

Abstract
Cholangiocarcinoma (CC), the malignant tumor of the epithelial cells lining the biliary ducts, has undergone a worldwide increase in incidence and mortality. The malignant transformation of the biliary cells originates from a multistep process evolving through chronic inflammation of the biliary tract to CC. In the last few years several advances have been towards understanding and clarifying the molecular mechanisms implicated in the cholangiocarcinogenesis process. However, many pathophysiologic aspects governing the growth of CC are still undefined. The poor prognosis of this tumor underlines the urgent need to codify the underlying molecular mechanisms involved in the growth and progression of CC in order to design effective preventive measures and valid treatment regimens. This review reports on progresses made in the last few years in clarifying the molecular pathways involved in the process of cholangiocarcinogenesis.
Keywords: Cholangiocarcinoma, Molecular mechanism, Cholangiocarcinogenesis, Genetic, Invasion, Apoptosis.
INTRODUCTION
The malignant transformation of cholangiocytes, the epithelial cells lining the biliary tree, gives rise to cholangiocarcinoma (CC). These tumors may arise from any part of the biliary tree and are anatomically classified as intrahepatic or extrahepatic. 50% of CCs, also called Klatskin tumors, develops in the liver hilum, 42% within extrahepatic biliary ducts and only 8% originate in the intrahepatic biliary ducts. In more of 90% of cases this tumor is histologically classified as an adenocarcinoma[1,2].
Epidemiological data show that the incidence, prevalence and mortality of intrahepatic CC are increasing worldwide[3]. The high mortality and poor outcome of this disease is certainly due to the lack of tools for early diagnosis and treatment[3]. Surgery represents the only curative treatment for CC[1]. However, surgery is only feasible at an early stage of this neoplasia and is characterized by a high rate of recurrence[1]. Unfortunately, because of the lack of specific symptoms coupled with the high level of invasiveness and easy involvement of critical anatomical structures[1,2], in the majority of cases the tumor is usually very advanced at the time of diagnosis. For this reason surgery assumes a palliative more than curative role[1]. Generally, the survival rate is very poor, with less than 5% of the patients surviving up to 5 years[2,3]. Chemotherapy and radiation therapies have been used in an attempt to control this disease and improve the survival and the quality of life of patients with advanced CC. However, these therapeutic strategies are not effective in prolonging long-term survival[4] and their role is only palliative. Recent therapeutic options include brachytherapy and photodynamic therapy, although their effect is not yet established[1,2].
Some risk factors for CC development have been identified[1]. However, it is a common experience for clinicians in western countries that none of the known specific risk factors are detectable in patients affected by this malignancy.
Recent studies in this field of research have focused on the identification of the molecular mechanisms that regulate CC development. These studies have described many aspects of the intracellular mechanisms associated with the malignant transformation of cholangiocytes that had remained obscure for many years. In particular, several studies have helped to clarify the link between chronic bile duct inflammation and acquisition of a malignant phenotype by cholangiocytes[5]. In this review, we have summarized what is currently known about the mechanisms implicated in the multistep process of cholangiocarcinogenesis.
PROCESS OF CHOLANGIOCARCINOGENESIS
Analogously to other malignant tumors, CC develops as a multistep process of cellular transformation. Biliary epithelium cells undergo genetic and epigenetic alterations in regulatory genes, which accumulate and lead to the activation of oncogenes and the dysregulation of tumor suppressor genes (TSGs)[6-9]. A multitude of mutated genes and pathways have been described in malignant cholangiocytes.
Even if a small number of CCs arise in normal liver, CCs generally develop in a background of chronic inflammation of bile ducts and the consequent cholangiocyte injury associated with cholestasis[1,6].
Some risk factors related to the development of CC are well known[10]. Among them age greater than 65 years, liver fluke infestation by Opistorchis viverrini and Clonorchis sinensis, primary sclerosing cholangitis (PSC), hepatolithiasis, Caroli’s disease, congenital choledochal cysts, bile duct adenoma, anomalous pancreaticobiliary-junction malformations, thorotrast[10]. A number of other factors predisposing to CC development have also been described: smoking, papillomatosis, liver cirrhosis, diabetes mellitus, dioxin and vinyl chloride intoxication, HIV, HBV and HCV infections[10]. However, none of these specific conditions is often detectable in patients affected by this cancer. Independently of the existence of risk factors, malignant transformation of cholangiocytes arises against a background of chronic inflammation[4,6,9,11]. The network of cytokines and molecules present in high concentration during this chronic inflammatory process triggers and maintains the multistep process of cholangiocarcinogenesis[1,6,9,11] (Figure 1). This process is characterized by a progressive accumulation of chromosomal, genetic and epigenetic alterations[7,12,13] (Figure 1). The final result is a sustained overproduction of cytokines, stimulatory or inhibitory growth factors and hormones that drive cholangiocytes to irreversible changes in cell physiology, i.e. (1) dysregulated growth, (2) high capacity of invasiveness; and (3) capacity to metastasize[6,9,11] (Figure 1). Two other particular characteristics of cholangiocarcinoma cells are the epigenetic changes[14] and the process of epithelial-to-mesenchymal transition (EMT)[15] associated with the malignant transformation.
Figure 1.
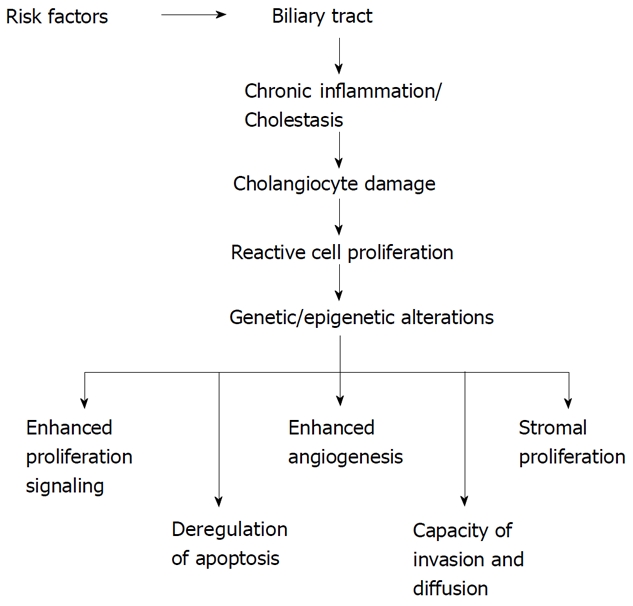
Cholangiocarcinogenesis model. Proposed mechanisms and cellular events linked to malignant transformation of biliary epithelia.
GENETIC ALTERATIONS
Genetic alterations are followed by specific changes in cell physiology such as: (1) stimulation of growth induced by autocrine signals; (2) dysregulated of the mechanisms of replication; (3) insensitivity to growth inhibitory mechanisms; (4) escape from cell apoptosis; and (5) neo-angiogenesis, tissue invasiveness and metastasis[16]. The final result of these altered processes is an uncontrolled cell growth. The principal genes altered during the development of CC, such as K-ras, p53, p14ARF, p16INK4a and β-catenin, are also altered in other cancer types[7].
K-ras and p53
Mutations of K-ras and p53 oncogenes are described in several epithelial carcinomas. Moreover, K-ras and p53 oncogenes are also often mutated in CC cells[7,17-19]. Such point mutations are located in codon 12 and consist of changes from glycine (GGT) to aspartic acid (GAT) or, less frequently, to valine. Furthermore, mutations have been located at codon 13, involving GGT to GAT, and codon 61, involving CAA to CAC changes[20]. In addition, some Authors described that the expression of K-ras correlates with the gross morphology and location of the tumor in the liver[7,20]. In summary, these differences in K-ras mutations could be due to the existence of different subtypes of cancers, racial and geographical differences of the patients or use of different assay techniques[7].
p53 is a fundamental tumor suppressor gene with two important functions: the induction of cell cycle arrest and suppression of Bcl-2 protein expression with consequent blockage of apoptosis. The incidence of p53 gene mutation is high and varies from 20 to 80% of cases[7]. The evidence that this mutation is more frequent in the mass-forming type tumors means that p53 mutation could be related to the development of intrahepatic CCs of the peripheral small bile ducts[7,21,22].
In many cases, the p53 protein forms complexes with other molecules such as WAF-1 and mdm-2, which favor its inactivation[23].
p14ARF and p16INK4a are cell cycle regulator genes implicated in the genesis of CCs. However, their mutation or deletion is not frequent in CCs[24,25].
NKG2D
Natural killer (NK) cells are implicated in tumor surveillance by cell-mediated cytotoxicity[26]. The natural killer group 2, member D cell receptor, also known as NKG2D, is expressed by NK cells and T-lymphocytes and is involved in their cytotoxic activity[27]. The role of these cells is highly relevant since some studies have suggested that high levels of cytotoxicity protect PSC patients from the development of CCs in a background of chronic inflammation of the biliary tract[28]. Melum et al[28] recently evaluated the NKG2D gene in PSC-affected patients and showed that two single nucleotide polymorphisms (SNPs) of the gene were associated with an increased risk of CCs. In addition, a homozygous condition for the non-risk alleles is linked to an extremely low risk of CCs[28]. This finding could be helpful in identifying low risk CC-patients. The development of cancer is a complex biological process, and other yet unknown polymorphisms are likely to be associated with CC risk. Combining NKG2D SNPs with other polymorphisms in a panel of markers may be useful in creating a test to assess CC risk[29].
