

Abstract
In humans, heterozygous mutations in the ATP-dependent chromatin remodeling gene CHD7 cause CHARGE syndrome, a common cause of deaf-blindness, balance disorders, congenital heart malformations, and olfactory dysfunction with an estimated incidence of approximately 1 in 10,000 newborns. The clinical features of CHARGE in humans and mice are highly variable and incompletely penetrant, and most mutations appear to result in haploinsufficiency of functional CHD7 protein. Mice with heterozygous loss of function mutations in Chd7 are a good model for CHARGE syndrome, and analyses of mouse mutant phenotypes have begun to clarify a role for CHD7 during development and into adulthood. Chd7 heterozygous mutant mice have postnatal delayed growth, inner ear malformations, anosmia/hyposmia, and craniofacial defects, and Chd7 homozygous mutants are embryonic lethal. A central question in developmental biology is how chromodomain proteins like CHD7 regulate important developmental processes, and whether they directly activate or repress downstream gene transcription or act more globally to alter chromatin structure and/or function. CHD7 is expressed in a wide variety of tissues during development, suggesting that it has tissue-specific and developmental stage-specific roles. Here we review recent and ongoing analyses of CHD7 function in mouse models and cell based systems. These studies explore tissue-specific effects of CHD7 deficiency, known CHD7 interacting proteins, and downstream target sites for CHD7 binding. CHD7 is emerging as a critical regulator of important developmental processes in organs affected in human CHARGE syndrome.
Keywords: CHARGE syndrome, CHD7, chromodomain helicase DNA binding gene 7
Introduction
Regulation of eukaryotic genes is essential for normal tissue and organ development and maintenance. Chromatin structure has a vital role in gene regulation, via its effects on cellular proliferation and maintenance of the differentiated state. A central goal in current developmental biology is to identify molecular pathways that regulate chromatin structure and gene expression, and to understand how this regulation influences organogenesis. In humans, haploinsufficiency for the chromodomain helicase DNA binding gene 7 (CHD7) causes CHARGE syndrome, a multiple anomaly condition characterized by ocular coloboma, heart defects, atresia of the choanae, retarded growth and development, genital hypoplasia and ear abnormalities including deafness and vestibular disorders (1). The identification of CHD7 as the causative gene for CHARGE syndrome has led to several recent advancements in our understanding of this complex disorder. Furthermore, with the establishment of mouse models for CHARGE, it is now possible to begin detailed molecular analyses of how CHD7 functions in specific tissues and cell types both during development and into adulthood. Here we review recent studies which explore the underlying molecular genetic mechanisms by which CHD7 regulates the development and maintenance of various tissues. Researchers are now poised to charge into the future with a well equipped toolbox for analyzing CHD7 function.
CHD7 mutations cause CHARGE syndrome
In humans, heterozygous CHD7 mutations cause CHARGE syndrome, a clinically variable, multiple congenital anomaly condition affecting development of the inner ear, central nervous system, olfactory system, eyes, heart, choanae (the region between the oropharynx and nasal passages), genitalia, and craniofacial structures including the hard and soft palates, lip, external ear, and midface (2–6). CHARGE is a common cause of deaf-blindness, balance disorders, olfactory dysfunction, and congenital heart malformations, with an estimated incidence of 1:10,000 in newborns (7–9). The human CHD7 gene spans 188 kb on chromosome 8q12.1, with a 8994 bp open reading frame and transcription start site in exon 2. Heterozygosity for nonsense, deletion, or missense CHD7 mutations occurs in 60–80% of patients with CHARGE (2–5, 10, 11). Human CHD7 mutations are distributed throughout the coding sequence and do not appear to be correlated with specific aspects of the clinical phenotype (2–5, 10, 11). Most human CHD7 mutations identified thus far are de novo; however, evidence for germline mosaicism has been reported in families with multiple affected siblings (3, 12–14).
The clinical features of CHARGE in humans and mice are highly variable and incompletely penetrant. We recently reviewed all reports of CHD7-mutation positive CHARGE individuals, and found that the most commonly affected organ in humans with CHD7 mutations is the ear (2–6, 12–35). Ear defects in CHARGE include temporal bone abnormalities, external ear malformations, and hearing loss (2–6, 12–35). In addition to ear abnormalities, a majority of CHARGE patients are also affected by some combination of the following: ocular coloboma, heart defects, delayed growth and development, and genital hypoplasia (2–6, 12–35). Although olfactory function is less commonly analyzed in CHARGE patients, a majority of CHARGE patients analyzed have some form of olfactory defect including olfactory bulb hypoplasia and/or aplasia, and impaired olfaction ranging from mild hyposmia to anosmia (16, 24, 26, 28, 36–40). Less commonly reported clinical features associated with CHARGE and CHD7 mutations include choanal atresia, facial nerve palsy, cleft lip and/or plate, and tracheoesophageal fistula (2–6, 12–35). The variability in CHARGE features suggests that mutations in CHD7 lead to pleiotropic developmental defects; however, the mechanisms underlying these defects have not yet been determined.
Mouse models of CHARGE and tissue specific defects
Mouse CHD7 protein is comprised of 2985 amino acids, and has a predicted molecular weight of 334 kDa. Both human and mouse Chd7 genes contain 38 exons, and have similar exon-intron structures. At the protein level, there is 94.7% sequence identity between mouse and human CHD7, which corresponds to 89.7% nucleotide sequence identity. Heterozygous Chd7 mutant mice were originally identified by ethylnitrosourea (ENU) mutagenesis, with each of nine different lines carrying a single nonsense Chd7 mutation (41). These nine Chd7 mutant mouse lines are viable, with phenotypes that include head bobbing, circling behaviors, disrupted lateral semicircular canals, hyperactivity, reduced postnatal growth, variable cleft palate, choanal atresia, cardiac septal defects, hemorrhage, prenatal death, genital abnormalities, keratoconjunctivitis sicca (dry eye), and olfactory defects (41). Our laboratory generated heterozygous loss of function Chd7Gt/+ mice using Chd7 gene trap embryonic stem (ES) cells (42). Chd7Gt/+ mice have phenotypic features similar to those generated by ENU mutagenesis (42). Chd7Gt/Gt mice are embryonic lethal by E11, and exhibit developmental growth delays such as reduction in size of multiple organs including brain, eyes, ears, and craniofacial structures (42). The intrauterine lethality of homozygous Chd7Gt/Gt embryos and the severity of developmental malformations in Chd7Gt/+ mice indicate a lack of redundancy for prenatal Chd7 function in the mouse genome, and suggest important roles for CHD7 in organogenesis (42). Chd7 is expressed in specific tissues during mouse and human embryogenesis including the ear, brain, cranial nerves, olfactory epithelium, olfactory bulb, pituitary, heart, liver, eye, gut, kidney, and craniofacial structures (Figure 1) (5, 39, 41, 42). Chd7 is also expressed in the adult mouse olfactory epithelium, olfactory bulb, and in the rostral migratory stream (39).
