] with 2 equivalents of peracetic acid (CH3CO3H) in CH3CN at 15°C produces within 20 minutes a novel species (2) with a UV-vis spectrum showing a band at λmax = 750 nm (ε = 200 M−1cm−1) and a weaker shoulder around 900 nm (ε ~ 100 M−1cm−1) (Figure 1). 2 is relatively stable in solution, with a half-life time (t1/2) of 2.4 hours at 15°C. The nature of the transient species 2 has been determined by a combination of Mössbauer, 1H-NMR spectroscopy, resonance Raman, ESI-MS spectrometry and DFT calculations. We have studied the Mössbauer spectra of 2 in acetonitrile between 4.2 K and 100 K in parallel applied magnetic fields up to 8.0 T. Two representative spectra are shown in Figure 2. In zero applied magnetic field (Figure 2A), 2 exhibits at 4.2 K a quadrupole doublet, representing 85(5)% of the total iron, with quadrupole splitting ΔEQ = 0.73 mms−1 and isomer shift δ = 0.05 mm.s−1. These parameters, together with the fact that 2 has integer electron spin (as witnessed by the observation that 2 displays a doublet rather than magnetically split spectra in zero field), show that 2 is an FeIV complex. Spectra recorded in applied magnetic fields, e. g. Figure 2B, have features very similar to those reported for S = 1 FeIV(O) complexes; S = 2 FeIV(O) complexes display quite different spectra.[6, 8, 10, 24] We have analyzed the spectra of 2 with the commonly used S = 1 spin Hamiltonian[23]; the parameters obtained are very similar to those obtained for other S = 1 FeIV(O) complexes and are listed in the caption of Figure 2. The spectrum of Figure 2A indicates the presence of an S = 0 diferric contaminant with ΔEQ = 1.60 mm.s−1 and δ = 0.45 mm·s−1, representing ca. ≈ 15% of Fe. For the present chemistry such complexes are decomposition products that frequently represent the thermodynamic sink.[13]
Scheme 3.

Schematic view of formation and decay of 2, along with the proposed chemical structures of 1, 2 and 3.
Figure 1.

UV-vis spectrum of 2 along with its ESI-MS spectrum.
Figure 2.
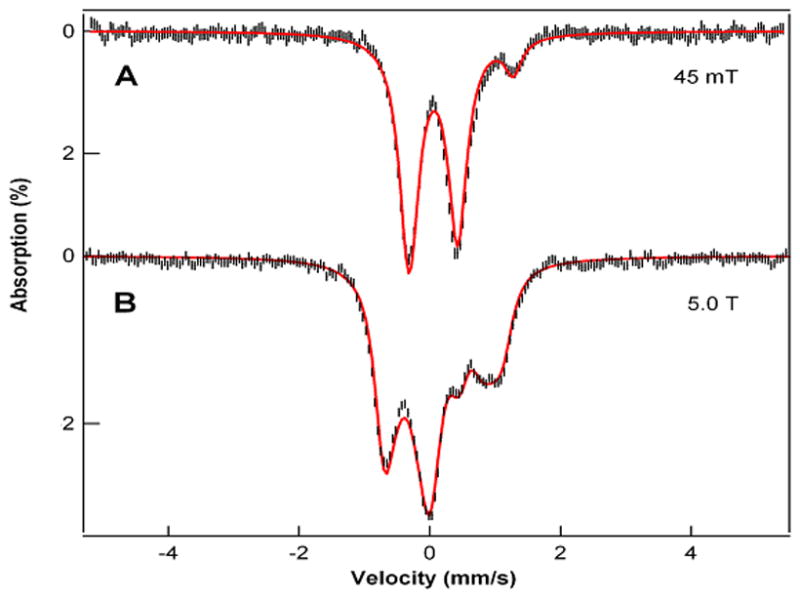
Mössbauer spectrum of 2 in acetonitrile, recorded at 4.2 K in parallel field as indicated. Solid lines are spectral simulations based on an S = 1 spin Hamiltonian[23] using the zero-field splitting parameters D = +27 cm−1, E/D = 0, 57Fe magnetic hyperfine tensor Ax,y,z/gnβ = (−23.7,−20.5, −4.5) T, quadrupole parameters ΔEQ = 0.73 mm·s−1, η = 1, and isomer shift δ = 0.05 mm·s−1.
High-resolution ESI-MS definitively proves the nature of 2 (Figure 1, inset) as it reveals a clean spectrum with a major peak with a mass value at m/z = 469.08 and an isotopic pattern fully consistent with the ion {[FeIV(O)(CF3SO3)(Me,HPytacn)]}+. Evidence for the presence of the oxo ligand was provided by vibrational spectroscopy. Resonance Raman analysis on frozen acetonitrile solutions of 2 with laser excitation at λexc = 407.9 nm shows a resonance enhanced feature at 831 cm−1 that downshifts to 788 cm−1 when 2 is allowed to react with H218O. The energy of the band falls in the range of reported synthetic[11, 15, 22, 24, 46] and enzymatic[47] nonheme Fe=O stretching vibrations, and the −43 cm−1 shift is in reasonable agreement with the shift of −37 cm−1 predicted by Hooke’s law for a diatomic Fe-O stretch and −40 cm−1 predicted by DFT calculations.[48,49] In conclusion, the spectroscopic data allow us to formulate 2 as a new iron(IV)-oxo species [FeIV(O)(Me,HPytacn)(S)]2+, where S = CH3CN or H2O. The nature of the sixth ligand could not be experimentally determined, but insight into its nature can be obtained from DFT calculations described in the next section. Acetonitrile binding was previously documented in the crystallographic characterization of [FeIV(O)(tmc)(CH3CN)]2+.[11] However, in the present case, the presence of a water ligand is proposed on the basis of DFT calculations (vide infra), which indicated that these species are thermodynamically more stable than the corresponding analogue containing an acetonitrile ligand.[13, 14]
Because of the inequivalence of the two binding positions not occupied by Me,HPytacn, two isomeric forms are possible for 2 (2a and 2b, Scheme 3). These two isomers differ not only in the group disposed trans to the oxo ligand but also in the relative orientation of the pyridine ring with respect to the Fe-O axis. However, the 1H-NMR spectrum of 2 (Figure 3) showed a unique set of sharp signals that could be assigned to the pyridine β (42 ppm), γ (10 ppm) and β′ (−13 ppm) protons. The shift pattern for these signals is reminiscent of that found in the 1H-NMR spectrum of [FeIV(O)(N4Py)]2+ in which all four pyridine rings are disposed parallel to the Fe-O axis, and different from the pattern associated with pyridine rings oriented perpendicular to the Fe=O axis.[27] Thus, in the present case, the NMR spectrum indicates that only isomer 2a is present in solution, with the pyridine ring disposed parallel to the Fe-O axis.
Figure 3.
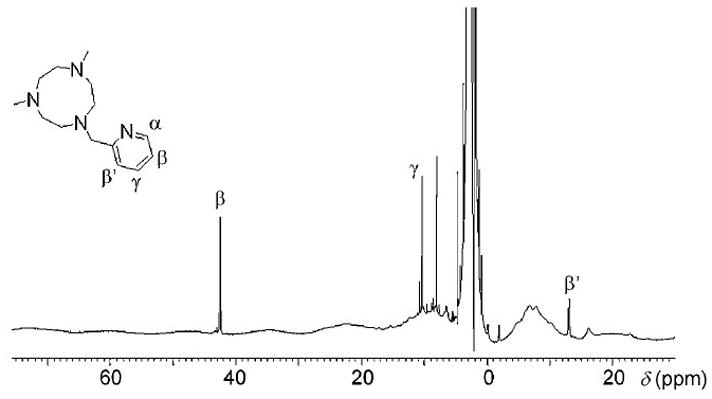
1H NMR spectra of 2 at 298 K in CD3CN, along with selected signal identifications.
Despite the inherently low stability of high valent oxo-iron(IV) species, the 1H-NMR spectrum of 2 lacks signals corresponding to 1[50] and 3 (the dinuclear Fe(III) decomposition product, vide infra, Sup Info), suggesting that 2 is prepared with good purity, in accord with the Mössbauer analysis (vide supra).
DFT analysis of the structures of the two isomers of 2
DFT calculations were employed to help clarifying the molecular and electronic structure of 2.[48, 49] The nature of the sixth ligand that completes the octahedral coordination sphere of the iron center was first studied by comparing the relative energies of the corresponding [FeIV(O)(Me,HPytacn)(S)]2+ (S = CH3CN and H2O) complexes. Computational analysis for S = H2O indicates that hydrogen bonding between the water ligand and a second, external water molecule is very strong because of the dicationic nature of 2. This interaction stabilizes [FeIV(O)(Me,H Pytacn)(H2O)]2+ relative to [FeIV(O)(Me,HPytacn)(CH3CN)]2+ by ΔG = 16.1 kcal·mol−1. Acetonitrile binding has been previously demonstrated in the X-ray structure of [FeIV(O)(tmc)(CH3CN)]2+,[11] for which the hydrophobic pocket created by the N-Me groups of tmc isolates the acetonitrile ligand. This is likely to prevent intermolecular interactions with additional water molecules. In contrast, the tripodal tetradentate Me,HPytacn ligand lacks such a sterically restricted hydrophobic pocket, which may constitute the key structural feature that favors water binding over acetonitrile in 2.
Optimized molecular structures of [FeIV(O)(Me,HPytacn)(H2O)]2+ (2a and 2b) obtained by DFT calculations are shown in Figure 4, with selected structural parameters given in the figure caption.[51] The relative DFT energies of the two isomeric forms of 2 indicate that 2a is energetically favored by 2.1 kcal·mol−1, thus supporting our structural assignment based on NMR spectroscopy. Energies corresponding to singlet (S = 0) and quintet states (S = 2) of 2a and 2b were also computed (Table S7). The substantially higher energies of these states, when compared with the triplet (S = 1) state further support the Mössbauer-based spin state assignment of 2. Structural parameters for 2a and 2b are very similar. The computed Fe-O distances in 2a and 2b are 1.62 Å and 1.63 Å, respectively, in good agreement with structurally characterized examples of synthetic[11, 17, 22, 24–27, 52] and enzymatic[53, 54] non-heme iron(IV)-oxo species. The oxo ligand exerts a trans effect that is indicated by a ~ 0.1 Å lengthening of the corresponding trans Fe-N distance (compare dFe1-N4 = 2.19 Å in 2b with 2.06 Å in 2a, and dFe1-N3 = 1.99 Å in 2b with 2.11 Å in 2a). The Fe-Npy distance is 2.03 Å in 2a and 2.02 Å in 2b, values that appear somewhat intermediate between the short dFe-Npy~ 1.95 Å of the pyridine rings cis to the oxo ligand in [FeIV(O)(N4Py)](ClO4)2[27] and the longer dFe-Npy = 2.118(3) Å of [FeIV(O)(Pytmc)](CF3SO3)2[22] for which the pyridine is bound trans to the oxo ligand.