Activation-induced cytidine deaminase
Activation-induced cytidine deaminase (AID) represents a member of the DNA/RNA-editing cytidine deaminase, apolipoprotein B mRNA-editing enzyme catalytic-polypeptide family. Recently, it has been shown that proinflammatory cytokines, abundant in clinical conditions such as PSC and CCs, significantly stimulated AID production in cholangiocytes[30]. Specifically, the DNA mutator AID gene is targeted from the IKK-β-dependent NF-κB activation pathway[30] and it is aberrantly expressed during chronic inflammation[30]. Consequently, aberrant expression of AID in biliary cells results in the generation of somatic mutations in tumor-related genes, including p53, c-myc, and the promoter region of the INK4A/p16 sequences[30]. Komori et al[30] speculated that proinflammatory cytokine stimulation is responsible for the aberrant AID gene expression in human cholangiocytes, thus providing a possible link between chronic biliary inflammation and the development of CC. However, further studies are necessary to clarify the significance and the role of AID production in stimulating precancerous cells to develop a critical number of genetic changes.
MOLECULAR STEPS OF CHOLANGIOCARCINOGENESIS
Enhanced proliferation signaling
Deregulation of several molecular mechanisms such as ErbB-2, MUC-1, Met, β-catenin, interleukin-6 (IL-6), transforming growth factor-β (TGF-β), signal transducer and activator of transcription-3 (STAT-3), Bcl-2, DCP4/Smad4, hepatocyte growth factor (HGF), reduced glutathione (GSH), Notch-1, tumor necrosis factor-related apoptosis-inducing ligand (TRAIL), p16INK4a, Ras/Raf and WISP1v[6,9] have been described in CCs as a consequence of the activation of cellular oncogenes or the inactivation of TSGs.
The network of cytokines released into the biliary microenvironment in the course of chronic inflammatory processes is responsible for the induction of malignant transformation of cholangiocytes. The trigger of the cholangiocarcinogenesis process is the activation of an autonomous proliferative signal in the biliary epithelium[6,9]. Cytokines are abundant in the course of chronic inflammation and they are secreted into the liver by a multitude of cell types such as hepatocytes, hepatic stellate cells, sinusoidal endothelial, and Kupffer cells. In addition, recent studies have demonstrated that cholangiocytes themselves produce and release cytokines[31] such as IL-6, TGF-β, IL-8, TGF-α and platelet-derived growth factor B chain[4], which all interact with biliary epithelium in an autocrine/paracrine manner. All these factors are able to regulate biliary cell pathophysiology and several studies have demonstrated that they also play a fundamental role in the development and growth of biliary tract cancers[12]. This suggestion is favored by the fact that cholangiocyte intracellular signaling in response to specific cytokine stimuli is altered during malignant processes[5]. The constitutive activation of cellular receptors and the mitogenic factors abundant in proximity to the biliary epithelium, stimulates the uncontrolled growth of malignant cholangiocytes[1,7].
The development of autonomous proliferative signaling is the principal step in the cholangiocarcinogenesis process (Figure 1). Mitogenic factors are locally secreted by a network of liver cells such as tumoral cells and by the constitutive activation of cellular receptors or various components of the intracellular signaling pathway. They stimulate and enhance the growth of malignant cholangiocytes[1,5].
IL-6 and myeloid cell leukemia-1 (Mcl-1): IL-6 and TGF-β are the two major cytokines involved in the regulation of CC growth[32]. IL-6 is a cytokine secreted in the liver in the course of inflammatory processes by several cell types, including cholangiocytes[21]. Such a cytokine both mediates the immune responses and stimulates the growth of normal and tumoral cells. Several pieces of data demonstrate a fundamental role of IL-6 in the pathogenesis and growth of CC[21].
IL-6 content is increased in the course of chronic inflammation and the neoplastic processes in biliary ducts. It is also found at increased levels in bile and serum of CC affected patients[33]. This cytokine stimulates cell proliferation by an autocrine/paracrine mechanism[34] and specifically promotes the activation of p44/p42 and p38 mitogen-activated protein kinase (MAPK) pathways[35] which, in turn, decrease the expression of p21 (WAF1/CIP1), a cell cycle controller protein[36]. Several studies have shown that this mitogenic property of IL-6 is mediated by the up-regulation of Mcl-1, a key antiapoptotic Bcl-2 family member protein[37]. More specifically, this effect is dependent upon IL-6 induced increased activation of STAT-3 (constitutively activated in malignant cholangiocytes), which regulates Mcl-1 transcription[22]. These data directly link the inflammatory mediator IL-6 to a potent antiapoptotic signaling mechanism in CC and suggest a critical role of STAT-3 in malignant transformation.
Furthermore, Mcl-1 increases cancer cell resistance to TRAIL[38,39] and inhibition of IL-6 induced expression of Mcl-1 restores sensitivity to TRAIL[40].
JAK/STAT: The activation of the Janus kinase (JAK)/STAT-3 pathway[22] by IL-6 enhances SOCS-3 which, through recruiting Tyr759 of gp130, turns off IL-6 signaling by a negative feed-back to JAK-1. The importance of this pathway in maintaining tumor growth is demonstrated by the fact that this inhibitory loop is inactivated in several human CC cell lines, and re-expression of SOCS-3 reduces STAT-3 activation with consequent inhibition of its target gene Mcl-1. This reduction in Mcl-1 sensitizes CC cells to TRAIL-induced cytotoxicity[39]. In summary, SOCS-3 re-expression can predispose CC cells to TRAIL cytotoxicity by inhibiting IL-6/STAT-3 pathways, with consequent down-regulation of Mcl-1. These findings suggest that the up-regulation of SOCS-3, obtained for example by demethylating agents, could have an important role in inhibiting CC cell growth[39]. The effect of IL-6 has been associated with epigenetic silencing of genes (see other review of this series)[41]. This represents one of the possible links between inflammation and the growth and survival of tumors[41].
Thus, the JAK/STAT pathway is one of the key signaling pathways mediating the resistance of CC cell to apoptosis. In a recent study Blechacz et al[42] showed that JAK/STAT signaling can be inhibited by the multikinase inhibitor sorafenib, which could therefore have a therapeutic role in reducing CC growth. The Authors demonstrated that sorafenib induces STAT-3 dephosphorylation by stimulating phosphatase SHP-2 activity and consequently sensitizes CC cells to TRAIL-mediated apoptosis[42].
TGF-β: Transforming growth factor-β, or TGF-β, is a cytokine implicated in a network of functions such as cell growth, differentiation, migration, apoptosis, adhesion, survival and immunity. Several liver cell types synthesize TGF-β[6], whereas cholangiocytes express this cytokine only in pathological conditions such as cholestasis[43]. In general, TGF-β exerts inhibitory effects. For example, it reduces human CC cell proliferation through regulation of the p21 cyclin dependent kinase inhibitor[44]. However, this antiproliferative effect in CC cells is hidden because of the mutations of its receptor (deletion of TβR1) and the alterations of intracellular signaling mediators (e.g. Smad4), together with the intracellular over-expression of cyclin D1[32,45,46]. The lack of TGF-β signaling also stimulates the deposition of fibrotic tissue, abundantly expressed by CC[1]. In summary, the dysregulation of TGF-β signaling leads to an increase of cell proliferation and formation of fibrosis, both hallmarks of CC[46].
DCP4/Smad4: DCP4/Smad4 is a tumor suppressor gene and is also a downstream of TGF-β cascade[47]. It has been postulated that the TGF-β/Smad pathway could play an important role in the regulation of CC growth[48]. In addition, loss of Smad4 correlates with the pTNM stage of intrahepatic CC[48]. Moreover, Smad4 interacts with the other tumor suppressor gene PTEN to maintain a physiologic cellular balance and block the process of cholangiocarcinogenesis[49]. This explains why the lack of functions of these TSGs favors the development of biliary malignancies[49].
c-Met/HGF: c-Met is a proto-oncogene located on chromosome 7q that codes for a tyrosine kinase growth factor receptor, highly expressed on the surface of CC cells[50,51]. The ligand for Met is HGF/scatter factor (SF). HGF/SF-Met pathways are implicated in embryonic development. However, abnormal Met signaling has been strongly implicated in the process of tumorigenesis, particularly in the growth of aggressive and metastatic cancers[52].
