

Abstract
We recently described a Venezuelan equine encephalitis virus (VEEV)-specific human monoclonal antibody (MAb), F5 nIgG, that recognizes a new neutralization epitope on the VEEV E2 envelope glycoprotein. In this study, we investigated the ability of F5 nIgG given prophylactically or therapeutically to protect mice from subcutaneous or aerosolized VEEV infection. F5 nIgG had potent ability to protect mice from infection by either route when administered 24 h before exposure; however, mice treated 24 h after aerosol exposure developed central nervous system infections but exhibited no clinical signs of disease. Infectious virus, viral antigen and RNA were detected in brains of both treated and untreated mice 2–6 days after aerosol exposure but were cleared from the brains of treated animals by 14–28 days after infection. This fully human MAb could be useful for prophylaxis or immediate therapy for individuals exposed to VEEV accidentally in the laboratory or during a deliberate release.
Keywords: Venezuelan equine encephalitis virus, Alphavirus, human monoclonal antibody, envelope glycoprotein, antibody prophylaxis, antibody therapy
Introduction
The Venezuelan equine encephalitis virus (VEEV) serocomplex is composed of arboviruses in the genus Alphavirus, family Togaviridae. VEEV is an emerging zoonotic pathogen that has caused several serious epizootics and epidemics in the last 20 years, largely in Central and South America (Weaver et al., 2004). It is an NIAID Category B priority pathogen, due principally to its growth to high titers in cell cultures, stability in nature, high infectivity when aerosolized, high attack rate, and its historical development as a bioweapon by both the US and the former Soviet Union (Bronze et al., 2002; Hawley and Eitzen, 2001; Weaver and Reisen, 2010). VEEV has also been responsible for a high number of laboratory infections due to parenteral inoculation or airborne exposure and was thus designated as a biocontainment level 3 pathogen (Subcommittee on Arbovirus Laboratory Safety, 1980).
In humans, VEEV can cause neurological disease, including convulsions, disorientation, drowsiness, and mental depression in up to 15% of infections, with a higher rate in children (Johnson & Martin, 1974). A mouse model for VEEV encephalitis has been widely used for studies of pathogenesis and testing of vaccines and therapeutics (Jackson et al., 1991; Kundin, 1966; Paessler et al., 2006). Infection of mice by either the subcutaneous (SC) or intranasal (IN) routes leads to invasion of the central nervous system (CNS), resulting in 100% mortality (Jackson et al., 1991; Ryzhikov et al., 1995). In the immunocompetent mouse, both the immune response to VEEV replication in the brain and direct cytopathology in the CNS contribute to encephalitis, which is the ultimate cause of death (Charles et al., 2001).
Currently there is no FDA-approved human vaccine for VEEV; TC-83, an attenuated, live virus vaccine, is available only under Investigational New Drug (IND) status to protect laboratory and military personnel. It has been associated with poor immunogenicity and relatively high reactogenicity (Pittman et al., 1996). Formalin-inactivated TC-83, known as C-84, has been used as a vaccine but requires frequent boosting and may not protect from aerosol infection (Jahrling and Stephenson, 1984). Recently a live-attenuated vaccine, V3526, was developed using site-directed mutagenesis of a virulent VEEV infectious cDNA clone. Although it was effective in animal studies, adverse events in phase 1 clinical trials resulted in its withdrawal (Fine et al., 2008; Pratt et al., 2003; Reed et al., 2005). In the absence of an approved vaccine or specific treatment for VEEV infections, there is a need for development of antiviral therapeutics that could penetrate the blood-brain barrier and limit viral replication in the CNS.
Using Sindbis virus (SINV) in SCID mice as a model of alphaviral encephalitis, Griffin and colleagues showed that adoptive transfer of hyperimmune mouse serum or monoclonal antibodies (MAb) to epitopes on the E2 envelope glycoprotein resulted in clearance of infectious virus and viral RNA from the CNS (Levine et al., 1991). Recovery was dependent on immune-mediated, non-cytolytic control and clearance of infectious virus from neurons, which are long-lived, terminally differentiated cells (Griffin, 2010); viral clearance was not accomplished by transfer of purified SINV-sensitized T-cells or dependent on host cytolytic factors. Phillpotts and colleagues showed that intraperitoneal (IP) administration of murine anti-VEEV E2 glycoprotein MAbs either 24 h before or immediately after aerosol challenge with virulent virus could protect mice from lethal disease (Phillpotts, 2006; Phillpotts et al., 2002). Prophylactic MAbs 1A3A-9 or 1A4A-1 protected 90–100% of virus-challenged mice; post-exposure treatment with MAb 1A3A-9 protected 47% of mice. VEEV-infected mice treated therapeutically 24 h post-infection (PI) had significant reductions in virus titers in peripheral organs by 5 days PI but titers were not reduced in brains (Phillpotts et al., 2002). Paessler et al. (2006) found that immunization of mice with avirulent, chimeric SIN/VEE viruses resulted in protection from lethal encephalitis following SC, IN or intracranial (IC) challenge with virulent VEEV. Although vaccination of mice elicited neutralizing antibodies, high levels of infectious challenge virus were found in the brains of mice 3 days PI (regardless of the challenge route), but were undetectable by 28 days PI.
In past studies we used murine (m) MAbs to analyze the antigenic structure of the VEEV E1 and E2 glycoproteins and determined that epitopes on the E2 protein located between amino acids (AA) 182–207 constituted a major neutralization domain (Roehrig et al., 1982; Roehrig and Mathews, 1985). MAbs specific for this domain have potent protective act ivity in a murine model when administered 24 h before or 24 h after peripheral or aerosol challenge with virulent VEEV (Mathews and Roehrig, 1982; Mathews et al., 1985; Phillpotts, 2006; Phillpotts et al., 2002). However, neutralizing anti-VEEV mMAbs are limited in their clinical application due to the human anti-mouse antibody response. Recently we reported the isolation of a human (h) MAb, F5 nIgG, which is specific for a newly recognized neutralization epitope (AA 115–119) on the VEEV E2 protein (Hunt et al., 2010). In this report we document the efficacy of this hMAb in protecting mice prior to or following either peripheral or airborne VEEV challenge.