Figure 4.
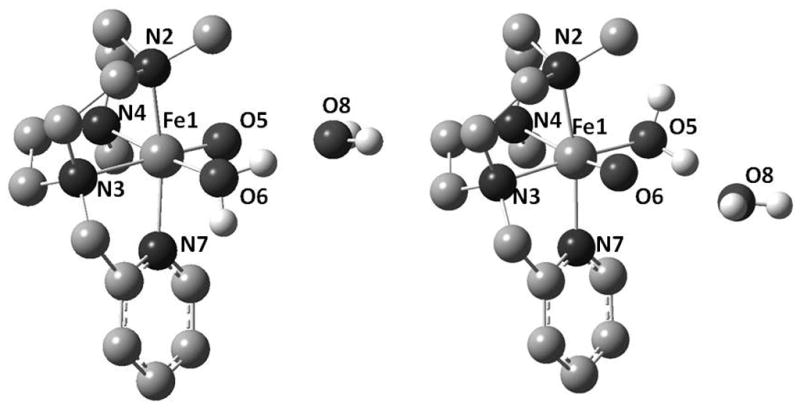
DFT-calculated structures of 2a (left) and 2b (right). Selected distances (Å) for 2a; Fe1-N2 2.05, Fe1-N3 2.11, Fe1-N4 2.06, Fe1-O5 2.06, Fe1-O6 1.62, Fe1-N7 2.03 and 2b; Fe1-N2 2.07, Fe1-N3 1.99, Fe1-N4 2.19, Fe1-O5 1.63, Fe1-O6 2.03, Fe1-N7 2.02.
Relative thermal stability and decay of 2
[FeIV(O)(Me,HPytacn)(S)]2+ (2) joins the growing family of iron(IV)-oxo species formed with tetradentate ligands tmc,[11] tpa[16], bqen[17], bpd[35] and bpmcn[14] (Scheme 2). In the case of tmc, the ligand topology gives rise to an iron center with two available trans sites, one of which is occupied by an oxo ligand in the FeIV-oxo compound. Instead, ligands like Me,HPytacn, tpa, bpd, bqen and bpmcn wrap around the metal ion, so that two sites in a cis relative disposition are ready for interaction with exogenous ligands. Thus, in the corresponding iron(IV)-oxo compounds, the oxo group binds to one of these positions and the other site is presumably occupied by a neutral labile molecule, such as an acetonitrile or water (Scheme 2). Despite the similar coordination spheres around the metal center, the resulting iron(IV)-oxo compounds exhibit remarkably different stabilities. [FeIV(O)(bpmcn)(S)]2+, [FeIV(O)(tpa)(S)]2+ and [FeIV(O)(bpd)(S)]2+ (S = CH3CN or H2O) can only be prepared in good yields at −40 °C because they easily decompose at higher temperatures.[13, 14, 35] [FeIV(O)(bqen)(S)]2+ has a lifetime of t1/2~ 30 min at 0 °C.[17] In contrast, 2 can be generated and remains relatively stable at 15 °C.
Thermal decay of 2 leads to a new species 3 that has been identified as [FeIII2(μ-O)(μ-CH3CO2)(Me,HPytacn)2]3+ by means of UV-vis spectroscopy, ESI-MS and comparison with an independently prepared sample characterized by X-ray crystallography (Figure 5).[55] The relatively poor stability observed for iron(IV)-oxo species with tripodal tetradentate ligands, in comparison with those that contain pentadentate or tetradentate ligands with trans- topologies, can be understood by considering that only the latter two offer effective steric protection of the oxo-ligand against decomposition into a diiron complex. In addition, these “stable” iron(IV) compounds lack a cis-labile site adjacent to the reactive oxo ligand, precluding pre-binding of a putative substrate. Based on these arguments, we suggest that the surprising thermal stability of 2 derives from a combination of the oxidatively robust nature of the Me,HPytacn ligand and some steric hindrance imposed by the N-Me groups of the ligand that project into the space surrounding the Fe=O unit. Consistent with this notion, in the absence of acetate ion, which facilitates dimerization, the oxidation of 1 with H2O2 in an aqueous:acetonitrile solvent mixture does not result in the common rapid formation of oxo-bridged ferric dimers,[56] but instead in the formation of the mononuclear species formulated as {[FeIII(OH)(Me,HPytacn)](CF3SO3)}+ on the basis of its ESI-MS spectra. The remarkable stability of 2 has allowed us to study some aspects of its biologically relevant reactivity which are detailed below.
Figure 5.
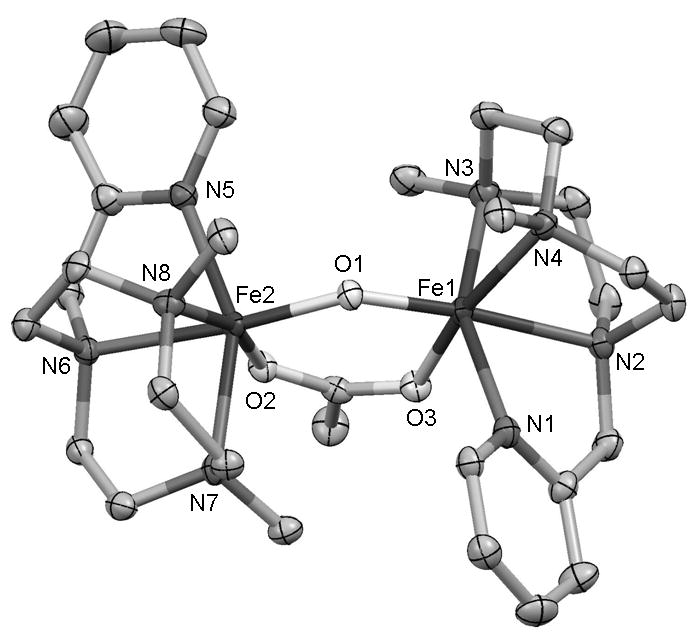
ORTEP plot (50% probability ellipsoids) of [FeIII2(μ-O)(μ-CH3CO2)(Me,HPytacn)2](ClO4)3·CH3CN, (3·CH3CN). Selected distances in Å: Fe1-O1 1.801(3), Fe1-O3 2.062(4), Fe1-N1 2.167(4), Fe1-N3 2.180(4), Fe1-N4 2.197(4), Fe1-N2 2.225(4), O1-Fe2 1.805(3), Fe2-O2 2.017(3), Fe2-N5 2.163(4), Fe2-N8 2.184(4), Fe2-N7 2.196(4), Fe2-N6 2.247(4). ∠ O1-Fe1-O3 95.42(15)o, Fe2-O1-Fe1 135.2(2)o.
Oxygen-atom exchange with water
Because of the biological relevance of water-exchange reactions at metal centers,[57] the ability of 2 to exchange its oxo ligand with H218O was studied by taking advantage of ESI-MS spectrometry. 2 was generated by reaction of 1 with 2 equivalents of CH3CO3H in acetonitrile. Subsequently, 600 equivalents of H218O were added to the reaction solution of 2, and several ESI-MS spectra were taken over time. The initial single peak at m/z = 469.1 associated with {[FeIV(16O)(CF3SO3)(Me,HPytacn)]}+ decreased gradually, while a new peak at m/z = 471.1 corresponding to {[FeIV(18O)(CF3SO3)(Me,HPytacn)]}+ appeared. Exchange of the oxygen atom with labeled water was complete in approximately 3 minutes at room temperature. Addition of pyridine-N-oxide, a strong σ-donating ligand, inhibited the water exchange exhibited by 2 suggesting that the available coordination site in a cis configuration with respect to the oxo group plays an important role in facilitating the water-exchange process.
Kinetic analyses of water exchange in 2 were performed by monitoring 18O incorporation into 2 directly via ESI-MS, and indirectly by quenching of a solution of 2 and H218O with thioanisole at different reaction times and analyzing the percentage of labeled sulfoxide generated by GC-MS. Within error, the two methods afforded identical reaction rates, supporting the validity of the second approach. In any case, exchange rate constants were obtained by non-linear regression fitting of the data to a single exponential function.
The rate of water incorporation into 2 (kobs) was found to be linearly dependent on [H218O], and independent of [2]. The data are consistent with the rate law in equation 1, where the use of a relative large [H218O] allows for its analysis as a pseudo-first order reaction;
A bimolecular rate constant of kexc = 0.028 ± 0.003 M−1 s−1 at 295K was extracted from this analysis. Activation parameters for water exchange were also determined by measuring exchange rates between 275K and 305K, affording ΔH≠ = 10.2 ± 0.8 kcal·mol−1 and ΔS≠ = −32 ± 3 cal·K−1·mol−1 (Figure 6). The negative activation entropy is consistent with an associative event in the transition state, most likely a bimolecular collision between the FeIV(O) species and a water molecule. Second-order rate constants for water exchange at [FeIV(O)(tpa)(S)]2+ were also measured (Table 1). The accumulated data indicate that water exchange rate for 2 is substantially faster than those for [FeIV(O)(tmc)(CH3CN)]2+ and [FeIV(O)(N4Py)]2+, which lack cis-available sites to the oxo ligand, and also somewhat faster than [FeIV(O)(tpa)(S)]2+. Nevertheless, considering the pentadentate nature of N4Py, the higher reaction rate observed for 2 represents a rather modest enhancement. Furthermore, the smaller reaction rate measured for [FeIV(O)(tpa)(S)]2+ suggests that reaction rates are not solely determined by the presence of a cis-labile site. Analysis of the enthalpy and entropy activation parameters indicates that the origin of the faster exchange rate in 2 has an entropic origin, which compensates for its higher activation enthalpy.
