

Abstract
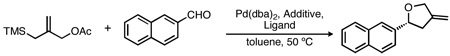
The palladium-catalyzed [3+2] cycloaddition of trimethylenemethane (TMM) with aldehydes is a direct and efficient route to methylenetetrahydrofurans. Herein we describe the first asymmetric synthesis of methylenetetrahydrofurans utilizing a palladium-TMM complex in the presence of a novel phosphoramidite ligand possessing a stereogenic phophorus. The method allows for the formation of chiral disubstituted tetrahydrofurans in good yields and enantioselectivies.
The prevalence of tetrahydrofurans in natural and synthetic products has stimulated a variety of efforts towards their construction.1 Among these methods, those that utilize cycloadditions offer a distinct advantage due to their ability to generate multiple bonds in a single step.2 While enantioselective cycloadditions generating tetrahydrofurans are known,3 few reports utilize aldehydes as a coupling partner despite the benefits of these ubiquitous and readily-available substrates.4 One such strategy would involve the [3+2] cycloaddition of aldehydes with palladium-trimethylenemethane (TMM), generated in situ from 3-acetoxy-2-trimethylsilylmethyl-1-propene (1) and catalytic palladium, provided that a suitable chiral ligand could be developed.
Recently, our group disclosed a new class of chiral phosphoramidites (I) that has led to the development of an asymmetric version of the TMM cycloaddition. An advantage of modular ligands such as I is their potential for steric and electronic modification. Indeed, in our previous work we effectively adjusted the pyrrolidine of I in order to achieve highly enantioselective syntheses of cyclopentanes (using Ar = phenyl),5 pyrrolidines (Ar = 2-naphthyl),6 and bicyclo[4.3.1]decadienes (Ar = p-biphenyl)7. In related efforts, the Hayashi group has described the analogous 1,4-zwitterion in enantioselective cycloadditions with 1,1-dicyanocyclopropanes and N-tosyl aziridines (Ar = phenyl),8 as well as cycloadditions using phosphoramidites with acyclic amines.9
 |
(1) |
Despite these advances, extension of the TMM cycloaddition towards the synthesis of tetrahydrofurans using these ligands has remained a challenge, generally affording products with only modest ee (eq 1). Conspicuously absent from the previous work, however, is any modification of the BINOL component of the phosphoramidite. While 3,3’-disubstituted BINOL derivatives are common in chiral catalysis, mono-substituted derivatives are rare.10 Additionally, such ligands possess added complexity in that they bear a stereogenic phosphorus, mandating efficient stereocontrol during ligand synthesis. Herein, we report our efforts to prepare such C1-symmetric ligands and their utilization in the palladium-catalyzed TMM cycloaddition with aldehydes, allowing for the synthesis of methylenetetrahydrofurans in high yield and enantiopurity.
We have previously demonstrated that the Pd-TMM cycloaddition is a direct and efficient method for the generation of highly substituted tetrahydrofurans.11 A critical breakthrough in this pioneering work was the discovery that Lewis acid co-catalysts such as tin or indium12 salts facilitate cycloaddition. For example, reaction of cinnamaldehyde in the absence of a Lewis acid affords a mixture of the desired cycloadduct, an acyclic by-product, and oligomeric impurities in 65% yield (eq 2). In the presence of catalytic Me3SnOAc, however, the desired product is formed in 95% yield (eq 3).11a We attributed the change in reactivity to activation of the carbonyl group and coordination of the intermediate alkoxide (III) by the Lewis acid, resulting in a softer nucleophile that is more suitable for the intramolecular cyclization with the Pd-π-allyl.
 |
Our initial efforts to develop the asymmetric aldehyde cycloaddition focused on the use of ligand L1 with 2-naphthaldehyde as a test substrate. In light of the above mechanistic discussion, we were surprised to see comparable yields of the desired cycloadduct when the reaction was run with or without In(acac)3 (Table 1, entries 1 and 2). When other substrates were examined, however, yields were generally 20–30% lower without In(acac)3. Based on these results, a survey of Lewis acids seemed appropriate. To better evaluate potential Lewis acids, we switched to ligand L2, which gave only a 33% yield in the absence of a co-catalyst (entry 3). Indium (entry 4) or tin (entry 5) salts gave significantly better yields, although the toxicity of the latter makes this option unattractive. B-Methoxy-9-borabicyclo[3.3.1]nonane gave the best yield with 2-naphthaldehyde (entry 6), but when this additive was tested with other substrates the results were comparable or worse than In(acac)3. We briefly examined chiral boranes such as 2-methyl-CBS-oxazaborolidine, but were unable to improve enantioselectivity (entries 7 and 8). We therefore adopted a standard procedure employing 10 mol% In(acac)3 for further optimization. Returning our attention to the effect of the ligand, we explored bis-1-naphthyl L3 and the bis-p-biphenyl L4, but neither was competitive with L1. Ligands bearing acyclic amines such as Feringa ligand L5 resulted in near-racemic product.
