

Summary
PKA holoenzymes contain two catalytic (C) and a regulatory (R) subunit dimer where the two R-subunits are linked by an N-terminal Dimerization/Docking (D/D) domain. Cooperative binding of four cAMP molecules induces major structural changes in the R-subunits that cause kinase activation. While cooperativity exists between the two tandem cAMP binding domains, additional levels of cooperativity are associated with the tetramer. This allostery cannot be appreciated by studying heterodimers formed between C-subunit and deletion mutants of R that lack the D/D domain. Of critical importance is the flexible linker in the R-subunit that contains the Inhibitor Site (IS) that mimics the PKA substrate sequence and binds to the active site of the C-subunit. Two flexible linkers connect the IS to the D/D domain (N-Linker) and to the cAMP binding domains (C-Linker). In the cAMP-bound conformation IS and the C-Linker are disordered but become structured at the R:C interface when RIα binds to the C-subunit. The overall conformation of the tetramer, however, is mediated in large part by the N-Linker. To probe the function of the N-Linker in RIα and specifically to determine how the N-Linker contributes to assembly of the tetrameric holoenzyme, we engineered a monomeric form of RIα that contains most of the N-Linker, RIα(73-244), and crystallized a holoenzyme complex. Our previous RIα constructs began with the inhibitor site. RIα(73-244):C holoenzyme does not form stable dimers in solution without the dimerization domain; however, part of the extended linker is now ordered by interactions with a symmetry-related-dimer in the crystal. This complex of two symmetry-related dimers forms a tetramer that not only reveals novel mechanisms for allosteric regulation but also has many features that are consistent with known properties of the full-length holoenzyme. A model of the tetrameric RIα holoenzyme, based on this structure, is also consistent with small angle X-ray and neutron scattering data reported earlier. The model has been validated with new SAXS data and with a mutant of RIα that is localized to a novel interface that is unique to the tetramer.
Introduction
The regulatory (R) subunits of cAMP-dependent protein kinase (PKA) are modular and highly dynamic. In the absence of cAMP the R-subunit dimer is attached to two PKA catalytic (C) subunits and maintains the enzyme in an inactive tetramer. Each R-subunit contains a dimerization/docking (D/D) domain at the N-terminus that is joined by a flexible linker to two cyclic nucleotide binding domains at the C-terminus (CNB-A and CNB-B). The linker contains an inhibitor site (IS) that docks to the active site cleft in the C-subunit in the inactive holoenzyme but is disordered in the dissociated free R-subunits (Li et al., 2000). The linker, as summarized in Figure 1, can be divided into three segments, the consensus inhibitor site (P-3 to P+1), the N-Linker that joins the inhibitor site to the D/D domain, and the C-Linker that becomes ordered in the heterodimeric holoenzyme complex. While much has been learned from the structures of the cAMP binding domains of the RIα and RIIβ subunits (Diller et al., 2001; Su et al., 1995), and the C-subunit has been crystallized in many different conformational states (Akamine et al., 2003; Knighton et al., 1991a; Madhusudan et al., 2002; Zheng et al., 1993), it was not until we solved structures of heterodimeric holoenzyme complexes that we could appreciate for the first time how the C-subunit was actually inhibited by the R-subunits, and how the complex was then activated by cAMP (Brown et al., 2009; Kim et al., 2007; Kim et al., 2005; Wu et al., 2007). The challenge now is to understand how the full length tetrameric holoenzymes are assembled as these represent the true physiological state of PKA. This is essential if we are to appreciate the full allosteric potential of this key signaling enzyme. An additional goal is to determine how the N-Linker contributes to the assembly of the tetrameric holoenzyme.
Figure 1. Extended N-Linker in RIα(73-244):C holoenzyme complex.

A. Domain organization of different RIα constructs. Sequence of the linker between Dimerization Domain (D/D) and cAMP binding domain A (CNB-A) is shown. Constructs that were crystallized in holoenzyme complexes are marked with asterisks. B. Sequence alignment of the linkers in different R-subunits. Possible sites of phosphorylation are shown as red dots. C. RIα(73-244):C structure overlaid with the previously solved RIα(91-244):C (dark grey). Catalytic subunit is colored white (N-lobe) and tan (C-lobe). CNB-A is colored teal, the linker is colored red. N-terminal Cα atoms of the RIα constructs are shown as spheres.
Mammalian genomes typically code for four separate and functionally non-redundant R-subunit isoforms (RIα, RIβ, RIIα, and RIIβ). Although it has been difficult to crystallize full length R-subunits, presumably because the linkers are flexible, structures of the CNB domains and the D/D domains have been solved (Diller et al., 2001; Kinderman et al., 2006; Sarma et al., 2010; Su et al., 1995). To understand how the tetrameric holoenzymes are spatially organized, we initially used small angle X-ray and neutron scattering (SAXS/SANS). This revealed surprisingly that the shapes of the various R-subunits and holoenzymes are quite different (Heller et al., 2004; Vigil et al., 2004b; Vigil et al., 2006). The RIα subunit and its corresponding holoenzyme are Y-shaped while the RII subunits are more rod-like and dumb-bell shaped. The RIIα holoenzyme remains extended while the RIIβ holoenzyme compacts into a globular protein (Vigil et al., 2006). These different architectures are due primarily to differences in the N-Linker regions (Figure 1) and suggest that the allosteric signaling in each holoenzyme will be distinct. The Inhibitor Site and the C-Linker become organized upon association with the C-subunit; however, the role and ordering of the N-Linker, which contains many putative binding motifs, remains unknown.
It is known that cAMP activation of tetrameric holoenzyme is a cooperative process, that is reflected by the increased Hill coefficients (Herberg et al., 1994). Other evidence further supports the importance of the N-Linker and the tetrameric configuration of the holoenzyme. Limited proteolysis of free RIα, for example, cleaves at Arg92 just before the inhibitor site whereas trypsin cleaves at Arg72 in the holoenzyme suggesting that at least part of the N-Linker is protected in the holoenzyme (Cheng et al., 2001). In addition, using cysteine mutagenesis coupled with fluorescence anisotropy, we explored the flexibility of residues that lie in different regions of RIα (Li et al., 2000). We found that residues that flank the D/D domain, Thr6 and Leu66, were quite flexible independent of whether cAMP or catalytic subunit was bound. Other residues such as Ser99 were flexible in the dissociated R-subunit but immobilized in the holoenzyme. Two residues that lie in the N-Linker, Ser75 and Ser81, were flexible in the free RIα subunit but became much less mobile in the holoenzyme suggesting that they might be interacting with another part of the protein and could possibly be contributing to forming the tetramer. A final piece of evidence that supported the importance of the N-Linker for holoenzyme formation came from mutagenesis of two residues in the C-subunit that are predicted to lie on the surface where the N-Linker would bind (Cheng et al., 2001). Mutation of Arg133 selectively interfered with binding of RIIα whereas replacing Asp328 with Ala interfered with binding of RIα. These results led us to predict that the orientation of the N-Linker was not only important for formation of the tetrameric holoenzyme but also that the orientation of the N-Linker would be different in RI and RII (Cheng et al., 2001).
