

Abstract
Lung fibrosis is an ultimate consequence of pulmonary oxygen toxicity in human and animal models. Excessive production and deposition of extracellular matrix proteins, e. g. collagen-I, is the most important feature of pulmonary fibrosis in hyperoxia-induced lung injury. In the present study, we investigated the role of RhoA and reactive oxygen species (ROS) in collagen-I synthesis in hyperoxic lung fibroblasts and in a mouse model of oxygen toxicity. Exposure of human lung fibroblasts to hyperoxia resulted in RhoA activation and increase in collagen-I synthesis and cell proliferation. Inhibition of RhoA by C3 transferase CT-04, dominant-negative RhoA mutant T19N, or RhoA siRNA prevented hyperoxia-induced collagen-I synthesis. Constitutively active RhoA mutant Q63L mimicked the effect of hyperoxia on collagen-I expression. Moreover, Rho kinase (ROCK) inhibitor Y27632 inhibited collagen-I synthesis in hyperoxic lung fibroblasts and fibrosis in mouse lungs after oxygen toxicity. Furthermore, ROS scavenger tiron attenuated hyperoxia-induced increases in RhoA activation and collagen-I synthesis in lung fibroblasts and mouse lungs after oxygen toxicity. More importantly, we found that hyperoxia induced separation of guanine nucleotide dissociation inhibitor (GDI) from RhoA in lung fibroblasts and mouse lungs. Further, tiron prevented the separation of GDI from RhoA in hyperoxic lung fibroblasts and mouse lungs with oxygen toxicity. Together, these results indicate that ROS-induced separation of GDI from RhoA leads to RhoA activation with oxygen toxicity. ROS-dependent RhoA activation is responsible for the increase in collagen-I synthesis in hyperoxic lung fibroblasts and mouse lungs.
Keywords: oxygen toxicity, lung, fibroblasts, collagen, RhoA, reactive oxygen species
Introduction
Oxygen therapy is an important element in the management of various conditions such as neonatal or adult respiratory distress syndrome, primary and secondary pulmonary hypertension, circulatory shock, infection, and multiple-organ failure syndrome. However, prolonged exposure to higher levels of oxygen leads to lung injury resulting in diffuse alveolar damage, intense cellular infiltration, and deposition of interstitial collagen fibers [1,2]. Lung fibrosis is a life threatening consequence of pulmonary oxygen toxicity in human and animal models [3,4]. Excessive production and deposition of extracellular matrix proteins is key process in the pulmonary fibrosis occurring in hyperoxia-induced lung injury [5]. Collagen is the major extracellular matrix (ECM) component of the lungs and is vital for maintaining the normal lung architecture. The increase in collagen synthesis from lung alveolar interstitial fibroblasts is correlated with changes in the viscoelastic behavior and impairs lung function in hyperoxia-induced lung injuries [6]. Exposure of lung fibroblasts to hyperoxia stimulates fibroblasts proliferation and increases collagen protein [7,8]. In the present study, we also found that hyperoxia increases the levels of collagen-I mRNA and protein. These data suggest that hyperoxia directly affects lung fibroblast function in collagen-I synthesis.
The molecular mechanisms for hyperoxia-induced collagen synthesis have not been clarified. During hyperoxic exposure, large amount of reactive oxygen species (ROS) are generated from the mitochondrial electron transport [9] or NADPH oxidase-catalyzed reaction [10–12]. ROS cause injuries to pulmonary cells including epithelia, endothelia, and macrophages, which lead to extensive inflammatory responses with leukocyte infiltration, injury, and death of lung cells [13]. Emerging evidence suggests that ROS are involved in the pathogenesis of idiopathic pulmonary fibrosis (IPF) and the animal model of bleomycin-induced lung fibrosis [14–18]. However, it remains unclear whether and how ROS is associated to hyperoxia-induced pulmonary fibrosis and collagen synthesis from lung fibroblasts. In this study, we found that ROS scavenger tiron prevents collagen-I synthesis from hyperoxic lung fibroblasts and hyperoxia-induced lung fibrosis in mice. These data indicate that ROS play an important role in the fibrogenesis of pulmonary oxygen toxicity.
Small GTPases including RhoA, Rac and CDC42 are essential in the regulation of various cellular functions including formation of F-actin stress fibers and focal adhesion complexes and transcription of genes containing serum response element [19]. These GTPase exist in active GTP-bound state and inactive GDP-bound state. Rho GTPase activity is regulated by guanine nucleotide exchange factor (GEF) and guanine nucleotide dissociation inhibitor (GDI). GEF facilitates the GTP-bound active RhoA state by promoting the exchange of GDP for GTP, whereas association of GDI to RhoA promotes GDP-bound inactive state [20]. The downstream events for Rho activation include activation of Rho kinase (ROCK) and increased formation of actin stress fibers. Recently, Hemnes and colleagues [21] reported that RhoA activation is involved in the formation of pulmonary fibrotic lesion. Inhibition of RhoA by simvastatin or sildenafil prevents bleomycin-induced pulmonary fibrosis [21,22]. However, it is not clear whether RhoA is activated in lung fibrosis induced by oxygen toxicity. Interestingly, Knock et al [23] and Chi et al [24] recently reported that ROS causes RhoA activation in vascular smooth muscle cells and endothelial cells through an unknown mechanism. In the present study, we examined the role of RhoA activation in hyperoxia-induced collagen-I synthesis in lung fibroblasts and mouse lungs. We demonstrated for the first time that ROS-dependent decrease in the binding of RhoA to GDI leads to RhoA activation at oxygen toxicity. ROS-dependent RhoA activation plays critical role in the increase in collagen-I synthesis in hyperoxic lung fibroblasts and mouse lungs.
Materials and methods
Reagents and materials
Mouse anti-collagen-I antibody and GDI polyclonal antibody were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-RhoA antibody and cell permeable Rho inhibitor C-3 transferase (CT-04) were from Cytoskeleton (Denver, CO). Antibody against GAPDH was from Cell Signaling (Beverly, MA). Dihydroethidine (DHE) was from Invitrogen (Carlsbad, CA). Mammalian expression plasmids pcDNA3-EGFP containing cDNA of wild type of RhoA, constitutively active Rho-A with Q63 substituted with L (Q63L), and dominant-negative RhoA with T19 substituted with N containing GFP cDNA (T19N) were obtained from Addgene, (Cambridge, MA). Masson Trichrome kit was obtained from Richard-Alan Scientific (Kalamazoo, MI). Other reagents were purchased from Sigma (St. Louis, MO).
Cell culture and hyperoxic exposure
Human lung fibroblasts HFL-1 were obtained from ATCC. Third- to eighth-passage cells were maintained in F12K medium containing 10% fetal bovine serum and antibiotics (10 u/ml penicillin, 100 μg/ml streptomycin, 20 μg/ml gentamicin, and 2 μg/ml Fungizone) and were used 2 or 3 days after confluence. For hyperoxic exposure, confluent HFL-1 fibroblasts were incubated to 95% O2 and 5% CO2 at 37°C. Normoxia was air and 5% CO2. In some experiments, confluent HFL-1 fibroblasts were exposed to 80% O2 and 5% CO2 for 12 h and then 50% O2 and 5%CO2 for 12 h at 37°C.