Results
Prophylactic protection from virulent VEEV challenge by passive transfer of hMAb F5 nIgG
To determine prophylactic efficacy of neutralizing hMAb F5 nIgG, we challenged mice by either SC or aerosol exposure to virulent VEEV 24 h after passive transfer of hMAb F5. Six groups of 10 mice each were inoculated IP with varying doses (0.01–100 µg) of purified hMAb F5 and challenged 24 h later by SC injection of 100sc 50% morbidity doses (MD50)(20 PFU/0.1 ml) of virulent VEEV Trinidad donkey (TrD) strain (Table 1). The survival rate of the groups given 0.1–100 µg hMAb was 80–100% (P<0.001, two-tailed Fisher’s exact test). None of the mice in either the control (PBS-treated) group or the group treated with 0.01 µg MAb survived virus challenge. Both 500 and 50 µg passively transferred hMAb F5 protected 90–100% of mice challenged 24 h later with 100aeroMD50 aerosolized VEEV (P<0.0002; Table 1). High titers of passively transferred hMAb persisted 14 days after challenge in sera of survivors inoculated with 500 µg of antibody prior to aerosol infection or 100 µg of antibody prior to SC challenge. A murine antibody response to VEEV was undetectable in animals given ≥10 µg hMAb, indicating that sterilizing immunity had been established in these animals. Mice inoculated prophylactically with 1 or 0.1 µg hMAb mounted an immune response following SC challenge, resulting in the production of murine anti-viral antibody that might have contributed to protection from virus challenge (Table 1).
Table 1.
Prophylactic efficacy of human (h) MAb F5 nIgG for mice infected with virulent Venezuelan equine encephalitis virus (VEEV) Trinidad Donkey (TrD) strain by subcutaneous (SC) or aerosol routes.
| HMAb prophylaxis for infected mice | ||||||
|---|---|---|---|---|---|---|
| SC VEEV infectiona | Aerosol VEEV infectionb | |||||
| µg hMAb | % Survivorsc |
hIgGd | mIgGe | % Survivorsc |
hIgGd | mIgGe |
| 500 | -- | -- | -- | 90 | 4,299 | <5 |
| 100 | 100 | 3715 | <10 | |||
| 50 | 100 | -- | -- | 100 | 479 | <5 |
| 10 | 100 | 89 | <10 | -- | -- | -- |
| 1 | 80 | <10 | 29 | -- | -- | -- |
| 0.1 | 100 | <10 | 912 | -- | -- | -- |
| 0.01 | 0 | -- | -- | -- | -- | -- |
| PBS | 0 | -- | -- | 0 | -- | -- |
100scMD50, 20 PFU/0.1 ml.
100,000 PFU/5 ml.
10 mice per group.
hIgG (F5 nIgG) geometric mean anti-VEEV ELISA titer of survivors’ sera.
Murine (m) IgG geometric mean anti-VEEV ELISA titer of survivors’ sera.
--, not done.
To assess the ability of hMAb F5 to prevent virus replication, groups of 3 mice were inoculated IP with 50 µg hMAb F5 24 h prior to aerosolized VEEV challenge and euthanized on days 1, 3, and 5 PI. Brain tissue and serum samples were obtained from each treated animal as well as from 3 untreated controls for titration of infectious VEEV by plaque assay (Table 2). The limits of virus detection were 100 PFU/g for brain tissue and 100 PFU/ml for sera. Little to no virus was detected in tissues of hMAb F5-treated mice compared to the untreated controls, which had brain titers of 109.8 PFU/g. As expected, serum samples from untreated mice contained no detectable infectious virus at 6.5 days PI; however, a geometric mean titer (GMT) of 105.7 PFU/ml serum was determined for samples from 11 untreated animals collected on days 1 to 4 PI (based on data used to construct Fig. 1).
Table 2.
Virus replication in mouse tissues following prophylaxis with 50 µg hMAb F5 nIgG 24 h prior to aerosol infection with Venezuelan equine encephalitis virus (VEEV).
| VEEV titer | ||||
|---|---|---|---|---|
| Mouse # |
50 µg F5 nIgGa |
Euthanasia and tissue collection, DPIb |
PFU/g brain tissue |
PFU/ml serum |
| 1 | + | 1 | <100 | <100 |
| 2 | + | 1 | <100 | <100 |
| 3 | + | 1 | <100 | 2200 |
| 4 | + | 3 | <100 | <100 |
| 5 | + | 3 | <100 | <100 |
| 6 | + | 3 | <100 | <100 |
| 7 | + | 5 | 700 | <100 |
| 8 | + | 5 | <100 | <100 |
| 9 | + | 5 | <100 | <100 |
| 10 | -- | 6.5 | 5.5 X 109 | <100 |
| 11 | -- | 6.5 | 2.1 X 109 | -- |
| 12 | -- | 6.5 | 9.8 X 109 | <100 |
intraperitoneal administration 24 h prior to infection.
DPI, days post-infection.
--, not done
Fig. 1.

Titer of Venezuelan equine encephalitis virus (VEEV) in various tissues for 6 or 7 days following aerosol infection in both untreated mice and mice treated intraperitoneally with 50 µg of human MAb F5 nIgG 24 h post-infection. Geometric mean titers (GMT) of VEEV per g tissue from untreated mice are shown with solid lines and from hMAb-treated mice with dotted lines. For untreated mice, GMTs were determined from n = 3 mice for days 1, 2, 3, 5, and 6 and n=3 for day 4; for hMAb-treated mice, GMTs were determined from n = 3 mice for days 2 and 3, n = 2 for days 4 and 5, and n = 1 for day 6. Tissues: serum (◆, red), brain (●, gray), lung (▲, pink), spleen (■, green), heart (x, blue), liver (−, brown), and kidney (+, cyan).
Therapeutic efficacy of passively transferred hMAb F5 nIgG following virulent VEEV challenge
Based on preliminary experiments, doses of 500 or 50 µg of passively transferred hMAb F5 were chosen to determine therapeutic efficacy following SC or aerosol challenge with virulent VEEV. Groups of 10 mice were infected by SC inoculation of 100scMD50 or aerosol exposure to 65aeroMD50 VEEV TrD, 24 h prior to IP hMAb transfer. Treatment with a 500 µg dose of hMAb F5 within 24 h of infection resulted in 90% to 100% survival of mice infected by either route (Table 3). Significant recovery (P<0.0002) after SC infection was provided by a dose of 500 µg but not 50 µg of hMAb F5. Surviving mice that received either treatment dose mounted an anti-viral immune response that generated high murine antibody titers in sera collected on day 14 PI. However, hMAb was not detectable in survivor sera, although it had been observed when 500 µg or 100 µg hMAb was administered prophylactically (Table 1).