Figure 6.
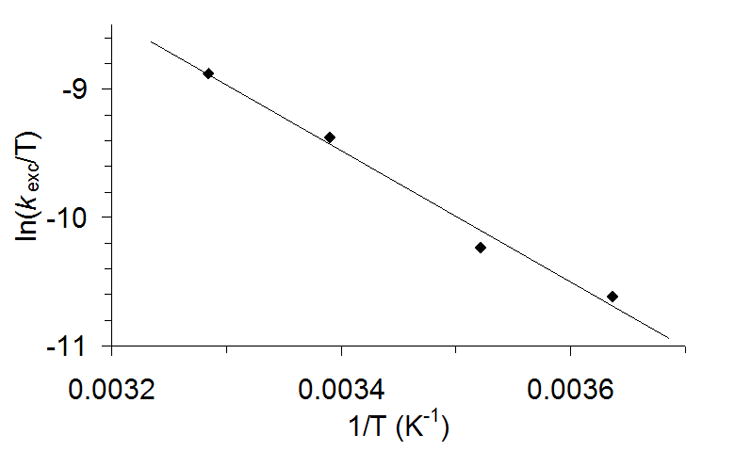
Eyring plot for the determination of the activation parameters of the oxygen atom exchange of 2 with H218O ([H218O] = 0.2 M).
Table 1.
Kinetic parameters for the water exchange reaction at selected oxo-iron(IV) complexes
| Compound | akexc ×10−3 M−1 s−1 | ΔH≠ kcal mol−1 | ΔS≠ cal K−1 mol−1 | Ref |
|---|---|---|---|---|
| 2 | 28±3 | 10.2±0.8 | −32±3 | - |
| [Fe(O)(tmc)(CH3CN)]2+ | 2.0b | 4.1±0.6 | −57±8 | [44] |
| [Fe(O)(N4Py)]2+ | 2.4b | 4.0±0.6 | −57±8 | [44] |
| [Fe(O)(tpa)(S)]2+ | 8±2 | c | c | - |
At 295K
Calculated from experimental data in ref. 44.
Eyring analysis could not be performed because of limited thermal stability.
Computational analysis of the water exchange mechanism
An obvious goal arising from these studies is to establish the fundamental chemical steps by which 2 can exchange its oxygen atom with water. The answers to this question may be of fundamental interest due to the structural similarity between 2 and biologically relevant non-heme oxo-iron(IV) species, in which the oxo group binds in a cis configuration with respect to one or more available coordination sites. In the heme paradigm, the incorporation of oxygen from water is explained by the so-called oxo-hydroxo tautomerism proposed by Meunier and co-workers,[41] which entails the binding of labeled water trans to the oxo group and then tautomerization of this species to a symmetric trans-dihydroxoiron(IV) intermediate that scrambles the label (Scheme 1). This pathway clearly cannot be operative for 2, since there is no binding site trans to the oxo group available for coordination with water, and an alternative mechanism must apply in the present case. The mechanism depicted in Scheme 4 starts with the iron(IV)-oxo complex 2a, with a coordinated water molecule in a cis position with respect to the oxo group. In the DFT study[49,58] of the water exchange mechanism, the solvent effects and the dispersion correction have been taken into account in the single point calculations. Solvent effects for acetonitrile were computed using the Polarizable Continuum Model (PCM)[59] as implemented in Gaussian09.[50] The dispersion energy has been computed using the DFT-D3 method and program of S. Grimme et al.[60] According to our theoretical calculations,[58] a key aspect of the reaction is the presence of an exogenous water molecule at close distance of the oxo and the water ligands. In the first step the exogenous water assists the transfer of one H+ from the bound H2O to the oxo ligand, via a transition state (TS1), with a barrier of ΔG≠ = 9.3 kcal·mol−1. After the first proton transfer event, an FeIV(OH)2 species (I1) is formed which is energetically favored compared to the starting complex 2a by 2.2 kcal·mol−1. Our spectroscopic data do not provide evidence for a significant accumulation of I1 in solution, but a dihydroxomanganese(IV) species has recently been described for a related macrocyclic N4-ligand,[61] and analogous LFeIV(OH)2 species have been proposed on the basis of DFT methods for a bispidine-based complex (Scheme 2).[32, 62] It is possible that the energy of I1 is underestimated in our calculations, and that it is slightly higher in energy than the corresponding oxo-aqua isomeric species (see also below for a discussion based on computed Mössbauer parameters). In I1, the exogenous water molecule has a H-bond interaction with the O5H hydroxide ligand (oxo group in 2a). In the next step, the exogenous water changes its interaction from the O5H hydroxo group to the second hydroxo ligand (O6H, aqua in 2a) and the intermediate I2 is formed. This step is endothermic with a free energy cost of 4.2 kcal·mol−1. When the free energy corrections are included TS2 and I2 are essentially isoenergetic. Close inspection reveals that TS2 and I2 differ in the orientation of the O5-H bond, which is not involved in hydrogen bonding with the external water molecule. I2 evolves via a second H+ transfer, passing through TS3 and forming 2b, where the oxo ligand is cis to the NCH2py ligand. Considering that TS2 and I2 are nearly isoenergetic, the larger barrier along the overall process is TS2 + TS3 with ΔG≠ = 13.0 kcal·mol−1, and corresponds to the second proton transfer. Proton transfer not assisted by the second water molecule has an activation enthalpy barrier about three times higher. Isomer 2b could not be experimentally observed because it is 2.11 kcal·mol−1 higher in energy than 2a, and because the activation barriers associated to each of the elemental steps that connect the backwards conversion of 2a to 2b are small. In this scenario, water ligand exchange at 2b and microscopic microreversibility leads to the formation of 2a, where the oxygen atom of the oxo group (trans to a NCH2Py unit) comes from water. Interestingly, the mechanism for water exchange at 2 bears resemblance to the DFT-computed conversion of [FeIV(OH)2L]2+ (where L stands for a tetradentate bispidine ligand related to bpd, Scheme 2) to [FeIV(O)(OH2)L]2+.[32, 62] Calculations indicate that this formal tautomerization takes place with a small energy barrier of 8.3 kcal·mol−1. Unlike in 2, the reaction involves a spin-crossover from the S = 1 FeIV(OH)2 to the thermodynamically more stable (4.4 kcal.mol−1) S = 2 FeIV(O)(OH2) tautomer, without assistance of a second water molecule. In contrast, all iron(IV) species in our calculations remain on the S = 1 surface.
Scheme 4.
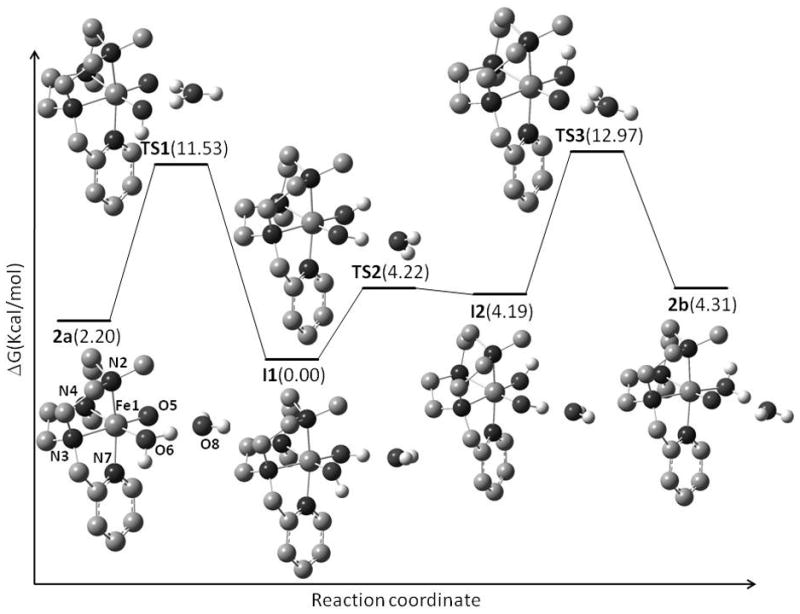
DFT mechanism for water exchange at 2.
Taking this computed mechanism into consideration, the first order kinetic dependence on water concentration is attributed to the initial equilibrium step involving the binding of a labeled water molecule, which replaces the initially bound unlabeled water. Nam, Que and coworkers have proposed that a seven-coordinate cis-oxo-aqua intermediate is formed in the first step of the reaction between [Fe(O)(N4Py)]2+ and[Fe(O)(tmc)(CH3CN)]2+ with water (Scheme 1).[34] Such mechanism may be also possible for 2 and [Fe(O)(tpa)(S)]2+, but owing to the presence of the labile water/acetonitrile ligand, a six-coordinate intermediate 2b formed via a ligand exchange reaction is favored. The breaking of the Fe–OH2 bond is likely to be the origin of the higher activation enthalpy which however, is entropically compensated because of the dissociative nature of the event.
An obvious consequence of the present mechanism is that, owing to the asymmetry of the two cis-labile sites, oxygen incorporation from water into the oxo ligand requires two oxo-hydroxo tautomerism reactions. That constitutes a fundamental mechanistic difference from oxygen atom incorporation from water in metalloporphyrins.[41]
DFT computation of the Mössbauer parameters of the species implicated in the water exchange mechanism
Considering that the DFT-computed water-exchange mechanism predicts that tautomeric bis-hydroxo-iron(IV) (I1 and I2) and oxo-iron(IV) (2a and 2b) are close in energy, the corresponding Mössbauer parameters were calculated with DFT methods and they are collected in Table 2. (See experimental section for details on the DFT analysis). The calculated isomer shifts for 2a and 2b are a bit too high relative to the experimental values. The Pittsburgh group, using Gaussian with the B3LYP functional (calibration of Vrajmasu et al.[14a]), has observed that the calculated δ values for TMC come out quite well while those of some most FeIV=O complexes with pyridine and amine ligands are generally too high, by as much as 0.09 mm/s. For the question at hand it is significant that the ΔEQ values of I1 and I2 are much larger than those of 2a and 2b. In agreement, large ΔEQ values have been reported for [FeIV(OH)(OOtBu)(bpmcn)]2+,[14b] ΔEQ = 1.75 mm·s−1 and the FeIV-OH site of [(OH)(LOMe)FeIV(μ-O)FeIV(O)(LOMe)]3+, L = tris((4-methoxy-3,5-dimethylpyridin-2-yl)d2-methyl)amine),ΔEQ = 1.96 mm·s−1.[14c] We conclude that, despite I1 being the lower energy species in the DFT-calculated mechanism, the Mössbauer parameters favor the oxo-iron(IV) formulation, in agreement with the resonance Raman evidence for an Fe=O unit.