Determination of RhoA activation
The activity of RhoA GTPase was detected using Rho activation assay from Millipore according to manufacturer protocol. Briefly, HFL-1 fibroblasts were exposed to hyperoxia for 1–24 h and cells were lysed in MLB buffer containing 25 mM HEPES, 150mM NaCl, 1% Igepal CA-630, 1mM Mg Cl2 1 mM EDTA and 10% glycerol. Lysates were incubated with recombinant Rhotekin bound to glutathione-agarose at 4° for 45 minutes. Immunoprecipitates were collected by centrifugation and washed three times with MLB buffer. Proteins were eluted from agarose beads by boiling the samples in 30 μl of SDS immunoblotting sample buffer. Agarose beads were pelleted by centrifugation at 10,000 g and supernatants were analyzed for RhoA contents by Western blot.
Knocking-down of RhoA protein
RhoA protein was knocked down using siRNA against RhoA mRNA. The siRNA was obtained from Santa Cruz Biotechnology (#sc-29471). A negative control siRNA (#AM4611, Applied Biosystems) was used as control. The sequences of these siRNAs are not disclosed by the companies. Pre-confluent HFL-1 fibroblasts were transfected with 1 μg of siRNA against RhoA mRNA or a control siRNA using RNAiFest transfection reagent (Qiagen, Valencia, CA) in F12K medium containing 4% FBS according to the manufacturer’s protocol. The ratio of siRNA to transfection reagent was 1:3. 48 h after transfection, HFL-1 cells were exposed to hyperoxia or normoxia. Then RhoA activity and protein levels of RhoA and collagen-I were analyzed.
RNA isolation and real-time PCR
Total RNA was isolated from lung fibroblasts and lung tissue homogenates using RNAesy kit (Qiagen, Valencia, CA) according to manufacturer’s protocol. To measure COL1A1 mRNA and COL1A2 mRNA, quantitative real time RT-PCR was performed by using TaqMan® gene expression assay from Applied Biosystems (Foster City, CA). The assay IDs are Mm01302043_g1 for mouse COL1A1, Mm01165187_m1 for mouse COL1A2, Hs00164004_m1 for human COL1A1, Hs00164099_m1 for human COL1A2, and Hs03003631_g1 for 18s rRNA. The primer sequences were not disclosed by the company. ABI 7500 Sequence Detector (Perkin-Elmer Applied Biosystem, Foster City, CA) was programmed for the PCR conditions: 95°C for 10 min, 40 cycles of 95°C for 15 s and 60°C for 1 min. For the relative quantification of COL1A1 and COL1A2 mRNA contents, the comparative threshold cycle (CT) method was employed. CT shows the PCR cycles at which an increase in reporter fluorescence above a background signal can first be detected. CT values of endogenous control (18s rRNA) were first subtracted from CT values of collagen genes to derive a ΔCT value. The relative COL1A1 and COL1A2 mRNA contents were then calculated using the expression 2−ΔΔCT, where the values for ΔΔCT were obtained by subtracting ΔCT of treatment groups from ΔCT of control group.
Transfection of fibroblasts with plasmid encoding wild type RhoA, constitutively active RhoA mutant, or with dominant-negative RhoA mutant
Mammalian expression vectors containing wild type RhoA cDNA or constitutively active RhoA (Q63L), or dominant-negative RhoA mutants cDNA (T19N) (Addgene, Cambridge, MA) were transfected into HFL-1 cells using Lipofectamine LTX with PLUS reagent (Invitrogen, Carlsbad, CA) according to manufacturer protocol. 48 h after transfection, cells were exposed to hyperoxia or normoxia and then levels of RhoA and collagen-I and RhoA activity were analyzed.
Co-immunoprecipition of RhoA and GDI
The fibroblasts HFL-1 lysates or lung homogenates were incubated with anti-RhoA antibody at 4°C overnight. 30 μl of protein A sepharose was added and samples were further incubated for 2 h at 4°C. Immunoprecipitates were collected by centrifugation ad washed three times in buffer containing 50 mM Tris-HCl pH 7.5, 150 mM NaCl, and 0.1% Triton X-100. Proteins were eluted from sepharose beads by boiling the samples in 30 μl of SDS immunoblotting sample buffer. Sepharose beads were pelleted by centrifugation at 10,000 g and supernatants were analyzed for RhoA and GDI by Western blot.
Western blot analysis
The lysate proteins from cell or lung homogenates (20 to 40 μg) were separated on a 4-20% Tris-glycline SDS-PAGE and electrophoretically transferred onto nitrocellulose membranes. The membranes were incubated in blocking solution at room temperature for 1–2 h and then hybridized with primary antibodies against collagen-I, RhoA, GDI and GAPDH overnight at 4°C. The bands were detected by an immunochemiluminescence method. The density was quantitated by Bio-Rad Quantity One Software.
Measurement of superoxide radicals
HFL-1 fibroblasts and frozen sections of mouse lungs were loaded with 10 μM DHE for 30 min. After washing, fluorescence images were acquired using a confocal laser scanning microscope LSM 510. The excitation and emission wavelengths were 510 nm and 590 nm.
Cell proliferation assay
Proliferation of HFL-1 lung fibroblasts was assayed by using a kit from Roche (Indianapolis, IN, USA) that monitors the incorporation of bromodeoxyuridine (BrdU) into newly synthesized DNA. The BrdU was detected using anti-BrdU-peroxidase conjugate in accordance with the manufacturer’s instructions. After reactions were stopped, A450 was measured by SpectraMax M2e microplate reader.
Animals and hyperoxic exposure
Male C57BL/6 mice with ages between 8 and 10 weeks were purchased from the Jackson Laboratory (Bar Harbor, ME). All experiments were performed in accordance with the guiding principles of the Guide for the Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee (IACUC) of the Georgia Health Sciences University. For hyperoxic exposure, mice were kept in a clear plastic polypropylene chamber (30”×20”×20”) ad libitum with free access to food and water. The oxygen concentration in the chamber was maintained using Proox Oxygen Controller (BioSpherix, Lacona, NY) at 80% for 5 days and then changed to 50% for another 10 days (15 days in total). The oxygen mixture was humidified, and the concentration of CO2 in the chamber was lower than 0.3%. Starting at the time when being changed to 50% oxygen, the hyperoxic mice were given ROCK inhibitor Y27632 (5 mg/kg, daily by IP injection) or ROS scavenger tiron (1.5 g/kg, daily by IP injection) or same volume of PBS. The normoxic mice were kept in room air and also received daily IP injection of same amount of tiron, or Y27632, or PBS at same time as the hyperoxic mice.