CC cells express high levels of HGF, both in vitro and in vivo, together with the up-regulation and hyper-phosphorylation of c-Met, its specific surface receptor[53]. These data suggest the existence of an autocrine-loop that governs the growth of malignant cholangiocytes[53]. Increased expression of the c-Met gene in CC cells is associated with increased cell migration and invasion. Conversely, inhibition of c-Met expression is followed by reduced cellular growth and invasiveness63. HGF binds to c-Met and induces the autophosphorylation of an intracellular tyrosine kinase on the β-subunit of the receptor[52]. This process is followed by the activation of a network of signaling molecules such as Src, P13K, Gab1, SOS, Grb2, and MEK1/2[54], all involved in the regulation of cell invasion, angiogenesis and tumor differentiation/proliferation[55,56].
ErbB-2: Several CC cell lines express large amounts of the protein ErbB-2[57,58]. This molecule is involved in the development and progression of biliary cancer[59] and its over-expression maintains growth and survival of CC cells. In addition to the direct effect on tumoral cells, ErbB-2 stimulates the production of Cyclooxygenase-2 (COX-2), which forms a complex with a subunit of the IL-6 receptor[60]. This effect suggests a close link between IL-6 and ErbB-2 signaling[1,60].
Recently, Lai and colleagues showed that normal cholangiocytes transfected with the neu (the rat homologue of ErbB-2) oncogene undergo a malignant transformation that closely resembles the molecular features of human CC[61].
GSH: Reduced GSH is the principal intracellular defense against oxidative stress during inflammation[62]. The role of GSH is to favor the reduced state of intracellular molecules and to participate in the detoxification of a multitude of substances[62]. Several studies have demonstrated that GSH is produced by cholangiocytes, but it can also be secreted by hepatocytes and thus be absorbed from bile[9]. An increase of intracellular GSH is associated with decreased cholangiocyte apoptosis and increased Bcl-2 protein expression due to decreased Bcl-2 degradation[62]. Conversely, GSH reduction predisposes cholangiocytes to apoptosis.
Nitric oxide (NO): Inducible nitric oxide synthase (iNOS) is an enzyme found in high concentration during the course of inflammations and malignancies of the biliary tract[63]. Similarly to COX-2, this protein is also involved in a variety of events such as cell proliferation, survival, and angiogenesis[64]. Several pieces of evidence show a strong link between iNOS and COX-2 in the generation of CC[63]. Even if these two proteins could be induced independently[65], iNOS stimulates COX-2 expression through NO production. In particular, iNOS enhances cholangiocyte COX-2 expression, probably through activation of p38 MAPK and JNK1/2[63].
The malignant transformation of cholangiocytes is correlated with an increase of iNOS[13], which stimulates NO. NO counteracts the mechanisms of DNA repair, thus favoring its damage and mutagenesis[9]. Similar to what happens in pancreatic cancer, the effect of NO in promoting CC development is due to the up-regulation of Notch-1[32]. To strengthen this hypothesis, evidence shows that Notch-1 is hyper-expressed in cholangiocytes of patients affected by PSC as well as in CC cells and it co-localizes with iNOS[66]. Notch-1 is stimulated by NO and its expression can be inhibited after cell transfection with iNOS antisense constructs[66]. Inflammatory cytokines represent a stimulus for activation of iNOS. After its activation, the consequent increase of intracellular NO exerts a double effect in the process of cholangiocarcinogenesis: allowing accumulation of DNA mutations by inhibiting DNA repair mechanism[67] and enhancing COX-2 expression[63,66,68].
COX-2: COX is the key enzyme involved in the process of inflammation since it is implicated in the genesis of prostaglandins. COX-1 and COX-2 are the two specific isoforms of COX. COX-1 is constitutively present in several cell types and participates in the homeostatic functions of prostaglandins, while the inducible COX-2 can be stimulated by many molecules, such as cytokines and lipopolysaccharides[69].
COX-2 is increased during the process of inflammation and it stimulates and maintains CC cell growth[70]. Evidence suggests a strong link between COX-2 and the cholangiocarcinogenesis process. For example, in a murine model of biliary adenocarcinoma induced by over-expression of ErbB-2, an increase of COX-2 was initially observed[71] and over-expression of COX-2 enhances rat CC cell growth[72]. Selective COX-2 inhibitors (e.g. celecoxib) have been positively tested to block CC cell proliferation by inducing the apoptotic process[57,72-74]. This antiproliferative effect is accompanied by an inhibition of PDK1 and PTEN, followed by a consequent decrease of Akt phosphorylation[75]. Furthermore, celecoxib treatment also inhibits CC cell proliferation through activation of cyclin-dependent kinase inhibitors p21waf1/cip1 and p27kip1, with consequent cell cycle arrest at G1/S phase[76]. However, not all COX-2 inhibitors are able to inhibit CC cell growth[72].
Recent studies have aimed to codify molecules able to induce the production of COX-2 during the inflammatory process. Oxisterols derive from cholesterol. The involvement of oxisterols in the process of cholangiocarcinogenesis was suggested by the observation that they are present in bile during cholestasis, one of the conditions predisposing to CC development[77]. Moreover, oxisterols enhance COX-2 expression in human CC cells in vitro[78].
COX-2 expression was moderately low in cholangiocytes obtained from patients affected by primary biliary cirrhosis (PBC), whereas cholangiocytes from patients affected by PSC showed a very high immunoreactivity[41]. In a recent review, Sirica et al[4] speculated that such a discrepancy between PBC and PSC might explain the higher incidence of CC in PSC affected patients[57].
It has been demonstrated that COX-2 is a downstream of iNOS-NO mediated pathway in the promotion of biliary carcinogenesis[43]. In fact, in immortalized murine cholangiocytes, iNOS inhibition markedly diminishes COX-2 mRNA and protein expression[43]. This event is reversed by the introduction of NO donors[43]. These data acquire a major potential clinical relevance since high immunoreactivity of both iNOS and COX-2 has been demonstrated in bile ducts of patients affected by PSC[41,43].
Bile acids: Bile acids accumulate in the course of cholestasis and regulate CC cell growth. Specifically, deoxycholic acid (DA) activates the epidermal growth factor receptor (EGFR), which is known to stimulate pro-survival and pro-proliferative signaling such as the PI3-kinase[79,80]. The mechanism by which this occurs is very complex as demonstrated by Werneburg et al. These Authors showed that the bile acid-dependent activation of EGFR is blocked by Tumor Necrosis Factor-α (TNF-α) antiserum and by metalloproteinase inhibitor[79]. The hypothesis, therefore, is that bile acid induced activation of EGFR is secondary to metalloproteinase activation, which is required for TNF-α release from the membrane and which consequently activates EGFR[79]. In addition to the PI3-kinase signaling, the bile acid dependent activation of EGFR elicits CC cell escape from apoptosis also by promoting the expression of anti-apoptotic molecules[79]. The exposure of CC cells to DA markedly increases the cellular Mcl-1 and Raf-1 inhibitors block this increase of Mcl-1, rendering the cells much more sensitive to Fas-induced apoptosis[37,38].
However, the actions of bile acids on CC development are diverse. For example, bile acid-induced EGFR activation also leads to the stimulation of the MAPK cascade with activation of ERK1/2, p38 and JNK proteins[78] thus having a pro-carcinogenic effect. In addition, the activation of ERK1/2 and p38 is then responsible for COX-2 expression[78]. This effect could suggest that bile acids may play an important role in CC development in a non-inflammatory condition. Therefore, bile acids affect a multitude of intracellular events implicated in CC and members of the wide family of bile acids may exert different, even opposite, effects on malignant cholangiocytes. In fact, we recently demonstrated that tauroursodeoxycholate (TUDCA) inhibits human cholangiocarcinoma growth via Ca2+, PKC-alpha and the MAPK-dependent pathways[81]. Previous data showed that TUDCA stimulates hepatocyte regeneration[82]. It is plausible that different cell types could respond to the same stimulus in an opposite way. Similarly, gastrin stimulates the growth of several cells such as colonic epithelial[83] and pancreatic adenocarcinoma[84], but inhibits cholangiocarcinoma cell growth[85].
Dysregulation of apoptosis
Apoptosis is an essential event employed by the organism to eliminate cells not able to repair DNA damage. A reduction or dysregulation of the apoptotic process permits the survival of mutated cholangiocytes, which could accumulate a series of mutations, thus favoring malignant cell transformation. The inhibition of apoptosis is associated with an increase of expression of Bcl-2, mutation of K-ras and/or dysregulation of p53[9].
Bcl-2, the prototype of the homonym family of antiapoptotic proteins[34], is overexpressed in CC cells[36]. These tumoral cells possess an apoptotic threshold significantly higher than normal cholangiocytes[86]. Several studies show that Bcl-2 antiapoptotic activity is exerted by preventing cytochrome-c release from the mitochondria, thereby preventing caspase-3 activation[86].
Many other factors can influence and regulate the apoptotic process of cholangiocytes. Besides being involved in the induction of cholangiocarcinogenesis mechanisms[9], NO inhibits apoptosis of malignant cholangiocytes. Indeed, Torok et al demonstrated that nitric-oxide-synthase (NOS)-cDNA leads to a resistance to etoposide-induced apoptosis through the nitrosylation of caspase-9, when transfected to CC cells[39].