Table 2.
DFT-Calculated Mössbauer parametersa of oxo-iron(IV) (2a and 2b) and bis-hydroxo-iron(IV) (I1 and I2) tautomeric species.
| Compound | ΔEQ (mm·s−1)a | δ (mm·s−1) |
|---|---|---|
| 2a | 0.57 | 0.18 |
| I1 | 2.28 | 0.12 |
| I2 | 2.44 | 0.18 |
| 2b | 0.83 | 0.17 |
| Exp | 0.73 | 0.05 |
See experimental section for details on the DFT analysis.
Oxidation of sulfides: oxygen atom transfer
The oxygen atom transfer ability of 2 was evaluated for the oxidation of sulfides. Complex 2 reacted rapidly at 273 K in CH3CN with 10 equiv of thioanisole affording the corresponding sulfoxide (methylphenylsulfoxide) in 98% yield. The reaction was monitored with UV-vis spectroscopy by following the decrease of the band at 750 nm characteristic of the iron(IV)-oxo complex 2. Under conditions of excess substrate (5 – 50 equiv with respect to 2) the reactions showed pseudo first-order behavior so that the observed reaction rates (kobs) were linearly dependent on substrate concentration. From this analysis, a second-order rate constant (kH) of 1.0 ± 0.1 M−1s−1 was obtained for the oxidation of thioanisole (see Supp. Info). Additional information about the mechanism of this transformation was gained through the initial observation that the reaction rates were highly dependent on the para substituent of the aromatic ring. Thus, we measured the second-order rate constants (kX) for a series of para-substituted methyl phenyl sulfides, p-X-thioanisoles (X = CN, Cl, CH3, OCH3). Plotting of log(kX/kH) for the different substrates against the corresponding Hammett parameters (σp) afforded a good linear correlation, and a Hammett value of ρ = − 1.5 was obtained (Figure 7). The negative ρ value reflects the electrophilicity of the oxo group in 2. On the other hand, a plot of the log(kX) against the one-electron oxidation potentials of each p-X-thioanisoles (Eoox) afforded a linear correlation with a slope of −3.0 (Supporting Information). As slope values around 10 would have been expected for a process initiated by 1-electron transfer,[63, 64] the observed slope of 3 indicates that the oxidation of sulfides by 2 does not occur by such a process but rather via a direct oxygen atom transfer mechanism. Further evidence for a direct oxygen atom transfer between the terminal oxo ligand and the sulfide came from labeling of the oxo group with 18O. This was achieved by oxygen atom exchange of 2 with H218O and subsequent reaction of the resulting [FeIV(18O)(Me,HPytacn)(H2O)]2+ with thioanisole. Analysis of the resulting sulfoxide by GC-MS indicated that 80% of the product was 18O-labeled.
Figure 7.
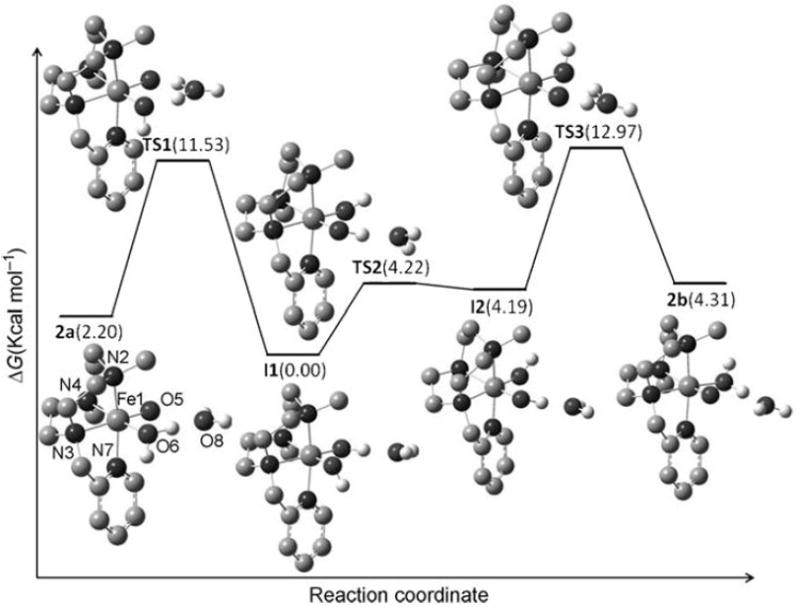
Hammett plot representing log(kX/kH) against the Hammett parameter (σp) for the reaction of 2 with p-X-thioanisoles at 273 K.
Despite the similarities between the oxygen atom transfer process in 2 and in other synthetically prepared iron(IV)-oxo species, reactions rates are highly dependent on the specific structure of the ligand. In particular, the oxidation of thioanisole by [FeIV(O)(N4Py)]2+ and [FeIV(O)(Bn-tpen)]2+ (Scheme 2)afforded second-order rate constants of 0.065 M−1s−1 (273 K) and 0.075 M−1s−1 (253 K), respectively, and this rate was even smaller for [FeIV(O)(tmc)(CH3CN)]2+ for which a value of 0.029 M−1s−1 at 35°C was obtained.[31] Comparison with the reactivity of [FeIV(O)(tpa)(S)]2+ is hampered due to its inherent instability which prevents proper measurement of the second-order rate constant at relatively high temperatures (a value of 0.44 M−1s−1 corresponding to the reaction with thioanisole at 228 K has been reported).[31] Nevertheless, from these studies it can be concluded that 2 and [FeIV(O)(tpa)(S)]2+, both of which bear tripodal tetradentate based ligands, are much more reactive in the oxygen atom transfer reaction than the corresponding complexes bearing tmc (tetradentate planar) or pentadentate (N4Py and Bn-tpen) ligands.
Oxidation of activated C-H bonds: hydrogen atom abstraction
The reaction of 2 with a series of substrates for H-atom abstraction was studied. 2 reacted with 5 equiv 9,10-dihydroanthracene (DHA) at 258 K affording anthracene as the only detected product by GC-MS. When the reaction was run under N2, 0.45 mmol anthracene/mmol of 2 was measured. The UV-vis spectra at the end of the reactions did not show formation of 1. In addition, ESI-MS spectrum of the resulting solution was dominated by ions corresponding to mononuclear FeIII species. Therefore, considering that Mössbauer spectra indicate a 85(5)% FeIV content in our preparations, on the basis of stoichiometric considerations we conclude that in this reaction 2 acts as a 1e− oxidant (equation 2), and that the yield of the reaction with DHA is nearly quantitative.
 |
(2) |
The kinetics of reactions of 2 with a series of substrates were studied by UV-vis following the decay of the band at 750 nm characteristic of the iron(IV)-oxo species 2. The decay of 2 followed good first-order kinetics under conditions of excess substrate and kobs values were linearly dependent on substrate concentration in all cases, thus allowing us to determine the corresponding second order rate constants, k2 (see supporting information). These values are collected in Table 3. Reaction of 2 with alkylaromatic substrates (toluene and ethylbenzene) caused rapid formation of brown-dark blue intense chromophores, indicative of alkylphenolate-bound iron(III) species. Considering the high bond dissociation energy (BDE) of the C-H bond in aromatic substrates, such reactions could not occur via H-atom abstraction but instead, they originate from electrophilic attack of the aromatic ring by 2.[37] For this reason, these substrates were not further addressed in the present study.
Table 3.
C-H Bond dissociation energies and reaction rates for the reaction of 2 with different substrates.a
| Substrate | BDE (kcal·mol−1)[67],[68] | k2 (M−1s−1) |
|---|---|---|
| 10-methyl-9,10- | 73.7 | 86 ± 19 |
| dihydroacridine (AcrH2) xanthene | 75.5 | 8.1 ± 0.8 |
| 9,10-dihydroanthracene (DHA) | 77 | 5.7 ± 0.9 |
| 1,4-cyclohexadiene (CHD) | 78 | 4.2 ± 0.3 |
| fluorene | 80 | 1.2 ± 0.2 |
| 2,3-dimethyl-2-butene | 84 | (1.6 ± 0.2) × 10−2 |
| tetrahydrofuran (THF) | 93 | (2.3± 0.3) × 10−3 |
| cyclohexaneb | 99.3 | (4 ± 1) × 10−4 |
Reactions were run in acetonitrile at 258 K, using 2 generated in situ from the reaction of 1 (1 mM) with 2 equiv of peracetic acid at 15°C.
298 K
2 proved to be a very powerful oxidant in C-H oxidation reactions (Tables 3 and 4). Compared to related dicationic iron(IV)-oxo complexes, under analogous conditions, reaction rates obtained with 2 are somewhat higher than those obtained with [FeIV(O)(N4Py)]2+ and nearly two orders of magnitude faster than with [FeIV(O)(tmc)(CH3CN)]2+ [24, 34] but somewhat lower than that found for [FeIV(O)(bpd)(CH3CN)]+2 (Scheme 2).[35] Furthermore, the second order rate constant for the oxidation of DHA with 2 appears to be several orders of magnitude higher than those reported for low-spin FeIIIFeIV and FeIVFeIV oxo-dimers, and approaches the remarkably high rate (k2 = 28 M−1s−1 at 193 K) recently measured for the high-spin [(HO)(LOMe)FeIII-O-FeIV(O)(LOMe)]2+, LOMe = tris((4-methoxy-3,5-dimethyl-pyridin-2-yl)methyl)amine (Table 4).[65] The high reactivity of the latter has been rationalized on the basis of the high spin (S = 2) state of the iron ions, and the compound has limited stability even at −80 °C. On the other hand, as already discussed, the metal ion found in 2 has a S = 1 spin state, and it is moderately stable at room temperature. As expected from its higher oxidation state, reaction rates with 2 are 2–3 orders of magnitude faster than those obtained with the lipoxygenase model [FeIII(OMe)(Py5)]2+ (Py5 = 2,6-bis(bis(2-pyridyl)methoxymethane)pyridine).[66] Indeed, 2 proved to be competent for performing the oxidation of strong C-H bonds such as THF (BDE = 93 kcal·mol−1) with a reaction rate k2 = (2.3 ± 0.3) × 10−3 M−1 s−1 at 258 K.[67] Most remarkably, 2 is also competent to perform the oxidation of cyclohexane (BDE = 99.3 kcal·mol−1) at room temperature with a second order rate constant, after correction for the number of C-H bonds, of k2′ = 3.6 × 10−5 M−1 s−1. GC analysis of the reactions reveals the formation of 0.12 mol cyclohexanone/mol of 2 and 0.28 mols cyclohexanol/mol of 2. Nevertheless, this reaction rate is relatively very slow when compared with the fast oxidation observed in catalytic 1/H2O2 cyclohexane oxidations, where a [FeV(O)(OH)(Me,HPytacn)]2+ species has been inferred from isotopic labeling experiments and DFT analysis.[45, 50]
Table 4.