Mouse lung experiments
Mice were anesthetized (pentobarbital, 90 mg/kg, i.p.) and the trachea was intubated. The mice were then euthanized by using thoracotomy. The blood in pulmonary circulation was rinsed by infusing PBS through pulmonary artery and the heart and lungs were removed. The right lungs were separated and snap-frozen in liquid nitrogen for preparing homogenates and the left lungs were filled with 4% paraformaldehyde (PFA) solution at 25 cm H2O and fixed in 4% PFA for 24 h. The assays of RhoA activity, protein contents of collagen-I and RhoA, mRNA contents of COL1A1 and COL1A2 were determined in lung homogenates.
Lung morphology analysis
After fixation, the left lungs were sliced midsagittally and embedded in paraffin. Twenty slides of 7 μm thickness were sectioned and were then subjected to Masson trichrome staining for collagen and morphological analysis. Ten slides were randomly selected for each lung and were examined by Olympus BX41 microscope. Olympus DP72 digital camera and DP2-BSW software were used to record the images. A minimum of 10 microscopic fields were examined for each slide. The collagen stain intensity was measured by color histogram mode of software ImageJ and expressed as ratio of blue color to green color as described previously [25].
Statistical analysis
In each experiment, experimental and control cells were matched for cell line, age, seeding density, number of passages, and number of days post confluence to avoid variation in tissue culture factors that can influence measurements of RhoA and collagen-I. Results are shown as the mean ± SE for n experiments. One-way ANOVA and post t-test analyses were used to determine the significance of differences between the means of different groups. P < 0.05 was considered statistically significant.
Results
Exposure of lung fibroblasts to hyperoxia results in increased expression of collagen-I
The effect of hyperoxia on the production of collagen-I by human lung fibroblasts was studied. We found that exposure of HFL-1 human lung fibroblasts to hyperoxia (95% oxygen) resulted in a time-dependent increase in collagen-I protein (figures 1A and 1B). Increased collagen-I protein was evident at 6 h and reached maximal level at 12 h. Meanwhile hyperoxia (95% oxygen) also increased the mRNA levels of COL1A1 and COL1A2 in HFL-1 cells (Figure 1C). The alteration of COL1A1 and COL1A2 mRNA was in a same time course as collagen-I protein, suggesting that the increased synthesis of collagen-I is due to increased expression of collagen mRNA COL1A1 and COL1A2. The increases in the protein contents and mRNA of collagen-I are not due to increases in the amount of cells caused by increased proliferation, as these results have been normalized to internal control levels of GAPDH and 18S RNA.
Figure 1.

Hyperoxia increases collagen-I synthesis in lung fibroblasts. Human lung HFL-1 fibroblasts were exposed to 95% oxygen for 3–24 h after which protein contents of collagen-I (A and B) and mRNA levels of COL1A1 and COL1A2 (C) were determined as described in Materials and Methods. (A) is representative blots of 4 separate experiments. (B) is a bar graph depicting the increase in collagen-I protein. Results are expressed as mean ± SE; n=4 experiments. * P < 0.05 vs. 0 time point.
Hyperoxia induces RhoA activation in HFL-1 lung fibroblasts
To determine whether hyperoxia activates RhoA, HFL-1 human lung fibroblasts were exposed to hyperoxia (95% oxygen) for 3–24 h and RhoA activation then was measured by using a pull-down assay. As shown in Figure 2, exposure of fibroblasts to 95% oxygen results in a time-dependent increase in the protein contents of active RhoA whereas total RhoA protein did not change. The increase in active RhoA was evident at 6 h of hyperoxic exposure and reached maximum at 12 h. These data indicate that hyperoxia induces RhoA activation in HFL-1 lung fibroblasts.
Figure 2.

Hyperoxia induces RhoA activation in lung fibroblasts. HFL-1 cells were exposed to 95% oxygen for 3–24 h after which the active and total RhoA in cell lysates was determined as described in Materials and Methods. (A) is representative blots of 4 separate experiments. (B) is a bar graph depicting the increase in active RhoA protein. Results are expressed as mean ± SE; n=4 experiments. * P < 0.05 vs. 0 time point.
Inhibitors of RhoA and ROCK prevent hyperoxia-induced increases in collagen synthesis and cell proliferation
To study whether RhoA activation is involved in hyperoxia-induced collagen synthesis, HFL-1 lung fibroblasts were exposed to hyperoxia (95% oxygen) for 24 h in the presence and absence of cell-permeable Rho inhibitor, C-3 transferase (CT-04, 1 μg/ml) or ROCK inhibitor, Y27632 (10 μM). We found that both CT-04 and Y27632 prevented hyperoxia-induced increase in collagen-I protein content (Figures 3A and 3B). Moreover, CT-04 and Y27632 blocked the increase in mRNA levels of COL1A1 and COL1A2 in hyperoxic HFL-1 cells (Figure 3C). Additionally, as shown in supplemental figure 1, exposure of lung fibroblasts to 80% oxygen for 12 h and then 50% oxygen for another 12 h caused an increase in cell proliferation. C-3 transferase (CT-04, 1 μg/ml) attenuated hyperoxia-induced increase in cell proliferation. These results suggest that RhoA activation is associated to hyperoxia-induced expression of collagen mRNA COL1A1 and COL1A2, collagen-I synthesis, and cell proliferation.
Figure 3.

RhoA inhibitor CT-04 and ROCK inhibitor Y27632 attenuate hyperoxia-induced increase in collagen-I synthesis in lung fibroblasts. HFL-1 cells were exposed to 95% oxygen in the presence and absence of cell-permeable Rho inhibitor C-3 transferase (CT-04, 1 μg/ml) or ROCK inhibitor Y27632 (10 μM) for 24 h after which protein contents of collagen-I (A and B) and mRNA levels of COL1A1 and COL1A2 (C) were determined as described in Materials and Methods. (A) is representative blots of 4 separate experiments. (B) is a bar graph depicting the changes in collagen-I protein. Results are expressed as mean ± SE; n=4 experiments. * P < 0.05 vs. normoxia group.
Over-expression of constitutively active RhoA mutant Q63L increases collagen-1 protein contents
To examine whether active RhoA levels are positively related to the synthesis of collagen, HFL-1 lung fibroblasts were transfected with plasmids containing wild type RhoA and constitutively active Rho-A mutant Q63L [26]. As shown in Figures 4A and 4B, the collagen-I protein contents were much higher in HFL-1 cell with constitutively active Rho-A mutant Q63L, suggesting that over-expression of constitutively active Rho-A mutant increased collagen-I synthesis. This result provides additional evidence that active RhoA is positively related to the collagen synthesis.
Figure 4.