Table 1.
Initial Reaction Optimizationa
 | ||||
|---|---|---|---|---|
| entry | Ligand | Additive | % yield | % ee |
| 1 | L1 | None | 63 | 79 |
| 2 | L1 | In(acac)3 | 67 | 67 |
| 3 | L2 | None | 33 | 56 |
| 4 | L2 | In(acac)3 | 61 | 51 |
| 5 | L2 | n-Bu3Sn(OAc) | 60 | 50 |
| 6 | L2 | B-OMe-9-BBN | 76 | 52 |
| 7 | L2 | (S)-Me-CBS | 55 | 46 |
| 8 | L2 | (R)-Me-CBS | 69 | 52 |
| 9 | L3 | In(acac)3 | 75 | 47 |
| 10 | L4 | In(acac)3 | 81 | 39 |
| 11 | L5 | In(acac)3 | 25 | 6 |
All reactions were conducted at 0.15 M in toluene at 50 °C, with 1.6 equivalents 1, 5% Pd(dba)2, 10% additive and 10% ligand. Yields are isolated values; ee’s were determined by chiral HPLC.

Establishing the bis-2-naphthyl pyrrolidine as the optimal amine for the phosphoramidite, we next examined the effect of derivatizing the BINOL (Scheme 1). Surprisingly, bis-3,3’-methylated L6 resulted in a completely inactive catalyst. However, mono-methylated L7 not only gave excellent conversion but also an increase in ee. Owing to the loss of C2 symmetry, such ligands bear a stereogenic phosphorus center that is set during the phosphoramidite synthesis. Interestingly, we observed no change in enantioselecitivity when using batches of ligands with different diastereomeric ratios, although the yield did change. This result, confirmed by selective preparation of both diastereomers, indicates that only a single diastereomer of the phosphoramidite is catalytically active.13 While the ethyl analog L8 gave a comparable result to that of L7, use of the i-propyl ligand L9 resulted in a substantial drop in both yield and selectivity. Such a drastic change suggests that both the steric demands and conformation of the substituent are important factors affecting the enantiocontrol of the ligand. We hypothesized that an ortho-substituent tethered to the 4-position of BINOL would be conformationally constrained and therefore might lead to better ee. Indeed, naphthofuran L10 led to a further improvement and dihydrofuran L11 gave the best result with an 80% yield and 87% ee.14
Scheme 1.

Selected BINOL Derivatization Resultsa
aAll reactions were conducted at 0.15 M in toluene at 50 °C, with 1.6 equivalents 1, 5% Pd(dba)2, 10% In(acac)3 and 10% ligand. Yields are isolated values; ee’s were determined by chiral HPLC.
With optimized conditions in place, the reaction scope was examined (Table 2). In most cases, the temperature could be lowered to 23 °C, resulting in slight improvements in ee. However, all attempts at 4 °C resulted in no reaction. Comparing the optical rotation of the known benzaldehyde cycloadduct (entry 2)15 allowed for the determination of absolute configuration, and all other examples are proposed by analogy. In large-scale experiments (1.0 mmol) using either 2-naphthaldehyde or benzaldehyde, catalyst loading could be lowered to 2.5% without erosion of yield or ee.16 Good to excellent yields and enantioselectivities were obtained with electron-rich benzaldehydes bearing various substitution patterns, including mono- (entries 3–6) and disubstituted (entry 7) substrates. While the reaction of 4-chlorobenzaldehyde proceeded with good yield and selectivity (entry 8), p-nitrobenzaldehyde gave only 36% ee under the optimized conditions (entry 9). Further testing with other Lewis acids suggested that coordination of the nitro group was interfering with the enantiodetermining event. However, employing ligand L1 in this case resulted in a 78% ee. Heterocyclic aldehydes also gave cycloadducts in good optical purity (entries 10–11). Chemoselective cycloaddition with the carbonyl group of cinnamaldehyde derivatives was also possible. While the reaction of cinnamaldehyde shows a 3:1 mixture of aldehyde and olefin cycloadducts with moderate ee for the oxacycle (entry 12), alpha-alkylated cinnamaldehydes showed exclusive reaction with the carbonyl group, giving methylenetetrahydrofurans in good ee (entries 13–14). At this time, the success of α,β-unsaturated aldehydes as substrates allows them to serve as a surrogate for saturated aliphatic aldehydes by simple hydrogenation of the double bond.