Table 3.
Therapeutic efficacy of human (h) MAb F5 nIgG inoculated intraperitoneally (IP) 24 h post-infection for mice infected with virulent Venezuelan equine encephalitis virus (VEEV) Trinidad Donkey (TrD) strain by subcutaneous (SC) or aerosol routes.
| MAb therapy for infected mice | ||||||
|---|---|---|---|---|---|---|
| SC VEEV infectiona | Aerosol VEEV infectionb | |||||
| µg hMAb | % Survivorsc | hIgGd | mIgGe | % Survivorsc | hIgGd | mIgGe |
| 500 | 90 | <10 | 6,813 | 100 | 107 | ≥74 |
| 50 | 30 | <10 | 12,589 | 80 | <10 | 79 |
| 10 | 0 | -- | -- | -- | -- | -- |
| PBS | 0 | -- | -- | 0 | -- | -- |
100scMD50, 20 PFU/0.1 ml.
100,000 PFU/5 ml.
10 mice per group.
hIgG (F5 nIgG) geometric mean anti-VEEV ELISA titer of survivors’ sera.
Murine (m) IgG geometric mean anti-VEEV ELISA titer of survivors’ sera.
--, not done.
Surprisingly, recovery of mice infected with VEEV by the aerosol route was achieved more efficiently than those infected SC; both 500 and 50 µg doses of hMAb protected mice in this study (Table 3; P<0.001). Sera from aerosol-infected groups of surviving mice also had much lower mIgG ELISA titers than mice infected by the SC route.
To determine the ability of a 50 µg therapeutic dose of hMAb F5 to control VEEV replication following aerosol exposure, groups of hMAb F5-treated and untreated mice were euthanized on days 1 to 6 PI and serum, brain, lung, spleen, heart, liver, and kidney samples were assayed for infectious virus (Fig. 1; Table S1). Untreated mice developed maximum virus GMTs of 106.0 to 109.4 PFU/g tissue as early as 1 day p.i. for lung tissue (106.5 PFU/g) and by 4 to 5 days PI for other tissues. Virus titers in all tissue samples except brain from hMAb F5-treated mice ranged from below the minimum detection limit (<102 PFU/g) to 103 PFU/g. Unexpectedly, hMAb-treated mice that exhibited no clinical signs of disease had virus titers in brain samples equivalent to those from untreated animals.
In our initial experiment, hMAb-treated mice were observed for 6 days following infection—the average survival time of naïve mice infected with VEEV TrD. Following prophylactic administration, only two mice had low levels of detectable infectious virus in brain or serum at any time PI (Table 2). In contrast, untreated mice had in excess of 109 PFU/g brain tissue when they were euthanized at day 6.5 PI. VEEV infection of the brain following aerosolized delivery was thus prevented by prophylactic administration of 50 µg hMAb F5. Despite survival of mice treated therapeutically 24 h PI, virus titers in brains of treated mice were equivalent to those of untreated mice sampled on days 3 to 6 after infection (Fig. 1). We therefore conducted an additional trial to determine how long infectious virus and viral RNA persisted in brains after VEEV aerosol challenge followed 24 h later by hMAb F5 treatment (Table 4). In this experimental protocol the untreated mice became severely ill on days 6 or 7 PI and were euthanized. Three hMAb F5-treated mice were euthanized on each of days 7, 14, and 28 PI, and brain tissues were assayed for infectious virus by plaque titration and for viral RNA by qRT-PCR. The untreated mice had a GMT of 109.6 PFU/g brain tissue following euthanasia on days 6 or 7 PI. The virus titer on day 7 PI in brain tissue from mice treated at 24 h PI with 50 µg hMAb varied from undetectable to109.4 PFU/g. By 14 days PI, no virus could be demonstrated in the F5-treated mouse brains, although 2 of 3 brains contained low levels of viral RNA as determined by qRT-PCR. By 28 days PI, neither infectious virus nor viral RNA could be detected in the any of the hMAb-treated mouse brain samples (Table 4).
Table 4.
Persistence of infectious virus and viral RNA in mouse brains following infection with aerosolized Venezuelan equine encephalitis virus (VEEV) and treatment with 50 µg human (h) MAb F5 nIgG 24 h post-infection (PI).
| qRT-PCR for viral RNA | |||||
|---|---|---|---|---|---|
| Mouse brain no. |
50 µg F5 nIgGa |
Euthanasia and tissue collection DPIb |
VEEV PFU/g tissue |
mean CTc | CT Std. Dev. |
| MB C-1d | − | 6 | 4.3 X 109 | 16.06 | 0.32 |
| MB C-2 | − | 6 | 3.6 X 108 | 18.04 | 0.25 |
| MB C-3 | − | 7 | 6.8 X 109 | 17.27 | 0.50 |
| MB-1 | + | 7 | 2.4 X 106 | 19.91 | 0.23 |
| MB-2 | + | 7 | <100 | 31.73 | 0.16 |
| MB-3 | + | 7 | 800 | 25.12 | 0.20 |
| MB-4 | + | 14 | <100 | 32.37 | 0.26 |
| MB-5 | + | 14 | <100 | 31.33 | 0.14 |
| MB-6 | + | 14 | <100 | NAe | – |
| MB-7 | + | 28 | <100 | NA | – |
| MB-8 | + | 28 | <100 | NA | – |
| MB-9 | + | 28 | <100 | NA | – |
| qRT-PCR controls | |||||
| VEEV RNA 1:5 | – | – | – | 17.39 | 0.09 |
| VEEV RNA 1:50 | – | – | – | 21.09 | 0.34 |
| Water only | – | – | – | NA | – |
| No template | – | – | – | NA | – |
24 h PI, intraperitoneally.
DPI, days post-infection.
CT, threshold cycle; values ≤38.5 interpreted as positive.
C-1, -2, -3, virus-infected, untreated mice.
NA, no amplification.
–, not done.
Detection of VEEV antigen by IHC in brain tissue from untreated and hMAb F5 nIgG-treated mice
To determine if the same brain regions and cell types were infected in hMAb-treated and untreated mice, tissue samples were harvested at 5 days PI from several areas of the CNS including olfactory bulb, brainstem, cerebrum and cerebellum, fixed, embedded in paraffin, and sectioned for IHC detection of VEEV antigen. Similar intensities and patterns of virus antigen were detected in neurons from all areas of the CNS examined in both untreated and hMAb-treated mice (Fig. 2). These findings were in agreement with the results from plaque assays demonstrating the presence of high viral titers in the brains of both hMAb F5-treated and untreated VEEV-infected mice (Fig.1).