Figure 1. Chd7 is expressed in CHARGE related tissues during development.

Frozen sections from E14.5 Chd7Gt/+ embryos demonstrate β-galactosidase (X-gal staining) in CHARGE-related organs including (A) olfactory bulbs (ob), (B) olfactory epithelium (oe), (C) limb, (D) heart (hrt), (E) kidney (kd), (F) enteric neurons of the gut (en), (G) crista (ca) and cochlea (coc) of the inner ear and (H) lens (le) and retina (re) of the eye. All sections are in the coronal orientation.
Inner ear abnormalities in humans and mice with CHD7 deficiency
The most consistent clinical feature associated with CHARGE syndrome is inner ear defects, including semicircular canal dysplasia that typically affects all three canals, and a Mondini form of cochlear hypoplasia (35, 43–45). In addition, both facial and vestibulocochlear nerve abnormalities are also reported (44, 46). Cochlear implants are a successful treatment for some CHARGE patients, provided there are no underlying nerve abnormalities. CHARGE patients also display vestibular dysfunction, including delayed postural development, abnormal vestibular testing, and balance/motor problems (47, 48). Although absence of the semicircular canals is believed to be a major cause of balance dysfunction, other factors such as ocular malformations, CNS defects, and skeletal abnormalities are also likely contributors.
Heterozygous Chd7 mice display circling and head-bobbing behaviors consistent with vestibular dysfunction phenotypes (41, 42). Detailed examination of Chd7 mutant mouse ears indicates a variety of lateral semicircular canal malformations, smaller posterior semicircular canals, and defects in innervation of the posterior crista (Figure 2) (42, 49–52). During embryonic development, semicircular canals are formed by sequential outpocketing, fusion of the otic epithelium, and tightly controlled processes to form three canals (53). Semicircular canal dysgenesis can be caused by mutations in a variety of transcription factors and signaling molecules (Table 1). Semicircular canal abnormalities observed in Chd7 mutant mice are postulated to be the result of smaller posterior and lateral canal outpocketings and delayed fusion of the epithelium (Figure 2) (51). Perturbations in signaling cascades both within the inner ear epithelium and surrounding mesenchyme can also result in semicircular canal defects (Table 1) (54). In summary, the inner ear phenotypes observed in heterozygous Chd7 mice are similar to those reported in CHARGE patients and include semicircular canal defects, innervation defects, and vestibular dysfunction. The precise molecular mechanisms of Chd7 function within the ear are still not understood, but likely involve complex interactions with essential transcription factors expressed during ear development.
Figure 2. Model of inner ear development in wild type and Chd7 heterozygous mutant mice.

The E11.5 otocyst extends dorsally to form the endolymphatic duct (ed, grey) and ventrally to form the cochlea (coc), and neuroblasts delaminate from the ventral epithelium to occupy the statoacoustic ganglion (sag, shown in blue). By E12.5, the epithelium of the canal pouches fuses to form the anterior (asc), posterior (psc) and lateral (lsc) semicircular canals. Between E13.5 and adulthood, the vestibular apparatus continues to mature, the cochlea completes its coiling, and the sensory epithelium becomes completely innervated. In Chd7 heterozygous mutant ears the lateral and posterior semicircular canals are truncated (lt), small, or misshapen. In addition, the nerve to the posterior ampulla (npa) is disrupted. Normal developmental structures are shown by a dashed line in Chd7 heterozygous mutants.
Table 1.
Mouse models of human semicircular canal dysgenesis
| Gene | Type of protein | Mouse inner ear defects | Human inner ear defects | References |
|---|---|---|---|---|
| Brn4 (Pou3f4) | Pou-domain transcription factor | ASC abnormalities | X-linked hereditary deafness type 3: Dysplasia of cochlea and semicircular canals and deafness | (69, 70) |
| Chd7 | Chromodomain helicase DNA binding protein | LSC, PSC and associated crista abnormalities | CHARGE Syndrome: Outer and inner ear abnormalities including semicircular canal defects and deafness | (41, 42, 49) |
| Eya1 | Transcription factor | ASC, PSC, LSC and associated crista absent | Branchio-oto-renal syndrome: Outer, Middle and Inner ear abnormalities and deafness | (71, 72) |
| Fgf3 | Fibroblast growth factor | ASC, PSC and LSC abnormalities | Syndromic deafness: Inner ear agenesis and deafness | (73–75) |
| Fgf10 | Fibroblast growth factor | ASC, PSC, LSC and associated crista abnormalities | LADD syndrome: Outer ear abnormalities, Inner ear abnormalities and deafness | (76, 77) |
| Jag1 | Notch receptor ligand | ASC, PSC and LSC abnormalities | Alagille syndrome: Posterior semicircular canal defects | (51, 78) |
| Six1 | Transcription factor | ASC, PSC and LSC absent | Branchio-oto-renal syndrome: Outer, Middle and Inner ear abnormalities and deafness | (79–81) |
Abbreviations: ASC, anterior/superior semicircular canal; LSC, lateral semicircular canal; PSC, posterior semicircular canal.
Impaired olfaction in humans and mice with CHD7 deficiency
Genital hypoplasia, delayed puberty, and delayed growth are common in CHARGE individuals (16, 24, 26, 28, 40). These features often occur in conjunction with olfactory defects, including hypoplastic olfactory bulbs and reduced olfaction (16, 24, 26, 28, 40). Previous studies of endocrine dysfunction in CHARGE patients reported growth delays, but only 9% of CHARGE patients had growth hormone deficiency, and growth delay was not associated with thyroid-stimulating hormone or adrenocorticotropic hormone deficiency (40). Analysis of female CHARGE patients over the age of 12 showed lack of spontaneous puberty and no response to stimulation with gonadotropin-releasing hormone (GnRH) (40). A majority of male CHARGE patients had low testosterone levels and cryptorchidism and/or micropenis. In contrast to females, male response to GnRH stimulation was variable and did not always correlate with testosterone levels (40). Endocrine dysfunction in CHARGE individuals is therefore likely to be multifactorial, with variable influence on olfaction, somatic growth, puberty, and fertility.
Mutations in CHD7 have been reported in individuals with Kallmann syndrome with and without a CHARGE syndrome diagnosis (24, 26). Kallmann syndrome is primarily characterized by idiopathic hypogonadotropic hypogonadism and anosmia (26, 55, 56). Idiopathic hypogonadotropic hypogonadism is characterized by impaired or absent sexual development due to sex steroid hormone deficiency, with low serum levels of the pituitary gonadotropins follicle-stimulating hormone (FSH) and luteinizing hormone (LH) as well as infertility (26, 55, 56). Kallmann-like features in humans are often associated with impairment of embryonic gonadotropin releasing hormone (GnRH) neuronal migration along olfactory neuronal tracts from the olfactory placode to the hypothalamus (26, 55, 56). Kallmann syndrome is genetically heterogeneous, with mutations reported in a variety of genes including KAL1, FGFR1, FGF8, PROKR2, PROK2, and CHD7 (26, 55–59). Mouse models exist for several of the genes associated with Kallmann syndrome (hypogonadotropic hypogonadism and reduced olfaction) and are listed in Table 2.
