

Abstract
This review article covers a concise account on fludeoxyglucose (18F–FDG) synthesis and quality control procedures with emphasis on practical synthesis Currently, 18F–FDG is the most successful PET radiopharmaceutical so far. The advancement in synthesis and quality control of 18F–FDG, together with its approval by the US FDA and the availability of reimbursement, are probably the main reasons for the flourish of clinical PET over the last 20 years. 18F–FDG can be synthesised by either electrophilic fluorination or nucleophilic fluorination reaction. Nucleophilic fluorination using mannose triflate as precursor and Kryptofix or tetrabutylammonium salts (TBA) is widely used because of higher yield and shorter reaction time. The quality control requirements of 18F–FDG can be found in United States Pharmacopeia (USP), British Pharmacopeia (BP), European Pharmacopeia (EP) and the Chemistry, Manufacturing, and Controls (CMC) section from United States Food and Drug Administration (US FDA) PET draft guidance documents. Basic requirements include radionuclidic identity, radiochemical purity, chemical purity, pH, residual solvent, sterility, and bacterial endotoxin level. Some of these tests (sterility, endotoxins and radionuclidic purity) can be finished after the 18F–FDG has been released. Although USP, BP and EP do not require filter membrane integrity test, many laboratories perform this test as an indirect evident of the product sterility. It is also interesting to note that there are major differences in 18F–FDG quality requirements among USP, BP, and CMC.
INTRODUCTION
18F–FDG is a glucose analogue in which the hydroxyl group on the 2–carbon of a glucose molecule is replaced by a fluoride atom. Like glucose, 18F–FDG is taken up into living cells by facilitated transport and then phosphorylated by hexokinase. Unlike glucose, 18F–FDG cannot undergo further metabolism because the hydroxyl group at the 2–carbon is a requirement for the process [1–2]. Nevertheless, 18F–FDG is a good indicator of glucose uptake and cell viability.
The uptake of glucose analogues into living cells also depends on modifications of various carbons at different positions. It has been shown that the specificity of 3–deoxyglucose (3–DG) and 4 deoxyglucose (4–DG) towards hexokinase reduced by 100–fold [3], hence 3–DG and 4–DG were not retained inside the cells. Similary, 3–fluoro–deoxyglucose and 4–fluoro-deoxyglucose do not accumulate in living cells as much as 18F–FDG. Although the nucleophilic substitution reaction is more widely used nowadays, the electrophilic fluorination reaction has an important place in the synthesis of 18F–FDG.
SYNTHESIS OF 18F-FDG BY ELECTROPHILIC FLUORINATION
The first synthesis of 18F–FDG was carried out in Brookhaven National Laboratory by Wolf et al in 1976 by electrophilic fluroination [4]. As shown in Figure 1, electrophilic fluorination refers to the addition of fluorine atoms across a double bond, producing a difluoro derivative of the parent compound. The electrophilic fluorination by Wolf et al involved the use of 3, 4,6–tri–O–acetyl–D–glucal as precursor. The glucal was treated with 18F–F2 to produce a 3:1 mixture of 18F labeled difluoro–glucose and difluoro–mannose derivatives. The difluoro–glucose derivative was separated and hydrolysed with hydrochloric acid to form 2–fluoro–2–deoxyglucose (Figure 2). The yield was 8% and the synthesis time was 2 hours [4].
Figure 1.
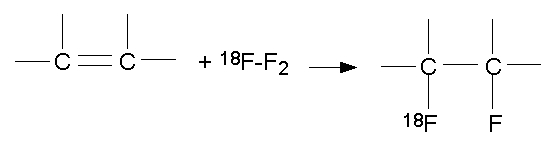
Electrophilic fluorination.
Figure 2.
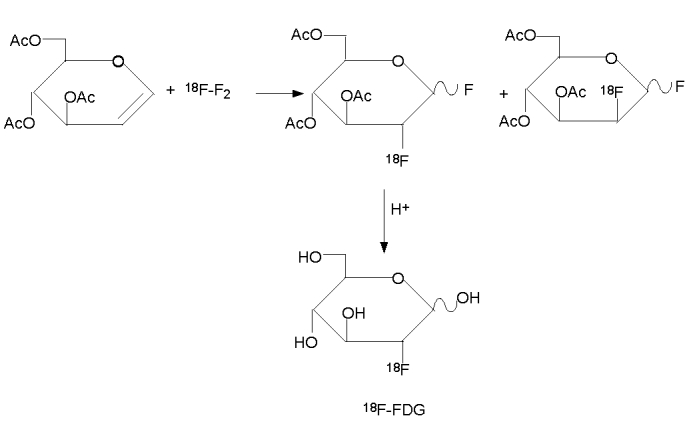
Synthesis of 18F-FDG by electrophilic fluorination.
Despite the low yield and long synthesis time, the Brookhaven team was able to collaborate with The Hospital of the University of Pennsylvania to map glucose metabolism in human brain [4]. This was the first 18F–FDG trial in human.