Fig. 2.
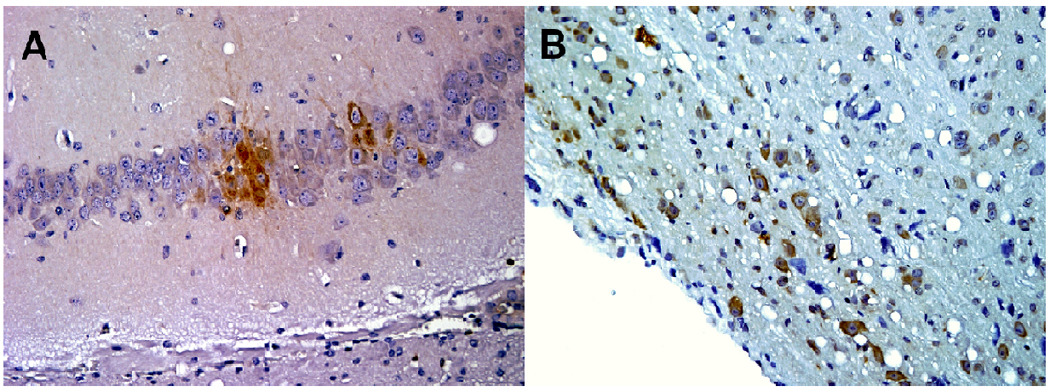
Immunohistochemistry of Venezuelan equine encephalitis virus-infected mouse brains harvested at 5 days post-infection (PI) from an untreated mouse (A) or a mouse treated with hMAb F5 nIgG 24 h PI (B).
Discussion
We previously described the antigenic structure of both the VEEV E1 and E2 glycoproteins using mMAbs, as well as identifying viral epitopes most important in the murine protective response (Mathews and Roehrig, 1982; Mathews et al., 1985; Roehrig and Mathews, 1985; Roehrig et al.,1982). More recently, we mapped the VEEV epitopes recognized by hMAbs (Hunt et al., 2010) and selected hMAb F5 nIgG, which has significant neutralizing ability and is specific for a unique E2 protein epitope, AA 115–119, for evaluation of its potential to provide either pre- or post-exposure protection from both SC and aerosol challenge with virulent VEEV TrD in a murine model.
The association of in vitro neutralizing activity with in vivo protection using anti-VEEV mMAbs or humanized mMAbs is well-documented (Hunt et al., 2006; Mathews and Roehrig, 1982; Phillpotts, 2006; Phillpotts et al., 2002). F5 nIgG had a VEEV TC-83 70% plaque-reduction neutralization endpoint of 12.5 ng/ml, which compares favorably to average endpoints of 29 and 100 ng/ml for humanized Hy4 IgG and mMAb 3B4C-4, respectively (Hunt et al., 2006, 2010). Prophylactic administration of 100 µg or 500 µg of Hy4 resulted in significant protection from IP or IN VEEV challenge, respectively (Hunt et al., 2006); thus, we expected F5 nIgG to provide prophylactic protection from SC or aerosol challenge with VEEV TrD in a similar manner. We found that prophylactic administration of 50 µg of F5 nIgG resulted in similar levels of protection from aerosol challenge as 500 µg of Hy4 IgG provided against IN VEEV challenge (Hunt et al., 2006; Table 1). As little as 100 ng of either Hy4 or F5 protected 90–100% of mice from lethal IP or SC challenge, and survivor sera contained significant murine anti-VEEV titers but little to no residual human antibody (Hunt et al., 2006; Table 1). Administration of 50 µg of prophylactic F5 nIgG resulted in complete protection and almost complete sterilizing immunity in mice that survived subsequent challenge with aerosolized VEEV, as evidenced by both the lack of a murine anti-VEEV humoral response in challenge survivors and the inability to detect infectious virus in most serum and brain samples collected on days 1 to 5 post-challenge (Tables 1,2). These data support results of previous studies with anti-VEEV neutralizing MAbs 1A4A-1, 1A3A-9, 1A3B-7 and 3B2A-9, which documented the protective capacity of mMAbs from SC and aerosol VEEV challenge (Phillpotts, 2006; Phillpotts et al., 2002).
We found that a dose of 500 µg F5 nIgG was effective in protecting mice 24 h after either SC or aerosol infection with VEEV TrD (Table 3). In our study, murine anti-VEEV titers in mice treated after SC virus infection were significantly higher than in mice infected by the aerosol route; no hIgG could be detected after 14 days in mice that survived SC challenge (Table 3). Phillpotts et al. (2002) reported that 100 µg mMAb 1A3A-9 provided significant post-exposure protection to mice when administered 2 or 24 h, but not 72 h, following aerosol VEEV infection. They also showed that 24 h-post-exposure treatment of VEEV-infected mice with mMAb led to significant reductions in virus titers in peripheral organs for 5 days PI, but not in brains of about half of treated mice. This finding led the investigators to suggest that although MAb given prophylactically could prevent virus replication in the brain, therapeutic activity depended on both rapid clearance from the periphery and prevention of virus infection of the brain, and that treatment would have little effect once a CNS infection was established. We followed VEEV titers in tissue and serum samples from F5 nIgG-therapeutically treated and untreated mice for 6 days after aerosol infection and also found that infectious virus replication was controlled in the periphery, but not in the brain (Fig. 1). Demonstration of similar intensity and cell specificity of virus antigen expression in the brains of hMAb-treated and untreated mice was confirmed by detection of virus antigen in neurons by IHC (Fig. 2). Subsequently, we followed hMAb-treated mice for 28 days PI and found that infectious virus as well as viral RNA was cleared from brains by 14–28 days PI (Table 4). Despite initial high titers of virus in brains, none of the mice euthanized on days 7, 14, or 28 following infection showed any clinical signs of disease.
We have not yet investigated a possible increase in pharmacologic potency by administering a mixture of MAbs Hy4 IgG and F5 nIgG. Mixing of mMAbs 1A4A-1 and 1A3A-9 demonstrated no enhanced efficacy when used for treatment (Phillpotts et al., 2002). Because F5 and Hy4 bind to different regions on the E2 protein it is feasible that this mixture might show enhanced efficacy. Additionally, cocktails of therapeutically relevant antibodies could reduce the probability of selecting resistant strains or neutralization-escape variants in vivo (Sanna et al., 2000).