Table 2.
Mouse models of human Kallmann syndrome (idiopathic hypogonadotropic hypogonadism and olfactory dysfunction)
Odorant detection is a complex process that requires normal function of several different tissues and organs. Odorants are first detected in the nasal olfactory epithelium by odorants binding to specific odorant receptors located on the surface of olfactory cilia (Figure 3). Bound odorants activate olfactory sensory neurons, which are bipolar neurons that project axons to the glomeruli in the olfactory bulb. Olfactory bulb glomeruli contain dendrites of mitral cells and a variety of interneurons and periglomerular cells. Neuronal signals are then relayed to higher brain regions in the central nervous system (CNS).
Figure 3. Model of olfactory function in wild type and Chd7Gt/+ mice.

Following inhalation, odorants are detected in the olfactory epithelium by binding to specific odorant receptors located on the surface of olfactory cilia. Bound odorants activate olfactory sensory neurons, which are bipolar neurons that project axons to the glomeruli in the olfactory bulb. Each olfactory sensory neuron (ex. blue, red, green, and purple) contains one type of odorant receptor in the cilia and each neuron with that type of odorant receptor projects an axon to the same glomerulus in the olfactory bulb. Electrical signals sent by olfactory sensory neurons are detected by mitral cell dendrites in the glomeruli. These signals are then sent to higher brain regions in the central nervous system. Wild type mouse olfactory epithelium contains densely packed olfactory sensory neurons which project to the olfactory bulb. However, Chd7Gt/+ young adult mice have 30% fewer olfactory sensory neurons and olfactory bulb hypoplasia. We hypothesize that aged Chd7Gt/+ mice have a greater reduction in both olfactory sensory neurons and size of the olfactory bulb.
Since olfactory ability is reliant upon a number of components working in a coordinated fashion, we chose to look at olfactory function in mice utilizing methods that are not influenced by behavioral issues such as circling and hyperactivity. We observed, using electro-olfactogram of the olfactory epithelium, a lack of functional responses in olfactory sensory neurons to a variety of odorants in young (6 week old) adult Chd7Gt/+ mice (39). Additionally, we found that young adult Chd7Gt/+ mice have fewer olfactory sensory neurons and olfactory bulb hypoplasia (Figure 3) (39). In order to identify the underlying mechanisms by which CHD7 regulates olfactory sensory neurons, we analyzed cell-type specific expression of Chd7 in the young adult olfactory epithelium (39). CHD7 is present in 97% of proliferating neural stem cells and in Ascl1-positive (Mash1) and NeuroD-positive pro-neuronal cells in the adult mouse olfactory epithelium by immunofluorescence (39). Chemical ablation of the adult mouse olfactory epithelium in Chd7Gt/+ mice results in reduced regenerative capacity of olfactory sensory neurons, indicating ongoing requirements for CHD7 in adulthood (39). Consequently, we hypothesize that with age or insult, further reductions in olfactory sensory neurons and olfactory bulb size could occur, but this remains to be formally tested (Figure 3). In a concurrent study, Chd7Whi/+ mice had reduced olfaction (assayed by sniff response to mouse urine compared to water), olfactory bulb hypoplasia, reproductive dysfunction, and decreased fertility compared to wild type mice (60). Taken together, these studies suggest that CHD7 has critical functions in olfactory sensory neuron integrity and odorant detection.
CHD7 and other CHD proteins: classification and characteristics
CHD proteins are a part of the large group of ATP-dependent chromatin remodelers. The mammalian genome encodes approximately 30 such genes that appear to be non-redundant and vital for normal embryonic development (61). Many ATP-dependent chromatin remodeling genes demonstrate haploinsufficiency, indicating that their products may be involved in rate-limiting steps during development (61). CHD7 is one of nine ATP-dependent chromatin remodeling CHD enzymes that are characterized by the presence of two chromodomains, centrally located helicase domains, and less well defined carboxy terminal domains (Figure 4) (61–63). CHD proteins use ATP hydrolysis to regulate access to DNA by altering nucleosomes (61–63). The nine CHD proteins are broadly classified into three subfamilies based upon their amino acid sequence and functional protein domains (61–63). CHD7 is a member of the class III chromodomain proteins together with CHD5, CHD6, CHD8, and CHD9 (61–63). CHD7 contains a SANT (SWI3, ADA2, N-COR, and TFIIB) domain, which is conserved among many regulators of transcription and chromatin structure, and is believed to function as a histone tail binding module (64). CHD7, CHD8, and CHD9 contain two BRK domains, the function of which appears to be specific to higher eukaryotes since it is not present in yeast chromatin remodeling factors (63). CHD7 and the other eight CHD proteins are an interesting class of novel ATP-dependent chromatin remodelers with unique protein motifs that are incompletely characterized. To date, CHD7 is the only member of this class of proteins for which mutations have been associated with a well described human syndrome.
Figure 4.
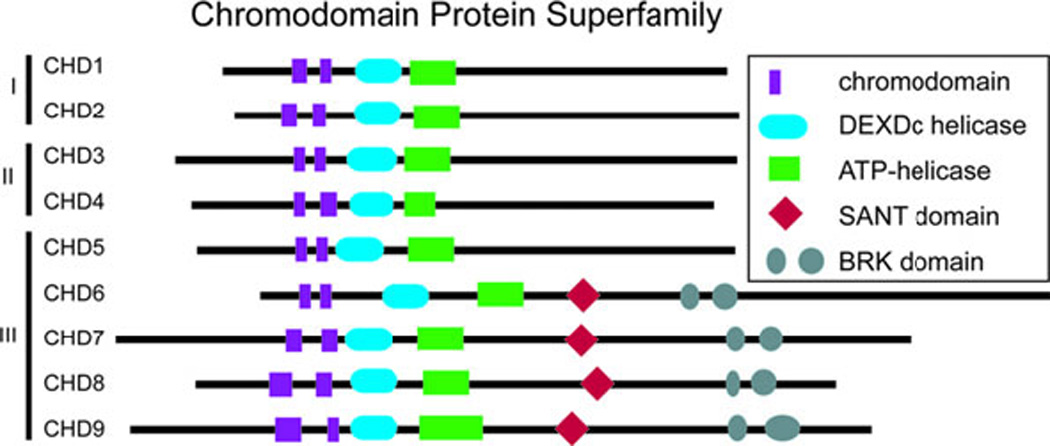
The chromodomain family of proteins contains 9 members that are subdivided into 3 classes on the basis of shared protein motifs. CHD7 is a member of the third class together with CHD5, CHD6, CHD8, and CHD9.