Several improvements to the electrophilic fluorination described above were made thereafter. One of the most useful modifications was the use of acetylhypofluorite 18F–CH3CO2F. The acetylhypofluorite can be produced in situ from 18F–F2. The yield was higher and the synthesis reaction was easier to control [4–6 ].
The major limitation of electrophilc fluorination was that only 50% of the radioactive fluorine atoms were incorporated into the precursors. In addition, the 18F–F2 was produced from a Neon gas target with 0.1% to 1% of fluorine gas via a 20Ne(d,α)18F reaction. The specific activity is lower due to the presence of the non–radioactive fluorine gas. The maintenance and operation of a Neon target is troublesome and the yield of 18F– was much lower than with the 20Ne(d,α)18F reaction than with the 18O(p,n)18F– reaction. [4, 7–8]
SYNTHESIS OF 18F-FDG BY NUCLEOPHILIC FLUORINATION
Many attempts have been made to develop a nucleophilic substitution for the synthesis of 18F–FDG. This included the use of 18F–CsF, 18F–Et4NF, and 18F–KHF [4, 9–14]. But the major breakthrough was reported in 1986 by Hamacher et al who had used Kryptofix 222TM as a catalyst [15]. The reaction had a consisten yield of over 50% and the reaction time was shortened to 50 min.
Nucleophilic substitution is a chemical reaction involving the addition of a nucleophilic molecule (highly negatively charged molecule) into a molecule with a leaving group (electron drawing group attached to the parent molecule through an unstable chemical bond). Figure 3 is a general scheme for an SN2 nucleophilic substitution reaction. The nucleophilic molecule has a high affinity towards the relatively electron deficient center in the parent molecule created by the electron pulling leaving group. As a result, the nucleophilic molecule forms a covalent bond with the parent molecule and displaces the leaving group. The stereo–configuration of the parent molecule is also changed.
Figure 3.
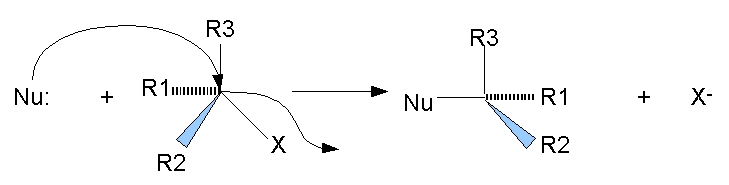
Nucleophilic substitution: Nu = nucleophilic molecule, X = leaving group.
In the synthesis of 18F–FDG, 18F ion is the nucleophile. The precursor is mannose triflate in which the 1,3,4,6 position carbons of a mannose moleucle are protected with an acetyl group and triflate is the leaving group at the 2–carbon. In the presence of Kryptofix 222TM as catalyst and acetonitrile as solvent, 18F ion approaches the mannose triflate at the 2–carbon, while the triflate group leaves the protected mannose molecule to form 18F–FDG (Figure 4).
Figure 4.
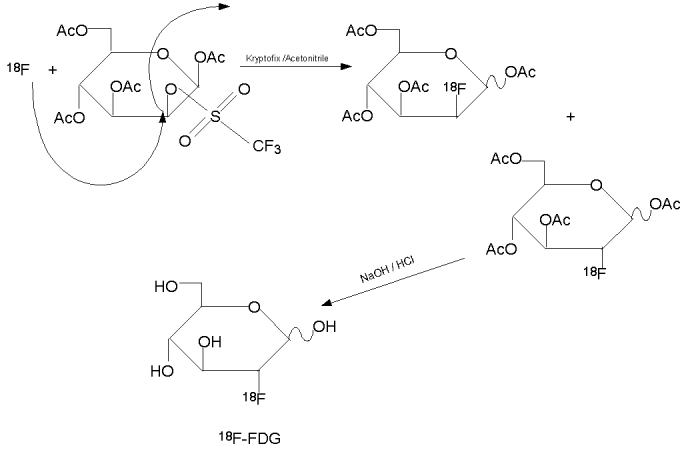
Synthesis of 18F-FDG by nucleophilic substitution.
Although synthesis of 18F–FDG can be carried out in different computer controlled automatic synthesizers, the nucleophilic process proceeds in roughly same stages:
Removal of 18F from the 18O- water coming out from the cyclotron target.
Fluorine has a high hydration energy, so water is not a suitable solvent in this synthesis. Polar aprotic solvent such as acetonitile should be used in an SN2 nucleophilic substitution reaction. Since 18F– is produced by a 18O(p,n)18F– reaction, it is necessary to isolate the 18F ion from its aqueous environment. The most convenient way to isolate is to use a light QMA (Quaternary ammonium anion exchange) Sep–Pak column (Accell Plus QMA Sep–PakTM). The 18F– is retained by or via an ion–exchange reaction and allowed the 18O–water to flow through. The retained 18F– is then eluted with an acetonitrile solution of Kryptofix and potassium carbonate (Figure 5).