Notch-1 and COX-2 reduce TRAIL-mediated apoptosis[16,40]. Recent studies showed that the high quantity of COX-2 in CC cells inhibits Fas-induced apoptosis[40] and the selective COX-2 inhibitor celecoxib enhances cell death by apoptosis, through inhibition of PI3-kinase signaling[24]. However, COX-2 expression correlates with tumor differentiation and is increased in highly differentiated biliary cancers[23].
TRAIL exerts its functions in malignant cholangiocytes[44]. The activation this ligand selectively stimulates apoptosis only in neoplastic and not in normal cholangiocytes[44]. It has been described that high level expression of Mcl-1 in CC cells blocks TRAIL-induced apoptosis[44]. This implies that if Mcl-1 expression is reduced by specific small-interfering mRNA or stable transfection with Mcl-1-small-hairpin-RNA, CC cells become sensitive to TRAIL-induced apoptosis[44]. The expression of Mcl-1 is also modulated by bile acids, which accumulate in the condition of cholestasis. DA, for example, is able to increase the cellular Mcl-1 by blocking protein degradation via activation of an EGFR/Raf-1 pathway. Furthermore, Raf-1 inhibitors antagonize the increase of Mcl-1, rendering the cells much more sensitive to Fas-dependent apoptosis[44,87].
Growth and invasion of CC
The high capacity for invasion and metastasis are two other important features of CC cells. Malignant cholangiocytes stimulate the development of a rich vascular structure, which supports the metabolic needs and ensures an adequate source of oxygen and nutrients to the same cells[1]. Tumor angiogenesis is enhanced by high levels of vascular endothelial growth factor (VEGF)[1,7], which is stimulated by β-catenin[53] and TGF-β, and which is expressed by the surrounding mesenchymal cells and the malignant cholangiocytes themselves. This complex system of pathways delineates an autocrine/paracrine mechanism, which supports the production of VEGF necessary for tumor development and growth[54].
A recent study showed that CC expresses a high level of matrix metalloproteinases (MMP) and a correlation between this large amount of MMP and major clinical invasiveness was reported[55].
Human aspartyl (asparaginyl) β-hydroxylase and proteins related to the connective tissue growth factor family are also highly expressed in CC cells[56]. Their concentration is proportional to the increased motility and invasiveness of tumoral cells[56].
“Cell adhesion molecules” represent a network of factors that play a critical role in enhancing cancer invasion and metastasis[57]. Expression of E-cadherin, α-catenin, and β-catenin is reduced in a majority of biliary tumors and their down-regulation correlates with a high grade malignancy of the tumor. On the contrary, their reduction does not correlate with vascular invasion, metastasis and p53 expression[57].
WISP1v is a member of the connective tissue growth factor family[60]. It has been reported that the expression of WISP1v is significantly associated with high lymphatic and perineural invasion of tumor cells, as well as a poor clinical prognosis[61]. Furthermore, WISP1v stimulates the invasive phenotype of CC cells with activation of both p38 and p42/p44 MAPKs[61].
Cellular senescence
Cellular senescence represents a physiologic process leading to growth arrest due to telomere shortening. Malignant cholangiocytes express high levels of the enzyme telomerase[21], which blocks telomere shortening, thus maintaining chromosomal length. This permits the cells to preserve their replication activity. The expression of human telomerase was homogeneously detected in intrahepatic CC cells, whereas its expression was heterogeneous in the dysplastic biliary lesions[51]. Furthermore, this enzyme was not detected in nondysplastic biliary epithelia, in hepatolithiasis or in normal livers. This indicates that malignant cholangiocytes acquire telomerase activities in the dysplastic condition, thus triggering processes favoring malignant transformation[51]. Recent studies have shown that IL-6 enhances telomerase activity[52].
MICRO-RNA AND CC
Micro-RNAs (miRNAs) are single-stranded RNA molecules of 21-24 nucleotides in length, able to regulate gene expression. miRNAs are encoded by genes from whose DNA they are transcribed but miRNAs are not translated into protein. They are complementary to mRNA molecules, and their main function is to down-regulate gene expression. The miRNA gene expression profile in CC cells[88] has been described only recently. However, only a limited number of genes in CC have been analyzed so far[88]. Interestingly, the oncomiRs miR-141, miR-21, miR-23a, miR-27a, let-7a and miR-200b are up-regulated, while the tumor suppressor miRNAs miR-29b and miR-370 are down-regulated in malignant cholangiocytes[14,89-92].
The miRNAs possess specific functions in CC cells. For example, the up-regulated miR-141 may target the CLOCK gene, which modulates circadian rhythms and suppresses tumor growth[91]. To strengthen this concept, the inhibition of miR-141 reduces CC cell growth[91].
The involvement of other miRNAs in stimulating CC proliferation has also been described. For example, miR-21 modulates PTEN and is an anti-apoptotic and pro-survival factor[14] which is inhibited by gemcitabine, the chemotherapic drug used for CC. When dysregulated, PTPN12 stimulates tumor cell survival and the carcinogenesis process[91]. It has been suggested that PTPN12 could represent a target gene for miR-200b[91].
Moreover, the same MiR-200b could inhibit ZFHX1B, which is involved in the TGF-β signaling pathway and in the processes of EMT via regulation of E-Cadherin[93]. Similar to a variety of cancers where the expression of MiR-29b is reduced[94-96], CC express low levels of this molecule[89]. Mott et al[89] showed an inverse relationship between miR-29b and Mcl-1 expression in CC cells. In fact, the reduction of miR-29b is accompanied with an increase of the antiapoptotic Mcl-1.
MiR-370 expression is reduced in malignant compared to normal cholangiocytes[90]. Some evidence shows that MiR-370 targets MAP3K8, which is consequently up-regulated in CC cell lines as well as in tumor cell xenografts in vivo[14,90]. Epigenetic regulation of miR-370 occurs by hypermethylation and through IL-6[14,90].
IN VIVO ANIMAL MODELS OF CC
The evaluation of therapeutic molecules using in vivo animal models of disease is of fundamental importance in testing specific antitumoral drugs[97]. Many studies evaluating the effect of substances or compounds on CC cells have been conducted using xenograft systems in murine animals[98,99]. However, although these studies produced exciting and encouraging results, these results correlate only poorly with clinical outcomes. This finding underlines the need to recreate and use organ-specific in vivo cancer models[100-102].
Several in vivo animal models of biliary malignancies have been described. Hamsters and rats develop CC after treatment with carcinogenetic chemical compounds such as [N-nitrosobis (2-oxopropyl) amine][103], methylazoxymethyl acetate[104], dimethylnitrosamine[105], furan[106] and thioacetamide[107,108]. Furthermore, animals infected with O viverrini[109], are able to develop biliary malignancies. Recently, several new genetic CC models have been described. Liver-specific combined deletion of the TSGs Smad4 plus PTEN results in formation of CC in mice[49] and p53-deficient mice treated with carbon tetrachloride develop intrahepatic mass-forming CC[110]. Moreover, Sirica and colleagues have developed two models of CC in which malignant transformation of explanted rat cholangiocytes followed by direct biliary inoculation of these cells result in CC formation in 56% to 100% of animals[61,111].
In summary, models of CC have been developed in recent years which resemble human CC in many characteristics. The majority of these models represent intrahepatic CC, but genetic models of hilar CC are still missing[97].
CONCLUSION
The multitude of factors released in the environment during the course of cholestasis and chronic inflammation trigger genomic and epigenetic damage leading to malignant transformation and uncontrolled proliferation of cholangiocytes. CC is a highly lethal disease with an extremely poor response to conventional anticancer therapies and a poor survival rate. In the last few years, research has made important steps in clarifying the intracellular pathways of malignant cholangiocytes. Only the complete identification of molecular pathways involved in the pathogenesis of malignant changes of cholangiocytes will permit the discovery of novel tools for early diagnosis, and the detection of specific molecular targets for therapies.