CHD7 binding sites and interacting proteins
Recent studies have begun to clarify a role for CHD7 in regulating gene expression during tissue development and maintenance. In a study using chromatin immunoprecipitation followed by microarray based sequence analysis (ChIP-chip), CHD7 has been shown to bind in a cell type specific manner to methylated histone H3 lysine 4 in enhancer regions of numerous genes in human colorectal carcinoma cells, human neuroblastoma cells, and mouse embryonic stem cells before and after differentiation, indicating that CHD7 may have temporal and tissue-specific functions (65). CHD7 has also been implicated to regulate multipotent neural crest-like cells by binding to PBAF components including BRG1, BAF170, BAF155, BAF57, PB1, ARID2, and BRD7 (66). In mesenchymal stem cells, CHD7 regulates cell fate specification during osteoblast and adipocyte differentiation (67). CHD7 forms a complex with NLK, SETDB1, and PPAR-γ, and binds to methylated lysine 4 and lysine 9 residues on histone H3 at PPAR-γ target promoters, which suppresses ligand-induced transactivation of PPAR-γ target genes (67). Additionally, the CHD7 Drosophila orthologue, Kismet, is involved in transcriptional elongation by RNA polymerase II through recruitment of ASH1 and TRX and may help maintain stem cell pluripotency by regulating methylation of histone H3 lysine 27 (68). Together, these data indicate multiple roles for CHD7 in regulating transcription, potentially affecting tissue and developmental stage-specific processes (Figure 5).
Figure 5. Model for CHD7 transcriptional regulation.

CHD7 binds to enhancer regions of target genes together with a tissue-specific complex of proteins. The CHD7 tissue-specific complex either activates or represses downstream target gene expression in a developmental stage specific manner.
Summary and future studies
In summary, CHARGE syndrome is a monogenic disease which exhibits highly variable expressivity. Organs affected in CHARGE during development include neural tube, neural crest, and placodal derivatives. Certain tissues such as the inner ear and olfactory system appear to be highly sensitive to CHD7 dosage, whereas other organs such as the heart, kidney, and skeletal systems are more variably affected. CHD7 haploinsufficiency occurs in 60–80% of CHARGE patients and mutations in CHD7 are distributed throughout the coding sequence. Further studies are necessary to determine whether the remaining 20–40% of CHARGE patients have mutations in CHD7 regulatory elements or in other genes. It is also possible that unidentified environmental factors contribute to the phenotype. Heterozygous Chd7 mutant mice and in vitro ES cell analyses have begun to clarify a role for CHD7 in the development and maintenance of a variety of tissues (39, 41, 42, 60, 65–68). These studies suggest that CHD7 has roles in stem cell maintenance and cell fate specification (39, 65–68). In vitro analyses have implicated that CHD7 is a member of the PBAF complex in neural crest-like cells (66) and that CHD7 forms a complex with NLK, SETDB1, and PPAR-γ (67). These studies open the door for further in vivo assays which should identify important downstream effectors of CHD7. Taken together, these observations suggest that CHD7 is likely to participate in combinatorial protein complexes which bind DNA in enhancer regions and regulate gene transcription in a temporal and tissue-specific manner.
To date, much has been learned about CHARGE syndrome, but several important questions remain unanswered. Is there a core set of CHD7 binding partners that regulate CHD7 function, and if so are they tissue and age-specific? What are the upstream regulators of CHD7? Finally, can we use information about CHD7 function in cells to help design therapies for patients diagnosed with CHARGE syndrome?
Acknowledgements
The authors thank Dr. Jirair K. Bedoyan for critical reading of the manuscript. W.S.L. is supported by a training grant from the National Institute of Deafness and Other Communication Disorders (T32DC00011). D.M.M. is supported by grants from the National Institutes of Health (R01DC009410 and R01NS054784).
Footnotes
Conflict of Interest: W.S.L., E.A.H., and D.M.M. declare no conflict of interest.
References
- 1.Hall BD. Choanal atresia and associated multiple anomalies. J Pediatr. 1979;95:395–398. doi: 10.1016/s0022-3476(79)80513-2. [DOI] [PubMed] [Google Scholar]
- 2.Aramaki M, Udaka T, Kosaki R, Makita Y, Okamoto N, Yoshihashi H, Oki H, Nanao K, Moriyama N, Oku S, Hasegawa T, Takahashi T, Fukushima Y, Kawame H, Kosaki K. Phenotypic spectrum of CHARGE syndrome with CHD7 mutations. J Pediatr. 2006;148:410–414. doi: 10.1016/j.jpeds.2005.10.044. [DOI] [PubMed] [Google Scholar]
- 3.Jongmans MC, Admiraal RJ, van der Donk KP, Vissers LE, Baas AF, Kapusta L, van Hagen JM, Donnai D, de Ravel TJ, Veltman JA, Geurts van Kessel A, De Vries BB, Brunner HG, Hoefsloot LH, van Ravenswaaij CM. CHARGE syndrome: the phenotypic spectrum of mutations in the CHD7 gene. J Med Genet. 2006;43:306–314. doi: 10.1136/jmg.2005.036061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lalani SR, Safiullah AM, Fernbach SD, Harutyunyan KG, Thaller C, Peterson LE, McPherson JD, Gibbs RA, White LD, Hefner M, Davenport SL, Graham JM, Bacino CA, Glass NL, Towbin JA, Craigen WJ, Neish SR, Lin AE, Belmont JW. Spectrum of CHD7 Mutations in 110 Individuals with CHARGE Syndrome and Genotype-Phenotype Correlation. Am J Hum Genet. 2006;78:303–314. doi: 10.1086/500273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sanlaville D, Etchevers HC, Gonzales M, Martinovic J, Clement-Ziza M, Delezoide AL, Aubry MC, Pelet A, Chemouny S, Cruaud C, Audollent S, Esculpavit C, Goudefroye G, Ozilou C, Fredouille C, Joye N, Morichon-Delvallez N, Dumez Y, Weissenbach J, Munnich A, Amiel J, Encha-Razavi F, Lyonnet S, Vekemans M, Attie-Bitach T. Phenotypic spectrum of CHARGE syndrome in fetuses with CHD7 truncating mutations correlates with expression during human development. J Med Genet. 2006;43:211–217. doi: 10.1136/jmg.2005.036160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vissers LE, van Ravenswaaij CM, Admiraal R, Hurst JA, de Vries BB, Janssen IM, van der Vliet WA, Huys EH, de Jong PJ, Hamel BC, Schoenmakers EF, Brunner HG, Veltman JA, van Kessel AG. Mutations in a new member of the chromodomain gene family cause CHARGE syndrome. Nat Genet. 2004;36:955–957. doi: 10.1038/ng1407. [DOI] [PubMed] [Google Scholar]