Based on extensive studies with SINV, as well as VEEV, the complex association of the immune response in CNS disease pathogenesis and recovery is well established (Griffin, 2010). Unlike SINV, VEEV is directly cytopathic for cells of the CNS in the absence of an immune response (Charles et al., 2001). Although T cells and their related cytokines, especially IFN-γ, have a prominent role in clearing virus from the CNS, mice deficient in mature B cells are not able to clear virus from the brain or prevent persistent virus replication (Griffin, 2010). For protection by passive antibody transfer, viral clearance is dependent on the amount and specificity of antibody transferred; antibody to the E2 protein is most effective (Johnston and Peters, 1996). In the current study the amount of IP-administered anti-E2 F5 nIgG required for effective prophylaxis was as little as 100 ng for subsequent SC VEEV challenge and 50 µg for aerosol challenge (Table 1). The effective dose of post-exposure F5 nIgG, delivered 24 h PI, was 500 µg for SC VEEV infection or 50 µg for aerosol infection (Table 3). Local production of antibody in the CNS or nasal mucosa may also play a role in protection and continued suppression of viral replication due to persistant viral RNA (Charles et al., 1997; Elvin et al., 2002; Griffin, 2010). Although SINV RNA was detected by RT-PCR for 12 months after infection in mice effectively treated with passive antibody (Levine and Griffin, 1992; Levine et al., 1991), we found no detectable viral RNA 14–28 days PI in brain tissue from mice infected by aerosolized VEEV and treated 24 h PI with 50 µg F5 nIgG. The mechanism of antibody suppression of intracellular virus replication is not completely understood, but appears to require bivalent antibody and is independent of IgG isotype (Ubol et al., 1995).
Human exposure to VEEV in nature is usually mosquito-transmitted and related to the occurrence of epizootic disease in equines and can probably be best controlled by routine vaccination of equine populations (Johnson and Martin, 1974). In addition, humans can be occupationally (laboratorians, veterinarians or field workers) exposed to VEEV via injection or aerosol routes (Slepushkin, 1959; Zarate et al., 1968) or through an act of bioterrorism (intentional release). Because no FDA-licensed, human vaccine or specific antiviral drugs are currently available, an antiviral, protective hMAb for passive immunization has a number of advantages: 1) provides a state of immediate immunity and can be used prophylactically, 2) is highly specific and 3) has minimal toxicity or reactogenicity for the human host. Technological developments in antibody engineering and recombinant DNA technology, as well as antibiotic resistance, have created more interest in passive antibody immunization for prevention and treatment of infectious diseases (Casadevall, 2002; Casadevall et al., 2004; Krause et al., 1997; Weltzin and Monath, 1999; Zeitlin et al., 1999). Several antibody-based therapies are being developed for viral encephalitides. A phase I clinical study of the safety, pharmacokinetics, and immunogenicity of a neutralizing, recombinant humanized mMAb (MGAWN1) targeting the E glycoprotein of the encephalitogenic flavivirus West Nile virus has recently been reported (Beigel et al., 2010). A hMAb cocktail that neutralizes rabies virus has been evaluated in phase I studies as an alternative strategy to human and equine rabies immunoglobulin for post-exposure prophylaxis in humans (Bakker et al., 2008).
Although the mouse model for VEEV disease is considered to closely reflect the disease process in humans, studies in different immunocompetent inbred mouse strains (Balb/c, C3H/HeN, A/J) have revealed some variability in response to vaccines or passive antibody immunization (Elvin et al., 2002; Hart et al., 2000; Kinney et al., 1988; Phillpotts et al., 2002). We have used outbred mice (ICR or Swiss Webster) in our evaluations of the protective capacity of human or humanized anti-VEEV E2 MAbs in an effort to assess an overall species response (Hunt et al., 2006). Our results suggest that hMAb F5 nIgG is a valuable antiviral, capable of providing both pre- and post-exposure protection from peripheral and aerosol VEEV challenge, if used at appropriate doses. It is also effective in reducing or eliminating persistent viral RNA replication. Since passively-administered humanized antibody titer in mouse serum was fairly stable over a two-week period, prophylactic protection for a similar period is theoretically possible (Hunt et al., 2006); however, therapeutic doses must be given within 24 h of infection for significant survival rates since the average survival time of VEEV-infected mice is only about six days. The VEEV-infected, passively immunized mouse model will also provide a good platform for future studies of the contributions of other components of the immune response to disease or survival of animals. In addition, further evaluation of the potential of anti-VEEV passive human antibody is needed in nonhuman primates.
Materials and methods
Viruses and antibodies
VEEVs used in this study were subtype 1AB, strains TC-83 and TrD, obtained from the Centers for Disease Control and Prevention (CDC), Division of Vector-Borne Diseases (DVBD), Fort Collins, CO. Viruses were grown in Vero or BHK-21 cells and purified by equilibrium density gradient centrifugation (Obijeski et al, 1976). Outbred ICR mice were exposed to serial 10-fold dilutions of VEEV TrD to determine the MD50 for both SC and aerosol routes of infection. MD50 were calculated by the method of Reed and Muench (Reed and Muench, 1938) MAbs Hy4 IgG (clone 26C) and F5 nIgG (clone 1E9) have been previously described (Hunt et al., 2006, 2010). Both MAbs were expressed from stable 293-EBNA cell lines and purified on a protein A column using FPLC.
Enzyme immunoassays
Indirect ELISAs were performed essentially as previously described (Hunt et al., 2006; Roehrig et al., 1980), using native TC-83 virus as antigen (1 µg/well). Specific binding of passively administered hMAbs and murine antiviral antibodies in murine sera were detected by goat anti-human Fc-specific- or goat anti-mouse IgG-alkaline phosphatase (AP) conjugates, respectively. An absorbance ratio (A405 sample/A405 negative control) > 2 was considered to be positive.
Passive antibody transfer and virus challenge in mice
Outbred adult ICR mice, 6–8 weeks old, were used for all passive antibody protection studies. The use of animals for research purposes complied with relevant federal guidelines and specific protocols were approved by the Colorado State University Institutional Animal Care and Use Committee.