RhoA activation is responsible for hyperoxia-induced increase in collagen-I synthesis in lung fibroblasts. (A) and (B): HFL-1 fibroblasts were transfected with or without plasmids containing wild-type RhoA gene and constitutively active RhoA mutant Q63L. 48 h after transfection, protein contents of collagen-I and GFP (a tag in the plasmids) were assayed. (A) is a representative blots of 4 separate experiments. (B) is a bar graph depicting the changes in collagen-I and GFP protein. Results are expressed as mean ± SE; n=4 experiments. * P < 0.05 vs. the group of wild-type plasmids (WT plasmids). (C) and (D): HFL-1 fibroblasts were transfected with plasmids containing wild-type RhoA gene and dominant negative RhoA mutant T19N gene. 48 h after transfection, cells were exposed normoxia or hyperoxia (95% oxygen) for 24 h. The protein contents of collagen-I were assayed. (C) is representative blots of 4 separate experiments. (D) is a bar graph depicting the changes in collagen-I protein. Results are expressed as mean ± SE; n=4 experiments. * P < 0.05 vs. normoxia group. (E) and (F): HFL-1 fibroblasts were transfected with control siRNA or RhoA siRNA. 48 h after transfection, cells were exposed normoxia or hyperoxia (95% oxygen) for 24 h. The protein contents of collagen-I and total RhoA were assayed. (E) is representative blots of 4 separate experiments. (F) is a bar graph depicting the changes in collagen-I protein. Results are expressed as mean ± SE; n=4 experiments. * P < 0.05 vs. normoxia group.
Over-expression of dominant-negative RhoA mutant T19N and RhoA siRNA prevent hyperoxia-induced increase in collagen synthesis
To further study whether RhoA activation is responsible for hyperoxia-induced collagen synthesis, we determined the collagen-I synthesis in HFL-1 lung fibroblasts transfected with plasmids containing wild type RhoA and dominant-negative RhoA mutant T19N [26] as well as control siRNA and RhoA siRNA under normoxic and hyperoxic condition. As shown in Figures 4C and 4D, hyperoxia increased collagen-I protein contents in HFL-1 cells transfected with plasmids containing wild type RhoA. However, hyperoxia-induced elevation of collagen-I protein was prevented in HFL-1 cells transfected with plasmids containing dominant-negative RhoA mutant T19N. Furthermore, knocking-down RhoA expression using RhoA siRNA prevented the increase in collagen-I protein in hyperoxic lung fibroblasts (Figures 4E and 4F). Together, these results indicated that RhoA activation mediates hyperoxia-induced collagen-I synthesis.
ROS scavenger tiron prevents hyperoxia-induced RhoA activation and collagen synthesis
In order to study the role of ROS in hyperoxia-induced RhoA activation and collagen synthesis, lung fibroblasts were exposed to hyperoxia (95% oxygen) for 24 h in the presence and absence of tiron, a ROS scavenger. As shown in supplemental figure 4, hyperoxia significantly increased ROS production and tiron (5 mM) prevented the increase in ROS in hyperoxic HFL-1 cells. Importantly, we found that hyperoxia induced increases in active RhoA and collagen-I protein contents in HFL-1 cells (Figure 5). Moreover, increases in active RhoA and collagen-I protein were prevented in hyperoxic HFL-1 cells treated with tiron (5 mM) (Figure 5), indicating that RhoA activation and RhoA-mediated collagen-I synthesis in lung fibroblasts are ROS-dependent.
Figure 5.

ROS scavenger tiron prevents hyperoxia-induced RhoA activation and collagen-I expression in fibroblasts. HFL-1 cells were exposed to 95% oxygen or normoxia in the presence and absence of membrane-permeable ROS scavenger tiron (5 mM) for 24 h after which RhoA activity and collagen-I protein content were measured as described in Materials and Methods. (A) is representative blots of 4 separate experiments. (B) is a bar graph depicting the changes in active RhoA protein. (C) is a bar graph depicting the changes in collagen-I protein. Results are expressed as mean ± SE; n=4 experiments. * P < 0.05 vs. normoxia group.
ROS scavenger tiron prevents the decrease in association of RhoA and GDI in hyperoxic HFL-1 fibroblasts
To study the mechanism for hyperoxia-induced activation of RhoA, we analyzed the association of RhoA and GDI using co-immunoprecipitation. As shown in Figure 6, exposure of HFL-1 fibroblasts to hyperoxia (95% oxygen) for 6 h resulted in a decrease in the amount of GDI pulled down by immunoprecipitation using anti-RhoA antibody. In addition, tiron prevented the decrease in the amount of GDI pulled down by RhoA antibody (Figure 6). These data indicated that hyperoxia inhibits the association between RhoA and GDI. Increase in ROS production in hyperoxic lung fibroblasts causes the dissociation of RhoA from GDI leading to RhoA activation.
Figure 6.

The effect of hyperoxia and tiron on RhoA-GDI association in lung fibroblasts. HFL-1 fibroblasts were exposed to 95% oxygen or normoxia in the presence and absence of membrane-permeable ROS scavenger tiron (5 mM) for 24 h after which RhoA-GDI association were assayed by co-immunoprecipitation using anti-RhoA antibody. (A) is representative blots of 5 separate experiments. (B) is a bar graph depicting changes in the amount of GDI protein pulled down by using anti-RhoA antibody. Results are expressed as mean ± SE; n=5 experiments. * P < 0.05 vs. normoxia group.
Effects of tiron and hyperoxia (80% and then 50% oxygen) on cell proliferation, collagen-I protein, RhoA activation, and RhoA-GDI association in lung fibroblasts
We determined cell proliferation, collagen-I protein, and RhoA activation in lung fibroblasts exposed to 80% oxygen for 12 h and then 50% oxygen for another 12 h in the presence and absence of ROS scavenger tiron. As shown in supplemental figures 2 and 3, exposure of lung fibroblasts to 80% oxygen for 12 h and then 50% oxygen for another 12 h caused increases in collagen-I protein and RhoA activation and a decrease in RhoA-GDI association. Tiron (5 mM) attenuated increases in cell proliferation (supplemental figure 1), collagen-I protein, and RhoA activation and the decrease in RhoA-GDI association (supplemental figures 2 and 3).
ROCK inhibitor Y27632 attenuates hyperoxia-induced collagen synthesis in mouse lungs
To confirm our in vitro observation of collagen synthesis mediated by activated RhoA/ROCK pathway in hyperoxic lung fibroblasts, we utilize an animal model of hyperoxia-induced lung fibrosis. Male C57BL/6 mice were exposed to 80% oxygen for 5 days, and then exposed to 50% oxygen for another 10 days. Starting at the time when being changed to 50% oxygen, the hyperoxic mice were given daily injection of ROCK inhibitor Y27632 (5 mg/kg, daily by IP injection) or same volume of PBS. The normoxic mice were kept in room air and also received daily IP injection of same amount of Y27632 or PBS at same time. We found that the hyperoxic lungs showed remarkable fibrosis with Masson’s trichrome staining (Figures 7A and 7B). Y27632 attenuated collagen staining in hyperoxic lungs (Figures 7A and 7B). The collagen synthesis was further confirmed by Western blotting analysis of lung homogenates. As shown in Figures 7C, 7D and 7E, the homogenates from hyperoxic mouse lungs contained much higher levels of collagen-I protein and COL1A1 and COL1A2 mRNA. Y27632 inhibited increases in collagen-I protein and COL1A1 and COL1A2 mRNA in hyperoxic mouse lungs (Figures 7C, 7D and 7E). These results indicated that ROCK activation is responsible for hyperoxia-induced collagen-I synthesis and lung fibrosis.