Figure 5.
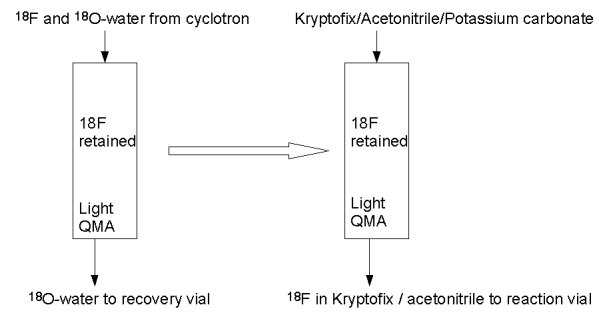
(A) Retention of 18F-FDG in light QMA ion exchange column (B) elution of 18F from light QMA ion exchange column.
In an aqueous environment, any negatively charged ions must be accompanied by positively charged counterparts. Usually, the 18F– washed out from the cyclotron target is accompanied by traces of metal ions from the surface of the target body. When passing through the light QMA anion exchange ion, the 18F – is retained and the metal ions will be lost in the 18O– water. Hence, it is necessary to introduce a positively charged counter ion to restore the 18F– reactivity before evaporation of residual 18O– enriched water [16].
Several types of positively charged counter ions have been used, including large metal ions such as rubidium or cesium; potassium ion complexed by a large ring structure such as Kryptofix 222TM and tetrabutylammonium salts [16–17]. Kryptofix 222TM is a cyclic crown ether (Figure 6), which binds the potassium ion, preventing the formation of 18F–KF. Thus, potassium acts as the counter ion of 18F– to enhance its reactivity but does not interfere with the synthesis.
Figure 6.
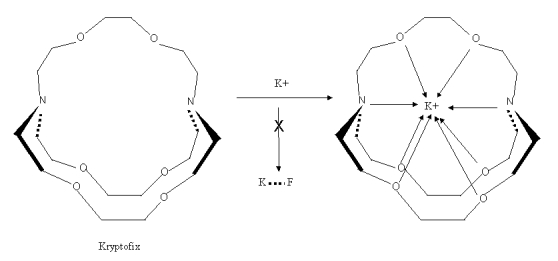
Kryptofix 222 ™ and K+.
Since Kryptofix 222TM causes apnea and convulsion, all automatic synthesis modules have multiple removal steps so that there is only negligible amount of Kryptofix in the final 18F–FDG products. Tetrabutylammonium salts (TBA) are also widely used as catalyst in place of Kryptofix 222TM [18].
Logically, the addition of a counter cation also includes the addition of another anion. The carbonate anion is most widely used because it is less likely to interfere with the synthesis [16].
Evaporation of residual 18O- water from the 18F with acetonitrile
After the 18F– is eluted into reaction vessel, it is necessary to evaporate any residual water from the solution. The advantage of using acetonitile as the eluting solvent is that it forms an azeotropic mixture with water. Evaporation of the acetonitrile in a nitrogen atmosphere will at the same time remove any residual 18O– water escaped into the reaction vessel together with the 18F. Most of the 18F–FDG automatic synthesizers perform the acetonitrile evaporation step several times to ensure all the residual 18O– water is removed. All components of the synthesis system are also rinsed with acetonitrile to remove moisture. Dry nitrogen (moisture content less than 3 ppm) should be used in the synthesis
Addition of mannose triflate into the 18F- with acetonitrile.
The nucleophilic substitution takes place in this stage. After the evaporation of any residual water, the precursor is added to the 18F–. The choice of precursor depends on the ease of preparation, ease of producing the final product, consistency, yields, and so on. The most commonly used precursor molecule in synthesis of 18F–FDG is 1,3,4,6–O–Acetyl–2–O–trifluoro-methanesulfonyl–beta–D–mannopyranose (mannose triflate). Its structure (Figure 7) is similar to that of FDG, except with a triflate group at the 2 carbon position and acetyl groups at 1,3,4,6 position carbons via ester bonds, which can be readily broken at a higher or lower pH. The use of acetyl groups is to protect the hydroxy groups so that fluorination would not occur at these positions. The 18F ion approaches the mannose triflate at the 2 position carbon, while the triflate group leaves the protected mannose molecule to form 18F–FDG (Figure 4). After the nucleophilic replacement of the triflate group by 18F–, the acetyl groups can be easily removed by hydrolysis to give rise to 18F–FDG
Figure 7.
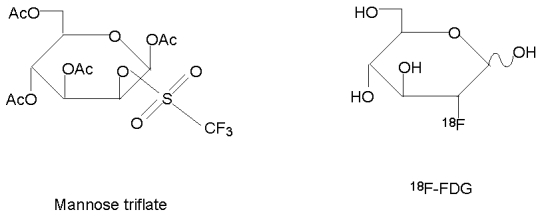
Structures of Mannose triflate and 18F-FDG.