For studies using prophylactic antibody treatment, groups of 10 mice were inoculated IP with 100 µl containing specified amounts of purified MAb F5 nIgG approximately 24 h prior to virus challenge. For therapeutic treatment, mice were inoculated IP with MAb 24 h after virus infection. Mice were challenged either SC with 100scMD50 (20 PFU/0.1 ml) of VEEV TrD or by aerosol route with a dose of approximately 65aeroMD50 (100,000 PFU/5 ml). Aerosols containing VEEV were produced using an Inhalation Exposure System (GlasCol, Inc., Terre Haute, IN) in a BSL-3 suite. A wire-mesh basket with 5 compartments held the mice during the aerosolization process. Both MAb-treated and control groups of mice, located in separate compartments, were exposed to virus at the same time. Five ml of virus stock solution diluted in PBS (20,000 PFU/ml) were injected into the nebulizer and timers were set for a 20-min nebulizing cycle, a 20-min cloud-decay time, and a 15-min decontamination period. Although virus concentrations within the aerosol stream were not monitored, the MD50 values were determined by repeated trials in which serial dilutions of stock virus were placed in the nebulizer, and mouse morbidity/mortality recorded. Additionally, untreated, VEEV-infected animals were included in every experimental trial using hMAb treatment of infected mice. Over years of use, we have found this system to deliver consistent concentrations of organisms to mouse lungs. All mice were monitored for signs of illness for 2 weeks and survivors were bled 14 days post-challenge. Although some animals died from VEEV infection, death was not a required endpoint, and animals were euthanized when they exhibited severe illness or paralysis.
Replication and clearance of virus from the brains of untreated and MAb F5-treated mice
Studies to monitor infectious virus and viral RNA were conducted using smaller groups of mice (n = 3) infected with aerosolized VEEV. Mice were either prophylactically or therapeutically treated with hMAb F5 or untreated, and serum and tissue samples were collected for virus titration on days 1 through 6 PI. Tissue samples were frozen at −80°C until they were processed by homogenizing in BA1 diluent [1X M199 with Hanks’ balanced salt solution, 0.05 M Tris buffer (pH 7.6), 1% bovine serum albumin, 0.35 g/liter sodium bicarbonate, 100 mg/liter streptomycin, 1 mg/ml Fungizone] as 10% suspensions using a mixer mill. The suspensions were clarified by centrifugation and supernatants and sera were assayed by plaque titration on Vero cell monolayers.
A longer-term study for 28 days evaluated the presence of infectious virus and viral RNA in the brains of mice exposed to aerosolized VEEV and treated with MAb F5 24 h post-exposure. Brain tissue samples were collected from 3 mice on days 6/7, 14, and 28 and were processed and assayed for virus as described above. In addition, 0.4 ml of each 10% brain homogenate supernatant was added to 2 ml of Trizol Reagent (Invitrogen, Carlsbad, CA) and frozen at −80°C for subsequent RNA extraction.
RNA extraction and quantitative (q) RT-PCR
Viral RNA was extracted from mouse brain homogenate supernatants (0.4 ml), processed as described above, using a PureLink RNA Mini Kit (Invitrogen) according to the manufacturer’s instructions. RNA was eluted in 50 or 100 µl RNase-free water, quantitated using an ND1000 NanoDrop spectrophotometer (ThermoScientific, West Palm Beach, FL), and stored at −80°C until used. Quantitative RT-PCR assays were performed with 50 ng of extracted RNA, 50 pmol of each VEEV TC-83 primer, and 10 pmol of a dual-labeled VEEV TC-83 probe containing a FAM fluorescent reporter at the 5’ end and TAMRA quencher molecule at the 3’ end, in a total volume of 50 µl using the Quantitect Probe RT-PCR Kit (Qiagen, Valencia, CA) according to the manufacturer’s instructions. The VEEV TC-83 primers and probe were kindly provided by Dr. R. S. Lanciotti of DVBD, CDC, Ft. Collins, CO. Amplification and fluorescence detection were performed on the iCycler (Bio-Rad Laboratories, Hercules, CA). As recommended b the manufacturer, 45 amplification cycles were performed. Results were determined by the amplification cycle at which fluorescence increased above the threshold value (set at 104 relative fluorescence units) by use of the PCR baseline-subtracted curve fit analysis mode. Samples with a threshold cycle (CT) value of ≤38.5 were considered positive.
Immunohistochemistry (IHC)
Tissues from mice infected with VEEV TrD were fixed in buffered formalin, embedded in paraffin and sectioned. For IHC the sections were deparaffinized, treated with proteinase K for 10 min and blocked by sequential treatment with hydrogen peroxide (10 min), glycine buffer (15 min), and blocking solution (overnight). The sections were then treated overnight with mouse anti-VEEV (TC-83) ascitic fluid (CDC VS0119) diluted 1:200 in blocking solution, washed, and incubated with horseradish peroxidase-labeled anti-mouse IgG. The sections were washed again, exposed to the substrate diaminobenzidene-tetrahydrochloride and counterstained with Lillie’s modified hematoxylin.
Supplementary Material
Acknowledgments
We thank Drs. Amy Lambert and Robert Lanciotti of the Centers for Disease Control, Fort Collins, CO, for providing VEEV-specific primers, probe, control RNA and technical assistance for qRT-PCR, Dr. Nicole Marlenee for technical expertise with all phases of studies involving mice, and Airn Tolnay for performing the immunohistochemistry.
Role of funding source
This research was supported by NIH/NIAID grant U54AI-065357 to the Rocky Mountain Regional Center of Excellence for Biodefense and Emerging Infectious Disease Research (http://www.rmrce.colostate.edu/). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Richard A. Bowen, Email: Richard.Bowen@colostate.edu.
John T. Roehrig, Email: jtr1@cdc.gov.
Carol D. Blair, Email: Carol.Blair@colostate.edu.