Figure 7.


ROCK inhibitor Y27632 attenuates collagen-I synthesis in mouse lungs of oxygen toxicity. Male C57BL/6 mice were exposed to 80% oxygen for 5 days, and then exposed to 50% oxygen for another 10 days. Starting at the time when being changed to 50% oxygen, the hyperoxic mice were given daily injection of ROCK inhibitor Y27632 (5 mg/kg, daily by IP injection) or same volume of PBS. The normoxic mice were kept in room air and also received daily IP injection of same dose of tiron or PBS at same time. (A) is representative images of lung sections of Masson trichrome staining. (B) is a bar graph depicting changes in the intensity of collagen staining in lung tissues expressed as ratio of bue to green color. (C) is representative blots of 8 experiments. (D) is a bar graph depicting changes in collagen-I protein in lung tissues. (E) is a bar graph depicting changes in COL1A1 and COL1A2 mRNA levels in lung tissues. Results are expressed as mean ± SE; n=8 experiments. * P < 0.05 vs. normoxia group; ** P<0.05 vs. normoxia + Y27632 group; # P<0.05 vs. hyperoxia group.
ROS scavenger tiron prevents hyperoxia-induced RhoA activation and the dissociation of RhoA and GDI in mouse lungs
Male C57BL/6 mice were exposed to 80% oxygen for 5 days, and then exposed to 50% oxygen for another 10 days (15 days in total). The hyperoxic mice were given daily injection of ROS scavenger tiron (1.5 g/kg, daily by IP injection) or same volume of PBS starting at the time when being changed to 50% oxygen. The normoxic mice were kept in room air and also received daily IP injection of same dose of tiron or PBS at same time. We found that exposure of mice to 80% oxygen for 5 days (day 5) caused an increase in RhoA activity in lungs (Figures 8A and 8B). RhoA activity continued to increase after being changed to 50% oxygen for 10 days (day 15) (Figures 8A and 8B). There was a lower amount of GDI protein pulled down by immunoprecipitation using RhoA antibody in the homogenates from hyperoxic lungs compared with that from normoxic lungs (Figures 8E and 8F). More importantly, tiron prevented the increase in ROS production in hyperoxic lungs (supplemental figure 5). Scavenging ROS by tiron prevented the increase in RhoA activation and the decrease in the association of RhoA and GDI in hyperoxic lungs (Figures 8C, 8D, 8E, and 8E). These data indicate that the activation of RhoA and the dissociation of RhoA and GDI are attributed to the increase in ROS production in hyperoxia-induced lung fibrosis.
Figure 8.


The effect of hyperoxia and tiron on RhoA activation and RhoA-GDI association in mouse lungs of oxygen toxicity. Male C57BL/6 mice were exposed to 80% oxygen for 5 days, and then exposed to 50% oxygen for another 10 days (15 days in total). Starting at the time when being changed to 50% oxygen, the hyperoxic mice were given daily injection of ROS scavenger tiron (1.5 g/kg, daily by IP injection) or same volume of PBS. The normoxic mice were kept in room air and also received daily IP injection of same dose of tiron or PBS at same time. (A) is representive blots of 5 experiments. (B) is a bar graph showing time-dependent changes in RhoA activity. (C) is representative blots of 8 experiments. (D) is a bar graph depicting changes in collagen-I protein in lung tissues (day 15). (E) is representative blots of co-immunoprecipitation of RhoA and GDI of 8 exprements. (F) is a bar graph depicting changes in the amount of GDI protein pulled down by using anti-RhoA antibody in lung homogenates (day 15). Results are expressed as mean ± SE; n=8 experiments. # P < 0.05 vs. day 0; * P < 0.05 vs. normoxia group.
Tiron prevents hyperoxia-induced collagen synthesis in mouse lungs
To confirm our in vitro observation that collagen synthesis mediated by activated RhoA is dependent on ROS production in hyperoxic lung fibroblasts, we performed Masson’s trichrome staining on the lung slides from normoxic and hyperoxic mice treated with or without tiron in a same schedule as in the experiments described above. As shown in Figures 9A and 9B, the hyperoxic lungs showed remarkable fibrosis. Tiron attenuated collagen staining in hyperoxic lungs (Figures 9A and 9B). Furthermore, tiron inhibited the increases in collagen-I protein and COL1A1 and COL1A2 mRNA in hyperoxic mouse lungs (Figures 9C, 9D and 9E). Together, these results showed that collagen-I synthesis and lung fibrosis mediated by activated RhoA is due to the increase in ROS production in hyperoxic lungs.
Figure 9.
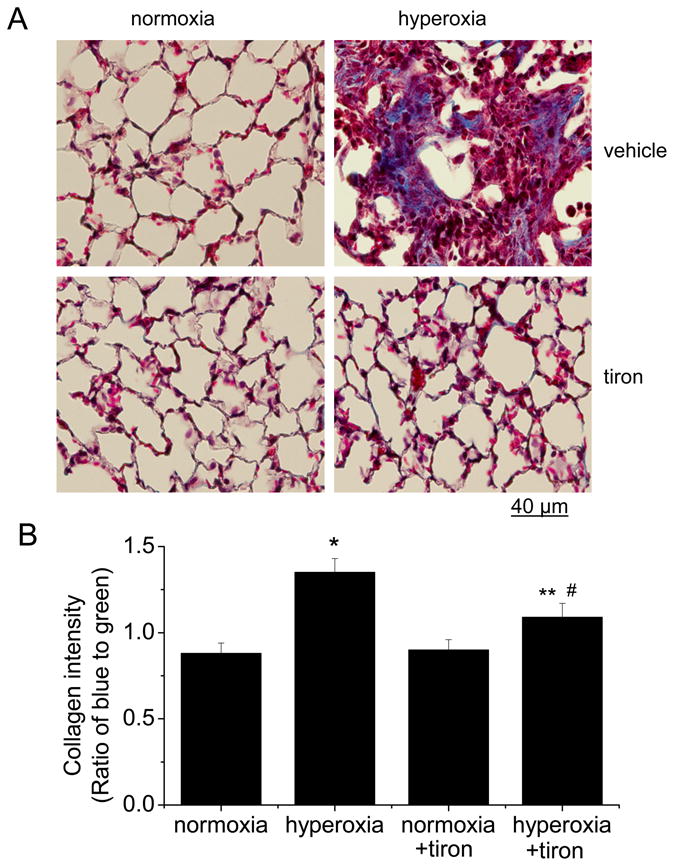

Tiron attenuates collagen-I synthesis in mouse lungs of oxygen toxicity. Male C57BL/6 mice were exposed to 80% oxygen for 5 days, and then exposed to 50% oxygen for another 10 days. The hyperoxic mice were given daily injection of ROS scavenger tiron (1.5 g/kg, daily by IP injection) or same volume of PBS starting at the time when being changed to 50% oxygen. The normoxic mice were kept in room air and also received daily IP injection of same dose of tiron or PBS at same time. (A) is representative images of lung sections of Masson trichrome staining. (B) is a bar graph depicting changes in the intensity of collagen staining in lung tissues expressed as ratio of bue to green color. (C) is representative blots of 8 experiments. (D) is a bar graph depicting changes in collagen-I protein in lung tissues. (E) is a bar graph depicting changes in COL1A1 and COL1A2 mRNA levels in lung tissues. Results are expressed as mean ± SE; n=8 experiments. * P < 0.05 vs. normoxia group; ** P<0.05 vs. normoxia + tiron group; # P<0.05 vs. hyperoxia group.