The choice of leaving a group is an important consideration. A good leaving group should have the properties of leaving the parent molecule readily. Once it departs from the parent molecule, its negative charge is stabilised by delocalisation and it will not re–enter the parent molecule.
Commonly available leaving groups includes triflates, tosylates, mesylates among others. The choice of leaving a group depends on the nature of the reaction, the solvent, the stability of the precursor, and so on. All of the leaving groups listed in Table 1 except chlorides have been used in radio–fluorination reactions. In the synthesis of 18F–FDG, trifaltes produces a higher and more consistent yield at about 50 to 60% [16].
Table 1.
A comparison of various leaving groups
| Leaving group Properties | |
| Trifates | very good leaving group |
| Toyslate | |
| Mesylate | |
| Iodide | moderate leaving group |
| Bromide | |
| Chloride | poor leaving group |
Hydrolysis to remove the protective acetyl groups to form 18F-FDG
The final step of the synthesis is to remove the protective acetyl groups on the 1,3,4,6 position carbons. This can be accomplished by either using hydrochloric acid (acid hydrolysis) or sodium hydroxide (base hydrolysis). Acid hydrolysis requires a longer time and higher temperature. Base hydrolysis, which is more commonly used currently, is faster and takes place at room temperature. One of the improved base hydrolysis is to adsorb the 1,3,4,6 acetyl protected 18F labeled 2 dexoyglucose on to a C–18 reverse phase column. All other impurities can be removed by rinsing heavily with water. Sodium hydroxide is added to the column so that the base hydrolysis occurs on the column surface. The final 18F–FDG product can be eluted with water while the unhydrolysed or partially hydrolysed 1,3,4,6 acetyl protected 18F labeled 2 dexoyglucose remains on the column [19].
Purification of the final 18F-FDG product.
Purification of the final 18F–FDG can be performed with a series of anion exchange column, C–18 reverse phase column and alumina column. Most automatic synthesizers can produce 18F–FDG of over 95% routinely.
QUALITY CONTROL OF 18F-FDG
The quality requirements of 18F–FDG are set out in various pharmacopeia including the USP [20], BP [21], EP [22], etc. The US FDA has also published a draft Chemistry, Manufacturing and Controls (CMC) document concerning 18F–FDG [23]. It should be noted that the quality control requirements of 18F–FDG differ among these references. An excellent comparison between them can be found elsewhere [24]. In Asia, Taiwan has established an official guidelines for the compounding of PET drug products, as well as for the quality control of 18F–FDG.
Different countries my adopt a different set of standards. The BP is described in this article solely because this is the standard adopted by the author’s country. Table 2 lists the quality control tests required by BP [21]. Due to short half–life of 18F–FDG, not all the listed tests can be completed before release of the 18F–FDG product. The BP allows the 18F–FDG to be released before the radionuclidic purity test, bacterial endotoxin test, and sterility test are finished.
Table 2.
quality control tests of 18F-FDG listed in BP
| Test | Method | Acceptance Criteria |
| Character | Not specified | Clear, colourless or slightly yellow liquid |
| Identification | Gama- Gamma-spectrum | Photon energy of 0.511Mev or 1.022MeV |
| Half-life measurement | 105 to 115 min | |
| Examine the chromatogram peak from radiochemical purity test | The product should have the same retention time as the reference solution | |
| pH | Not specified | 4.5 to 8.5 |
| Chemical Purity: | ||
| 2-FDG | HPLC | The area of the 2-FDG peak is not greater than the area of the reference peak (10mg/maximum injected dose in mL) |
| Kryptofix | Colourimetirc | The test spot should not be darker than the reference spot |
| tetra-alky ammonium salt | HPLC | The area of the product peak is not greater that the area of the reference peak (2.75 mg / maximum injected dose in mL) |
| Residual solvent: | ||
| Acetonitrile | Not specified | Less than 4.1mg per maximum dose volume injected |
| Radionuclidic purity | Gama-spectrum | Photon energy of 0.511Mev or 1.022MeV |
| measurement of half-life | 105 to 115 min | |
| Radiochemical Purity | HPLC TLC |
Not less than 95% of the total radioactivity |
| Sterility | Standard sterility test according to BP | No bacterial growth |
| Bacterial Endotoxin | Not specified | 175 international unit/maximum dose in mL |
| Radioactivity | Measurement in calibrated dose calibrator | ---- |
There are other tests not listed in the BP, but may be of significance. The BP does not list a test for ethanol, which is widely used in the synthesis of 18F–FDG. Both USP and BP do not list the membrane filter integrity test. However, the test is essential as an indirect evidence of the 18F–FDG product sterility because the sterility test result will not be available until much later.