References
- Bakker ABH, Python C, Kissling CJ, Pandya P, Marissen WE, Brink MF, Lagerwerf F, Worst S, van Corven E, Kostense S, Hartmann K, Weverling G, Jl, Uytdehaag F, Herzog C, Briggs DJ, Rupprecht CE, Grimaldi R, Goudsmit J. First administration to humans of a monoclonal antibody cocktail against rabies virus: Safety, tolerability, and neutralizing activity. Vaccine. 2008;26:5922–5927. doi: 10.1016/j.vaccine.2008.08.050. [DOI] [PubMed] [Google Scholar]
- Beigel JH, Nordstrom JL, Pillemer SR, Roncal C, Goldwater DR, Li H, Holland PC, Johnson S, Stein K, Koenig S. Safety and pharmacokinetics of single intraveneous dose of MGAWM1, a novel monoclonal antibody to West Nile virus. Antimicrob. Agents Chemother. 2010;54:2431–2436. doi: 10.1128/AAC.01178-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronze MS, Huycke MM, Machado LJ, Voskuhl GW, Greenfield RA. Viral agents as biological weapons and agents of bioterrorism. Am. J. Med. Sci. 2002;323:316–325. doi: 10.1097/00000441-200206000-00004. [DOI] [PubMed] [Google Scholar]
- Casadevall A. Passive antibody administration (immediate immunity) as a specific defense against biological weapons. Emerg. Infect. Dis. 2002;8:333–341. doi: 10.3201/eid0808.010516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casadevall A, Dadachova E, Pirofski L-A. Passive antibody therapy for infectious diseases. Nature Rev. 2004;2:695–703. doi: 10.1038/nrmicro974. [DOI] [PubMed] [Google Scholar]
- Charles PC, Brown KW, Davis NL, Hart MK, Johnston RE. Mucosal immunity induced by parenteral immunization with a live attenuated Venezuelan equine encephalitis virus vaccine candidate. Virology. 1997;228:153–160. doi: 10.1006/viro.1996.8381. [DOI] [PubMed] [Google Scholar]
- Charles PC, Trgovcich J, Davis NL, Johnston RE. Immunopathogenesis and immune modulation of Venezuelan equine encephalitis virus-induced disease in the mouse. Virology. 2001;284:190–202. doi: 10.1006/viro.2001.0878. [DOI] [PubMed] [Google Scholar]
- Elvin SJ, Bennett AM, Phillpotts RJ. Role for mucosal immune responses and cell-mediated immune functions in protection from airborne challenge with Venezuelan equine encephalitis virus. J. Med. Virol. 2002;67:384–393. doi: 10.1002/jmv.10086. [DOI] [PubMed] [Google Scholar]
- Fine DL, Roberts BA, Terpening SJ, Mott J, Vasconcelos D, House RV. Neurovirulence evaluation of Venezuelan equine encephalitis (VEE) vaccine candidate V3526 in nonhuman primates. Vaccine. 2008;26:3497–3506. doi: 10.1016/j.vaccine.2008.04.044. [DOI] [PubMed] [Google Scholar]
- Griffin DE. Recovery from viral encephalomyelitis: immune-mediated noncytolytic virus clearance from neurons. Immunol. Res. 2010;47:123–133. doi: 10.1007/s12026-009-8143-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart MK, Caswell-Stephan K, Bakken R, Tamariello R, Pratt W, Davis N, Johnston RE, Smith J, Steele K. Improved mucosal protection against Venezuelan equine encephalitis virus is induced by the molecularly defined, live-attenuated v3526 vaccine candidate. Vaccine. 2000;18:3067–3075. doi: 10.1016/s0264-410x(00)00042-6. [DOI] [PubMed] [Google Scholar]
- Hawley RJ, Eitzen EM., Jr Biological weapons—a primer for microbiologists. Annu. Rev. Microbiol. 2001;55:235–253. doi: 10.1146/annurev.micro.55.1.235. [DOI] [PubMed] [Google Scholar]
- Hunt AR, Frederickson S, Hinkel C, Bowdish KS, Roehrig JT. A humanized murine monoclonal antibody protects mice either before or after challenge with virulent Venezuelan equine encephalomyelitis virus. J. Gen. Virol. 2006;87:2467–2476. doi: 10.1099/vir.0.81925-0. [DOI] [PubMed] [Google Scholar]
- Hunt AR, Frederickson S, Maruyama T, Roehrig JT, Blair CD. The first human epitope map of the alphaviral E1 and E2 proteins reveals a new E2 epitope with significant virus neutralizing activity. PLoS Negl. Trop. Dis. 2010;4:e739. doi: 10.1371/journal.pntd.0000739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson AC, SenGupta SK, Smith JF. Pathogenesis of Venezuelan equine encephalitis virus infection in mice and hamsters. Vet. Pathol. 1991;28:410–418. doi: 10.1177/030098589102800509. [DOI] [PubMed] [Google Scholar]
- Jahrling PB, Stephenson EH. Protective e fficacies of live attenuated and formaldehyde-inactivated Venezuelan equine encephalitis virus against aerosol challenge in hamsters. J. Clin. Microbiol. 1984;19:429–431. doi: 10.1128/jcm.19.3.429-431.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson KM, Martin DH. Venezuelan equine encephalitis. Adv. Vet. Sci. Comp. 1974;18:79–116. [PubMed] [Google Scholar]
- Johnston RE, Peters CJ. Alphaviruses. In: Fields BN, Knipe DM, Howley PM, Channock RM, Melnick JL, Monath TP, Roizman B, editors. Virology. New York: Lippincott-Raven Press; 1996. pp. 843–898. [Google Scholar]
- Kinney RM, Esposito JJ, Johnson BJB, Roehrig JT, Mathews JH, Barrett ADT, Trent DW. Recombinant vaccinia-Venezuelan equine encephalitis (VEE) virus expresses VEE structural proteins. J. Gen. Virol. 1988;69:3005–3013. doi: 10.1099/0022-1317-69-12-3005. [DOI] [PubMed] [Google Scholar]
- Krause RM, Dimmock NJ, Morens DM. summary of antibody workshop: The role of humoral immunity in the treatment and prevention of emerging and extant infectious diseases. J. Infect. Dis. 1997;176:549–559. doi: 10.1086/514074. [DOI] [PubMed] [Google Scholar]
- Kundin WD. Pathogenesis of Venezuelan equine encephalomyelitis virus. II. Infection in young and adult mice. J. Immunol. 1966;96:49–58. [PubMed] [Google Scholar]
- Levine B, Griffin DE. Persistence of viral RNA in mouse brains after recovery from acute alphavirus encephalitis. J. Virol. 1992;66:6429–6435. doi: 10.1128/jvi.66.11.6429-6435.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B, Hardwick JM, Trapp BD, Crawford TO, Bollinger RC, Griffin DE. Antibody-mediated clearance of alphavirus infection from neurons. Science. 1991;254:856–860. doi: 10.1126/science.1658936. [DOI] [PubMed] [Google Scholar]