Discussion
The goal of the present study was to determine the signal transduction mechanism which is responsible for fibrotic lesion and extracellular matrix deposition in lungs of oxygen toxicity. To this end, we utilized two models: a cell model of human lung fibroblasts exposed to 95% oxygen, and an animal model of mice exposed to 80% hyperoxia for 5 days and then to 50% oxygen for 10 days. We found that hyperoxia induces collagen-I synthesis and RhoA activation in both lung fibroblasts and mouse lungs. Inhibition of RhoA activation using RhoA inhibitor CT04, ROCK inhibitor Y27632, RhoA siRNA, or dominant-negative mutant T19N attenuates hyperoxia-induced collagen-I synthesis from lung fibroblasts. Moreover, expression of constitutively active RhoA mutant Q63L increases collagen-I synthesis in lung fibroblasts. More importantly, we also showed that ROCK inhibitor Y27632 attenuates collagen synthesis and fibrosis in mouse lungs exposed to hyperoxia. These results indicate that RhoA activation mediates hyperoxia-induced collagen-I synthesis and lung fibrosis.
Exposure of cultured lung fibroblasts to high concentrations of oxygen leads to increased formation of ROS [27]. In animal models, lungs exposed to high concentrations of oxygen contain large amount of ROS [28]. The source of hyperoxia-induced ROS production might be the mitochondrial electron transport [9] or NADPH oxidase-catalyzed reaction [10,11]. It has been reported that ROS are involved in the pathogenesis of idiopathic pulmonary fibrosis (IPF) and the animal model of bleomycin-induced lung fibrosis [14–18]. To investigate whether ROS are responsible for lung fibrosis induced by oxygen toxicity, we determined the effects of ROS scavenger tiron on collagen synthesis and cell proliferation of lung fibroblasts and collagen deposition in mouse lungs exposed to hyperoxia. Our data indicate that scavenging ROS using tiron prevents the increase in collagen synthesis and cell proliferation of hyperoxic lung fibroblasts and attenuates collagen deposition in mouse lungs of oxygen toxicity, indicating that ROS are responsible for collagen synthesis and lung fibrosis induced by oxygen toxicity.
Several mechanisms might be involved in ROS-induced collagen synthesis in hyperoxic lung fibroblasts. First, ROS may induce activation and release TGF-β1 from lung fibroblasts. It has been well-documented that TGF-β1 induces collagen synthesis from lung fibroblasts which is critical in the formation of fibroproliferative lung lesion [29,30]. However, our data shows that the levels of both active and total TGF-β1 in the medium of normoxic and hyperoxic fibroblasts are comparable (supplemental figure 6), suggesting that TGF-β1 is not responsible for ROS-induced collagen-I synthesis in hyperoxic lung fibroblasts. Second, having proven in the present study that RhoA activation mediates hyperoxia-induced collagen-I synthesis and lung fibrosis, we evaluated whether ROS-induced collagen-I synthesis in hyperoxic lung fibroblasts occurs via RhoA pathway. We found that scavenging ROS using tiron prevents the increase in RhoA activation in hyperoxic lung fibroblasts and mouse lungs of oxygen toxicity, indicating that RhoA activation is required in ROS-induced collagen-I synthesis in hyperoxic lung fibroblasts. These data are consistent with the reports by Knock et al [23] and Chi et al [24] that ROS cause RhoA activation in vascular smooth muscle cells and endothelial cells.
RhoA activity is regulated by guanine nucleotide exchange factor (GEF) and guanine nucleotide dissociation inhibitor (GDI). GEF facilitates GTP-bound active RhoA state by promoting the exchange of GDP for GTP, whereas association of GDI to RhoA promotes GDP-bound inactive state [20]. We found that exposure of lung fibroblasts and mouse lung to hyperoxia causes a decrease in the association of GDI to RhoA. Furthermore, tiron prevents the decrease in the association of GDI to RhoA in hyperoxic lung fibroblasts and lungs of oxygen toxicity. These data provide the first evidence that dissociation of GDI from RhoA contributes to ROS-induced RhoA activation in hyperoxia-induced lung fibrosis.
The exact mechanisms of RhoA-mediated collagen-I synthesis in hyperoxic lung fibroblasts have not been completely understood. Activation of RhoA and Rho kinase leads to increased F-actin assembly. The resulting depletion of G-actin triggers activation of the transcription factor serum response factor (SRF) which initiate the expression of genes involving motility, differentiation and proliferation [31–33]. However, there is no direct evidence showing that collagen-I is a direct transcriptional target of SRF. In our experiments, treatment of lung fibroblasts with F-actin disrupter, swinholide A, prevented hyperoxia-induced collagen-I synthesis (supplemental figure 7), suggesting that actin cytoskeleton is involved in RhoA-mediated collagen-I synthesis and that collagen-I might be a direct transcriptional target of SRF. Further studies are necessary to confirm this speculation.
Exposure to hyperoxia is a well-recognized cause of lung injury [34,35]. In patients with neonatal or adult respiratory distress syndrome, however, high levels of oxygen are often administered with mechanical ventilation. Volutrauma or barotrauma due to ventilation with high tidal volumes causes lung injury and induces acute and chronic inflammatory responses [36]. Hyperoxia can exacerbate ventilator-induced lung injury [37–39]. Moreover, preexposure to hyperoxia causes increased lung injury and epithelial apoptosis in mice ventilated with high tidal volumes [40]. Thus, interaction between hyperoxia and mechanical stretch orchestrates the pathogenesis of acute lung injury [41].
In summary, our data provide the evidence for the first time that ROS-dependent decrease in the binding of RhoA to GDI leads to RhoA activation in hyperoxic lung fibroblasts and mouse lungs with oxygen toxicity. Our whole animal and cellular studies demonstrate that activation of RhoA/ROCK pathway contributes to increased collagen-I synthesis in hyperoxic lung fibrosis (Figure 10). Therefore, ROS scavengers and RhoA/ROCK inhibitors have therapeutic potential for the prevention and treatment of lung fibrosis caused by oxygen toxicity.
Figure 10.

A schematic pathway illustrating the role of ROS-dependent RhoA activation in collagen synthesis and lung fibrosis in oxygen toxicity. Hyperoxia induces the formation of ROS which causes RhoA-GDI dissociation and RhoA activation. RhoA activation leads to the increase in collagen synthesis in lung fibroblasts and lung fibrosis caused by oxygen toxicity.