Table 2.
Palladium-Catalyzed [3+2] Reactions of Aldehydesa
| entry | Substrate | Product |
T, °C |
% yield |
% ee |
|---|---|---|---|---|---|
| 1b | 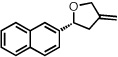 |
23 | 72 | 90 | |
| 2 | 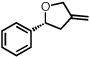 |
23 | 80 | 81 | |
| 3b | 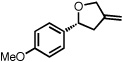 |
23 | 86 | 88 | |
| 4b | 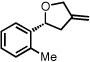 |
23 | 93 | 89 | |
| 5 | 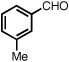 |
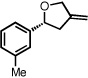 |
23 | 76 | 82 |
| 6 | 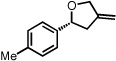 |
23 | 69 | 84 | |
| 7b | 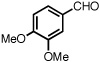 |
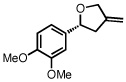 |
23 | 100 | 91 |
| 8b | 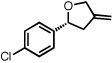 |
50 | 72 | 83 | |
| 9 | 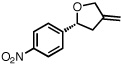 |
50 | 78 | 36 | |
| 50c | 62 | 78 | |||
| 10d | 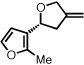 |
23 | 63 | 88 | |
| 11 | 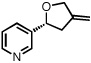 |
50 | 86 | 74 | |
| 12b | 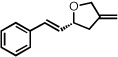 |
23 | 78e | 70 | |
| 13b | 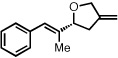 |
23 | 69 | 78 | |
| 14b | 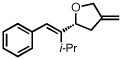 |
23 | 79 | 80 | |
All reactions were conducted for 3–24 hours at 0.15 M in toluene with 1.6 equivalents 1, 5% Pd(dba)2, 10% ligand and 10% In(acac)3 unless noted otherwise. Yields are isolated values; ee’s were determined by chiral HPLC.
Reaction conducted without In(acac)3.
Used ligand L1.
Used CpPd(η3-C3H5).
Isolated as a 3:1 mixture of carbonyl and olefin cycloadducts; ee is for the methylenetetrahydrofuran only.
Interestingly, when several substrates were tested without In(acac)3, the reaction still gave excellent yields of the desired cycloadduct (entries 1, 3, 4, 7, 8, 12–14). In fact, of those substrates tested without a Lewis acid, only in the case of 3-pyridinecarboxaldehyde did the absence of In(acac)3 lead to a clear reduction in yield (54% vs. 86% with In(acac)3). We note, also, that ligand L1 was capable of facilitating cycloaddition in the absence of a Lewis acid, albeit only in the reaction of 2-naphthaldehyde. We hypothesize that the more electron-rich L11 may enhance the nucleophilicity of the Pd-TMM intermediate (II) and stabilize the resultant Pd-π-allyl intermediate (III) towards cyclization. Furthermore, the increased steric bulk of ligand L11 may constrain the conformation to favor the desired intramolecular cyclization as well as inhibit approach of an external silylating agent.
In summary, we have developed a new phosphoramidite ligand bearing a stereogenic center at phosphorus. Interestingly, we find that only a single diastereomer of the ligand is catalytically active, allowing even mixtures diastereomeric at phosphorus to function in the reaction. Furthermore, the high reactivity combined with the steric demands of these ligands mitigate the need for the Lewis acid co-catalysts that proved essential for the previous achiral phosphite ligands employed for these cycloadditions. Utilizing the novel phosphoramidites in the palladium-catalyzed [3+2] cycloaddition of TMM with aldehydes allows for the formation of methylenetetrahydrofurans in good to excellent yields and enantioselectivities. Investigations into the full scope of this reaction, including the use of other carbonyl acceptors and substituted TMM donors, are currently underway and will be reported in due time.
Supplementary Material
Acknowledgement
We thank the NSF and the NIH (GM 033049) for their generous support of our programs. Palladium salts were a generous gift from Johnson-Matthey.