- 7.Harris J, Robert E, Kallen B. Epidemiology of choanal atresia with special reference to the CHARGE association. Pediatrics. 1997;99:363–367. doi: 10.1542/peds.99.3.363. [DOI] [PubMed] [Google Scholar]
- 8.Issekutz KA, Graham JM, Jr, Prasad C, Smith IM, Blake KD. An epidemiological analysis of CHARGE syndrome: preliminary results from a Canadian study. Am J Med Genet A. 2005;133:309–317. doi: 10.1002/ajmg.a.30560. [DOI] [PubMed] [Google Scholar]
- 9.Kallen K, Robert E, Mastroiacovo P, Castilla EE, Kallen B. CHARGE Association in newborns: a registry-based study. Teratology. 1999;60:334–343. doi: 10.1002/(SICI)1096-9926(199912)60:6<334::AID-TERA5>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 10.Sanlaville D, Verloes A. CHARGE syndrome: an update. Eur J Hum Genet. 2007;15:389–399. doi: 10.1038/sj.ejhg.5201778. [DOI] [PubMed] [Google Scholar]
- 11.Vuorela P, Ala-Mello S, Saloranta C, Penttinen M, Poyhonen M, Huoponen K, Borozdin W, Bausch B, Botzenhart EM, Wilhelm C, Kaariainen H, Kohlhase J. Molecular analysis of the CHD7 gene in CHARGE syndrome: identification of 22 novel mutations and evidence for a low contribution of large CHD7 deletions. Genet Med. 2007;9:690–694. doi: 10.1097/gim.0b013e318156e68e. [DOI] [PubMed] [Google Scholar]
- 12.Delahaye A, Sznajer Y, Lyonnet S, Elmaleh-Berges M, Delpierre I, Audollent S, Wiener-Vacher S, Mansbach AL, Amiel J, Baumann C, Bremond-Gignac D, Attie-Bitach T, Verloes A, Sanlaville D. Familial CHARGE syndrome because of CHD7 mutation: clinical intra- and interfamilial variability. Clin Genet. 2007;72:112–121. doi: 10.1111/j.1399-0004.2007.00821.x. [DOI] [PubMed] [Google Scholar]
- 13.Jongmans MC, Hoefsloot LH, van der Donk KP, Admiraal RJ, Magee A, van de Laar I, Hendriks Y, Verheij JB, Walpole I, Brunner HG, van Ravenswaaij CM. Familial CHARGE syndrome and the CHD7 gene: a recurrent missense mutation, intrafamilial recurrence and variability. Am J Med Genet A. 2008;146:43–50. doi: 10.1002/ajmg.a.31921. [DOI] [PubMed] [Google Scholar]
- 14.Pauli S, Pieper L, Haberle J, Grzmil P, Burfeind P, Steckel M, Lenz U, Michelmann HW. Proven germline mosaicism in a father of two children with CHARGE syndrome. Clin Genet. 2009;75:473–479. doi: 10.1111/j.1399-0004.2009.01151.x. [DOI] [PubMed] [Google Scholar]
- 15.Alazami AM, Alzahrani F, Alkuraya FS. Expanding the "E" in CHARGE. Am J Med Genet A. 2008;146A:1890–1892. doi: 10.1002/ajmg.a.32376. [DOI] [PubMed] [Google Scholar]
- 16.Asakura Y, Toyota Y, Muroya K, Kurosawa K, Fujita K, Aida N, Kawame H, Kosaki K, Adachi M. Endocrine and radiological studies in patients with molecularly confirmed CHARGE syndrome. J Clin Endocrinol Metab. 2008;93:920–924. doi: 10.1210/jc.2007-1419. [DOI] [PubMed] [Google Scholar]
- 17.Bergman JE, de Wijs I, Jongmans MC, Admiraal RJ, Hoefsloot LH, van Ravenswaaij-Arts CM. Exon copy number alterations of the CHD7 gene are not a major cause of CHARGE and CHARGE-like syndrome. Eur J Med Genet. 2008;51:417–425. doi: 10.1016/j.ejmg.2008.03.003. [DOI] [PubMed] [Google Scholar]
- 18.Chopra C, Baretto R, Duddridge M, Browning MJ. T-cell immunodeficiency in CHARGE syndrome. Acta Paediatr. 2009;98:408–410. doi: 10.1111/j.1651-2227.2008.01077.x. [DOI] [PubMed] [Google Scholar]
- 19.Felix TM, Hanshaw BC, Mueller R, Bitoun P, Murray JC. CHD7 gene and non-syndromic cleft lip and palate. Am J Med Genet A. 2006;140:2110–2114. doi: 10.1002/ajmg.a.31308. [DOI] [PubMed] [Google Scholar]
- 20.Fujita K, Aida N, Asakura Y, Kurosawa K, Niwa T, Muroya K, Adachi M, Nishimura G, Inoue T. Abnormal basiocciput development in CHARGE syndrome. AJNR Am J Neuroradiol. 2009;30:629–634. doi: 10.3174/ajnr.A1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gennery AR, Slatter MA, Rice J, Hoefsloot LH, Barge D, McLean-Tooke A, Montgomery T, Goodship JA, Burt AD, Flood TJ, Abinun M, Cant AJ, Johnson D. Mutations in CHD7 in patients with CHARGE syndrome cause T-B + natural killer cell + severe combined immune deficiency and may cause Omenn-like syndrome. Clin Exp Immunol. 2008;153:75–80. doi: 10.1111/j.1365-2249.2008.03681.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hoover-Fong J, Savage WJ, Lisi E, Winkelstein J, Thomas GH, Hoefsloot LH, Loeb DM. Congenital T cell deficiency in a patient with CHARGE syndrome. J Pediatr. 2009;154:140–142. doi: 10.1016/j.jpeds.2008.07.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Johnson D, Morrison N, Grant L, Turner T, Fantes J, Connor JM, Murday V. Confirmation of CHD7 as a cause of CHARGE association identified by mapping a balanced chromosome translocation in affected monozygotic twins. J Med Genet. 2006;43:280–284. doi: 10.1136/jmg.2005.032946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jongmans MC, van Ravenswaaij-Arts CM, Pitteloud N, Ogata T, Sato N, Claahsen-van der Grinten HL, van der Donk K, Seminara S, Bergman JE, Brunner HG, Crowley WF, Jr, Hoefsloot LH. CHD7 mutations in patients initially diagnosed with Kallmann syndrome--the clinical overlap with CHARGE syndrome. Clin Genet. 