Supplementary Material
Acknowledgments
We thank Dr. William R. Caldwell for critical reading of the manuscript.
This work was supported by NIH grant R01HL088261 (to YS), Flight Attendants Medical Research Institute grant 072104 (to YS), and American Heart Association Greater Southeast Affiliate grant 0855338E (to YS).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Meyrick B. Pathology of the adult respiratory distress syndrome. Crit Care Clin. 1986;2:405–428. [PubMed] [Google Scholar]
- 2.Davis WB, Rennard SI, Bitterman PB, Crystal RG. Pulmonary oxygen toxicity. Early reversible changes in human alveolar structures induced by hyperoxia. N Engl J Med. 1983;309:878–883. doi: 10.1056/NEJM198310133091502. [DOI] [PubMed] [Google Scholar]
- 3.Riley DJ, Kerr JS, Yu SY. Effect of proline analogs on oxygen toxicity-induced pulmonary fibrosis in the rat. Toxicol Appl Pharmacol. 1984;75:554–560. doi: 10.1016/0041-008x(84)90192-3. [DOI] [PubMed] [Google Scholar]
- 4.Weinberger B, Laskin DL, Heck DE, Laskin JD. Oxygen toxicity in premature infants. Toxicol Appl Pharmacol. 2002;181:60–67. doi: 10.1006/taap.2002.9387. [DOI] [PubMed] [Google Scholar]
- 5.Chen N, Li JJ, Xue XD. Effect of losartan on lung fibrosis in neonatal rats with hyperoxia-induced chronic lung disease. Zhongguo Dang Dai Er Ke Za Zhi. 2007;9:591–594. [PubMed] [Google Scholar]
- 6.Ekekezie II, Thibeault DW, Rezaeikhaligh MH, Mabry SM, Norberg M, Reddy GK, Youssef J, Truog WE. High-dose inhaled nitric oxide and hyperoxia increases lung collagen accumulation in piglets. Biol Neonate. 2000;78:198–206. doi: 10.1159/000014271. [DOI] [PubMed] [Google Scholar]
- 7.Chen CM, Wang LF, Chou HC, Lang YD, Lai YP. Up-regulation of connective tissue growth factor in hyperoxia-induced lung fibrosis. Pediatr Res. 2007;62:128–133. doi: 10.1203/PDR.0b013e3180987202. [DOI] [PubMed] [Google Scholar]
- 8.Lang YD, Hung CL, Wu TY, Wang LF, Chen CM. The renin-angiotensin system mediates hyperoxia-induced collagen production in human lung fibroblasts. Free Radic Biol Med. 2010;49:88–95. doi: 10.1016/j.freeradbiomed.2010.03.022. [DOI] [PubMed] [Google Scholar]
- 9.Li J, Gao X, Qian M, Eaton JW. Mitochondrial metabolism underlies hyperoxic cell damage. Free Radic Biol Med. 2004;36:1460–1470. doi: 10.1016/j.freeradbiomed.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 10.Parinandi NL, Kleinberg MA, Usatyuk PV, Cummings RJ, Pennathur A, Cardounel AJ, Zweier JL, Garcia JG, Natarajan V. Hyperoxia-induced NAD(P)H oxidase activation and regulation by MAP kinases in human lung endothelial cells. Am J Physiol Lung Cell Mol Physiol. 2003;284:L26–L38. doi: 10.1152/ajplung.00123.2002. [DOI] [PubMed] [Google Scholar]
- 11.Usatyuk PV, Romer LH, He D, Parinandi NL, Kleinberg ME, Zhan S, Jacobson JR, Dudek SM, Pendyala S, Garcia JG, Natarajan V. Regulation of hyperoxia-induced NADPH oxidase activation in human lung endothelial cells by the actin cytoskeleton and cortactin. J Biol Chem. 2007;282:23284–23295. doi: 10.1074/jbc.M700535200. [DOI] [PubMed] [Google Scholar]
- 12.Brueckl C, Kaestle S, Kerem A, Habazettl H, Krombach F, Kuppe H, Kuebler WM. Hyperoxia-induced reactive oxygen species formation in pulmonary capillary endothelial cells in situ. Am J Respir Cell Mol Biol. 2006;34:453–463. doi: 10.1165/rcmb.2005-0223OC. [DOI] [PubMed] [Google Scholar]
- 13.Gore A, Muralidhar M, Espey MG, Degenhardt K, Mantell LL. Hyperoxia sensing: From molecular mechanisms to significance in disease. J Immunotoxicol. 2010 doi: 10.3109/1547691X.2010.492254. [DOI] [PubMed] [Google Scholar]
- 14.Lu Y, Azad N, Wang L, Iyer AK, Castranova V, Jiang BH, Rojanasakul Y. Phosphatidylinositol-3-kinase/akt regulates bleomycin-induced fibroblast proliferation and collagen production. Am J Respir Cell Mol Biol. 2010;42:432–441. doi: 10.1165/rcmb.2009-0002OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cho HY, Kleeberger SR. Genetic mechanisms of susceptibility to oxidative lung injury in mice. Free Radic Biol Med. 2007;42:433–445. doi: 10.1016/j.freeradbiomed.2006.11.021. [DOI] [PubMed] [Google Scholar]
- 16.Manoury B, Nenan S, Leclerc O, Guenon I, Boichot E, Planquois JM, Bertrand CP, Lagente V. The absence of reactive oxygen species production protects mice against bleomycin-induced pulmonary fibrosis. Respir Res. 2005;6:11. doi: 10.1186/1465-9921-6-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Teixeira KC, Soares FS, Rocha LG, Silveira PC, Silva LA, Valenca SS, Dal PF, Streck EL, Pinho RA. Attenuation of bleomycin-induced lung injury and oxidative stress by N-acetylcysteine plus deferoxamine. Pulm Pharmacol Ther. 2008;21:309–316. doi: 10.1016/j.pupt.2007.07.006. [DOI] [PubMed] [Google Scholar]
- 18.Yen HC, Chang HM, Majima HJ, Chen FY, Li SH. Levels of reactive oxygen species and primary antioxidant enzymes in WI38 versus transformed WI38 cells following bleomcyin treatment. Free Radic Biol Med. 2005;38:950–959. doi: 10.1016/j.freeradbiomed.2004.12.022. [DOI] [PubMed] [Google Scholar]
- 19.Raftopoulou M, Hall A. Cell migration: Rho GTPases lead the way. Dev Biol. 2004;265:23–32. doi: 10.1016/j.ydbio.2003.06.003. [DOI] [PubMed] [Google Scholar]
- 20.Faure J, Dagher MC. Interactions between Rho GTPases and Rho GDP dissociation inhibitor (Rho-GDI) Biochimie. 2001;83:409–414. doi: 10.1016/s0300-9084(01)01263-9. [DOI] [PubMed] [Google Scholar]
- 21.Hemnes AR, Zaiman A, Champion HC. PDE5A inhibition attenuates bleomycin-induced pulmonary fibrosis and pulmonary hypertension through inhibition of ROS generation and RhoA/Rho kinase activation. Am J Physiol Lung Cell Mol Physiol. 2008;294:L24–L33. doi: 10.1152/ajplung.00245.2007. [DOI] [PubMed] [Google Scholar]