The previous RIα monomers that were crystallized as a complex with the catalytic subunit began with the inhibitor site and contained no ordered residues from the N-Linker. To further explore the potential role of N-Linker residues and, in particular, to determine whether they contribute to formation of the tetramer, we engineered two longer constructs of RIα that contained 18 additional residues at the N-terminus compared to the constructs that had been crystallized previously as holoenzyme complexes (Figure 1). We also engineered dimeric forms of RIα that lacked the second CNB Domain, RIα(1-259) and RIα(1-244). Although all of these constructs readily formed holoenzyme, only one, RIα(73-244), crystallized as a holoenzyme complex with crystal packing that was distinct from any of our previous structures.
The crystal structure of RIα(73-244) bound to catalytic subunit and Mn2+ AMP-PNP, a non-hydrolysable analog of ATP, was solved to a resolution of 3.3Å. Although not all of the additional linker was ordered, residues 84-92 could be easily traced. While the inhibitor site docked to the active site cleft of the catalytic subunit as expected, the extended linker interacted primarily with the β4-β5 loop of the symmetry-related dimer. In addition, this β4-β5 loop docked onto the hydrophobic pocket of the catalytic subunit that is created by the αF-αG loop in the symmetry-related dimer, a site that is also used in different ways as a docking site for both PKI and RIIβ. While these two heterodimers do not have a high tendency to form a tetramer when they are not tethered to one another by the dimerization/docking domain, there are many features of this holoenzyme complex that are consistent with known features of the tetrameric holoenzyme. Forming the extended interface between the two heterodimers creates a highly allosteric surface that would explain the high Hill coefficient (1.6) that is associated with activation of the tetrameric holoenzyme compared to the heterodimer (Herberg et al., 1994). The two heterodimers are also oriented such that the linkers could be readily joined to the nearby D/D domain. From our previously solved structures, we are only missing 20 residues of the linker. Our model of the quaternary structure of the full-length tetramer, based on this structure, is also independently validated by our previous SAXS and SANS data where the C-subunits are well segregated from one another. To further validate this quaternary structure we also obtained SAXS data for an RIα(1-259):C complex, where the dimeric RIα is lacking the B-domain. This also is consistent with our model. Finally, we demonstrate that a mutation in the proposed interface between two heterodimers not only alters the cAMP induced activation of PKA but also reduces the Hill coefficient. This model allows us for the first time to appreciate the intricate ways in which the binding of a small messenger, cAMP, leads to the allosteric release of kinase inhibition.
Results
Overall structure of RIα(73-244):C Complex
To understand how the RIα N-Linker region contributes to the holoenzyme structure, we extended the N-terminus of the previous construct of RIα(91-244) by 18 residues. We also engineered the extended form of RIα(91-379). Both proteins were very stable, and both readily formed a high affinity complex with the C-subunit that could be isolated on a gel filtration column; however, only the RIα(73-244) holoenzyme complex gave high resolution crystals. The crystal structure of RIα(73-244) in complex with the C-subunit, AMP-PNP and two Mn++ ions was solved at 3.3Å. Although the overall structure of the complex is very similar to that of the previously solved RIα(91-244):C complex (Kim et al., 2005) (Figure 1), the crystal packing was different from other previous structures of free or complexed R- and C-subunits. The asymmetric unit contains a single heterodimer comprised of one R-subunit and one C-subunit. The C-subunit is generally unchanged and is in its closed conformation. The overall conformation of the R-subunit is also similar to what was seen in the RIα(91-244) complex although the β4-β5 loop is shifted about 2.5Å away from the C-subunit. All corresponding α-carbon atoms of the two complexes can be superimposed with an RMSD of 0.8Å (Maiti et al., 2004). In this structure, however, the electron density of the N-terminal linker region in RIα can now be clearly traced up to Pro84, nine residues more than in the previous RIα(91-244):C complex. To understand how the N-Linker is ordered, one must look to the symmetry-related dimer. Specifically, it is the β4-β5 loop of the symmetry-related dimer that orders the N-Linker. The consequences of this ordering creates a tetrameric configuration of the two heterodimers that not only orders the N-Linker of each dimer but also creates an allosteric symmetry-related interface that explains clearly and for the first time why the tetramer is essential for the highly cooperative activation of PKA by cAMP. The tetramer that is formed by the two symmetry mates is also consistent with our models of the full length tetrameric RIα holoenzyme based on small angle X-ray and neutron scattering (SAXS and SANS).
N-Linker is ordered by Inter-molecular interactions between symmetry-related holoenzymes
The Inhibitor Site is defined as residues P-3 to P+1 in the linker, and as RIα is a pseudosubstrate it has alanine in the P-site position (Ala97). While this segment docks to active site of the C-subunit in more-or-less the same conformation as described previously in the PKI complex and in the RIα and RIIα holoenzyme complexes, in this structure we can now trace the N-terminal linker up to Pro84. However, as seen in Figure 1C, most of this segment extruded out from the complex and did not make any interactions with its own R or C subunit. Analysis of the crystal packing showed that stabilization of this flexible linker is provided by a symmetry mate, designated here as R′:C′ (Figure 2). The two symmetry-related dimers have an extensive interface area: 829Å2 between the two R-subunits (R to R′) and two 243Å2 interfaces between C and R-subunits (C to R′ and C′ to R). The combined interface between C-R and C′-R′, thus, was 1316Å2, and this is comparable to the interface between the C and R subunits within each holoenzyme dimer (1524Å2). The interface was predominantly hydrophobic with desolvation energy estimated at -10.7 kcal/mol (Krissinel and Henrick, 2007).
Figure 2. Crystal packing of the RIα (73-244):C holoenzyme.

Color coding is the same as that used in Figure 1. Symmetry-related molecule is shown as surface.