- Mathews JH, Roehrig JT. Determination of the protective epitopes on the glycoproteins of Venezuelan equine encephalomyelitis virus by passive transfer of monoclonal antibodies. J. Immunol. 1982;129:2763–2767. [PubMed] [Google Scholar]
- Mathews JH, Roehrig JT, Trent DW. Role of complement and the Fc portion of immunoglobulin G in immunity to Venezuelan equine encephalomyelitis virus infection with glycoprotein-specific monoclonal antibodies. J. Virol. 1985;55:594–600. doi: 10.1128/jvi.55.3.594-600.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obijeski JF, Bishop DH, Murphy FA, Palmer EL. Structural proteins of La Crosse virus. J. Virol. 1976;19:985–997. doi: 10.1128/jvi.19.3.985-997.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paessler S, Ni H, Petrakova O, Fayzulin RZ, Yun N, Anishchenko M, Weaver SC, Frolov I. Replication and clearance of Venezuelan equine encephalitis virus from the brains of animals vaccinated with chimeric SIN/VEE viruses. J. Virol. 2006;80:2784–2796. doi: 10.1128/JVI.80.6.2784-2796.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillpotts RJ. Venezuelan equine encephalitis virus complex-specific monoclonal antibody provides broad protection, in murine models, against airborne challenge with viruses from serogroups I, II and III. Virus Res. 2006;120:107–112. doi: 10.1016/j.virusres.2006.02.003. [DOI] [PubMed] [Google Scholar]
- Phillpotts RJ, Jones LD, Howard SC. Monoclonal antibody protects mice against infection and disease when given either before or up to 24 h after airborne challenge with virulent Venezuelan equine encephalitis virus. Vaccine. 2002;20:1497–1504. doi: 10.1016/s0264-410x(01)00505-9. [DOI] [PubMed] [Google Scholar]
- Pittman PR, Makuch RS, Mangiafico JA, Cannon TL, Gibbs PH, Peters CJ. Long-term duration of detectable neutralizing antibodies after administration of live-attenuated VEE vaccine and following booster vaccination with inactivated VEE vaccine. Vaccine. 1996;14:337–343. doi: 10.1016/0264-410x(95)00168-z. [DOI] [PubMed] [Google Scholar]
- Pratt WD, Davis NL, Johnston RE, Smith JF. Genetically engineered, live attenuated vaccines for Venezuelan equine encephalitis: testing in animal models. Vaccine. 2003;21:3854–3862. doi: 10.1016/s0264-410x(03)00328-1. [DOI] [PubMed] [Google Scholar]
- Reed LJ, Muench H. A simple method of estimating fifty percent endpoints. Amer. J. Epidem. 1938;27:493–497. [Google Scholar]
- Reed DS, Lind CM, Lackemeyer MG, Sullivan LJ, Pratt WD, Parker MD. Genetically engineered, live, attenuated vaccines protect nonhuman primates against aerosol challenge with a virulent IE strain of Venezuelan equine encephalitis virus. Vaccine. 2005;23:3139–3147. doi: 10.1016/j.vaccine.2004.12.023. [DOI] [PubMed] [Google Scholar]
- Roehrig JT, Corser JA, Schlesinger M. Isolation and characterization of hybrid cell lines producing monoclonal antibodies directed against the structural proteins of Sindbis virus. Virology. 1980;101:41–49. doi: 10.1016/0042-6822(80)90481-x. [DOI] [PubMed] [Google Scholar]
- Roehrig JT, Day JW, Kinney RM. Antigenic analysis of the surface glycoproteins of a Venezuelan equine encephalomyelitis virus (TC-83) using monoclonal antibodies. Virology. 1982;118:269–278. doi: 10.1016/0042-6822(82)90346-4. [DOI] [PubMed] [Google Scholar]
- Roehrig JT, Mathews JH. The neutralization site on the E2 glycoprotein of Venezuelan equine encephalomyelitis (TC-83) virus is composed of multiple conformationally stable epitopes. Virology. 1985;142:347–356. doi: 10.1016/0042-6822(85)90343-5. [DOI] [PubMed] [Google Scholar]
- Ryzhikov AB, Ryabchikova EI, Sergeev AN, Tkacheva NV. Spread of Venezuelan equine encephalitis virus in mice olfactory tract. Arch. Virol. 1995;140:2243–2254. doi: 10.1007/BF01323243. [DOI] [PubMed] [Google Scholar]
- Sanna PP, Ramiro-Ibanez F, De Logu A. Synergistic interactions of antibodies in rate of virus neutralization. Virology. 2000;270:386–396. doi: 10.1006/viro.2000.0276. [DOI] [PubMed] [Google Scholar]
- Slepushkin AN. Epidemiological studies on case of Venezuelan equine encephalomyelitis in a laboratory. Vop. Virulsol. 1959;4:311–314. [PubMed] [Google Scholar]
- The Subcommittee on Arbovirus Laboratory Safety of the American Committee on Arthropod-borne Viruses. Laboratory safety for arboviruses and certain other viruses of vertebrates. Am. J. Trop. Med. Hyg. 1980;29:1359–1381. doi: 10.4269/ajtmh.1980.29.1359. [DOI] [PubMed] [Google Scholar]
- Ubol S, Levine B, Lee S-H, Greenspan NS, Griffin DE. Roles of immunoglobulin valency and the heavy-chain constant domain in antibody-mediated downregulation of Sindbis virus replication in persistently infected neurons. J. Virol. 1995;69:1990–1993. doi: 10.1128/jvi.69.3.1990-1993.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver SC, Ferro C, Barrera R, Boshell J, Navarro J-C. Venezuelan equine encephalitis. Annu. Rev. Entomol. 2004;49:141–174. doi: 10.1146/annurev.ento.49.061802.123422. [DOI] [PubMed] [Google Scholar]
- Weaver SC, Reisen WK. Present and future arboviral threats. Antiviral Res. 2010;85:328–345. doi: 10.1016/j.antiviral.2009.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weltzin R, Monath TP. Intranasal antibody prophylaxis for protection against viral disease. Clin. Microbiol. Rev. 1999;12:383–393. doi: 10.1128/cmr.12.3.383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarate ML, Scherer WF. Contact-spread of Venezuelan equine encephalitis virus among cotton rats via urine or feces and the naso- or oropharynx. A possible transmission cycle in nature. Am. J. Trop. Med. Hyg. 1968;17:894–899. doi: 10.4269/ajtmh.1968.17.894. [DOI] [PubMed] [Google Scholar]
- Zeitlin L, Cone RA, Whaley KJ. Using monoclonal antibodies to prevent mucosal transmission of epidemic infectious diseases. Emerg. Inf. Dis. 1999;5:54–64. doi: 10.3201/eid0501.990107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.