2009;75:65–71. doi: 10.1111/j.1399-0004.2008.01107.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jyonouchi S, McDonald-McGinn DM, Bale S, Zackai EH, Sullivan KE. CHARGE (coloboma, heart defect, atresia choanae, retarded growth and development, genital hypoplasia, ear anomalies/deafness) syndrome and chromosome 22q11.2 deletion syndrome: a comparison of immunologic and nonimmunologic phenotypic features. Pediatrics. 2009;123:e871–e877. doi: 10.1542/peds.2008-3400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim HG, Kurth I, Lan F, Meliciani I, Wenzel W, Eom SH, Kang GB, Rosenberger G, Tekin M, Ozata M, Bick DP, Sherins RJ, Walker SL, Shi Y, Gusella JF, Layman LC. Mutations in CHD7, encoding a chromatin-remodeling protein, cause idiopathic hypogonadotropic hypogonadism and Kallmann syndrome. Am J Hum Genet. 2008;83:511–519. doi: 10.1016/j.ajhg.2008.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee YW, Kim SC, Shin YL, Kim JW, Hong HS, Lee YK, Ki CS. Clinical and genetic analysis of the CHD7 gene in Korean patients with CHARGE syndrome. Clin Genet. 2009;75:290–293. doi: 10.1111/j.1399-0004.2008.01127.x. [DOI] [PubMed] [Google Scholar]
- 28.Ogata T, Fujiwara I, Ogawa E, Sato N, Udaka T, Kosaki K. Kallmann syndrome phenotype in a female patient with CHARGE syndrome and CHD7 mutation. Endocr J. 2006;53:741–743. doi: 10.1507/endocrj.k06-099. [DOI] [PubMed] [Google Scholar]
- 29.Sanka M, Tangsinmankong N, Loscalzo M, Sleasman JW, Dorsey MJ. Complete DiGeorge syndrome associated with CHD7 mutation. J Allergy Clin Immunol. 2007;120:952–954. doi: 10.1016/j.jaci.2007.08.013. [DOI] [PubMed] [Google Scholar]
- 30.Udaka T, Okamoto N, Aramaki M, Torii C, Kosaki R, Hosokai N, Hayakawa T, Takahata N, Takahashi T, Kosaki K. An Alu retrotransposition-mediated deletion of CHD7 in a patient with CHARGE syndrome. Am J Med Genet A. 2007;143:721–726. doi: 10.1002/ajmg.a.31441. [DOI] [PubMed] [Google Scholar]
- 31.Van de Laar I, Dooijes D, Hoefsloot L, Simon M, Hoogeboom J, Devriendt K. Limb anomalies in patients with CHARGE syndrome: an expansion of the phenotype. Am J Med Genet A. 2007;143A:2712–2715. doi: 10.1002/ajmg.a.32008. [DOI] [PubMed] [Google Scholar]
- 32.Wincent J, Holmberg E, Stromland K, Soller M, Mirzaei L, Djureinovic T, Robinson K, Anderlid B, Schoumans J. CHD7 mutation spectrum in 28 Swedish patients diagnosed with CHARGE syndrome. Clin Genet. 2008;74:31–38. doi: 10.1111/j.1399-0004.2008.01014.x. [DOI] [PubMed] [Google Scholar]
- 33.Wright EM, O'Connor R, Kerr BA. Radial aplasia in CHARGE syndrome: a new association. Eur J Med Genet. 2009;52:239–241. doi: 10.1016/j.ejmg.2009.03.017. [DOI] [PubMed] [Google Scholar]
- 34.Writzl K, Cale CM, Pierce CM, Wilson LC, Hennekam RC. Immunological abnormalities in CHARGE syndrome. Eur J Med Genet. 2007;50:338–345. doi: 10.1016/j.ejmg.2007.05.002. [DOI] [PubMed] [Google Scholar]
- 35.Zentner GE, Layman WS, Martin DM, Scacheri PC. Molecular and phenotypic aspects of CHD7 mutation in CHARGE syndrome. Am J Med Genet A. 152A:674–686. doi: 10.1002/ajmg.a.33323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Azoulay R, Fallet-Bianco C, Garel C, Grabar S, Kalifa G, Adamsbaum C. MRI of the olfactory bulbs and sulci in human fetuses. Pediatr Radiol. 2006;36:97–107. doi: 10.1007/s00247-005-0030-0. [DOI] [PubMed] [Google Scholar]
- 37.Blustajn J, Kirsch CF, Panigrahy A, Netchine I. Olfactory anomalies in CHARGE syndrome: imaging findings of a potential major diagnostic criterion. AJNR Am J Neuroradiol. 2008;29:1266–1269. doi: 10.3174/ajnr.A1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chalouhi C, Faulcon P, Le Bihan C, Hertz-Pannier L, Bonfils P, Abadie V. Olfactory evaluation in children: application to the CHARGE syndrome. Pediatrics. 2005;116:e81–e88. doi: 10.1542/peds.2004-1970. [DOI] [PubMed] [Google Scholar]
- 39.Layman WS, McEwen DP, Beyer LA, Lalani SR, Fernbach SD, Oh E, Swaroop A, Hegg CC, Raphael Y, Martens JR, Martin DM. Defects in neural stem cell proliferation and olfaction in Chd7 deficient mice indicate a mechanism for hyposmia in human CHARGE syndrome. Hum Mol Genet. 2009;18:1909–1923. doi: 10.1093/hmg/ddp112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pinto G, Abadie V, Mesnage R, Blustajn J, Cabrol S, Amiel J, Hertz-Pannier L, Bertrand AM, Lyonnet S, Rappaport R, Netchine I. CHARGE syndrome includes hypogonadotropic hypogonadism and abnormal olfactory bulb development. J Clin Endocrinol Metab. 2005;90:5621–5626. doi: 10.1210/jc.2004-2474. [DOI] [PubMed] [Google Scholar]
- 41.Bosman EA, Penn AC, Ambrose JC, Kettleborough R, Stemple DL, Steel KP. Multiple mutations in mouse Chd7 provide models for CHARGE syndrome. Hum Mol Genet. 2005;14:3463–3476. doi: 10.1093/hmg/ddi375. [DOI] [PubMed] [Google Scholar]
- 42.Hurd EA, Capers PL, Blauwkamp MN, Adams ME, Raphael Y, Poucher HK, Martin DM. Loss of Chd7 function in gene-trapped reporter mice is embryonic lethal and associated with severe defects in multiple developing tissues. Mamm Genome. 2007;18:94–104. doi: 10.1007/s00335-006-0107-6. [DOI] [PubMed] [Google Scholar]
- 43.Glueckert R, Rask-Andersen H, Sergi C, Schmutzhard J, Mueller B, Beckmann F, Rittinger O, Hoefsloot LH, Schrott-Fischer A, Janecke AR. Histology and synchrotron radiation-based microtomography of the inner ear in a molecularly confirmed case of CHARGE syndrome. Am J Med Genet A. 152A:665–673. doi: 10.1002/ajmg.a.33321. [DOI] [PubMed] [Google Scholar]
- 44.Morimoto AK, Wiggins RH, 3rd, Hudgins PA, Hedlund GL, Hamilton B, Mukherji SK, Telian SA, Harnsberger HR. Absent semicircular canals in CHARGE syndrome: radiologic spectrum of findings. AJNR Am J Neuroradiol. 2006;27:1663–1671. [PMC free article] [PubMed] [Google Scholar]