The major element that interacted with the extended linker in the interface was the β4-β5 loop in the cyclic nucleotide-binding (CNB) A domain from the R′-subunit in the symmetry-related heterodimer. This loop (Figure 3), which is highly conserved in an isoform-specific manner, is exposed to solvent in the absence of the N-Linker, but in the presence of the N-Linker, this loop from the symmetry-related dimer provides a mechanism for bridging the N-Linker and the C-subunit on the other dimer. Since the complex observed in the crystal had two-fold rotational symmetry, the β4-β5 loop in each R:C heterodimer was bound in exactly the same way to the symmetry-related heterodimer. We will consider first exactly how the β4-β5 loop from one dimer docks on the other dimer and then consider the consequences of this for creating a unique allosteric mechanism for activation of the tetrameric holoenzyme.
Figure 3. β4-β5 loop is a key element of the interface between symmetry-related heterodimers.

A. β4-β5 loop from the symmetry-related R′-subunit is marked by the yellow arrow. It docks to the αF-αG loop of the C-subunit and the extended N-Linker of the R-subunit. B. Sequence alignment of β4-β5 loops from different R-subunits. Four residues from the β4-β5 docking interface are shaded grey, universally conserved glycines are shaded yellow. C. A close-up view of the β4-β5 loop docking interface. Aliphatic parts of the interacting residues are shown as surfaces.
The N-Linker is ordered by the β4-β5 loop of the symmetry-related dimer
As shown in Figure 3A, the β4-β5 loop from the R′ subunit interacts with the N-Linker from the symmetry-related R-subunit and in addition docks onto the hydrophobic pocket in the symmetry-related C-subunit that is formed by the αF-αG loop. Three residues, Tyr183, Trp188 and Ser191, contribute prominently to this interface that is created by the β4-β5 loop, and this interface can be divided into two distinct but overlapping segments. We will consider first the interactions of the β4-β5 loop with the neighboring N-Linker and then its interactions with the neighboring C-subunit.
The two hydrophobic residues, Tyr183 and Trp188, are buttressed up against the N-Linker and provide the major interface with the N-Linker. In addition, two hydrogen bonds between Asn186 from the β4-β5 loop of the R′-subunit and backbone carbonyls of the N-terminal linker from the R-subunit (Pro87 and Val88) were detected, and these anchor the proline-rich segment that is a characteristic feature of the N-Linker in RI-subunits.
As seen in Figure 3 the β4-β5 loop also interacts with the αF-αG loop in the C-subunit of the symmetry-related dimer, and this site is known to be an important docking site for PKA and for other kinases as well. Specifically, Trp188 and Ser191 from the β4-β5 loop dock to the hydrophobic pocket on the C-subunit that is formed by Tyr235 and Phe239 from the αF-αG loop (Figure 4A). Although solvent exposed residues of this pocket are different in different kinases, its backbone geometry is absolutely conserved and is defined by a set of hydrophobic interactions between highly conserved residues: Trp222, Tyr229, Pro237, Phe238 and Ile250 (Figure 4a and b). This site is anchored to the central F-helix that serves as a general scaffold for most of the important residues in protein kinases (Kornev et al., 2008). This pocket often serves as an important protein/protein docking site. It is this pocket that is used as a tethering site for the amphipathic helix of PKI which binds with high affinity to the C-subunit (Knighton et al., 1991b). This pocket is also used in a very different way by the N-Linker of the RIIβ subunit when it is trapped in a complex with AMP-PNP (Brown et al., 2009) (Figure 4C). The αF-αG loop also links residues in the core that interact with P-3 and P-2 residues in the inhibitor peptide and the P+1 loop which provides the docking site for the P+1 residue. Finally, this loop positions the G Helix which is a critical docking site for the regulatory subunits and for other tethered substrate proteins (Figure 4A). This loop is thus a fundamental feature for peptide/protein recognition by every protein kinase.
Figure 4. αF-αG loop is a universal docking site.

A. Geometry of the αF-αG loop is conserved through all protein kinases. It is defined by conserved hydrophobic interactions of Pro237 and F238 with αF(Trp222 and Y229), αG(I250), which are connected by the universal APE-motif (colored red). The β4-β5 loop docking site is indicated by the yellow arrow. B. Structure based sequence alignment of αF-αG loops in different protein kinases. Universally conserved residues are shaded yellow. Residues that form hydrophobic interface are shaded grey. Two positions corresponding to Y235 and F239 which are exposed on the surface of the docking site are framed in red. C. Protein kinase A inhibitor PKI and N-terminal linker of RIIβ dock to the same groove formed by the αF-αG loop.
In addition to Tyr235 and Phe239 from the αF-αG loop, the hydrophobic interface was formed by two additional residues from the N-terminal linker, Val89 (P-7) and Arg92 (P-5) (Figure 3C). Arg92 in the P-5 position is strictly conserved in RIα subunits and is absent in RII subunits. Sequence alignment of the R subunits (Figures 1B and 3B) indicates that the β4-β5 loop and the N-Linker are among the most isoform distinct sequences given the high homology of the RI and RII subunits. Figure 3B compares the sequence in the β4-β5 loop region in various RI and RII subunits. All four residues of the interface between two heterodimers are highly conserved in RIα, less conserved in RIα, but are not conserved in RII subunits. In the RII subunits this segment is conserved differently suggesting that it will contribute in different ways to their holoenzyme structures, and this is consistent with our SAXS data (Vigil et al., 2004b).
N-linker contacts explain importance of Asp328 and Arg133 of the C-subunit for the holoenzyme formation
In the earlier work (Cheng et al., 2001) we demonstrated that sequence diversity in the N-linker of RIα and RIIβ is functionally important. Mutagenesis of two C-subunit residues, Arg133 and Asp328, showed that Arg133 is important for the formation of RIIβ holoenzyme whereas Asp328 was more important for RIα subunit. In all previously solved structures Arg133 interacts with Glu230 from the F-helix, while Asp328, is solvent exposed (Figure 5a). In this structure, however, the P-5 Arg92 binds to the Glu230 causing Arg133 to flip about 180° so that it is now contacting Asp328 (Figure 5b). As we pointed out, Arg92 is a characteristic feature of RIα subunits, that require ATP and two Mg++ ions for the full length holoenzyme formation. The observed flip of Arg133 further locks the ATP-bound holoenzyme into a closed conformation where the N- and C-lobes together bury the ATP. Here we can see why loss of Asp328 is uniquely detrimental to the formation RIα holoenzymes. Mutation of Glu230 to Gln led to disruption of the Arg133:Glu230 salt bridge and destabilization of the Arg133 side chain (1SYK (Wu et al., 2005)). The only other wild type PKA structure that did not have the Arg133:Glu230 salt bridge was the apo C-subunit with its open active site cleft and partly unstructured C-terminal tail (1J3H (Akamine et al., 2003)). Here we present the first evidence to show that Arg133 can form a salt bridge with Asp328 from the C-tail. It was also shown previously that mutagenesis of Asp328 can decrease the catalytic efficiency of PKA, but it was not clear why (Batkin et al., 2000).