DHA oxidation rates of various iron-oxo complexes.
| Complex | k2 (M−1s−1) | T (K) | Ref |
|---|---|---|---|
| 2 | 5.7 ± 0.9 | 258 | |
| [FeIV(O)(N4Py)]+2 | 2.8 | 258 | [75] |
| [FeIV(O)(bpd)(CH3CN)]+2 | 8.0 | 238 | [35] |
| [FeIV(O)(tmc)(CH3CN)]+2 | 0.016 | 243 | [24] |
| [FeIII(OMe)(Py5)]+2 | 5.0 × 10−3 | 298 | [66] |
| [(OH)(LOMe)FeIII (μ-O)FeIV(O)(LOMe)]+2 | 28 | 193 | [65] |
| [FeIIIFeIV(μ-O)2(LOMe)2]+3 | 10−5 | 193 | [65] |
| [FeIV2(μ-O)2(LOMe)2]+4 | 10−4 | 193 | [65] |
| [(OH)(LOMe)FeIV(μ-O)FeIV(O)(LOMe)]+3 | 0.027 | 193 | [65] |
| [FeIV(O)(tmp)] | 2.7 | 258 | [76] |
| [FeIV(O)(TMG3tren)]+2 | 0.090 | 243 | [24] |
LOMe = tris((4-methoxy-3,5-dimethylpyridin-2-yl)d2-methyl)amine, tmp = tetramesitylporphinate, TMG3tren = 1,1,1-tris{2-[N2-(1,1,3,3-tetramethylguanidino)]ethyl}amine.
Comparative reactivity profiles of the reaction of 2 against different substrates show that the rate constants decreased with the increase of the C-H BDE and more interestingly the log(k′2) values correlated linearly with the C-H BDE values of the substrates, giving a slope of approximately −0.2 (Table 3, Figure 8). The slope is comparable to those obtained for hydrogen abstraction reactions mediated by [FeIV(O)(N4Py)]2+[16] and [FeIII(OMe)(Py5)]2+,[66] and intermediate between the −0.4 and −0.1 observed for [RuIV(O)(bpy)2(py)]2+ and [MnIII(OMe)(Py5)]2+,[69, 70] respectively. Such a correlation strongly suggests that reactions take place via a H-atom abstraction mechanism, [69, 71–73] as early established for related FeIV compounds.[18, 34, 74]
Figure 8.
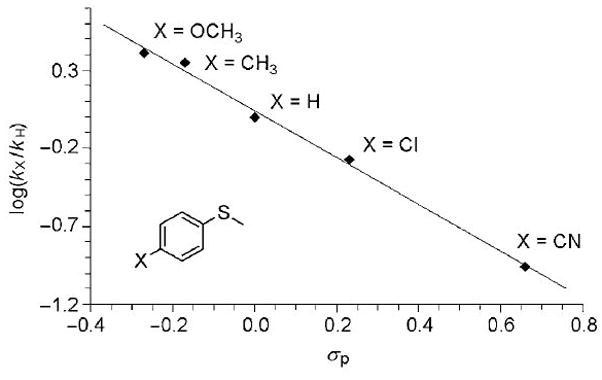
Plot of log(k′2) (determined at −15°C) against C-H BDE of different substrates.
Parallel reactions with deuterated 9,10-dihydroanthracene (d4-DHA) yielded a kinetic isotope effect (KIE) of 27, a value which is consistent with a C-H bond cleavage being the rate-determining step.[68] This large KIE value is well above the semi-classical limit of 7, suggesting a hydrogen atom transfer mechanism dominated by quantum mechanical tunneling.[74, 77, 78] For comparison, KIE values around 17 were obtained for the oxidation of DHA by [FeIV(O)(tmc)(X)]n+ (X = CH3CN, CF3COO− or N3−)[34] and even larger values were reported in the oxidation of benzyl alcohol by [FeIV(O)(N4Py)]+2 (KIE = 48) and [FeIV(O)(tpa)(S)]+2 (KIE = 58).[33] Moreover, such large isotope effects have also been observed in hydrogen atom abstraction reactions performed by the iron(IV)-oxo intermediate of TauD (KIE ~ 37)[79], the iron(III)-hydroxo active species of lypoxygenase (KIE~ 50)[80] and compound Q (FeIVFeIV) of the diiron enzyme soluble methane monooxygenase (sMMO) (KIE > 50).[81]
A particular case that deserves some comment is the reaction with the NADH analogue 10-methyl-9,10-dihydroacridine (AcrH2), which is considered a hydride donor.[82] The reaction of AcrH2 with 2 results in rapid formation (k2 = 86 ± 19 M−1 s−1) of the acridinium cation AcrH+, as evidenced by UV-vis spectroscopy. UV-vis quantification based on the characteristic spectral features of the cation accounts for 1 mmol AcrH+/mmol 2, indicating that 2 acts as a 2e− oxidant, and that the reaction is best described as a formal hydride transfer to the iron(IV)-oxo moiety. Interestingly, the corresponding oxidation rate constant (k2′) appears to be only slightly faster than expected from the log(k′2) vs BDE graph correlation (Figure 8) suggesting that a common rate determining step is operative for all the series of substrates plotted. In addition, the KIE evaluated from the reaction of 2 with AcrD2 at 258 K is 4.7 (Figure S11), which is intermediate between the values observed for [FeIV(O)(N4Py)]+2 (KIE = 13.5),[82] and those obtained recently with [FeIV(O)(bpd)(CH3CN)]2+, (KIE = 2.3) (Scheme 2).[35] On this basis, we propose that the reaction takes place via a rate determining hydrogen atom abstraction (HAT) that can be regarded as a proton coupled electron transfer (PCET), followed by a fast electron-transfer.[68, 76, 83, 84]
Considering that C-H bond oxidation by 2 occurs via a hydrogen atom abstraction mechanism,[73, 85, 86] the balance between the strength of the cleaved C-H bond and of the formed FeIII-OH bond must determine the observed reaction rates. Along this interpretation, we notice that reactions mediated by 2 are comparable to those associated with the most reactive FeIV(O) species containing nitrogen based pentadentate ligands such as [FeIV(O)(N4Py)]2+, which constitute one of the very rare examples of well defined metal-oxo species that are capable of breaking the strong C-H bond of cyclohexane at room temperature.[18, 87–89] The similar reaction rates observed for 2 and [FeIV(O)(N4Py)]2+ should be interpreted as both of them forming a FeIIIO-H bond of approximately the same energy. Previous literature reports on the estimation of the FeIIIO-H bond in [FeIV(O)(N4Py)]2+ have provided somewhat different values. Two independent theoretical studies yielded FeIIIO–H BDE’s of 84[90, 91] and 97 kcal·mol−1,[92] and some of us have experimentally estimated a more modest BDE = 78 kcal·mol−1 from its experimentally observed redox potential in water.[75] We can take the latter value as a lower limit estimate of the corresponding FeIIIO–H bond formed by H-abstraction in 2. Thus, as previously noted for [FeIV(O)(N4Py)]2+,[90] despite its rather surprising thermal stability, 2 is a very powerful oxidant. Besides the high oxidation state, one of the reasons at the origin of such a high reactivity is likely to be the positive charge of the [LFeIV(O)]2+ unit when L = neutral ligand (L = N4Py or Me,HPytacn), which results in a very electrophilic oxidant.
The remarkably high oxidative reactivity exhibited by 2 both in O-atom transfer and in H-abstraction reactions is unprecedented and deserves some comment. The factors that determine the reactivity of oxo-iron(IV) species have been the focus of intense debate. Accessibility of multiple spin states modulated by the nature of the ligand trans to the oxo group have been invoked to explain inverted tendencies in oxygen atom transfer vs H-abstraction reaction by series of FeIV compounds.[34] Basic anionic ligands X in a series of [FeIV(O)(X)(tmc)]+ complexes decrease their electrophilicity and O-atom transfer reactivity, but enhance their H-abstraction reactivity by populating a reactive quintet state. Population of an excited, highly reactive S = 2 state has also been proposed to account for the exceptional ability of [FeIV(O)(N4Py)]2+ to cleave strong C-H bonds via a H-atom abstraction mechanism.[90] The nature of the ligands cis to the oxo group is considered to have a more modest effect on the oxidative reactivity.[93] Computational studies have thus suggested that high-spin S = 2 FeIV(O) species could be much more reactive than their corresponding low spin S = 1 analogues. However, the recent preparation and characterization of the first synthetic S = 2 FeIV(O) species raises some questions about this prediction, because it exhibits reactivity only comparable to that of [FeIV(O)(N4Py)]2+.[24, 25] Steric effects were invoked by the authors to account for these observations. 2 contains a S = 1 spin state, and DFT computations show that it is well separated in energy from the S = 2 spin state (See table S7 in sup. Info). Therefore a two-state reactivity (TSR) scenario appears highly unlikely. In addition, high reactions rates are measured for both O-atom transfer and hydrogen-atom abstraction reactions. Its high oxidative reactivity may then add to the rationale of substrate accessibility to the reactive oxo ligand as a major factor dictating reactivity in these species. The high reactivity of 2 in C-H oxidation reactions finds a nice precedent in that recently described for [FeIV(O)(bpd)(CH3CN)]+2 (Scheme 2).[35] Both compounds have a N4-based ligand that enforces a cis-oxo-labile site. However, the bispidine is a weaker field ligand[32, 62] than Me,HPytacn,[50] and the TSR scenario is more likely. In this scenario, we propose that 2 is more reactive than most low spin iron(IV)-oxo species bearing pentadentate and/or planar tetradentate ligands because the oxo ligand in 2 is more exposed, and susceptible to interaction with small substrate molecules. However, compared with previously reported tetradentate ligands, Me,HPytacn affords a very significant degree of steric protection against bimetallic dimerization reactions.