Footnotes
Peer reviewers: Enzo Ierardi, Professor, Section of Gastroenterology, Department of Medical Sciences, University of Foggia, AOU Ospedali Riuniti, Viale Pinto, Foggia 71100, Italy; Hartmut Jaeschke, PhD, Professor, Department of Pharmacology, Toxicology & Therapeutics, University of Kansas Medical Center, 3901 Rainbow Blvd, MS 1018, Kansas City, KS 66160, United States
S- Editor Zhang HN L- Editor Hughes D E- Editor Liu N
References
- 1.Lazaridis KN, Gores GJ. Cholangiocarcinoma. Gastroenterology. 2005;128:1655–1667. doi: 10.1053/j.gastro.2005.03.040. [DOI] [PubMed] [Google Scholar]
- 2.Khan SA, Thomas HC, Davidson BR, Taylor-Robinson SD. Cholangiocarcinoma. Lancet. 2005;366:1303–1314. doi: 10.1016/S0140-6736(05)67530-7. [DOI] [PubMed] [Google Scholar]
- 3.Patel T. Worldwide trends in mortality from biliary tract malignancies. BMC Cancer. 2002;2:10. doi: 10.1186/1471-2407-2-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sirica AE. Cholangiocarcinoma: molecular targeting strategies for chemoprevention and therapy. Hepatology. 2005;41:5–15. doi: 10.1002/hep.20537. [DOI] [PubMed] [Google Scholar]
- 5.Berthiaume EP, Wands J. The molecular pathogenesis of cholangiocarcinoma. Semin Liver Dis. 2004;24:127–137. doi: 10.1055/s-2004-828890. [DOI] [PubMed] [Google Scholar]
- 6.Fava G, Marzioni M, Benedetti A, Glaser S, DeMorrow S, Francis H, Alpini G. Molecular pathology of biliary tract cancers. Cancer Lett. 2007;250:155–167. doi: 10.1016/j.canlet.2006.09.011. [DOI] [PubMed] [Google Scholar]
- 7.Sandhu DS, Shire AM, Roberts LR. Epigenetic DNA hypermethylation in cholangiocarcinoma: potential roles in pathogenesis, diagnosis and identification of treatment targets. Liver Int. 2008;28:12–27. doi: 10.1111/j.1478-3231.2007.01624.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tischoff I, Wittekind C, Tannapfel A. Role of epigenetic alterations in cholangiocarcinoma. J Hepatobiliary Pancreat Surg. 2006;13:274–279. doi: 10.1007/s00534-005-1055-3. [DOI] [PubMed] [Google Scholar]
- 9.Okuda K, Nakanuma Y, Miyazaki M. Cholangiocarcinoma: recent progress. Part 2: molecular pathology and treatment. J Gastroenterol Hepatol. 2002;17:1056–1063. doi: 10.1046/j.1440-1746.2002.02780.x. [DOI] [PubMed] [Google Scholar]
- 10.Lazaridis KN. Cholangiocarcinoma: Epidemiology, risk factors and molecular pathogenesis. In: De Morrow S, Glaser S, Alpini G, Marzioni M, Fava G, et al., editors. Pathophysiologiy of the Intrahepatic Biliary Epithelium. Kerala, India: Transworld Research Network; 2008. pp. 301–313. [Google Scholar]
- 11.Alpini G, Francis H, Marzioni M, Alvaro D, Gaudio E, Lorenzini I, Benedetti A, Fava G. New York: Springer; 2010. Molecular Pathology of Cholangiocarcinoma; p. in press. [Google Scholar]
- 12.Lee JH, Abraham SC, Kim HS, Nam JH, Choi C, Lee MC, Park CS, Juhng SW, Rashid A, Hamilton SR, et al. Inverse relationship between APC gene mutation in gastric adenomas and development of adenocarcinoma. Am J Pathol. 2002;161:611–618. doi: 10.1016/S0002-9440(10)64216-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chan AO, Broaddus RR, Houlihan PS, Issa JP, Hamilton SR, Rashid A. CpG island methylation in aberrant crypt foci of the colorectum. Am J Pathol. 2002;160:1823–1830. doi: 10.1016/S0002-9440(10)61128-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stutes M, Tran S, DeMorrow S. Genetic and epigenetic changes associated with cholangiocarcinoma: from DNA methylation to microRNAs. World J Gastroenterol. 2007;13:6465–6469. doi: 10.3748/wjg.v13.i48.6465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yoo HJ, Yun BR, Kwon JH, Ahn HS, Seol MA, Lee MJ, Yu GR, Yu HC, Hong B, Choi K, et al. Genetic and expression alterations in association with the sarcomatous change of cholangiocarcinoma cells. Exp Mol Med. 2009;41:102–115. doi: 10.3858/emm.2009.41.2.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 17.Sturm PD, Baas IO, Clement MJ, Nakeeb A, Johan G, Offerhaus A, Hruban RH, Pitt HA. Alterations of the p53 tumor-suppressor gene and K-ras oncogene in perihilar cholangiocarcinomas from a high-incidence area. Int J Cancer. 1998;78:695–698. doi: 10.1002/(sici)1097-0215(19981209)78:6<695::aid-ijc5>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 18.Kiba T, Tsuda H, Pairojkul C, Inoue S, Sugimura T, Hirohashi S. Mutations of the p53 tumor suppressor gene and the ras gene family in intrahepatic cholangiocellular carcinomas in Japan and Thailand. Mol Carcinog. 1993;8:312–318. doi: 10.1002/mc.2940080415. [DOI] [PubMed] [Google Scholar]
- 19.Wattanasirichaigoon S, Tasanakhajorn U, Jesadapatarakul S. The incidence of K-ras codon 12 mutations in cholangiocarcinoma detected by polymerase chain reaction technique. J Med Assoc Thai. 1998;81:316–323. [PubMed] [Google Scholar]
- 20.Ohashi K, Nakajima Y, Kanehiro H, Tsutsumi M, Taki J, Aomatsu Y, Yoshimura A, Ko S, Kin T, Yagura K. Ki-ras mutations and p53 protein expressions in intrahepatic cholangiocarcinomas: relation to gross tumor morphology. Gastroenterology. 1995;109:1612–1617. doi: 10.1016/0016-5085(95)90650-9. [DOI] [PubMed] [Google Scholar]
- 21.Meng F, Yamagiwa Y, Ueno Y, Patel T. Over-expression of interleukin-6 enhances cell survival and transformed cell growth in human malignant cholangiocytes. J Hepatol. 2006;44:1055–1065. doi: 10.1016/j.jhep.2005.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Isomoto H, Kobayashi S, Werneburg NW, Bronk SF, Guicciardi ME, Frank DA, Gores GJ. Interleukin 6 upregulates myeloid cell leukemia-1 expression through a STAT3 pathway in cholangiocarcinoma cells. Hepatology. 2005;42:1329–1338. doi: 10.1002/hep.20966. [DOI] [PubMed] [Google Scholar]
- 23.Lee S, Kim WH, Jung HY, Yang MH, Kang GH. Aberrant CpG island methylation of multiple genes in intrahepatic cholangiocarcinoma. Am J Pathol. 2002;161:1015–1022. doi: 10.1016/S0002-9440(10)64262-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tannapfel A, Benicke M, Katalinic A, Uhlmann D, Köckerling F, Hauss J, Wittekind C. Frequency of p16(INK4A) alterations and K-ras mutations in intrahepatic cholangiocarcinoma of the liver. Gut. 2000;47:721–727. doi: 10.1136/gut.47.5.721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ahrendt SA, Eisenberger CF, Yip L, Rashid A, Chow JT, Pitt HA, Sidransky D. Chromosome 9p21 loss and p16 inactivation in primary sclerosing cholangitis-associated cholangiocarcinoma. J Surg Res. 1999;84:88–93. doi: 10.1006/jsre.1999.5615. [DOI] [PubMed] [Google Scholar]
- 26.Eagle RA, Trowsdale J. Promiscuity and the single receptor: NKG2D. Nat Rev Immunol. 2007;7:737–744. doi: 10.1038/nri2144. [DOI] [PubMed] [Google Scholar]
- 27.Coudert JD, Held W. The role of the NKG2D receptor for tumor immunity. Semin Cancer Biol. 2006;16:333–343. doi: 10.1016/j.semcancer.2006.07.008. [DOI] [PubMed] [Google Scholar]
- 28.Melum E, Karlsen TH, Schrumpf E, Bergquist A, Thorsby E, Boberg KM, Lie BA. Cholangiocarcinoma in primary sclerosing cholangitis is associated with NKG2D polymorphisms. Hepatology. 2008;47:90–96. doi: 10.1002/hep.21964. [DOI] [PubMed] [Google Scholar]
- 29.Lazaridis KN. Dissecting the genetic susceptibility for cholangiocarcinoma in primary sclerosing cholangitis. Hepatology. 2008;47:8–10. doi: 10.1002/hep.22100. [DOI] [PubMed] [Google Scholar]