Figure 5. R133 flip in RIα (73-244):C holoenzyme.

A. In all previously known structures of PKA C-subunit R133 was bound to E230 from the αF-helix. B. In the new holoenzyme structure it forms a salt bridge with D328 from the conserved FDDY motif in the C-terminal tail.
Allosteric Interface is created in the tetrameric complex
The detailed features showing how the N- and C-Linkers are anchored in the tetrameric holoenzyme that is made up of two symmetry-related dimers are indicated in Figure 6. What is revealed here for the first time is how binding of cAMP to one site can simultaneously influence both the R:C interface in its own R:C partner as we saw in earlier structures but also how it can influence positioning of the N-Linker from the symmetry-related dimer. Such influence is provided by the multiple direct contacts of the β4-β5 loop region of each CNB-A domain to the N-terminal linker of the opposite Regulatory subunit (Figure 3c). This would explain for the first time why the Hill Coefficient is increased in the tetramer as compared to the dimer (Herberg et al., 1994). It also creates a unique symmetry that was never previously appreciated. This is the surface that faces towards the C-terminal surface of the helical D/D domain, and it remains to be determined whether there are additional interactions that are created between the D/D domain and the tetrameric configuration of the two dimers. In addition, as seen in Figure 1, there are two predicted phosphorylation sites in the missing 20 residues of the N-Linker, and these could significantly alter the configuration of the tetramer as well as the allosteric properties of the tetramer.
Figure 6. N-Linker mediates cross-talk between two heterodimers in PKA RIα holoenzyme.
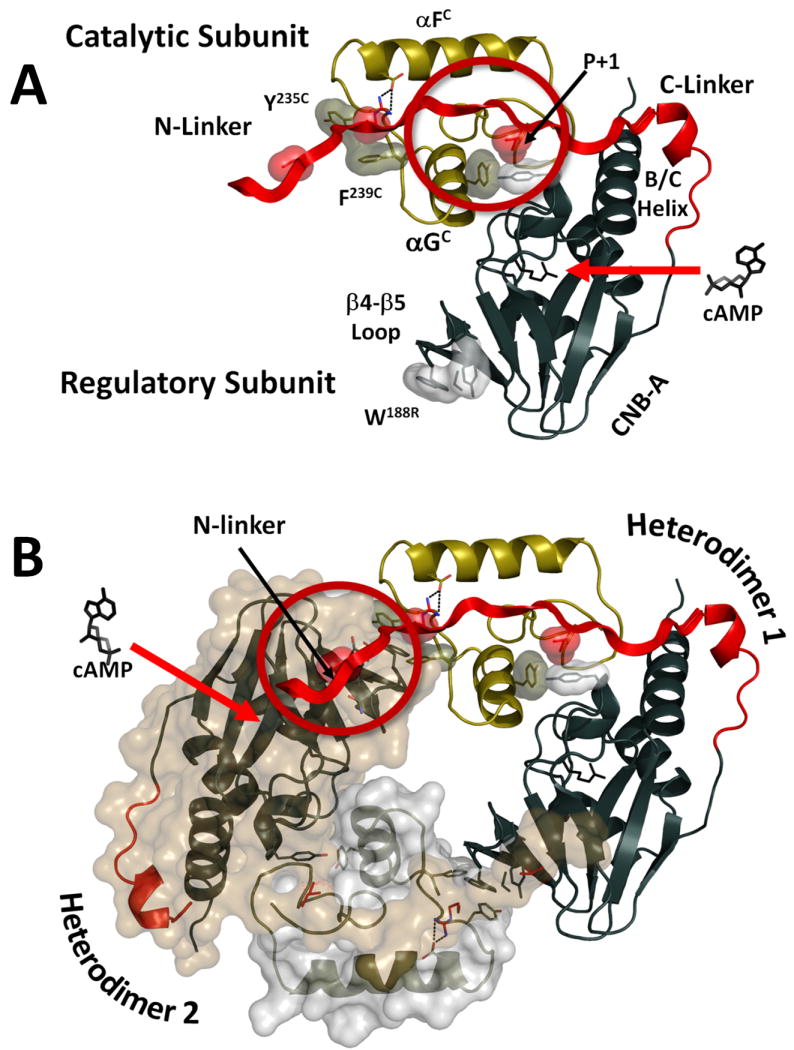
A. Binding cAMP molecule to CNB-A in a heterodimer (indicated by the red arrow) disrupts the R:C interface in the C-linker around the P+1 loop, αG-helix region (indicated by the red circle). B. Binding of the second cAMP molecule to the symmetry related R′-subunit disrupts the R′:C and R′:R interface in the N-linker area.
Changes in the Phosphate Binding Cassette
The signature motif for cAMP binding is the Phosphate Binding Cassette (PBC), which is embedded within the β subdomain of the CNB. As shown previously, the PBC is distorted in the holoenzyme and assumes a conformation that has a low affinity for cAMP (Kim et al., 2005; Wu et al., 2007). Figure 7 shows how the PBC is further distorted by the docking of the β4-β5 loop to the N-Linker of the symmetry-related dimer. In this figure the positions of the β4-β5 loop and PBC in the cAMP-bound conformation are compared with their conformation in the holoenzyme complexes that are formed with RIα(91-244) where the N-Linker is missing vs. RIα(73-244) where the N-Linker is included. From this alignment it is clear that the configuration of the β4-β5 loop is not significantly altered by its interactions with the symmetry-related dimer, although its position is slightly displaced. However, two changes are seen in the PBC. The tip of the PBC is different when the N-Linker is present and the region extending from Arg209 to Ala211 is moved slightly. This slight movement of Ala210 is significant, because this segment provides part of the hydrophobic packing for the nucleotide. This segment is referred to as the “Base Binding Region” (McNicholl et al., 2010; Rehmann et al., 2003). This distortion of the PBC:β4 interface causes the cAMP binding pocket to be occluded so that access of the nucleotide to the PBC is further restricted. This likely makes this complex even more resistant to activation by cAMP.
Figure 7. Structural changes in the Phosphate Binding Cassette (PBC) in RIα (73-244):C holoenzyme.

A. Overlay of PBCs in cAMP-bound RIα (1RGS) (white) and RIα(91-244):C holoenzyme (3FHI) (golden). B. C-terminal part of the PBC in RIα(73-244):C holoenzyme (red) is distorted. The β4-β5 loop position in the new holoenzyme is also shifted. C. V182 from the “Base Binding Region” interacts with the A210-A211 region of the PBC, occluding cAMP binding. cAMP is modeled from the cAMP-bound RIα structure.