Conclusion
In this work we have reported the preparation of a new S = 1 iron(IV)-oxo species (2) with a tripodal tetradentate ligand that has remarkable thermal stability, especially when compared with structurally related complexes like [FeIV(O)(bpmcn)(S)]2+, [FeIV(O)(tpa)(S)]2+, and [FeIV(O)(bpd)(CH3CN)]+2. 2 has a labile site cis to the oxo group, which constitutes a very common structural feature of non-heme iron oxygenases. 2 rapidly exchanges its oxygen atom with H218O following a mechanism in which an exogenous water molecule assists the hydrogen transfer from the coordinated water molecule to the oxo group. Despite its remarkable thermal stability, 2 is a very good oxidant both with respect to oxygen atom transfer to sulfides and hydrogen-atom abstraction of alkane C-H bonds, demonstrating a highly electrophilic character arising from the FeIV oxidation state in a neutral N-based ligand environment. The higher oxidative reactivity of 2 when compared with complexes containing pentadentate ligands is attributed to the tetradentate nature of the Me,HPytacn ligand, which does not provide steric encumbrance of the oxo ligand in 2, thus providing the basis of its remarkable oxidative reactivity.
Experimental Section
Materials and methods
Reagents and solvents used were of commercially available reagent quality unless otherwise stated. Preparation and handling of air-sensitive materials were done under an inert atmosphere either on a Schlenk line or in a glove box. Acetonitrile was purchased from Scharlau. H218O (95% 18O-enriched) was received from ICON Isotopes.
Synthesis of complexes
The starting complex[Fe(CF3SO3)2(Me,HPytacn)] (1) was prepared following the previously reported experimental procedure.[45]
Preparation of iron(IV)-oxo complex (2) was performed as follows: to a 1 mM solution of 1 in acetonitrile (2 mL total volume) 2 equiv of peracetic acid solution (100 μL of a 40 mM solution in CH3CN obtained by dilution of commercially available 32% w/w peracetic acid solution in acetic acid) were added at 288 K. The formation of 2 was followed by UV-vis spectroscopy showing the appearance of a band at 750 nm (λmax = 200 M−1cm−1) with a shoulder at 900 nm which developed in about 10 minutes. The resulting solution of 2 (1 mM) was directly used for subsequent reactivity studies.
Direct preparation of [FeIII2(μ-O)(μ-CH3COO)(Me,HPytacn)2](ClO4)3·CH3CN (3·CH3CN)
54.6 mg of compound 1 (90 μmols) were dissolved in MeCN (2 mL) under an inert atmosphere. 200 μL of an acetonitrile solution 0.30 M in CH3COOH and Et3N (60 μmols CH3COOH and Et3N) were added at once, which caused an immediate color change from dark pink to bright yellow. Immediately a balloon filled with O2 was connected into the reaction vessel and the solution became red-brown in a few seconds. After stirring for 3 h, the solvent from the resulting solution was removed under reduced pressure which afforded a brown oil. The resulting product was redissolved in acetonitrile and 19 mg NaClO4·H2O (135 μmols) were added. The solution was stirred for about 2 hours, filtered through Celite and diethyl ether was slowly diffused. 37 mg of brown crystals of 3·CH3CN suitable for X-ray diffraction were obtained (38 μmols, 84%). 1H-NMR (200 MHz, CDCl3, 300K) δ = 26.43, 18.27, 17.41, 14.47, 6.57 ppm. FT-IR (ATR) ν = 1611, 1523 (CH3COO), 1447 (C=Car), 1073, 621 cm−1 (ClO4).UV/Vis (CH3CN): λmax (ε/Fe) = 427 (660), 462 (690), 513 (500), 692 nm (58 mol−1·cm−1). ESI-MS: m/z: 227.6 [M-3ClO4]+3, 880.9 [M-ClO4]+.
Oxygen atom exchange with water
Kinetic studies on oxygen atom exchange of 2 with H218O were performed by quenching aliquots of the reaction mixture with thioanisole at different times, and analyzing the percentage of 18O-labeled methylphenyl sulfoxide generated by GC-MS. pH dependence of the reaction rates was not studied. In a typical experiment, to 2.5 mL of a stirred solution of 2 in acetonitrile (1 mM), the appropriate amount of H218O (from 6 to 60 μL) was added at once. At different reaction times, an aliquot of the reaction mixture (350 μL) was directly poured into a solution containing 7 μL of thioanisole in 300 μL of CH3CN. The resulting yellow solution was stirred for 30 minutes at room temperature, filtered through a short path of basic alumina and washed with 2 mL of ethyl acetate. The sample was analysed by GC-MS. The percentage of 18O-labeled sulfoxide was determined by the isotopic pattern showed by the peaks at m/z = 125 and 140. In a typical experiment, a total of 7 aliquots were taken out from the reaction mixture. The temperature of the reaction mixture (between 0 and 30 °C) was controlled by means of a cryostat or a thermostatized water bath. The percentage of 18O-labeled sulfoxide is directly related to the percentage of iron(IV)-oxo species (2) that has exchanged its oxygen atom with H218O. The percentage of 18O-labeled sulfoxide over time could be fitted to a pseudo-first-order kinetics that allowed us to measure the rate constants (kobs) corresponding to oxygen-atom exchange with 18O-labeled water in 2.
Oxidation of activated C-H bonds and sulfides
The appropriate amounts of substrates (diluted in acetonitrile) were added to a solution of 2 (1 mM) and the subsequent decay of the spectral changes corresponding to the iron(IV)-oxo were directly monitored by UV-vis spectroscopy. The kinetic studies were performed at specific temperatures: 273 K for the oxidation of sulfides and 258 K for the oxidation of activated C-H bonds. Unless specifically stated, reactions were run under air. Rate constants, kobs, were determined by pseudo-first-order fitting of the decrease of the absorption band at 750 nm. Product analyses for the oxidation of thioanisole and DHA were performed by gas chromatography. Prior to injection, an internal standard (biphenyl) was added to the solution which was further filtered through basic alumina and washed with ethyl acetate. Calibration curves with authentic products were generated to obtain quantitative determination of the oxidized products.
Details on the DFT calculations of the Mossbauer parameters
Details on the DFT calculations are collected in refs The FeIV DFT Mössbauer parameters were calculated at OPBE/TZP[94] level within the conductor-like screening (COSMO) solvation model[95] (with dielectric constant ε =37.5, Acetonitrile) using the Amsterdam Density Functional (ADF) suite of program.[96] The isomer shift (δ), which is proportional to the electron density [ρ(0)] difference at the Fe nuclei between the studied system and a reference system (normaly α-Fe at 300K), were calculated according to the procedure described in Refs.[97] with utility programs provided by Han and Noodleman. The isomer shift is given by:
where ρ(0) is obtained using ADF and Han and Noodleman code; A is a constant chosen close to the electron density at the Fe nucleus in the reference state; and α and C were determined by linear regression between the calculated ρ(0) and experimental δ, Han and Noodleman for their Fe2.5+,3+,3.5+,4+ training set obtained α = −0.312 ± 0.022 and C = 0.373 ± 0.014 mm s−1 for OPBE. The quadrupole splitting, which is proportional to the electric field gradient (EFG) at the Fe nucleus, is directly given by ADF.
Supplementary Material
Acknowledgments
Financial support for this work was provided by MEC-Spain (Project CTQ2009 08464/BQU and CTQ2008-06696/BQU), and the US National Institutes of Health (GM-33162 to L.Q. and EB-001475 to E.M.). M.C. acknowledges Generalitat de Catalunya for an ICREA-Academia Award and SGR 2009 SGR637. A.C., I. P. and M.G. thank MEC for FPU-PhD grants. We thank RahuCat for a generous gift of tritosyltacn. We thank W. Han and L. Noodleman for providing the utility programs to compute the Mössbauer parameters of the iron complexes and M. Swart for his useful comments about the Mössbauer calculations.