Character:
Although the BP does not specify the test method, it is obvious that a visual inspection of the 18F–FDG is implied. The product should be observed behind adequate shielding. While BP allows a sightly yellow colour, this may indicate the presence of impurities. An 18F–FDG product should only be clear and colourless.
Identity (radionuclidic and radiochemical)
In BP, the tests for radionuclidic identity and radiochemical identity are the same tests for radionuclidic purity and radiochemical purity. The radionuclidic identity can be confirmed either by obtaining a gamma spectrum or measuring the half life of the product. However, the photon energy of 0.511 MeV and the sum peak at 1.022 MeV are common features to positron emitters. Hence, obtaining a gamma spectrum may not be adequate in confirming the presence of 18F– [24].
Measurement of the half–life can be carried out by measuring the same test solution in the same dose calibrator at 2 or more time points. The half–life is then calculated by plugging the results into the radioactivity decay equation. The BP does not specify the time interval between each measurement, but it should be long enough to allow a significant decay. The author suggests a minimum decay period of of 20 to 30 min. Other experts have established that a minimum of 10 min is necessary [24]. The measurement of half–life is a more reliable method in confirming the presence of 18F–.
In BP, the radiochemical identity can be confirmed either by HPLC or TLC. The TLC is easier, but as accurate and reliable as the HPLC. However, TLC may take longer time. For the test of 18F–FDG, the TLC stationary phase is TLC–SG and the mobile phase is acetonitrile : water (95%:5% v/v). The Rf of the 18F–FDG, free 18F–, and acetylated 18F–FDG are abut 0.45, 0.0 and 0.8 to 0.95 respectively. It should be noted that TLC results can vary according to different brands of TLC plates and operation conditions. It is therefore important to use the same brand of TLC–SG plate and freshly prepared mobile phase if possible. When plates with a new batch number (from the same brand) are used, the Rf values should be confirmed as per the validation process. The spotting technique also has significant effects on the TLC results. The spot size should be about 2 to 5 µL. It should be dried and placed above the mobile phase level.
pH
The pH value of an injectable should be as close to the physiological pH as possible. The BP does not specify a method for testing the pH of the 18F–FDG. Some laboratories would use pH papers while others would use pH meters. It should be noted that the pH paper used should be verified with standard pH buffers, display a colour change for each 0.5 pH unit, and the pH value measured using pH paper is only an approximate [24]
Chemical purity
The BP specifies the chemical purity FDG and 2–chloro–deoxyglucose (for acid hydrolysis synthesis only) to be determined by HPLC with a strong basic anion exchange column. The author has used a Carbopac™ column with good results, however, other commercially available strong basic anion exchange columns can perform equally well. The mobile phase is 0.1M NaOH and the flow rate is 1ml/min. Since NaOH absorbs carbon dioxide from air readily, it should be protected from air, stored in plastic containers and freshly prepared if possible. The Carbopac™ column is also very sensitive to carbonate ions. This adds to the importance of protecting the NaOH from air.
The test protocol includes injecting and run the HPLC of a reference standard solution and then run the HPLC of the test solution. The acceptance criteria is the area under the FDG peak of the test solution should be less than that of the reference solution. In theory, the reference material used should be of pharmacopeia grade. The USP has listed three USP grade FDG reference standards, but so far it has not been available commercially. One can only obtain non pharmacopeia grade FDG or 2–chloro–deoxyglucose from commercial vendors for preparation of reference solutions.
It is interesting to note that the BP states the preparation of glucose reference solution in addition to FDG and 2–chloro–deoxyglucose but does not require the reporting of glucose quantity presence in an 18F–FDG product.
The test of Kryptofix involves spotting the test solution and the reference standard on a TLC–SG plate and then develop the plate in a mixture of methanol and ammonia (9:1 v/v). The developed plate is then exposed to iodine vapor. The test solution spot should have a colour lighter than the reference solution spot. However, this TLC method is unreliable. The spots can be indistinct [24]. Alternatively, Kryptofix can be determined by placing the TLC plate in an iodine chamber directly or by GC [25].
Residual Solvent
The BP lists only the determination of residual acetonitrile in the 18F–FDG product. But the BP does not specify the test method, although the description implies that a GC should be used.. The GC column should be used for aqueous solvent and the oven temperature should be constant. A flame ionisation detector is adequate. The actual temperature, carrier gas flow rate, and run time vary among different laboratories.
The BP does not mention any test of residual absolute ethanol. Since absolute ethanol is widely used in deferent 18F–FDG synthesis modules and GC test takes only a few minutes, it is better to measure the residual absolute ethanol concentration in the 18F–FDG product. Many laboratories adopt the USP limits of 0.05% or 5mg/mL
Radionuclidic purity
The BP lists recording gamma spectrum and measuring half–life as two methods to determine the radionuclidic purity of a 18F–FDG product. Measurement of half–life can only confirm the presence of 18F. It does not reveal the percentage purity of the 18F– present. The more accurate method is to obtain a gamma spectrum with a multi–channel analyzer after confirmation of 18F– by measuring its half–life The BP allows the 18F–FDG to be released before the completion of this test.