Model of the RIα tetrameric holoenzyme is consistent with SAXS/SANS data
Previously we assessed the general shape of PKA tetramers by using small angle X-ray and neutron scattering (SAXS/SANS) techniques (Heller et al., 2004; Vigil et al., 2006). Deuterium labeling of RIα subunits showed, in particular, that in the tetrameric holoenzyme they are in a close contact, while the two C-subunits are completely separated. To test if our present results are consistent with these data we created a model of the full length PKA tetramer based on the two symmetry-related dimers in our crystal (Figure 8A). To introduce the C-terminal CNB domain that was missing in the current RIα construct, we used structure of the C-subunit bound to the RIα(91-379) that also has an R333K mutation that prevents cAMP binding to the B-domain (2QCS (Kim et al., 2007)). Two copies of this heterodimer were positioned according to the packing depicted in Figure 2. As indicated earlier, the N-termini of the two RIα chains are positioned in a way that allows them to be readily linked to the D/D domain. Scattering data for this model were calculated by the CRYSOL program (Svergun et al., 1995) and fit to the experimental data for RIα holoenzyme with χ2=1.22 (Figure 8C). Ab intio reconstruction of the complex based on the previously reported SAXS data using the GASBOR program, gave us a butterfly shaped shell that also fits our model reasonably well (Figure 8B). Analysis of the distance distribution functions P(r) for the R-subunits and C-subunits showed that they correspond well to the previously reported SANS results: R-subunits form a compact object, while C-subunits are widely separated (Figure S1a,b, Supporting text).
Figure 8. Model of the RIα tetrameric holoenzyme.

A. The proposed positions of two PKA heterodimers correspond to the observed packing (Figure 2). Possible position of the Dimerization/Docking domain bound to the AKAP helix is illustrated using 3IM4 structure. Still unknown linkers between the DD-domain and RIα CNB domains are shown as dashed lines. B. Space filling model obtained from the previously reported SAXS data (Heller et al., 2004) (tan spheres) is overlaid with the proposed model. C-subunits are shown as tan colored surface. R-subunits are shown as tan cartoon. C. Small Angle X-ray scattering curve computed from the RIα(73-244):C tetrameric complex showed on the Figure 2. Dots represent experimental data for RIα(1-259):C holoenzyme. D. cAMP-induced activation of PKA monitored by FAP-IP20 binding. See also Figure S1.
Experimental validation of the model
To test this model of the tetramer, we used two approaches. One strategy was to characterize the tetrameric holoenzyme formed with RIα(1-259) (Figure 1A) that is only 15 residues longer than the crystallized construct. This dimer expresses well and the holoenzyme formed readily. The Ka (cAMP) had a Hill coefficient of less than 1.0 as reported previously (Herberg et al., 1994) confirming the importance of the B domain for allostery. The SAXS intensity profile of the RIα(1-259):C complex was close to the theoretical curve predicted by our model (Figure S1c, Supporting text).
To further validate the PKA holoenzyme model, we mutated Trp188 from the β4-β5 loop to Asp. As shown in Figure 3b the sequence of the β4-β5 loop is highly conserved in an isoform-specific manner. Nevertheless, there is no obvious role for this loop in either the cAMP bound structure or for the heterodimer with the C-subunit and the deletion mutant of RIa. In all of these earlier structures the β4-β5 is exposed to solvent. Previous studies of folding showed that the mutation of Trp188 had no effect on overall structure or unfolding of RIα (Leon et al., 2000). However, according to our model, Trp188 is one of the key contact residues in this loop. As seen in Figure 8d, the W188D mutation interfered with cAMP induced activation of PKA increasing the EC50 from to 29 to 95 nM. In addition the Hill coefficient decreased from 2.1 to 1.7. While the detailed mechanism for the complex and novel allosteric interactions between the A and B domains in the tetramer vs. the dimer are now being defined comprehensively by further analysis of a set of mutants in the β4-β5 loop, as well as in the linker, the results shown here demonstrate that modification of the β4-β5 loop is sufficient to perturb interactions between the two heterodimers in the tetrameric holoenzyme. Single particle image reconstruction of the full length RIa tetramer is also generating models that are quite consistent with the model proposed here (unpublished results).
Discussion
To probe the function of the N-Linker in the RIα subunit of PKA and specifically to determine whether the N-Linker contributes to assembly of the tetrameric holoenzyme and to allostery, we engineered a monomeric form of RIα that contains most of the N-Linker, RIα(73-244), and crystallized a holoenzyme complex. In this holoenzyme the N-Linker is extended by 18 residues compared to our previous structures (Kim et al., 2007; Kim et al., 2005). Although nine N-terminal residues were not resolved, the coordinates for an additional nine N-Linker residues were resolved for the first time. Surprisingly, they were not bound to the C-subunit of their own heterodimer (Figure 1C). Instead, the linker was docked onto the R-subunit of the symmetry-related heterodimer (Figure 2). Specifically, the β4-β5 loop from one RIα subunit was docked onto the N-Linker of a symmetry-related dimer thus creating a novel tetrameric interface. Analysis of the interface between the two heterodimers showed that it is mostly hydrophobic and can be relatively stable in solution. This led us to the suggestion that this interface can in fact represent interactions between R and C subunits in the full length PKA tetrameric complex. This suggestion is supported by several observations including enhanced cooperativity in the tetramer (Herberg et al., 1994), protection of the N-Linker from proteolytic cleavage in the holoenzyme compared to free RIα, and isoform-specificity for docking of this portion of the N-Linker (Cheng et al., 2001). It was also shown that mutation of Arg133 in the C-subunit is important for RIIβ holoenzyme formation, while mutation of Asp328 plays an essential role in RIα holoenzyme. Our model for the first time can provide explanation of these results. According to the model the RIα-specific Arg92 in P-5 position
The structure also explains for the first time why the β4-β5 loop and the segment of the N-Linker that immediately precedes the Inhibitor Site are conserved in such a unique way in each isoform. The importance of the β4-β5 loop for activation of the tetrameric holoenzyme was also confirmed by mutagenesis. Finally, a model of the tetrameric holoenzyme, based on this structure, is quite consistent with our previous SAXS and SANS results (Heller et al., 2004; Vigil et al., 2004a). To further confirm the model we carried out SAXS analysis of a mutant RIα tetramer that lacks the B domain.