- 45.Satar B, Mukherji SK, Telian SA. Congenital aplasia of the semicircular canals. Otol Neurotol. 2003;24:437–446. doi: 10.1097/00129492-200305000-00014. [DOI] [PubMed] [Google Scholar]
- 46.Arndt S, Laszig R, Beck R, Schild C, Maier W, Birkenhager R, Kroeger S, Wesarg T, Aschendorff A. Spectrum of hearing disorders and their management in children with CHARGE syndrome. Otol Neurotol. 31:67–73. doi: 10.1097/MAO.0b013e3181c0e972. [DOI] [PubMed] [Google Scholar]
- 47.Abadie V, Wiener-Vacher S, Morisseau-Durand MP, Poree C, Amiel J, Amanou L, Peigne C, Lyonnet S, Manac'h Y. Vestibular anomalies in CHARGE syndrome: investigations on and consequences for postural development. Eur J Pediatr. 2000;159:569–574. doi: 10.1007/s004319900409. [DOI] [PubMed] [Google Scholar]
- 48.Wiener-Vacher SR, Amanou L, Denise P, Narcy P, Manach Y. Vestibular function in children with the CHARGE association. Arch Otolaryngol Head Neck Surg. 1999;125:342–347. doi: 10.1001/archotol.125.3.342. [DOI] [PubMed] [Google Scholar]
- 49.Adams ME, Hurd EA, Beyer LA, Swiderski DL, Raphael Y, Martin DM. Defects in vestibular sensory epithelia and innervation in mice with loss of Chd7 function: Implications for human CHARGE syndrome. J Comp Neurol. 2007;504:519–532. doi: 10.1002/cne.21460. [DOI] [PubMed] [Google Scholar]
- 50.Hawker K, Fuchs H, Angelis MH, Steel KP. Two new mouse mutants with vestibular defects that map to the highly mutable locus on chromosome 4. Int J Audiol. 2005;44:171–177. doi: 10.1080/14992020500057434. [DOI] [PubMed] [Google Scholar]
- 51.Kiernan AE, Erven A, Voegeling S, Peters J, Nolan P, Hunter J, Bacon Y, Steel KP, Brown SD, Guenet JL. ENU mutagenesis reveals a highly mutable locus on mouse Chromosome 4 that affects ear morphogenesis. Mamm Genome. 2002;13:142–148. doi: 10.1007/BF02684018. [DOI] [PubMed] [Google Scholar]
- 52.Pau H, Hawker K, Fuchs H, De Angelis MH, Steel KP. Characterization of a new mouse mutant, flouncer, with a balance defect and inner ear malformation. Otol Neurotol. 2004;25:707–713. doi: 10.1097/00129492-200409000-00010. [DOI] [PubMed] [Google Scholar]
- 53.Martin P, Swanson GJ. Descriptive and experimental analysis of the epithelial remodellings that control semicircular canal formation in the developing mouse inner ear. Dev Biol. 1993;159:549–558. doi: 10.1006/dbio.1993.1263. [DOI] [PubMed] [Google Scholar]
- 54.Salminen M, Meyer BI, Bober E, Gruss P. Netrin 1 is required for semicircular canal formation in the mouse inner ear. Development. 2000;127:13–22. doi: 10.1242/dev.127.1.13. [DOI] [PubMed] [Google Scholar]
- 55.Bhagavath B, Podolsky RH, Ozata M, Bolu E, Bick DP, Kulharya A, Sherins RJ, Layman LC. Clinical and molecular characterization of a large sample of patients with hypogonadotropic hypogonadism. Fertil Steril. 2006;85:706–713. doi: 10.1016/j.fertnstert.2005.08.044. [DOI] [PubMed] [Google Scholar]
- 56.Kim HG, Bhagavath B, Layman LC. Clinical manifestations of impaired GnRH neuron development and function. Neurosignals. 2008;16:165–182. doi: 10.1159/000111561. [DOI] [PubMed] [Google Scholar]
- 57.Chung WC, Moyle SS, Tsai PS. Fibroblast growth factor 8 signaling through fibroblast growth factor receptor 1 is required for the emergence of gonadotropin-releasing hormone neurons. Endocrinology. 2008;149:4997–5003. doi: 10.1210/en.2007-1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Falardeau J, Chung WC, Beenken A, Raivio T, Plummer L, Sidis Y, Jacobson-Dickman EE, Eliseenkova AV, Ma J, Dwyer A, Quinton R, Na S, Hall JE, Huot C, Alois N, Pearce SH, Cole LW, Hughes V, Mohammadi M, Tsai P, Pitteloud N. Decreased FGF8 signaling causes deficiency of gonadotropin-releasing hormone in humans and mice. J Clin Invest. 2008;118:2822–2831. doi: 10.1172/JCI34538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hardelin JP, Dode C, et al. The complex genetics of Kallmann syndrome: KAL1, FGFR1, FGF8, PROKR2, PROK2. Sex Dev. 2008;2:181–193. doi: 10.1159/000152034. [DOI] [PubMed] [Google Scholar]
- 60.Bergman JE, Bosman EA, van Ravenswaaij-Arts CM, Steel KP. Study of smell and reproductive organs in a mouse model for CHARGE syndrome. Eur J Hum Genet. 2009 doi: 10.1038/ejhg.2009.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ho L, Crabtree GR. Chromatin remodelling during development. Nature. 463:474–484. doi: 10.1038/nature08911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hall JA, Georgel PT. CHD proteins: a diverse family with strong ties. Biochem Cell Biol. 2007;85:463–476. doi: 10.1139/O07-063. [DOI] [PubMed] [Google Scholar]
- 63.Marfella CG, Imbalzano AN. The Chd family of chromatin remodelers. Mutat Res. 2007;618:30–40. doi: 10.1016/j.mrfmmm.2006.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Boyer LA, Latek RR, Peterson CL. The SANT domain: a unique histone-tail-binding module? Nat Rev Mol Cell Biol. 2004;5:158–163. doi: 10.1038/nrm1314. [DOI] [PubMed] [Google Scholar]
- 65.Schnetz MP, Bartels CF, Shastri K, Balasubramanian D, Zentner GE, Balaji R, Zhang X, Song L, Wang Z, Laframboise T, Crawford GE, Scacheri PC. Genomic distribution of CHD7 on chromatin tracks H3K4 methylation patterns. Genome Res. 2009 doi: 10.1101/gr.086983.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bajpai R, Chen DA, Rada-Iglesias A, Zhang J, Xiong Y, Helms J, Chang CP, Zhao Y, Swigut T, Wysocka J. CHD7 cooperates with PBAF to control multipotent neural crest formation. Nature. 463:958–962. doi: 10.1038/nature08733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Takada I, Mihara M, Suzawa M, Ohtake F, Kobayashi S, Igarashi M, Youn MY, Takeyama K, Nakamura T, Mezaki Y, Takezawa S, Yogiashi Y, Kitagawa H, Yamada G, Takada S, Minami Y, Shibuya H, Matsumoto K, Kato S. A histone lysine methyltransferase activated by non-canonical Wnt signalling suppresses PPAR-gamma transactivation. Nat Cell Biol. 2007;9:1273–1285. doi: 10.1038/ncb1647. [DOI] [PubMed] [Google Scholar]