Some experts doubt the necessity of carrying out a radionuclidic purity determination since its outcome is not crucial to patient welfare and imagine quality[24]. In fact, many laboratories measure the half–life of their 18F–FDG, but they do not obtain gamma spectra of their 18F–FDG products routinely.
Radiochemical purity
The BP lists both HPLC method and TLC method for the determination of radiochemical purity. The method has been described in radiochemical identity under section “(2) Identity (radionuclidic and radiochemical)”
Sterility
Sterility is to be tested by incubating the test sample with both Soybean Casein Digest Medium(SCDM) and Fluid Thioglycollate (FTM) Medium for 14 days at 37°C. Soybean Casein Digest Medium is a culture media for aerobic bacteria and fungi while FTM is a media for anaerobic bacteria. Growth Promotion Tests should be performed simultaneously. This test is performed by incubating “reference bacteria” in SCDM and FTM. Bacterial growth should be visible within a specified period of incubation (Table 3). Results of the growth promotion would indicate that the SCDM and FTM are capable of supporting bacterial growth, hence results of the sterility test are reliable. However, the US FDA has recommended a 30–hr window for 18F–FDG within which the sterility test must be started.
Table 3.
Test microorganisms listed in BP suitable for Growth Promotion test
| Microorganism | Incubation | ||
| Species | Strain | Incubation temperature | Maximum duration within which bacterial growth is visible |
| Aerobic bacteria: | |||
| Staphylcoccus aureus | ATCC 6538 CIP4.83 NCTC 10788 NCIMB 9518 |
30 to 35°C in FTM | 3 days |
| Bacillus subtilis | ATCC6633 CIP52.62 NCIMB 8054 |
||
| Pseudomonas aeruginosa | ATCC 9027 NCIMB 8626 CIP82.118 |
||
| Anaerobic bacteria | |||
| Clostridium sporogenes | ATCC 19404 CIP79.3 |
30 to 35°C in SCDM | 3 days |
| Fungi | |||
| Candida albicans | ATCC 10231 IP48.72 ATCC 2091 IP1180.79 |
30 to 35°C in FTM | 5 days |
| Aspergillus niger | ATCC 16404 | ||
Most PET facility would forward their samples to other microbiology laboratories for sterility test. A period of decay is necessary to ensure that the radioactivity level is not excessive. In many cases, a 24–hr window may not be long enough. Individual laboratories should establish their own protocols in this matter.
Bacterial endotoxins (LAL test)
The bacterial endotoxins level is commonly tested using the gel–clot technique. The technique uses a lysate of amoebocytes from horseshoe crab, Limulus polyphemus. The addition of bacterial endotoxins to a lysate solution produces turbidity, precipitation or gelation of the mixture. Most commercially available endotoxin testing kits require an incubation period of 20 to 60 min. Hence, it is unlikely that the test can be completed before release of the product. The BP allows the release of the 18F–FDG before completion of the bacterial endotoxins test. Some PET facility would forward their samples to other microbiology laboratories for endotoxins test. As described earlier, a period of decay is necessary to ensure that the radioactivity level is not excessive.
Bacterial endotoxins level can also be determined by spectrophotmetry. The chromogenic method makes use of the colour change of a substrate produced by the formation of an enzyme which in turn results from the addition of endotxins to Limulus polyphemus lysate. Gram–negative bacterial endotoxins have been found to activate a proenzyme in Limulus polyphemus lysate. The rate of this activation reaction depends on the concentration of the endotoxins present. The activated proenzyme then catalyses the spitting of substrates added. The splitting of the substrates results in a colour change which can be monitored by spectrophotometry. Then time required for the appearance of the colour change is inversely proportional to the endotoxins concentration present. Hence, the endotoxins concentration can be determined by comparing the reaction time of a sample to a standard curve generated from a series of standards containing known concentrations of endotoxins [26, 27].
The endotoxins concentration in a sample can also be determined by measuring the turbidity change during the gel–clot formation using spectrophotometry. The time for onset of turbity is inversely related to the endotoxind concentration present. Endotoxind level in unknown sample can be determined by comparing the time required for turbidity onset to a standard curve generated from a series of standards with known endotoxins concentrations [28]. However, such method is extremely sensitive to interference from polysaccharide such as β–Glucans. Improved methods have been developed to reduce such interference [29].
Filter membrane integrity test
This test is not required by BP and USP, but is required in the CMC section of US FDA [23]. Many laboratories have also included this test as one of their routine quality control tests of 18F–FDG.
Since the 18F–FDG is released and injected into patients before the sterility results are available, there is virtually no assurance of the product sterility. Filter membrane integrity test provide an indirect evident that the product is sterile. The argument is that if the integrity of the filter membrane is not compromised, the filter would have performed its function of removing any bacteria present in the 18F–FDG product.