Two methods, limited proteolysis (Cheng et al., 2001) and cys-scanning mutagenesis of linker residues coupled with fluorescence polarization (Li et al., 2000), indicated that the N-Linker is flexible in free RIα but ordered in the holoenzyme. It was shown that limited proteolysis of cAMP-bound RIα with trypsin cleaves at Arg92 whereas in the holoenzyme cleavage occurs at Arg72. Cys scanning mutagenesis, on the other hand, allowed us to probe the flexibility of residues in this region (positions 75 and 81). These results also support the conclusion that the N-Linker is much more ordered in the holoenzyme. In this structure we begin to understand for the first time how the N-Linker can be ordered.
As summarized in Figure 9, direct docking of the linker region to the R-subunit of the symmetry-related heterodimer can explain the enhanced cooperativity of activation in the tetramer (Herberg et al., 1994). Although the entire linker region is disordered in the cAMP-bound RIα subunit, the Inhibitor Site through the C-Linker becomes ordered at the R:C interface when the RIα subunit binds to the C-subunit (Kim et al., 2007; Kim et al., 2005). Of particular interest here, is the holoenzyme complex of RIα(92-244) where the RI subunit begins with the Inhibitor Site. Extending the N-Linker segment by 18 residues causes the protein to crystallize in a completely different space group and an additional portion of the N-Linker can now be seen. Figure 9, in particular, shows how the N-Linker is ordered by the β4-β5 loop of the symmetryrelated dimer and also shows how cAMP binding to one dimer will not only release its own associated C-subunit through previously described interactions with its own C-Linker; it will also unleash the adjacent heterodimer through its interactions with the N-Linker. In our model, binding of cAMP to one of the R-subunits in the tetramer will directly affect the linker of the other R-subunit, thus promoting dissociation of both C-subunits and contributing to the enhanced allostery that is characteristic of the tetrameric holoenzyme.
Figure 9. Proposed model of Intra- and Inter-molecular regulation of PKA by cAMP.

C-linker of each R-subunits in the tetramer is controlled by its own CNB-A domain. N-linker is controlled by the symmetry related R′-subunit.
The interface between the two heterodimers in our model also highlights two regions that are highly conserved in RIα but are different in the RII subunits. This suggests that the observed interaction is not a result of random crystal packing but is biologically relevant. One such region is the β4-β5 loop, which is linked to a motif that is referred to as the “Base Binding Region” (McNicholl et al., 2010; Rehmann et al., 2003) because it makes a hydrophobic contact to the adenine ring (nucleobase) of cAMP. Specifically, Val182 in β4 together with Ala210 and Ala211 form one side of the hydrophobic cap that provide the docking site for the adenine ring of cAMP (Figure 7). The adjacent loop itself is not conserved through different CNB domains but is highly conserved in RIα subunits (Figure 3B). In all of our previous structures this loop is exposed to solvent. Here we see a molecular explanation for the conserved residues, as they bind to another RIα-specific region of the N-linker, the segment that precedes the Inhibitory Site (Figure 1). It is the complementarily between these two sites that provides an explanation for their conservation.
Convincing evidence that this tetrameric configuration of the two RIα heterodimers reflects the general conformation of the full length tetrameric holoenzyme comes from our previous analyses of the RIα holoenzyme conformation in solution using SAXS and SANS. The proposed model of the tetrameric holoenzyme that we built here based on the RIα(73-244) crystal structure is consistent with low resolution models based on SAXS/SANS data reported earlier (Heller et al., 2004) and additional SAXS experiments of the RIα(1-259):C complex. Even more convincing is the substantial separation of the C-subunits in the complex. This was first detected by SANS experiments and is also reflected by our model. Additionally this model is supported by mutagenesis of the β4-β5 loop demonstrating that alteration of the predicted interface in the tetramer perturbs cAMP dependent activation of PKA (Figure 8).
While details of this model obviously need to be confirmed by further biochemical studies as well as by further structures, it provides for the first time a framework for understanding how the tetramer can be allosterically regulated, potentially in both positive and/or negative ways, that would not be possible in the simple heterodimer. It also emphasizes how important it is to obtain structures of larger complexes that more accurately reflect full length proteins if we are to appreciate the full allosteric potential of the highly dynamic signaling proteins.
Materials and Methods
Protein Preparation
The catalytic subunit was expressed and purified as previously described (Gangal et al., 1998). The peak I of C-subunit which contains 4 phosphorylated residues (Ser10, Ser139, Thr197, and Ser338) was used for crystallography. Three regulatory subunit deletion mutants (RIα(73-379), RIα(73-379:R333K), and RIα(73-244) were generated by Quikchange site-directed mutagenesis according to the Stratagene protocol. These mutants contained most of the linker residues compared to the previous structure of RIα (91-244). All mutants were expressed in E. coli BL21 (DE3) cells (Novagen) and purified as described previously (Saraswat et al., 1988) with slight modification. The cells were lysed and the spin supernatant was filtered with 0.22mm filter and loaded onto a Profinia protein purification system. The sample was run over a cartridge containing cAMP bound to resin, washed with lysis buffer containing 0.7M NaCl and then eluted with lysis buffer containing 35mM cGMP at pH 5.8. Eluted protein was then run on S75 gel filtration column.
Complex Formation
Each of the RIα mutants were mixed separately with the wild type C-subunit in a 1:1.2 molar ratio and dialyzed at 4°C in 10 mM MOPS (pH 7.0), 2 mM MnCl2, 50 mM NaCl, 1 mM TCEP-HCl and 0.2 mM AMP-PNP.
Crystallization
The RIα(73-244):C complex was crystallized in 0.1M MES pH 6.0 and 12% PEG 20,000 by using a Douglas Instruments Oryx8 crystallography robot as 1:1 protein solution:crystallizing solution and incubated at room temperature. Crystals were flash frozen in a cryoprotectant solution (mother liquor containing 15% glycerol) and a data set was collected to 3.3Å on the Advanced Light Source beamline 8.2.1. Data was processed and scaled using HKL2000 (Table 1). The structure was solved using the RIα(91-244):C complex structure as the molecular replacement probe.