- 68.Srinivasan S, Dorighi KM, Tamkun JW. Drosophila Kismet regulates histone H3 lysine 27 methylation and early elongation by RNA polymerase II. PLoS Genet. 2008;4 doi: 10.1371/journal.pgen.1000217. e1000217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Phippard D, Lu L, Lee D, Saunders JC, Crenshaw EB., 3rd Targeted mutagenesis of the POU-domain gene Brn4/Pou3f4 causes developmental defects in the inner ear. J Neurosci. 1999;19:5980–5989. doi: 10.1523/JNEUROSCI.19-14-05980.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Vore AP, Chang EH, Hoppe JE, Butler MG, Forrester S, Schneider MC, Smith LL, Burke DW, Campbell CA, Smith RJ. Deletion of and novel missense mutation in POU3F4 in 2 families segregating X-linked nonsyndromic deafness. Arch Otolaryngol Head Neck Surg. 2005;131:1057–1063. doi: 10.1001/archotol.131.12.1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Abdelhak S, Kalatzis V, Heilig R, Compain S, Samson D, Vincent C, Weil D, Cruaud C, Sahly I, Leibovici M, Bitner-Glindzicz M, Francis M, Lacombe D, Vigneron J, Charachon R, Boven K, Bedbeder P, Van Regemorter N, Weissenbach J, Petit C. A human homologue of the Drosophila eyes absent gene underlies branchio-oto-renal (BOR) syndrome and identifies a novel gene family. Nat Genet. 1997;15:157–164. doi: 10.1038/ng0297-157. [DOI] [PubMed] [Google Scholar]
- 72.Xu PX, Adams J, Peters H, Brown MC, Heaney S, Maas R. Eya1-deficient mice lack ears and kidneys and show abnormal apoptosis of organ primordia. Nat Genet. 1999;23:113–117. doi: 10.1038/12722. [DOI] [PubMed] [Google Scholar]
- 73.Hatch EP, Noyes CA, Wang X, Wright TJ, Mansour SL. Fgf3 is required for dorsal patterning and morphogenesis of the inner ear epithelium. Development. 2007;134:3615–3625. doi: 10.1242/dev.006627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mansour SL. Targeted disruption of int-2 (fgf-3) causes developmental defects in the tail and inner ear. Mol Reprod Dev. 1994;39:62–67. doi: 10.1002/mrd.1080390111. discussion 67–68. [DOI] [PubMed] [Google Scholar]
- 75.Tekin M, Hismi BO, Fitoz S, Ozdag H, Cengiz FB, Sirmaci A, Aslan I, Inceoglu B, Yuksel-Konuk EB, Yilmaz ST, Yasun O, Akar N. Homozygous mutations in fibroblast growth factor 3 are associated with a new form of syndromic deafness characterized by inner ear agenesis, microtia, and microdontia. Am J Hum Genet. 2007;80:338–344. doi: 10.1086/510920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Milunsky JM, Zhao G, Maher TA, Colby R, Everman DB. LADD syndrome is caused by FGF10 mutations. Clin Genet. 2006;69:349–354. doi: 10.1111/j.1399-0004.2006.00597.x. [DOI] [PubMed] [Google Scholar]
- 77.Pauley S, Wright TJ, Pirvola U, Ornitz D, Beisel K, Fritzsch B. Expression and function of FGF10 in mammalian inner ear development. Dev Dyn. 2003;227:203–215. doi: 10.1002/dvdy.10297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Koch B, Goold A, Egelhoff J, Benton C. Partial absence of the posterior semicircular canal in Alagille syndrome: CT findings. Pediatr Radiol. 2006;36:977–979. doi: 10.1007/s00247-006-0230-2. [DOI] [PubMed] [Google Scholar]
- 79.Laclef C, Souil E, Demignon J, Maire P. Thymus, kidney and craniofacial abnormalities in Six 1 deficient mice. Mech Dev. 2003;120:669–679. doi: 10.1016/s0925-4773(03)00065-0. [DOI] [PubMed] [Google Scholar]
- 80.Ruf RG, Xu PX, Silvius D, Otto EA, Beekmann F, Muerb UT, Kumar S, Neuhaus TJ, Kemper MJ, Raymond RM, Jr, Brophy PD, Berkman J, Gattas M, Hyland V, Ruf EM, Schwartz C, Chang EH, Smith RJ, Stratakis CA, Weil D, Petit C, Hildebrandt F. SIX1 mutations cause branchio-oto-renal syndrome by disruption of EYA1-SIX1-DNA complexes. Proc Natl Acad Sci U S A. 2004;101:8090–8095. doi: 10.1073/pnas.0308475101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zheng W, Huang L, Wei ZB, Silvius D, Tang B, Xu PX. The role of Six1 in mammalian auditory system development. Development. 2003;130:3989–4000. doi: 10.1242/dev.00628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kawauchi S, Shou J, Santos R, Hebert JM, McConnell SK, Mason I, Calof AL. Fgf8 expression defines a morphogenetic center required for olfactory neurogenesis and nasal cavity development in the mouse. Development. 2005;132:5211–5223. doi: 10.1242/dev.02143. [DOI] [PubMed] [Google Scholar]
- 83.Meyers EN, Lewandoski M, Martin GR. An Fgf8 mutant allelic series generated by Cre- and Flp-mediated recombination. Nat Genet. 1998;18:136–141. doi: 10.1038/ng0298-136. [DOI] [PubMed] [Google Scholar]
- 84.Ng KL, Li JD, Cheng MY, Leslie FM, Lee AG, Zhou QY. Dependence of olfactory bulb neurogenesis on prokineticin 2 signaling. Science. 2005;308:1923–1927. doi: 10.1126/science.1112103. [DOI] [PubMed] [Google Scholar]
- 85.Zhang C, Ng KL, Li JD, He F, Anderson DJ, Sun YE, Zhou QY. Prokineticin 2 is a target gene of proneural basic helix-loop-helix factors for olfactory bulb neurogenesis. J Biol Chem. 2007;282:6917–6921. doi: 10.1074/jbc.C600290200. [DOI] [PubMed] [Google Scholar]
- 86.Matsumoto S, Yamazaki C, Masumoto KH, Nagano M, Naito M, Soga T, Hiyama H, Matsumoto M, Takasaki J, Kamohara M, Matsuo A, Ishii H, Kobori M, Katoh M, Matsushime H, Furuichi K, Shigeyoshi Y. Abnormal development of the olfactory bulb and reproductive system in mice lacking prokineticin receptor PKR2. Proc Natl Acad Sci U S A. 2006;103:4140–4145. doi: 10.1073/pnas.0508881103. [DOI] [PMC free article] [PubMed] [Google Scholar]