- 22.Ou XM, Feng YL, Wen FQ, Huang XY, Xiao J, Wang K, Wang T. Simvastatin attenuates bleomycin-induced pulmonary fibrosis in mice. Chin Med J (Engl ) 2008;121:1821–1829. [PubMed] [Google Scholar]
- 23.Knock GA, Snetkov VA, Shaifta Y, Connolly M, Drndarski S, Noah A, Pourmahram GE, Becker S, Aaronson PI, Ward JP. Superoxide constricts rat pulmonary arteries via Rho-kinase-mediated Ca(2+) sensitization. Free Radic Biol Med. 2009;46:633–642. doi: 10.1016/j.freeradbiomed.2008.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chi AY, Waypa GB, Mungai PT, Schumacker PT. Prolonged hypoxia increases ROS signaling and RhoA activation in pulmonary artery smooth muscle and endothelial cells. Antioxid Redox Signal. 2010;12:603–610. doi: 10.1089/ars.2009.2861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ukang S, Ampawong S, Kengkoom K. Collagen Measurement and Staining Pattern of Wound Healing Comparison with Fixations and Stains. J Microscopy Society of Tailand. 2008;22:37–41. [Google Scholar]
- 26.Subauste MC, Von HM, Benard V, Chamberlain CE, Chuang TH, Chu K, Bokoch GM, Hahn KM. Rho family proteins modulate rapid apoptosis induced by cytotoxic T lymphocytes and Fas. J Biol Chem. 2000;275:9725–9733. doi: 10.1074/jbc.275.13.9725. [DOI] [PubMed] [Google Scholar]
- 27.Conconi MT, Baiguera S, Guidolin D, Furlan C, Menti AM, Vigolo S, Belloni AS, Parnigotto PP, Nussdorfer GG. Effects of hyperbaric oxygen on proliferative and apoptotic activities and reactive oxygen species generation in mouse fibroblast 3T3/J2 cell line. J Investig Med. 2003;51:227–232. doi: 10.1136/jim-51-04-24. [DOI] [PubMed] [Google Scholar]
- 28.Singleton PA, Pendyala S, Gorshkova IA, Mambetsariev N, Moitra J, Garcia JG, Natarajan V. Dynamin 2 and c-Abl are novel regulators of hyperoxia-mediated NADPH oxidase activation and reactive oxygen species production in caveolin-enriched microdomains of the endothelium. J Biol Chem. 2009;284:34964–34975. doi: 10.1074/jbc.M109.013771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Salazar KD, Lankford SM, Brody AR. Mesenchymal stem cells produce Wnt isoforms and TGF-beta1 that mediate proliferation and procollagen expression by lung fibroblasts. Am J Physiol Lung Cell Mol Physiol. 2009;297:L1002–L1011. doi: 10.1152/ajplung.90347.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nonaka M, Pawankar R, Fukumoto A, Yagi T. Heterogeneous response of nasal and lung fibroblasts to transforming growth factor-beta 1. Clin Exp Allergy. 2008;38:812–821. doi: 10.1111/j.1365-2222.2008.02959.x. [DOI] [PubMed] [Google Scholar]
- 31.Morita T, Mayanagi T, Sobue K. Reorganization of the actin cytoskeleton via transcriptional regulation of cytoskeletal/focal adhesion genes by myocardin-related transcription factors (MRTFs/MAL/MKLs) Exp Cell Res. 2007;313:3432–3445. doi: 10.1016/j.yexcr.2007.07.008. [DOI] [PubMed] [Google Scholar]
- 32.Mulder J, Ariaens A, van Horck FP, Moolenaar WH. Inhibition of RhoA-mediated SRF activation by p116Rip. FEBS Lett. 2005;579:6121–6127. doi: 10.1016/j.febslet.2005.09.083. [DOI] [PubMed] [Google Scholar]
- 33.Saka M, Obata K, Ichihara S, Cheng XW, Kimata H, Noda A, Izawa H, Nagata K, Yokota M. Attenuation of ventricular hypertrophy and fibrosis in rats by pitavastatin: potential role of the RhoA-extracellular signal-regulated kinase-serum response factor signalling pathway. Clin Exp Pharmacol Physiol. 2006;33:1164–1171. doi: 10.1111/j.1440-1681.2006.04508.x. [DOI] [PubMed] [Google Scholar]
- 34.Otterbein LE, Kolls JK, Mantell LL, Cook JL, Alam J, Choi AM. Exogenous administration of heme oxygenase-1 by gene transfer provides protection against hyperoxia-induced lung injury. J Clin Invest. 1999;103:1047–1054. doi: 10.1172/JCI5342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nash G, Blennerhassett JB, Pontoppidan H. Pulmonary lesions associated with oxygen therapy and artifical ventilation. N Engl J Med. 1967;276:368–374. doi: 10.1056/NEJM196702162760702. [DOI] [PubMed] [Google Scholar]
- 36.Kaynar AM, Houghton AM, Lum EH, Pitt BR, Shapiro SD. Neutrophil elastase is needed for neutrophil emigration into lungs in ventilator-induced lung injury. Am J Respir Cell Mol Biol. 2008;39:53–60. doi: 10.1165/rcmb.2007-0315OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li LF, Liao SK, Ko YS, Lee CH, Quinn DA. Hyperoxia increases ventilator-induced lung injury via mitogen-activated protein kinases: a prospective, controlled animal experiment. Crit Care. 2007;11:R25. doi: 10.1186/cc5704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dos Santos CC. Hyperoxic acute lung injury and ventilator-induced/associated lung injury: new insights into intracellular signaling pathways. Crit Care. 2007;11:126. doi: 10.1186/cc5733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ehlert CA, Truog WE, Thibeault DW, Garg U, Norberg M, Rezaiekhaligh M, Mabry S, Ekekezie II. Hyperoxia and tidal volume: Independent and combined effects on neonatal pulmonary inflammation. Biol Neonate. 2006;90:89–97. doi: 10.1159/000092005. [DOI] [PubMed] [Google Scholar]
- 40.Makena PS, Luellen CL, Balazs L, Ghosh MC, Parthasarathi K, Waters CM, Sinclair SE. Preexposure to hyperoxia causes increased lung injury and epithelial apoptosis in mice ventilated with high tidal volumes. Am J Physiol Lung Cell Mol Physiol. 2010;299:L711–L719. doi: 10.1152/ajplung.00072.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sinclair SE, Altemeier WA, Matute-Bello G, Chi EY. Augmented lung injury due to interaction between hyperoxia and mechanical ventilation. Crit Care Med. 2004;32:2496–2501. doi: 10.1097/01.ccm.0000148231.04642.8d. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.