- 30.Komori J, Marusawa H, Machimoto T, Endo Y, Kinoshita K, Kou T, Haga H, Ikai I, Uemoto S, Chiba T. Activation-induced cytidine deaminase links bile duct inflammation to human cholangiocarcinoma. Hepatology. 2008;47:888–896. doi: 10.1002/hep.22125. [DOI] [PubMed] [Google Scholar]
- 31.Yasoshima M, Kono N, Sugawara H, Katayanagi K, Harada K, Nakanuma Y. Increased expression of interleukin-6 and tumor necrosis factor-alpha in pathologic biliary epithelial cells: in situ and culture study. Lab Invest. 1998;78:89–100. [PubMed] [Google Scholar]
- 32.Yamagiwa Y, Patel T. Cytokine Regulation of Cholangiocyte Growth. In: Alpini G, Alvaro D, Marzioni M, LeSage G, LaRusso NF, et al., editors. The Pathophysiology of Biliary Epithelia. Georgetown, TX: Landes Bioscience; 2004. pp. 227–234. [Google Scholar]
- 33.Akiyama T, Hasegawa T, Sejima T, Sahara H, Seto K, Saito H, Takashima S. Serum and bile interleukin 6 after percutaneous transhepatic cholangio-drainage. Hepatogastroenterology. 1998;45:665–671. [PubMed] [Google Scholar]
- 34.Okada K, Shimizu Y, Nambu S, Higuchi K, Watanabe A. Interleukin-6 functions as an autocrine growth factor in a cholangiocarcinoma cell line. J Gastroenterol Hepatol. 1994;9:462–467. doi: 10.1111/j.1440-1746.1994.tb01275.x. [DOI] [PubMed] [Google Scholar]
- 35.Park J, Tadlock L, Gores GJ, Patel T. Inhibition of interleukin 6-mediated mitogen-activated protein kinase activation attenuates growth of a cholangiocarcinoma cell line. Hepatology. 1999;30:1128–1133. doi: 10.1002/hep.510300522. [DOI] [PubMed] [Google Scholar]
- 36.Tadlock L, Patel T. Involvement of p38 mitogen-activated protein kinase signaling in transformed growth of a cholangiocarcinoma cell line. Hepatology. 2001;33:43–51. doi: 10.1053/jhep.2001.20676. [DOI] [PubMed] [Google Scholar]
- 37.Yoon JH, Higuchi H, Werneburg NW, Kaufmann SH, Gores GJ. Bile acids induce cyclooxygenase-2 expression via the epidermal growth factor receptor in a human cholangiocarcinoma cell line. Gastroenterology. 2002;122:985–993. doi: 10.1053/gast.2002.32410. [DOI] [PubMed] [Google Scholar]
- 38.Taniai M, Grambihler A, Higuchi H, Werneburg N, Bronk SF, Farrugia DJ, Kaufmann SH, Gores GJ. Mcl-1 mediates tumor necrosis factor-related apoptosis-inducing ligand resistance in human cholangiocarcinoma cells. Cancer Res. 2004;64:3517–3524. doi: 10.1158/0008-5472.CAN-03-2770. [DOI] [PubMed] [Google Scholar]
- 39.Isomoto H, Mott JL, Kobayashi S, Werneburg NW, Bronk SF, Haan S, Gores GJ. Sustained IL-6/STAT-3 signaling in cholangiocarcinoma cells due to SOCS-3 epigenetic silencing. Gastroenterology. 2007;132:384–396. doi: 10.1053/j.gastro.2006.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kobayashi S, Werneburg NW, Bronk SF, Kaufmann SH, Gores GJ. Interleukin-6 contributes to Mcl-1 up-regulation and TRAIL resistance via an Akt-signaling pathway in cholangiocarcinoma cells. Gastroenterology. 2005;128:2054–2065. doi: 10.1053/j.gastro.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 41.Isomoto H. Epigenetic alterations in cholangiocarcinoma-sustained IL-6/STAT3 signaling in cholangio- carcinoma due to SOCS3 epigenetic silencing. Digestion. 2009;79 Suppl 1:2–8. doi: 10.1159/000167859. [DOI] [PubMed] [Google Scholar]
- 42.Blechacz BR, Smoot RL, Bronk SF, Werneburg NW, Sirica AE, Gores GJ. Sorafenib inhibits signal transducer and activator of transcription-3 signaling in cholangiocarcinoma cells by activating the phosphatase shatterproof 2. Hepatology. 2009;50:1861–1870. doi: 10.1002/hep.23214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Saperstein LA, Jirtle RL, Farouk M, Thompson HJ, Chung KS, Meyers WC. Transforming growth factor-beta 1 and mannose 6-phosphate/insulin-like growth factor-II receptor expression during intrahepatic bile duct hyperplasia and biliary fibrosis in the rat. Hepatology. 1994;19:412–417. [PubMed] [Google Scholar]
- 44.Miyazaki M, Ohashi R, Tsuji T, Mihara K, Gohda E, Namba M. Transforming growth factor-beta 1 stimulates or inhibits cell growth via down- or up-regulation of p21/Waf1. Biochem Biophys Res Commun. 1998;246:873–880. doi: 10.1006/bbrc.1998.8712. [DOI] [PubMed] [Google Scholar]
- 45.Zen Y, Harada K, Sasaki M, Chen TC, Chen MF, Yeh TS, Jan YY, Huang SF, Nimura Y, Nakanuma Y. Intrahepatic cholangiocarcinoma escapes from growth inhibitory effect of transforming growth factor-beta1 by overexpression of cyclin D1. Lab Invest. 2005;85:572–581. doi: 10.1038/labinvest.3700236. [DOI] [PubMed] [Google Scholar]
- 46.Yazumi S, Ko K, Watanabe N, Shinohara H, Yoshikawa K, Chiba T, Takahashi R. Disrupted transforming growth factor-beta signaling and deregulated growth in human biliary tract cancer cells. Int J Cancer. 2000;86:782–789. doi: 10.1002/(sici)1097-0215(20000615)86:6<782::aid-ijc5>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 47.Chuang SC, Lee KT, Tsai KB, Sheen PC, Nagai E, Mizumoto K, Tanaka M. Immunohistochemical study of DPC4 and p53 proteins in gallbladder and bile duct cancers. World J Surg. 2004;28:995–1000. doi: 10.1007/s00268-004-7447-8. [DOI] [PubMed] [Google Scholar]
- 48.Kang YK, Kim WH, Jang JJ. Expression of G1-S modulators (p53, p16, p27, cyclin D1, Rb) and Smad4/Dpc4 in intrahepatic cholangiocarcinoma. Hum Pathol. 2002;33:877–883. doi: 10.1053/hupa.2002.127444. [DOI] [PubMed] [Google Scholar]
- 49.Xu X, Kobayashi S, Qiao W, Li C, Xiao C, Radaeva S, Stiles B, Wang RH, Ohara N, Yoshino T, et al. Induction of intrahepatic cholangiocellular carcinoma by liver-specific disruption of Smad4 and Pten in mice. J Clin Invest. 2006;116:1843–1852. doi: 10.1172/JCI27282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Terada T, Okada Y, Nakanuma Y. Expression of immunoreactive matrix metalloproteinases and tissue inhibitors of matrix metalloproteinases in human normal livers and primary liver tumors. Hepatology. 1996;23:1341–1344. doi: 10.1053/jhep.1996.v23.pm0008675149. [DOI] [PubMed] [Google Scholar]
- 51.Radaeva S, Ferreira-Gonzalez A, Sirica AE. Overexpression of C-NEU and C-MET during rat liver cholangiocarcinogenesis: A link between biliary intestinal metaplasia and mucin-producing cholangiocarcinoma. Hepatology. 1999;29:1453–1462. doi: 10.1002/hep.510290524. [DOI] [PubMed] [Google Scholar]
- 52.Furge KA, Zhang YW, Vande Woude GF. Met receptor tyrosine kinase: enhanced signaling through adapter proteins. Oncogene. 2000;19:5582–5589. doi: 10.1038/sj.onc.1203859. [DOI] [PubMed] [Google Scholar]
- 53.Lai GH, Radaeva S, Nakamura T, Sirica AE. Unique epithelial cell production of hepatocyte growth factor/scatter factor by putative precancerous intestinal metaplasias and associated “intestinal-type” biliary cancer chemically induced in rat liver. Hepatology. 2000;31:1257–1265. doi: 10.1053/jhep.2000.8108. [DOI] [PubMed] [Google Scholar]
- 54.Socoteanu MP, Mott F, Alpini G, Frankel AE. c-Met targeted therapy of cholangiocarcinoma. World J Gastroenterol. 2008;14:2990–2994. doi: 10.3748/wjg.14.2990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gao CF, Vande Woude GF. HGF/SF-Met signaling in tumor progression. Cell Res. 2005;15:49–5157. doi: 10.1038/sj.cr.7290264. [DOI] [PubMed] [Google Scholar]
- 56.Birchmeier C, Birchmeier W, Gherardi E, Vande Woude GF. Met, metastasis, motility and more. Nat Rev Mol Cell Biol. 2003;4:915–925. doi: 10.1038/nrm1261. [DOI] [PubMed] [Google Scholar]