A few filter membrane integrity testing devices are available commercially. Some of them rather complicated and some of them are simple hand–held types. The mechanisms behind them are similar. A stream of air is passed through the devices to the filter, then to a reservoir of water. An indicator will show the pressure exerted on the filter membrane by the air stream. The filter membrane should be able to stand the maximum pressure indicated in the specification of the filter. If the membrane is broken the air stream will pass through the membrane into the water. Air bubble will then be seen. (Figure 8).
Figure 8.

(A): Filter membrane is intact, no air passes through the membrane, no air bubble in water.
(B): Filter membrane is broken or at bubble point, air passes through the membrane, air bubble in water. The bubble point should be higher than or equal to maximum pressure listed in the specification of the filter.
The biggest disadvantage of performing filter membrane integrity test is that usually the filter membranes are highly radioactive immediately after production of 18F–FDG. But allowing a 24–hr decay would defeat the purpose of providing an evident of sterility before injecting the 18F–FDG. Individual laboratories would have to develop their own protocols in this matter.
CONCLUSION
Much of the current success in clinical PET can be attributed to the development of 18F–FDG. Synthesis of 18F–FDG is probably the most repeatable and highest yield in all PET radiopharmaceuticals synthesis. However, the future of PET would depend on the upcoming of new radiopharmaceuticals and the regulatory framework for the usage and approval of new PET drug products (e.g. NDA, IND etc). Synthesis, quality control and regulation of 18F–FDG become a model in the development new PET radiopharmaceuticals. Nucleophilic and electrophilic fluorinations are very common reactions to label compounds with 18F. The concept of using automatic synthesis modules is now a platform in PET radiopharmaceuticals synthesis. It is hope that this article will provide a brief review of 18F–FDG synthesis and quality control for those who are interested in development of PET radiopharmaceuticals. However, this article is only a concise review and not complete. Interested readers are encouraged to seek more detailed information
REFERENCES
- 1.Gallagher BM, Fowler JS, Gutterson NI, et al. Metabolic trapping as a principle of oradiopharmaceutical design: some factors resposible for the biodistribution of [18F] 2-deoxy-2-fluoro-D-glucose. J Nucl Med. 1978;19(10):1154–61. [PubMed] [Google Scholar]
- 2.Silverman M, Aganon MA, Chinard FP. Specificity of monosaccharide transport in dog kidney. Am J Physiol. 1970;218(3):743–50. doi: 10.1152/ajplegacy.1970.218.3.743. [DOI] [PubMed] [Google Scholar]
- 3.Bessell EM, Courtenay VD, Foster AB, et al. Some in vivo and in vitro antitumour effects of the deoxyfluoro-D-glucopyranoses. Eur J Cancer. 1973;9(7):463–70. doi: 10.1016/0014-2964(73)90128-x. [DOI] [PubMed] [Google Scholar]
- 4.Fowler JS, Ido T. Initial and subsequent approach for the synthesis of 18FDG. Semin Nucl Med. 2002;32(1):6–12. doi: 10.1053/snuc.2002.29270. [DOI] [PubMed] [Google Scholar]
- 5.Ehrenkaufer RE, Potocki JF, Jewett DM. Simple synthesis of F-18-labeled 2-fluoro-2-deoxy-D-glucose: concise communication. J Nucl Med. 1984;25(3):333–7. [PubMed] [Google Scholar]
- 6.Jewett DM, Potocki JF, Ehrenkaufer RE. A gas-solid phase microchemical method for the synthesis of acetyl hypofluorite. J Fluor Chem. 1984;24:477–84. [Google Scholar]
- 7.Casella V, Ido T, Wolf AP, et al. Anhydrous F-18 labeled eslemental flurine for radiopharmaceutical preparation. J Nucl Med. 1980;21(8):750–7. [PubMed] [Google Scholar]
- 8.Levy S, Elmaleh D, Livni E. A new method using anhydrous [18F] fluoride to radiolabel 2- [18F] fluoro-2-dexy-D-glucos. J Nucl Med. 1982;23:918–22. [PubMed] [Google Scholar]
- 9.Levy S, Elmaleh DR, Livni E. A new method using anhydrous [18F]fluoride to radiolabel 2-[18F]fluoro-2-deoxy-D-glucose. J Nucl Med. 1982;23(10):918–22. [PubMed] [Google Scholar]