Table 1. Data Collection and Refinement Statistics.
| RIα (73-244):C | |
|---|---|
| Data collection | |
|
| |
| Space group | P 322 1 |
|
| |
| Cell dimensions | |
| a (Å) | 116.7 |
| b (Å) | 116.7 |
| c (Å) | 140.1 |
| γ (°) | 120.0 |
|
| |
| No. of molecule per asymmetrical unit | 1 |
|
| |
| Resolution (Å) | 3.3 |
|
| |
| Rmerge | 0.088 (0.40)* |
|
| |
| Completeness (%) | 97.1 (93.7) |
|
| |
| I/sigma | 19.3 (3.4) |
|
| |
| No. reflections | 27860 |
|
| |
| Refinement | |
|
| |
| Resolution (Å) | 50.0-3.3 |
|
| |
| Rwork / Rfree (%) | 24.2/29.0 |
|
| |
| No. of protein residues | 522 |
|
| |
| No. of ligand/ion | 3 |
|
| |
| No. of water | 16 |
|
| |
| R.m.s. deviations | |
|
| |
| Bond lengths (Å) | 0.010 |
|
| |
| Bond angles (°) | 1.6 |
|
| |
| Ramachandran angles (%) | |
|
| |
| most favored (%) | 81.5 |
|
| |
| disallowed | none |
Values in parentheses are for the highest-resolution shell (3.30-3.39 Å)
RIα(73-379):C complex was initially crystallized in 30% PEG 400, 0.1M HEPES pH 7.5, 0.2M NaCl using Douglas Instruments Oryx8 crystallography robot. Drops were set up under silicone oil as 1:1 protein solution:crystallizing solution. However, the crystals can only diffraction to 8Å. RIα(75-379:R333K) with a mutation in the essential arginine in the PBC of the cAMP-binding domain B did not purify well and had many contaminating proteins, so it was not set up for crystallization.
Phasing for the structure was made by molecular replacement in AMORE using 3FHI as a search model. Refinement was made by REFMAC and CNS 1.2 programs. The model was manually built based on the density maps using Coot. The final model was evaluated by PROCHECK and had good geometry.
cAMP-induced activation
A fluorescence polarization assay was used to monitor PKA activity as described earlier (Saldanha et al., 2006). C-subunit concentration was 10 nM. R-subunit was added in 1.3:1 molar ratio. Solution contained 1 mM ATP, 10 mM MgCl2 and 2 nM FAM-IP20.
Small angle scattering evaluation
Evaluation of the of X-ray solution scattering curves was made by CRYSOL program (Svergun et al., 1995). Previously published SAXS data (Heller et al., 2004) were used for the space filling model for Figure 7b by GASBOR program (Svergun et al., 2001).
Accession numbers
The atomic coordinates and structure factors have been deposited in the Protein Data Bank (accession code 3PVB).
Supplementary Material
A: Comparison of 2D class averages of raw particle images(even columns) with corresponding reprojections from the 3D model of tetramer (odd colums). The side length of the individual panels is 24 nm. B. The negative stain EM density map of the R1a:C holoenzyme. 6,422 particles were used to reconstruct the density map by angular reconstitution approach using program EMAN and the the Fourier shell correlation (FSC) curve suggests a resolution of 22 A° with the FSC cut off at 0.5 criterion. We are now in the process of fitting our model into the density map.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akamine P, Madhusudan, Wu J, Xuong NH, Ten Eyck LF, Taylor SS. Dynamic features of cAMP-dependent protein kinase revealed by apoenzyme crystal structure. J Mol Biol. 2003;327:159–171. doi: 10.1016/s0022-2836(02)01446-8. [DOI] [PubMed] [Google Scholar]
- Batkin M, Schvartz I, Shaltiel S. Snapping of the carboxyl terminal tail of the catalytic subunit of PKA onto its core: characterization of the sites by mutagenesis. Biochemistry. 2000;39:5366–5373. doi: 10.1021/bi000153z. [DOI] [PubMed] [Google Scholar]
- Brown SH, Wu J, Kim C, Alberto K, Taylor SS. Novel isoform-specific interfaces revealed by PKA RIIbeta holoenzyme structures. J Mol Biol. 2009;393:1070–1082. doi: 10.1016/j.jmb.2009.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng CY, Yang J, Taylor SS, Blumenthal DK. Sensing domain dynamics in protein kinase A-I{alpha} complexes by solution X-ray scattering. J Biol Chem. 2009;284:35916–35925. doi: 10.1074/jbc.M109.059493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng X, Phelps C, Taylor SS. Differential binding of cAMP-dependent protein kinase regulatory subunit isoforms Ialpha and IIbeta to the catalytic subunit. J Biol Chem. 2001;276:4102–4108. doi: 10.1074/jbc.M006447200. [DOI] [PubMed] [Google Scholar]
- Diller TC, Madhusudan, Xuong NH, Taylor SS. Molecular basis for regulatory subunit diversity in cAMP-dependent protein kinase: crystal structure of the type II beta regulatory subunit. Structure. 2001;9:73–82. doi: 10.1016/s0969-2126(00)00556-6. [DOI] [PubMed] [Google Scholar]
- Gangal M, Cox S, Lew J, Clifford T, Garrod SM, Aschbaher M, Taylor SS, Johnson DA. Backbone flexibility of five sites on the catalytic subunit of cAMP-dependent protein kinase in the open and closed conformations. Biochemistry. 1998;37:13728–13735. doi: 10.1021/bi980560z. [DOI] [PubMed] [Google Scholar]
- Heller WT, Vigil D, Brown S, Blumenthal DK, Taylor SS, Trewhella J. C subunits binding to the protein kinase A RI alpha dimer induce a large conformational change. J Biol Chem. 2004;279:19084–19090. doi: 10.1074/jbc.M313405200. [DOI] [PubMed] [Google Scholar]
- Herberg FW, Dostmann WR, Zorn M, Davis SJ, Taylor SS. Crosstalk between domains in the regulatory subunit of cAMP-dependent protein kinase: influence of amino terminus on cAMP binding and holoenzyme formation. Biochemistry. 1994;33:7485–7494. doi: 10.1021/bi00189a057. [DOI] [PubMed] [Google Scholar]
- Kim C, Cheng CY, Saldanha SA, Taylor SS. PKA-I holoenzyme structure reveals a mechanism for cAMP-dependent activation. Cell. 2007;130:1032–1043. doi: 10.1016/j.cell.2007.07.018. [DOI] [PubMed] [Google Scholar]
- Kim C, Xuong NH, Taylor SS. Crystal structure of a complex between the catalytic and regulatory (RIalpha) subunits of PKA. Science. 2005;307:690–696. doi: 10.1126/science.1104607. [DOI] [PubMed] [Google Scholar]
- Kinderman FS, Kim C, von Daake S, Ma Y, Pham BQ, Spraggon G, Xuong NH, Jennings PA, Taylor SS. A dynamic mechanism for AKAP binding to RII isoforms of cAMP-dependent protein kinase. Mol Cell. 2006;24:397–408. doi: 10.1016/j.molcel.2006.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knighton DR, Zheng JH, Ten Eyck LF, Ashford VA, Xuong NH, Taylor SS, Sowadski JM. Crystal structure of the catalytic subunit of cyclic adenosine monophosphate-dependent protein kinase. Science. 1991a;253:407–414. doi: 10.1126/science.1862342. [DOI] [PubMed] [Google Scholar]