- 57.Sirica AE, Lai GH, Endo K, Zhang Z, Yoon BI. Cyclooxygenase-2 and ERBB-2 in cholangiocarcinoma: potential therapeutic targets. Semin Liver Dis. 2002;22:303–313. doi: 10.1055/s-2002-34507. [DOI] [PubMed] [Google Scholar]
- 58.Endo K, Yoon BI, Pairojkul C, Demetris AJ, Sirica AE. ERBB-2 overexpression and cyclooxygenase-2 up-regulation in human cholangiocarcinoma and risk conditions. Hepatology. 2002;36:439–450. doi: 10.1053/jhep.2002.34435. [DOI] [PubMed] [Google Scholar]
- 59.Aishima SI, Taguchi KI, Sugimachi K, Shimada M, Sugimachi K, Tsuneyoshi M. c-erbB-2 and c-Met expression relates to cholangiocarcinogenesis and progression of intrahepatic cholangiocarcinoma. Histopathology. 2002;40:269–278. doi: 10.1046/j.1365-2559.2002.00353.x. [DOI] [PubMed] [Google Scholar]
- 60.Qiu Y, Ravi L, Kung HJ. Requirement of ErbB2 for signalling by interleukin-6 in prostate carcinoma cells. Nature. 1998;393:83–85. doi: 10.1038/30012. [DOI] [PubMed] [Google Scholar]
- 61.Lai GH, Zhang Z, Shen XN, Ward DJ, Dewitt JL, Holt SE, Rozich RA, Hixson DC, Sirica AE. erbB-2/neu transformed rat cholangiocytes recapitulate key cellular and molecular features of human bile duct cancer. Gastroenterology. 2005;129:2047–2057. doi: 10.1053/j.gastro.2005.10.010. [DOI] [PubMed] [Google Scholar]
- 62.Celli A, Que FG, Gores GJ, LaRusso NF. Glutathione depletion is associated with decreased Bcl-2 expression and increased apoptosis in cholangiocytes. Am J Physiol. 1998;275:G749–G757. doi: 10.1152/ajpgi.1998.275.4.G749. [DOI] [PubMed] [Google Scholar]
- 63.Ishimura N, Bronk SF, Gores GJ. Inducible nitric oxide synthase upregulates cyclooxygenase-2 in mouse cholangiocytes promoting cell growth. Am J Physiol Gastrointest Liver Physiol. 2004;287:G88–G95. doi: 10.1152/ajpgi.00539.2003. [DOI] [PubMed] [Google Scholar]
- 64.Jenkins DC, Charles IG, Thomsen LL, Moss DW, Holmes LS, Baylis SA, Rhodes P, Westmore K, Emson PC, Moncada S. Roles of nitric oxide in tumor growth. Proc Natl Acad Sci USA. 1995;92:4392–4396. doi: 10.1073/pnas.92.10.4392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Salvemini D, Misko TP, Masferrer JL, Seibert K, Currie MG, Needleman P. Nitric oxide activates cyclooxygenase enzymes. Proc Natl Acad Sci USA. 1993;90:7240–7244. doi: 10.1073/pnas.90.15.7240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ishimura N, Bronk SF, Gores GJ. Inducible nitric oxide synthase up-regulates Notch-1 in mouse cholangiocytes: implications for carcinogenesis. Gastroenterology. 2005;128:1354–1368. doi: 10.1053/j.gastro.2005.01.055. [DOI] [PubMed] [Google Scholar]
- 67.Jaiswal M, LaRusso NF, Gores GJ. Nitric oxide in gastrointestinal epithelial cell carcinogenesis: linking inflammation to oncogenesis. Am J Physiol Gastrointest Liver Physiol. 2001;281:G626–G634. doi: 10.1152/ajpgi.2001.281.3.G626. [DOI] [PubMed] [Google Scholar]
- 68.Dean JL, Sarsfield SJ, Tsounakou E, Saklatvala J. p38 Mitogen-activated protein kinase stabilizes mRNAs that contain cyclooxygenase-2 and tumor necrosis factor AU-rich elements by inhibiting deadenylation. J Biol Chem. 2003;278:39470–39476. doi: 10.1074/jbc.M306345200. [DOI] [PubMed] [Google Scholar]
- 69.Núñez Martínez O, Clemente Ricote G, García Monzón C. [Role of cyclooxygenase-2 in the pathogenesis of chronic liver diseases] Med Clin (Barc) 2003;121:743–748. doi: 10.1016/s0025-7753(03)74082-2. [DOI] [PubMed] [Google Scholar]
- 70.Brown JR, DuBois RN. COX-2: a molecular target for colorectal cancer prevention. J Clin Oncol. 2005;23:2840–2855. doi: 10.1200/JCO.2005.09.051. [DOI] [PubMed] [Google Scholar]
- 71.Kiguchi K, Carbajal S, Chan K, Beltrán L, Ruffino L, Shen J, Matsumoto T, Yoshimi N, DiGiovanni J. Constitutive expression of ErbB-2 in gallbladder epithelium results in development of adenocarcinoma. Cancer Res. 2001;61:6971–6976. [PubMed] [Google Scholar]
- 72.Zhang Z, Lai GH, Sirica AE. Celecoxib-induced apoptosis in rat cholangiocarcinoma cells mediated by Akt inactivation and Bax translocation. Hepatology. 2004;39:1028–1037. doi: 10.1002/hep.20143. [DOI] [PubMed] [Google Scholar]
- 73.Wu T, Leng J, Han C, Demetris AJ. The cyclooxygenase-2 inhibitor celecoxib blocks phosphorylation of Akt and induces apoptosis in human cholangiocarcinoma cells. Mol Cancer Ther. 2004;3:299–307. [PubMed] [Google Scholar]
- 74.Lai GH, Zhang Z, Sirica AE. Celecoxib acts in a cyclooxygenase-2-independent manner and in synergy with emodin to suppress rat cholangiocarcinoma growth in vitro through a mechanism involving enhanced Akt inactivation and increased activation of caspases-9 and -3. Mol Cancer Ther. 2003;2:265–271. [PubMed] [Google Scholar]
- 75.Testa JR, Bellacosa A. AKT plays a central role in tumorigenesis. Proc Natl Acad Sci USA. 2001;98:10983–10985. doi: 10.1073/pnas.211430998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Han C, Leng J, Demetris AJ, Wu T. Cyclooxygenase-2 promotes human cholangiocarcinoma growth: evidence for cyclooxygenase-2-independent mechanism in celecoxib-mediated induction of p21waf1/cip1 and p27kip1 and cell cycle arrest. Cancer Res. 2004;64:1369–1376. doi: 10.1158/0008-5472.can-03-1086. [DOI] [PubMed] [Google Scholar]
- 77.Haigh WG, Lee SP. Identification of oxysterols in human bile and pigment gallstones. Gastroenterology. 2001;121:118–123. doi: 10.1053/gast.2001.25513. [DOI] [PubMed] [Google Scholar]
- 78.Yoon JH, Canbay AE, Werneburg NW, Lee SP, Gores GJ. Oxysterols induce cyclooxygenase-2 expression in cholangiocytes: implications for biliary tract carcinogenesis. Hepatology. 2004;39:732–738. doi: 10.1002/hep.20125. [DOI] [PubMed] [Google Scholar]
- 79.Werneburg NW, Yoon JH, Higuchi H, Gores GJ. Bile acids activate EGF receptor via a TGF-alpha-dependent mechanism in human cholangiocyte cell lines. Am J Physiol Gastrointest Liver Physiol. 2003;285:G31–G36. doi: 10.1152/ajpgi.00536.2002. [DOI] [PubMed] [Google Scholar]
- 80.Lipson KE, Pang L, Huber LJ, Chen H, Tsai JM, Hirth P, Gazit A, Levitzki A, McMahon G. Inhibition of platelet-derived growth factor and epidermal growth factor receptor signaling events after treatment of cells with specific synthetic inhibitors of tyrosine kinase phosphorylation. J Pharmacol Exp Ther. 1998;285:844–852. [PubMed] [Google Scholar]
- 81.Alpini G, Kanno N, Phinizy JL, Glaser S, Francis H, Taffetani S, LeSage G. Tauroursodeoxycholate inhibits human cholangiocarcinoma growth via Ca2+-, PKC-, and MAPK-dependent pathways. Am J Physiol Gastrointest Liver Physiol. 2004;286:G973–G982. doi: 10.1152/ajpgi.00270.2003. [DOI] [PubMed] [Google Scholar]
- 82.Barone M, Francavilla A, Polimeno L, Ierardi E, Romanelli D, Berloco P, Di Leo A, Panella C. Modulation of rat hepatocyte proliferation by bile salts: in vitro and in vivo studies. Hepatology. 1996;23:1159–1166. doi: 10.1053/jhep.1996.v23.pm0008621149. [DOI] [PubMed] [Google Scholar]
- 83.Yassin RR, Clearfield HR, Little KM. Gastrin’s trophic effect in the colon: identification of a signaling pathway mediated by protein kinase C. Peptides. 1993;14:1119–1124. doi: 10.1016/0196-9781(93)90164-c. [DOI] [PubMed] [Google Scholar]