Footnotes
Supporting Information Available: Experimental details and spectral data for all unknown compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.a Jalce G, Franck X, Figadere B. Tetrahedron: Asymmetry. 2009;20:2537. [Google Scholar]; b Wolfe JP, Hay MB. Tetrahedron. 2007;63:261. doi: 10.1016/j.tet.2006.08.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kobayashi S, Jorgensen KA, editors. Cycloaddition Reactions in Organic Synthesis. Germany: Wiley-VCH: Weinheim; 2002. [Google Scholar]
- 3.a Hodgson DM, Glen R, Grant GH, Redgrave AJ. J. Org. Chem. 2003;68:6153. doi: 10.1021/jo026307t. [DOI] [PubMed] [Google Scholar]; b Suga H, Inoue K, Inoue S, Kakehi A. J. Am. Chem. Soc. 2002;124:14836. doi: 10.1021/ja028676c. [DOI] [PubMed] [Google Scholar]; c Shimada N, Anada M, Makamua S, Nambu H, Tsutsui H, Hashimoto S. Org. Lett. 2008;10:3603. doi: 10.1021/ol8013733. [DOI] [PubMed] [Google Scholar]; d Trost BM, Jiang C. J. Am. Chem. Soc. 2001;123:12907. doi: 10.1021/ja012104v. [DOI] [PubMed] [Google Scholar]
- 4.Parsons AT, Johnson JS. J. Am. Chem. Soc. 2009;131:3122. doi: 10.1021/ja809873u. [DOI] [PubMed] [Google Scholar]
- 5.a Trost BM, Stambuli JP, Silverman SM, Schworer U. J. Am. Chem. Soc. 2006;128:13328. doi: 10.1021/ja0640750. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Trost BM, Cramer N, Silverman SM. J. Am. Chem. Soc. 2007;129:12396. doi: 10.1021/ja075335w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.a Trost BM, Silverman SM, Stambuli JP. J. Am. Chem. Soc. 2007;129:12398. doi: 10.1021/ja0753389. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Trost BM, Silverman SM. J. Am. Chem. Soc. 2010;132:8238. doi: 10.1021/ja102102d. [DOI] [PubMed] [Google Scholar]
- 7.Trost BM, McDougall PJ, Hartmann O, Wathen P. J. Am. Chem.Soc. 2008;130:14960. doi: 10.1021/ja806979b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shintani R, Murakami M, Tsuji T, Tanno H, Hayashi T. Org. Lett. 2009;11:5642. doi: 10.1021/ol902326s. [DOI] [PubMed] [Google Scholar]
- 9.a Shintani R, Murakami M, Hayashi T. J. Am. Chem. Soc. 2007;129:12356. doi: 10.1021/ja073997f. [DOI] [PubMed] [Google Scholar]; b Shintani R, Park S, Shirozu F, Murakami M, Hayashi T. J. Am. Chem. Soc. 2008;130:16174. doi: 10.1021/ja807662b. [DOI] [PubMed] [Google Scholar]; c Shintani R, Hayashi S-Y, Murakami M, Takeda M, Hayashi T. Org. Lett. 2009;11:3754. doi: 10.1021/ol901348f. [DOI] [PubMed] [Google Scholar]
- 10.For BINOL-derived C1-symmetric phosphoramidites, see: Reetz MT, Ma J-A, Goddard R. Angew. Chem. Int. Ed. 2005;44:412. doi: 10.1002/anie.200461624. Gavrilov KN, Benetsky EB, Boyko VE, Rastorguev EA, Davankov VA, Schaffner B, Borner A. Chirality. 2010;22:844. doi: 10.1002/chir.20845.
- 11.a Trost BM, King SA, Schmidt T. J. Am. Chem. Soc. 1989;111:5902. [Google Scholar]; b Trost BM, King SA. J. Am. Chem. Soc. 1990;112:408. [Google Scholar]; c Trost BM, King SA. Tetrahedron Lett. 1986;27:5971. [Google Scholar]
- 12.a Trost BM, Sharma S, Schmidt T. J. Am. Chem. Soc. 1992;114:7903. [Google Scholar]; b Trost BM, Sharma S, Schmidt T. Tetrahedron Lett. 1993;34:7183. [Google Scholar]
- 13.Monoalkylated derivatives L7, L8 and L9 were obtained in diastereomeric ratios of 9:1, 11:1 and 5:1, respectively, by coupling the aminodichlorophosphine with the BINOL derivatives at −78 °C in THF. Previous work suggests the active (major) diastereomer is the (RP,R,R,R) ligand since the the major signal in the 31P NMR spectrum is downfield of the minor signal. See ref. 10a.
- 14.Ligands L10 and L11 were prepared as single diastereomers by oupling the BINOL chlorophosphite with the pyrrolidine at −40 °C in oluene. We tentatively assign the ligands as the (RP,R,R,R)diastereomers by analogy to L7–L9, despite a relative shift of the 31P NMR signals wherein the isolated (active) diastereomer shows a 31P NMR signal upfield of the other diastereomer (see refs. 10a and 13). Efforts to verify this stereochemistry are ongoing.
- 15.Van der Heide TAJ, van der Baan JL, Bijpost EA, de Kanter FJJ, Bicklhaupt F, Klumpp GW. Tetrahedron Lett. 1993;34:4655. [Google Scholar]
- 16.See Supporting Information for details.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.