- Knighton DR, Zheng JH, Ten Eyck LF, Xuong NH, Taylor SS, Sowadski JM. Structure of a peptide inhibitor bound to the catalytic subunit of cyclic adenosine monophosphate-dependent protein kinase. Science. 1991b;253:414–420. doi: 10.1126/science.1862343. [DOI] [PubMed] [Google Scholar]
- Kornev AP, Taylor SS, Ten Eyck LF. A helix scaffold for the assembly of active protein kinases. Proc Natl Acad Sci U S A. 2008;105:14377–14382. doi: 10.1073/pnas.0807988105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- Leon DA, Canaves JM, Taylor SS. Probing the multidomain structure of the type I regulatory subunit of cAMP-dependent protein kinase using mutational analysis: role and environment of endogenous tryptophans. Biochemistry. 2000;39:5662–5671. doi: 10.1021/bi992819z. [DOI] [PubMed] [Google Scholar]
- Li F, Gangal M, Jones JM, Deich J, Lovett KE, Taylor SS, Johnson DA. Consequences of cAMP and catalytic-subunit binding on the flexibility of the A-kinase regulatory subunit. Biochemistry. 2000;39:15626–15632. doi: 10.1021/bi002196l. [DOI] [PubMed] [Google Scholar]
- Madhusudan, Akamine P, Xuong NH, Taylor SS. Crystal structure of a transition state mimic of the catalytic subunit of cAMP-dependent protein kinase. Nat Struct Biol. 2002;9:273–277. doi: 10.1038/nsb780. [DOI] [PubMed] [Google Scholar]
- Maiti R, Van Domselaar GH, Zhang H, Wishart DS. SuperPose: a simple server for sophisticated structural superposition. Nucleic Acids Res. 2004;32:W590–594. doi: 10.1093/nar/gkh477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNicholl ET, Das R, Sildas S, Taylor SS, Melacini G. Communication between tandem cAMP-binding domains in the regulatory subunit of protein kinase A (PKA)-I{alpha} as revealed by domain-silencing mutations. J Biol Chem. 2010 doi: 10.1074/jbc.M110.105783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehmann H, Prakash B, Wolf E, Rueppel A, de Rooij J, Bos JL, Wittinghofer A. Structure and regulation of the cAMP-binding domains of Epac2. Nature Structural Biology. 2003;10:26–32. doi: 10.1038/nsb878. [DOI] [PubMed] [Google Scholar]
- Saldanha SA, Kaler G, Cottam HB, Abagyan R, Taylor SS. Assay principle for modulators of protein-protein interactions and its application to non-ATP-competitive ligands targeting protein kinase A. Anal Chem. 2006;78:8265–8272. doi: 10.1021/ac061104g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saraswat LD, Filutowics M, Taylor S. Expression and mutagenesis of the regulatory subunit of cAMP-dependent protein kinase in Escherichia coli. Methods Enzymol. 1988;159:325–336. doi: 10.1016/0076-6879(88)59033-x. [DOI] [PubMed] [Google Scholar]
- Sarma GN, Kinderman FS, Kim C, von Daake S, Chen L, Wang BC, Taylor SS. Structure of D-AKAP2:PKA RI complex: insights into AKAP specificity and selectivity. Structure. 2010;18:155–166. doi: 10.1016/j.str.2009.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Y, Dostmann WR, Herberg FW, Durick K, Xuong NH, Ten Eyck L, Taylor SS, Varughese KI. Regulatory subunit of protein kinase A: structure of deletion mutant with cAMP binding domains. Science. 1995;269:807–813. doi: 10.1126/science.7638597. [DOI] [PubMed] [Google Scholar]
- Svergun D, Barberato C, Koch MH. CRYSOL- a Program to Evaluate X-ray Solution Scattering of Biological Macromolecules from Atomic Coordinates. Journal of Applied Crystallography. 1995;28:768–773. [Google Scholar]
- Svergun DI, Petoukhov MV, Koch MH. Determination of domain structure of proteins from X-ray solution scattering. Biophys J. 2001;80:2946–2953. doi: 10.1016/S0006-3495(01)76260-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigil D, Blumenthal DK, Brown S, Taylor SS, Trewhella J. Differential effects of substrate on type I and type II PKA holoenzyme dissociation. Biochemistry. 2004a;43:5629–5636. doi: 10.1021/bi0499157. [DOI] [PubMed] [Google Scholar]
- Vigil D, Blumenthal DK, Heller WT, Brown S, Canaves JM, Taylor SS, Trewhella J. Conformational differences among solution structures of the type Ialpha, IIalpha and IIbeta protein kinase A regulatory subunit homodimers: role of the linker regions. J Mol Biol. 2004b;337:1183–1194. doi: 10.1016/j.jmb.2004.02.028. [DOI] [PubMed] [Google Scholar]
- Vigil D, Blumenthal DK, Taylor SS, Trewhella J. Solution scattering reveals large differences in the global structures of type II protein kinase A isoforms. J Mol Biol. 2006;357:880–889. doi: 10.1016/j.jmb.2006.01.006. [DOI] [PubMed] [Google Scholar]
- Wu J, Brown SH, von Daake S, Taylor SS. PKA type IIalpha holoenzyme reveals a combinatorial strategy for isoform diversity. Science. 2007;318:274–279. doi: 10.1126/science.1146447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Yang J, Kannan N, Madhusudan, Xuong NH, Ten Eyck LF, Taylor SS. Crystal structure of the E230Q mutant of cAMP-dependent protein kinase reveals an unexpected apoenzyme conformation and an extended N-terminal A helix. Protein Sci. 2005;14:2871–2879. doi: 10.1110/ps.051715205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng J, Trafny EA, Knighton DR, Xuong NH, Taylor SS, Ten Eyck LF, Sowadski JM. 2.2 A refined crystal structure of the catalytic subunit of cAMP-dependent protein kinase complexed with MnATP and a peptide inhibitor. Acta Crystallogr D Biol Crystallogr. 1993;49:362–365. doi: 10.1107/S0907444993000423. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A: Comparison of 2D class averages of raw particle images(even columns) with corresponding reprojections from the 3D model of tetramer (odd colums). The side length of the individual panels is 24 nm. B. The negative stain EM density map of the R1a:C holoenzyme. 6,422 particles were used to reconstruct the density map by angular reconstitution approach using program EMAN and the the Fourier shell correlation (FSC) curve suggests a resolution of 22 A° with the FSC cut off at 0.5 criterion. We are now in the process of fitting our model into the density map.