- 10.Levy S, Livni E, Elmaleh D, et al. Direct displacement with anhydrous fluoride of the C-2 trifluoromethanesulfonate of methyl 4,6-O-benzylidene-3-O-methyl-2-O-trifluoromethyl-sulphonyl-â-D-mannoside. J Chem Soc Chem Commun. 1982:972–3. [Google Scholar]
- 11.Tewson TJ. Synthesis of no-carrier-added fluorine- 18 2-fluoro-2-deoxy-D-glucose. J Nucl Med. 1983;24(8):718–21. [PubMed] [Google Scholar]
- 12.Tewson TJ. Cyclic sulfur ester as substrates for nucleophilic substitution. A new synthesis of 2-deoxy-2-fluoro-D-glucose. J Org Chem. 1983;48:3507–10. [Google Scholar]
- 13.Szarek W, Hay GW, Perlmutter MM. A rapid stereospecific synthesis of 2-deoxy-2 fluoro-D-glucose using fluoride ion. J Chem Soc Chem Commun. 1982:1253–4. [Google Scholar]
- 14.Beeley PA, Szarek WA, Hay GW, et al. A synthesis od 2-deoxy-2- [18F] fluoro-D-glucose using accelerator-produced 18F-fluoride ion generated in a water target. Can J Chem. 1984;62:2709–11. [Google Scholar]
- 15.Hamacher K, Coenen HH, Stocklin G. Efficient stereospecific synthesis of no-carrier-added 2-[18F]-fluoro-2-deoxy-D-glucose using aminopolyether supported nucleophilic substitution. J Nucl Med. 1986;27(2):235–8. [PubMed] [Google Scholar]
- 16.Schlyer DJ. PET tracers and radiochemistry. Ann Acad Med Singapore. 2004;33(2):146–54. [PubMed] [Google Scholar]
- 17.Kiesewetter DO, Eckelman WC, Cohen RM. Syntheses and D2 receptor affinities of derivatives of Spiperone containing aliphatic halogen. Int J Appl Radiat. 1986;37:1181–8. doi: 10.1016/0883-2889(86)90003-1. [DOI] [PubMed] [Google Scholar]
- 18.Lemaire C, Damhaut PH, Lauricella B, et al. Fast [18F]FDG synthesis by alkaline hydrolysis on a low polarity solid phase support. J Labelled Comp Radiopharm. 2002;45(5):435–47. [Google Scholar]
- 19.Saha GB. Synthesis of PET radiopharmaceuticals. Basics of PET Imaging, Physics, Chemistry and Regulations. NY Springer Publishing. 2005:113. [Google Scholar]
- 20.The United States Pharmacopoeia, 25th ed, and The National Formulary, 20th ed. Rockvile, MD: United States Pharmacopeia Convention Inc; 2002. Fludeoxyglucose F 18 injection. pp. 752–3. [Google Scholar]
- 21.British Pharmacopoeia 2000. London: British Pharmacopoeia Commission; 2000. Monographs: Radiopharmaceutical preparation, Fludeoxyglucose [18F] injection. [Google Scholar]
- 22.European Pharmacopoeia. 4 edition. Strasbourg, France: European Directorate for the Quality of the Medicines; 2002. Fludeoxyglucose [18F] injection. pp. 1361–8. [Google Scholar]
- 23.Sample Formats: Application to Manufacture Ammonia N 13 Injection, Fluorodeoxyglucose F 18 Injection (FDG F 18) and Sodium Fluoride F 18 Injection Chemistry, Manufacturing, and Control Sections. Rockville, MD: USA FDA; 2000. Fludeoxyglucose F 18 injection. pp. 24–6. [Google Scholar]
- 24.Hung JC. Comparison of various requirements of the quality assurance procedures for (18)F-FDG injection. J Nucl Med. 2002;43(11):1495–506. [PubMed] [Google Scholar]
- 25.Ferrieri RA, Schlyer DJ, Alexoff DL, et al. Direct analysis of Kryptofix 2.2.2 in 18FDG by gas chromatography using a nitrogen-selective detector. Nucl Med Biol. 1993;20(3):367–9. doi: 10.1016/0969-8051(93)90061-x. [DOI] [PubMed] [Google Scholar]
- 26.Iwanaga S, Morita T, Harada T, et al. Chromogenic substrates for horseshoe crab clotting enzyme. Its application for the assay of bacterial endotoxins. Haemostasis. 1978;7(2-3):183–8. doi: 10.1159/000214260. [DOI] [PubMed] [Google Scholar]
- 27.Roth RI, Levin J, Behr S. A modified Limulus amebocyte lysate test with increased sensitivity for detection of bacterial endotoxin. J Lab Clin Med. 1989;114(3):306–11. [PubMed] [Google Scholar]
- 28.Yokota M, Kambayashi J, Tanaka T, et al. A simple turbidimetric time assay of the endotoxin in plasma. J Biochem Biophys Methods. 1989;18(2):97–104. doi: 10.1016/0165-022x(89)90071-7. [DOI] [PubMed] [Google Scholar]
- 29.Kambayashi J, Yokota M, Sakon M, et al. A novel endotoxin-specific assay by turbidimetry with Limulus amoebocyte lysate containing beta-glucan. J Biochem Biophys Methods. 1991;22(2):93–100. doi: 10.1016/0165-022x(91)90022-o. [DOI] [PubMed] [Google Scholar]