References
- 1.Abu-Omar MM, Loaiza A, Hontzeas N. Chem Rev. 2005;105:2227. doi: 10.1021/cr040653o. [DOI] [PubMed] [Google Scholar]
- 2.Costas M, Mehn MP, Jensen MP, Que L., Jr Chem Rev. 2004;104:939. doi: 10.1021/cr020628n. [DOI] [PubMed] [Google Scholar]
- 3.Solomon EI, Brunold TC, Davis MI, Kemsley JN, Lee S-K, Lehnert N, Neese F, Skulan AJ, Yang Y-S, Zhou J. Chem Rev. 2000;100:235. doi: 10.1021/cr9900275. [DOI] [PubMed] [Google Scholar]
- 4.Bollinger JM, Jr, Price JC, Hoffart LM, Barr EW, Krebs C. Eur J Inorg Chem. 2005;21:4245. [Google Scholar]
- 5.Price JC, Barr EW, Tirupati B, Bollinger JM, Jr, Krebs C. Biochemistry. 2003;42:7497. doi: 10.1021/bi030011f. [DOI] [PubMed] [Google Scholar]
- 6.Galoniæ DP, Barr EW, Walsh CT, Bollinger JM, Krebs C. Nat Chem Biol. 2007;3:113. doi: 10.1038/nchembio856. [DOI] [PubMed] [Google Scholar]
- 7.Hoffart LM, Barr EW, Guyer RB, Bollinger JJM, Krebs C. Proc Natl Acad Sci USA. 2006;103:14738. doi: 10.1073/pnas.0604005103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eser BE, Barr EW, Frantom PA, Saleh L, Bollinger JM, Jr, Krebs C, Fitzpatrick PF. J Am Chem Soc. 2007;129:11334. doi: 10.1021/ja074446s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Matthews ML, Krest CM, Barr EW, Vaillancourt FH, Walsh CT, Green MT, Krebs C, Bollinger JM., Jr Biochemistry. 2009;48:4331. doi: 10.1021/bi900109z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pestovsky O, Stoian S, Bominaar EL, Shan X, Münck E, Que L, Jr , Bakac A. Angew Chem. 2005;117:7031–7034. doi: 10.1002/anie.200502686. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2005;44:6871. [Google Scholar]
- 11.Rohde J-U, In J-H, Lim M-H, Brennessel WW, Bukowski MR, Stubna A, Münck E, Nam W, Que L., Jr Science. 2003;229:1037. doi: 10.1126/science.299.5609.1037. [DOI] [PubMed] [Google Scholar]
- 12.Ray K, England J, Fiedler AT, Martinho M, Munck E, Que L., Jr Angew Chem. 2008;120:8188. doi: 10.1002/anie.200802219. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2008;47:8068. [Google Scholar]
- 13.Lim MH, Rohde J-H, Stubna A, Bukowski MR, Costas M, Ho RYN, Münck E, Nam W, Que L., Jr Proc Acad Sci USA. 2003;100:3665. doi: 10.1073/pnas.0636830100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.a) Vrajmasu V, Münck E, Bominaar EL. Inorg Chem. 2003;42:5974. doi: 10.1021/ic0301371. [DOI] [PubMed] [Google Scholar]; b) Jensen MP, Costas M, Ho RYN, Kaizer J, Mairata i Payeras A, Münck E, Que L, Jr, Rohde J-U, Stubna A. J Am Chem Soc. 2005;127:10512. doi: 10.1021/ja0438765. [DOI] [PubMed] [Google Scholar]; c) Martinho M, Xue GQ, Fiedler AT, Que L, Jr, Bominaar EL, Munck E. J Am Chem Soc. 2009;131:5823. doi: 10.1021/ja8098917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martinho M, Banse F, Bartoli J-F, Mattioli TA, Battioni P, Horner O, Bourcier S, Girerd J-J. Inorg Chem. 2005;44:9592. doi: 10.1021/ic051213y. [DOI] [PubMed] [Google Scholar]
- 16.Paine TK, Costas M, Kaizer J, Que L., Jr J Biol Inorg Chem. 2006;11:272. doi: 10.1007/s00775-006-0089-6. [DOI] [PubMed] [Google Scholar]
- 17.Yoon J, Wilson SA, Jang YK, Seo MS, Nehru K, Hedman B, Hodgson KO, Bill E, Solomon EI, Nam W. Angew Chem. 2009;121:1283. doi: 10.1002/anie.200802672. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2009;48:1257. [Google Scholar]
- 18.Kaizer J, Klinker EJ, Oh NY, Rohde J-U, Song WJ, Stubna A, Kim J, Munck E, Nam W, Que L., Jr J Am Chem Soc. 2004;126:472. doi: 10.1021/ja037288n. [DOI] [PubMed] [Google Scholar]
- 19.Balland V, Charlot MF, Banse F, Girerd JJ, Mattioli TA, Bill E, Bartoli JF, Battioni P, Mansuy D. Eur J Inorg Chem. 2004;2:301. [Google Scholar]
- 20.Bautz J, Bukowski MR, Kerscher M, Stubna A, Comba P, Lienke A, Münck E, Que L., Jr Angew Chem. 2006;118:5810. doi: 10.1002/anie.200601134. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2006;45:5681. [Google Scholar]
- 21.Bukowski MR, Comba P, Lienke A, Limberg C, de Laorden CL, Mas-Balleste R, Merz M, Que L., Jr Angew Chem. 2006;118:3524. doi: 10.1002/anie.200504357. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2006;45:3446. [Google Scholar]
- 22.Thibon A, England J, Martinho M, Young VG, Jr, Frisch JR, Guillot R, Girerd J-J, Münck E, Que L, Jr, Banse F. Angew Chem. 2008;120:7172. doi: 10.1002/anie.200801832. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2008;47:7064. [Google Scholar]
- 23.Bukowski MR, Koehntop KD, Stubna A, Bominaar EL, Halfen JA, Münck E, Nam W, Que L., Jr Science. 2005;310:1000. doi: 10.1126/science.1119092. [DOI] [PubMed] [Google Scholar]
- 24.England J, Martinho M, Farquhar ER, Frisch JR, Bominaar EL, Münck E, Que L., Jr Angew Chem. 2009;121:3676. doi: 10.1002/anie.200900863. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2009;48:3622. [Google Scholar]
- 25.England J, Guo Y, Farquhar ER, Young VG, Jr, Munck E, Que L., Jr J Am Chem Soc. 2010;132:8635. doi: 10.1021/ja100366c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lacy DC, Gupta R, Stone KL, Greaves J, Ziller JW, Hendrich MP, Borovik AS. J Am Chem Soc. 2010;132:12188. doi: 10.1021/ja1047818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Klinker EJ, Kaizer J, Brennessel WW, Woodrum NL, Cramer CJ, Que L., Jr Angew Chem. 2005;117:3756. doi: 10.1002/anie.200500485. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2005;44:3690. [Google Scholar]
- 28.Que L., Jr Acc Chem Res. 2007;40:493. doi: 10.1021/ar700024g. [DOI] [PubMed] [Google Scholar]
- 29.Nam W. Acc Chem Res. 2007;40:522. doi: 10.1021/ar700027f. [DOI] [PubMed] [Google Scholar]
- 30.Sastri CV, Seo MS, Park MJ, Kim KM, Nam W. Chem Commun. 2005;11:1405. doi: 10.1039/b415507f. [DOI] [PubMed] [Google Scholar]
- 31.Park MJ, Lee J, Suh Y, Kim J, Nam W. J Am Chem Soc. 2006;128:2630. doi: 10.1021/ja055709q. [DOI] [PubMed] [Google Scholar]
- 32.Bautz J, Comba P, d Laorden CL, Menzel M, Rajaraman G. Angew Chem. 2007;119:8213. doi: 10.1002/anie.200701681. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2007;46:8067. [Google Scholar]
- 33.Oh NY, Suh Y, Park MJ, Seo MS, Kim J, Nam W. Angew Chem. 2005;117:4307. doi: 10.1002/anie.200500623. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2005;44:4235. [Google Scholar]
- 34.Sastri CV, Lee J, Oh K, Lee YJ, Lee J, Jackson TA, Ray K, Hirao H, Shin W, Halfen JA, Kim J, Que L, Jr, Shaik S, Nam W. Proc Natl Acad Sci USA. 2007;104:19181. doi: 10.1073/pnas.0709471104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Comba P, Fukuzumi S, Kotani H, Wunderlich S. Angew Chem. 2010;122:2679. doi: 10.1002/anie.200904427. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2010;49:2622. [Google Scholar]
- 36.Nam W. Acc Chem Res. 2007;40:465. doi: 10.1021/ar700027f. [DOI] [PubMed] [Google Scholar]
- 37.de Visser SP, Oh K, Han A-R, Nam W. Inorg Chem. 2007;46:4632. doi: 10.1021/ic700462h. [DOI] [PubMed] [Google Scholar]
- 38.Sastri CV, Oh K, Lee YJ, Seo MS, Shin W, Nam W. Angew Chem. 2006;118:4096. doi: 10.1002/anie.200504422. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2006;45:3992. [Google Scholar]
- 39.Ekkati AR, Kodanko JJ. J Am Chem Soc. 2007;129:12390. doi: 10.1021/ja075075i. [DOI] [PubMed] [Google Scholar]
- 40.Campanali AA, Kwiecien TD, Hryhorczuk L, Kodanko JJ. Inorg Chem. 2010;49:4759. doi: 10.1021/ic100439n. [DOI] [PubMed] [Google Scholar]
- 41.Bernadou J, Meunier B. Chem Commun. 1998:2167. [Google Scholar]
- 42.Wolfe MD, Lipscomb JD. J Biol Chem. 2003;278:829. doi: 10.1074/jbc.M209604200. [DOI] [PubMed] [Google Scholar]
- 43.Chen K, Que L., Jr Chem Commun. 1999:1375. [Google Scholar]
- 44.Seo MS, In J-H, Kim SO, Oh NY, Hong J, Kim J, Que L, Jr, Nam W. Angew Chem. 2004;116:2471. doi: 10.1002/anie.200353497. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2004;43:2417. [Google Scholar]
- 45.Company A, Gómez L, Güell M, Ribas X, Luis JM, Que L, Jr, Costas M. J Am Chem Soc. 2007;129:15766. doi: 10.1021/ja077761n. [DOI] [PubMed] [Google Scholar]
- 46.Sastri CV, Park MJ, Ohta T, Jackson TA, Stubna A, Seo MS, Lee J, Kim J, Kitagawa T, Munck E, Que L, Jr, Nam W. J Am Chem Soc. 2005;127:12494. doi: 10.1021/ja0540573. [DOI] [PubMed] [Google Scholar]
- 47.Proshlyakov DA, Henshaw TF, Monterosso GR, Ryle MJ, Hausinger RP. J Am Chem Soc. 2004;126:1022. doi: 10.1021/ja039113j. [DOI] [PubMed] [Google Scholar]
- 48.DFT geometries were optimized at the B3LYP level in conjunction with the SDD basis set and associated ECP for Fe, and 6-311G(d,p) basis set for the other atoms, as implemented in the Gaussian 09 program.[50] The energies were further refined by single point calculations using cc-pVTZ basis set for Fe, and the atoms bond to Fe, and cc-pVDZ basis set for the other atoms (B3LYP/cc-pVTZ&cc-pVDZ//B3LYP/SDD&6-311G(d,p)). Final free energies include energies computed at the B3LYP/cc-pVTZ&cc-pVDZ//B3LYP/SDD&6-311G(d,p) level of theory together with enthalpic and free energy corrections at the B3LYP/LANL2DZ&D95V level (i.e. B3LYP level in conjunction with the LANL2DZ basis set and associated ECP for Fe, and D95V basis set for the other atoms).
- 49.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Jr, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ. Gaussian 09, Revision A.02. Gaussian, Inc; Wallingford CT: 2009. [Google Scholar]
- 50.Company A, Gómez L, Fontrodona X, Ribas X, Costas M. Chem Eur J. 2008;14:5727. doi: 10.1002/chem.200800724. [DOI] [PubMed] [Google Scholar]
- 51.As above the final free energies include energies computed at the B3LYP/cc-pVTZ&cc-pVDZ//B3LYP/SDD&6-311G(d,p) level of theory together with enthalpic and free energy corrections at the B3LYP/LANL2DZ &D95V level. The cc-pVTZ basis set is also used for the H atoms bound to O.