- 84.Edwards BF, Redding TW, Schally AV. The effect of gastrointestinal hormones on the incorporation of tritiated thymidine in the pancreatic adenocarcinoma cell line (WD PaCa) Int J Pancreatol. 1989;5:191–201. doi: 10.1007/BF02924419. [DOI] [PubMed] [Google Scholar]
- 85.Kanno N, Glaser S, Chowdhury U, Phinizy JL, Baiocchi L, Francis H, LeSage G, Alpini G. Gastrin inhibits cholangiocarcinoma growth through increased apoptosis by activation of Ca2+-dependent protein kinase C-alpha. J Hepatol. 2001;34:284–291. doi: 10.1016/s0168-8278(00)00025-8. [DOI] [PubMed] [Google Scholar]
- 86.Harnois DM, Que FG, Celli A, LaRusso NF, Gores GJ. Bcl-2 is overexpressed and alters the threshold for apoptosis in a cholangiocarcinoma cell line. Hepatology. 1997;26:884–890. doi: 10.1002/hep.510260413. [DOI] [PubMed] [Google Scholar]
- 87.Foja S, Goldberg M, Schagdarsurengin U, Dammann R, Tannapfel A, Ballhausen WG. Promoter methylation and loss of coding exons of the fragile histidine triad (FHIT) gene in intrahepatic cholangiocarcinomas. Liver Int. 2005;25:1202–1208. doi: 10.1111/j.1478-3231.2005.01174.x. [DOI] [PubMed] [Google Scholar]
- 88.Varnholt H. The role of microRNAs in primary liver cancer. Ann Hepatol. 2008;7:104–113. [PubMed] [Google Scholar]
- 89.Mott JL, Kobayashi S, Bronk SF, Gores GJ. mir-29 regulates Mcl-1 protein expression and apoptosis. Oncogene. 2007;26:6133–6140. doi: 10.1038/sj.onc.1210436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Meng F, Wehbe-Janek H, Henson R, Smith H, Patel T. Epigenetic regulation of microRNA-370 by interleukin-6 in malignant human cholangiocytes. Oncogene. 2008;27:378–386. doi: 10.1038/sj.onc.1210648. [DOI] [PubMed] [Google Scholar]
- 91.Meng F, Henson R, Lang M, Wehbe H, Maheshwari S, Mendell JT, Jiang J, Schmittgen TD, Patel T. Involvement of human micro-RNA in growth and response to chemotherapy in human cholangiocarcinoma cell lines. Gastroenterology. 2006;130:2113–2129. doi: 10.1053/j.gastro.2006.02.057. [DOI] [PubMed] [Google Scholar]
- 92.Meng F, Henson R, Wehbe-Janek H, Smith H, Ueno Y, Patel T. The MicroRNA let-7a modulates interleukin-6-dependent STAT-3 survival signaling in malignant human cholangiocytes. J Biol Chem. 2007;282:8256–8264. doi: 10.1074/jbc.M607712200. [DOI] [PubMed] [Google Scholar]
- 93.Christoffersen NR, Silahtaroglu A, Orom UA, Kauppinen S, Lund AH. miR-200b mediates post-transcriptional repression of ZFHX1B. RNA. 2007;13:1172–1178. doi: 10.1261/rna.586807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Cummins JM, Velculescu VE. Implications of micro-RNA profiling for cancer diagnosis. Oncogene. 2006;25:6220–6227. doi: 10.1038/sj.onc.1209914. [DOI] [PubMed] [Google Scholar]
- 95.Calin GA, Ferracin M, Cimmino A, Di Leva G, Shimizu M, Wojcik SE, Iorio MV, Visone R, Sever NI, Fabbri M, et al. A MicroRNA signature associated with prognosis and progression in chronic lymphocytic leukemia. N Engl J Med. 2005;353:1793–1801. doi: 10.1056/NEJMoa050995. [DOI] [PubMed] [Google Scholar]
- 96.Iorio MV, Ferracin M, Liu CG, Veronese A, Spizzo R, Sabbioni S, Magri E, Pedriali M, Fabbri M, Campiglio M, et al. MicroRNA gene expression deregulation in human breast cancer. Cancer Res. 2005;65:7065–7070. doi: 10.1158/0008-5472.CAN-05-1783. [DOI] [PubMed] [Google Scholar]
- 97.Blechacz B, Gores GJ. Cholangiocarcinoma: advances in pathogenesis, diagnosis, and treatment. Hepatology. 2008;48:308–321. doi: 10.1002/hep.22310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Fava G, Marucci L, Glaser S, Francis H, De Morrow S, Benedetti A, Alvaro D, Venter J, Meininger C, Patel T, et al. gamma-Aminobutyric acid inhibits cholangiocarcinoma growth by cyclic AMP-dependent regulation of the protein kinase A/extracellular signal-regulated kinase 1/2 pathway. Cancer Res. 2005;65:11437–11446. doi: 10.1158/0008-5472.CAN-05-1470. [DOI] [PubMed] [Google Scholar]
- 99.Marienfeld C, Tadlock L, Yamagiwa Y, Patel T. Inhibition of cholangiocarcinoma growth by tannic acid. Hepatology. 2003;37:1097–1104. doi: 10.1053/jhep.2003.50192. [DOI] [PubMed] [Google Scholar]
- 100.Bibby MC. Orthotopic models of cancer for preclinical drug evaluation: advantages and disadvantages. Eur J Cancer. 2004;40:852–857. doi: 10.1016/j.ejca.2003.11.021. [DOI] [PubMed] [Google Scholar]
- 101.Sausville EA, Burger AM. Contributions of human tumor xenografts to anticancer drug development. Cancer Res. 2006;66:3351–3354, discussion 3354. doi: 10.1158/0008-5472.CAN-05-3627. [DOI] [PubMed] [Google Scholar]
- 102.Voskoglou-Nomikos T, Pater JL, Seymour L. Clinical predictive value of the in vitro cell line, human xenograft, and mouse allograft preclinical cancer models. Clin Cancer Res. 2003;9:4227–4439. [PubMed] [Google Scholar]
- 103.Iki K, Tsujiuchi T, Majima T, Sakitani H, Tsutsumi M, Takahama M, Yoshimoto M, Nakae D, Tsunoda T, Konishi Y. Increased telomerase activity in intrahepatic cholangiocellular carcinomas induced by N-nitrosobis(2-oxopropyl)amine in hamsters. Cancer Lett. 1998;131:185–190. doi: 10.1016/s0304-3835(98)00148-7. [DOI] [PubMed] [Google Scholar]
- 104.Imray CH, Newbold KM, Davis A, Lavelle-Jones M, Neoptolemos JP. Induction of cholangiocarcinoma in the Golden Syrian hamster using methylazoxymethyl acetate. Eur J Surg Oncol. 1992;18:373–378. [PubMed] [Google Scholar]
- 105.Thamavit W, Pairojkul C, Tiwawech D, Itoh M, Shirai T, Ito N. Promotion of cholangiocarcinogenesis in the hamster liver by bile duct ligation after dimethylnitrosamine initiation. Carcinogenesis. 1993;14:2415–2417. doi: 10.1093/carcin/14.11.2415. [DOI] [PubMed] [Google Scholar]
- 106.Maronpot RR, Giles HD, Dykes DJ, Irwin RD. Furan-induced hepatic cholangiocarcinomas in Fischer 344 rats. Toxicol Pathol. 1991;19:561–570. doi: 10.1177/019262339101900401. [DOI] [PubMed] [Google Scholar]
- 107.Yeh CN, Maitra A, Lee KF, Jan YY, Chen MF. Thioacetamide-induced intestinal-type cholangiocarcinoma in rat: an animal model recapitulating the multi-stage progression of human cholangiocarcinoma. Carcinogenesis. 2004;25:631–636. doi: 10.1093/carcin/bgh037. [DOI] [PubMed] [Google Scholar]
- 108.Fava G, Alpini G, Rychlicki C, Saccomanno S, DeMorrow S, Trozzi L, Candelaresi C, Venter J, Di Sario A, Marzioni M, et al. Leptin enhances cholangiocarcinoma cell growth. Cancer Res. 2008;68:6752–6761. doi: 10.1158/0008-5472.CAN-07-6682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Tesana S, Takahashi Y, Sithithaworn P, Ando K, Sakakura T, Yutanawiboonchai W, Pairojkul C, Ruangjirachuporn W. Ultrastructural and immunohistochemical analysis of cholangiocarcinoma in immunized Syrian golden hamsters infected with Opisthorchis viverrini and administered with dimethylnitrosamine. Parasitol Int. 2000;49:239–251. doi: 10.1016/s1383-5769(00)00052-0. [DOI] [PubMed] [Google Scholar]
- 110.Farazi PA, Zeisberg M, Glickman J, Zhang Y, Kalluri R, DePinho RA. Chronic bile duct injury associated with fibrotic matrix microenvironment provokes cholangiocarcinoma in p53-deficient mice. Cancer Res. 2006;66:6622–6627. doi: 10.1158/0008-5472.CAN-05-4609. [DOI] [PubMed] [Google Scholar]
- 111.Sirica AE, Zhang Z, Lai GH, Asano T, Shen XN, Ward DJ, Mahatme A, Dewitt JL. A novel “patient-like” model of cholangiocarcinoma progression based on bile duct inoculation of tumorigenic rat cholangiocyte cell lines. Hepatology. 2008;47:1178–1190. doi: 10.1002/hep.22088. [DOI] [PubMed] [Google Scholar]