- 52.Rohde J-U, Torelli S, Shan X, Lim MH, Klinker EJ, Kaizer J, Chen K, Nam W, Que L., Jr J Am Chem Soc. 2004;126:16750. doi: 10.1021/ja047667w. [DOI] [PubMed] [Google Scholar]
- 53.Riggs-Gelasco PJ, Price JC, Guyer RB, Brehm JH, Barr EW, Bollinger JM, Jr, Krebs C. J Am Chem Soc. 2004;126:8108. doi: 10.1021/ja048255q. [DOI] [PubMed] [Google Scholar]
- 54.Fujimori DG, Barr EW, Matthews ML, Koch GM, Yonce JR, Walsh CT, Martin Bollinger J, Jr, Krebs C, Riggs-Gelasco PJ. J Am Chem Soc. 2007;129:13408. doi: 10.1021/ja076454e. [DOI] [PubMed] [Google Scholar]
- 55.CCDC. 782986 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
- 56.Chen K, Que L., Jr J Am Chem Soc. 2001;123:6327. doi: 10.1021/ja010310x. [DOI] [PubMed] [Google Scholar]
- 57.Erras-Hanauer H, Clark T, vanEldik R. Coord Chem Rev. 2003:238–239. 233. [Google Scholar]
- 58.The final free energies include energies computed at the B3LYP-D3/cc-pVTZ&cc-pVDZ//B3LYP/LANL2DZ &D95V level of theory together with enthalpic and free energy corrections at the B3LYP/LANL2DZ &D95V level (see ref. 48 for more details). The single point calculations include the Acetonitrile solvent effect computed through Gaussian09 PCM approach and the London dispersion effects calculated using the S. Grimme DFT-D3 method.
- 59.Miertuš S, Scrocco E, Tomasi J. Chem Phys. 1981;55:117. [Google Scholar]
- 60.Grimme S, Antony J, Ehrlich S, Krieg H. J Chem Phys. 2010;132:154104. doi: 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]
- 61.Yin G, Danby AM, Kitko D, Carter JD, Scheper WM, Busch DH. J Am Chem Soc. 2007;129:1512. doi: 10.1021/ja0673229. [DOI] [PubMed] [Google Scholar]
- 62.Comba P, Rajaraman G. Inorg Chem. 2008;47:78. doi: 10.1021/ic701161r. [DOI] [PubMed] [Google Scholar]
- 63.Kim SO, Sastri CV, Seo MS, Kim J, Nam W. J Am Chem Soc. 2005;127:4178. doi: 10.1021/ja043083i. [DOI] [PubMed] [Google Scholar]
- 64.Goto Y, Matsui T, Ozaki S-i, Watanabe Y, Fukuzumi S. J Am Chem Soc. 1999;121:9497. [Google Scholar]
- 65.Xue G, Hont RD, Münck E, Que L., Jr Nat Chem. 2010;2:400. doi: 10.1038/nchem.586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Goldsmith CR, Jonas RT, Stack TDP. J Am Chem Soc. 2002;124:83. doi: 10.1021/ja016451g. [DOI] [PubMed] [Google Scholar]
- 67.Luo Y-R. Comprehensive Handbook of Chemical Bond Energies. CRCPress; 2007. [Google Scholar]
- 68.Matsuo T, Mayer JM. Inorg Chem. 2005;44:2150. doi: 10.1021/ic048170q. [DOI] [PubMed] [Google Scholar]
- 69.Bryant JR, Mayer JM. J Am Chem Soc. 2003;125:10351. doi: 10.1021/ja035276w. [DOI] [PubMed] [Google Scholar]
- 70.Goldsmith CR, Cole AP, Stack TDP. J Am Chem Soc. 2005;127:9904. doi: 10.1021/ja039283w. [DOI] [PubMed] [Google Scholar]
- 71.Larsen AS, Wang K, Lockwood MA, Rice GL, Won T-J, Lovell S, Sadílek M, Tureek F, Mayer JM. J Am Chem Soc. 2002;124:10112. doi: 10.1021/ja020204a. [DOI] [PubMed] [Google Scholar]
- 72.Lansky DE, Goldberg DP. Inorg Chem. 2006;45:5119. doi: 10.1021/ic060491+. [DOI] [PubMed] [Google Scholar]
- 73.Mayer JM. Acc Chem Res. 1998;31:441. [Google Scholar]
- 74.Klinker EJ, Shaik S, Hirao H, Que L., Jr Angew Chem. 2009;121:1317–1321. doi: 10.1002/anie.200804029. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2009;48:1291. [Google Scholar]
- 75.Wang D, Zhang M, Bühlmann P, Que L., Jr J Am Chem Soc. 2010;132:7638. doi: 10.1021/ja909923w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Fertinger C, Hessenauer-Ilicheva N, Franke A, van Eldik R. Chem Eur J. 2009;15:13435. doi: 10.1002/chem.200901804. [DOI] [PubMed] [Google Scholar]
- 77.Kohen A, Klinman JP. Acc Chem Res. 1998;31:397. [Google Scholar]
- 78.Pan Z, Horner JH, Newcomb M. J Am Chem Soc. 2008;130:7776. doi: 10.1021/ja802484n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Price JC, Barr EW, Glass TE, Krebs C, Martin Bollinger J., Jr J Am Chem Soc. 2003;125:13008. doi: 10.1021/ja037400h. [DOI] [PubMed] [Google Scholar]
- 80.Lewis ER, Johansen E, Holman TR. J Am Chem Soc. 1999;121:1395. [Google Scholar]
- 81.Nesheim JC, Lipscomb JD. Biochem. 1996;35:10240. doi: 10.1021/bi960596w. [DOI] [PubMed] [Google Scholar]
- 82.Fukuzumi S, Kotani H, Lee Y-M, Nam W. J Am Chem Soc. 2008;130:15134. doi: 10.1021/ja804969k. [DOI] [PubMed] [Google Scholar]
- 83.Jeong YJ, Kang Y, Han A-R, Lee Y-M, Kotani H, Fukuzumi S, Nam W. Angew Chem. 2008;120:7431. doi: 10.1002/anie.200802346. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2008;47:7321. [Google Scholar]
- 84.Arunkumar C, Lee Y-M, Lee JY, Fukuzumi S, Nam W. Chem Eur J. 2009;15:11482. doi: 10.1002/chem.200901362. [DOI] [PubMed] [Google Scholar]
- 85.Gardner KA, Mayer JM. Science. 1995;269:1849. doi: 10.1126/science.7569922. [DOI] [PubMed] [Google Scholar]
- 86.Cook GK, Mayer JM. J Am Chem Soc. 1995;117:7139. [Google Scholar]
- 87.Cook GK, Mayer JM. J Am Chem Soc. 1994;116:1855. [Google Scholar]
- 88.Wang D, Farquhar ER, Stubna A, Munck E, Que L., Jr Nat Chem. 2009;1:145. doi: 10.1038/nchem.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gunay A, Theopold KH. Chem Rev. 2010;110:1060. doi: 10.1021/cr900269x. [DOI] [PubMed] [Google Scholar]
- 90.Kumar D, Hirao H, Que L, Jr, Shaik S. J Am Chem Soc. 2005;127:8026. doi: 10.1021/ja0512428. [DOI] [PubMed] [Google Scholar]
- 91.Hirao H, Kumar D, Que L, Jr, Shaik S. J Am Chem Soc. 2006;128:8590. doi: 10.1021/ja061609o. [DOI] [PubMed] [Google Scholar]
- 92.De Visser SP. J Am Chem Soc. 2010;132:1087. doi: 10.1021/ja908340j. [DOI] [PubMed] [Google Scholar]
- 93.Zhou Y, Shan X, Mas-Ballesté R, Bukowski MR, Stubna A, Chakrabarti M, Slominski L, Halfen JA, Münck E, Que L., Jr Angew Chem. 2008;120:1922–1925. doi: 10.1002/anie.200704228. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2008;47:1896. [Google Scholar]
- 94.(a) Handy NC, Cohen AJ. Mol Phys. 2001;99:403. [Google Scholar]; b) Perdew JP, Burke K, Ernzerhof M. Phys Rev Lett. 1996;77:3865. doi: 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- 95.(a) Klamt A. J Phys Chem. 1995;99:2224. [Google Scholar]; b) Klamt A, Jonas V. J Chem Phys. 1996;105:9972. [Google Scholar]; c) Klamt A, Schüürmann G. J Chem Soc, Perkin Trans. 1993;2:799. [Google Scholar]
- 96.Baerends EJ, Autschbach J, Bérces A, Berger JA, Bickelhaupt FM, Bo C, de Boeij PL, Boerrigter PM, Cavallo L, Chong DP, Deng L, Dickson RM, Ellis DE, van Faassen M, Fan L, Fischer TH, Fonseca Guerra C, van Gisbergen SJA, Groeneveld JA, Gritsenko OV, Grüning M, Harris FE, van den Hoek P, Jacob CR, Jacobsen H, Jensen L, Kadantsev ES, van Kessel G, Klooster R, Kootstra F, van Lenthe E, McCormack DA, Michalak A, Neugebauer J, Nicu VP, Osinga VP, Patchkovskii S, Philipsen PHT, Post D, Pye CC, Ravenek W, Romaniello P, Ros P, Schipper PRT, Schreckenbach G, Snijders JG, Solà M, Swart M, Swerhone D, te Velde G, Vernooijs P, Versluis L, Visscher l, Visser O, Wang F, Wesolowski TA, van Wezenbeek EM, Wiesenekker G, Wolff SK, Woo TK, Yakovlev AL, Ziegler T. ADF 2009.01. SCM; Amsterdam, The Netherlands: 2009. [Google Scholar]
- 97.a) Han WG, Noodleman L. Inorg Chem. 2008;47:2975. doi: 10.1021/ic701194b. [DOI] [PubMed] [Google Scholar]; b) Han WG, Liu TQ, Lovell T, Noodleman L. J Comput Chem. 2006;27:1292. doi: 10.1002/jcc.20402. [DOI] [PubMed] [Google Scholar]; c) Han WG, Noodleman L. Inorg Chim Acta. 2008;361:973. doi: 10.1016/j.ica.2007.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.