

Abstract
The number of patients suffering from symptoms associated with gastrointestinal (GI) motility disorders is on the rise. GI motility disorders are accompanied by alteration of gastrointestinal smooth muscle functions. Currently available drugs, which can directly affect gastrointestinal smooth muscle and restore altered smooth muscle contractility to normal, are not satisfactory for treating patients with GI motility disorders. We have recently shown that ERK1/2 and p38MAPK signaling pathways play an important role in the contractile response not only of normal intestinal smooth muscle but also of inflamed intestinal smooth muscle. Here we discuss the possibility that ERK1/2 and p38MAPK signaling pathways represent ideal targets for generation of novel therapeutics for patients with GI motility disorders.
Keywords: Mitogen-activated protein kinase, P38MAPK, ERK1/2, Smooth muscle, Contractile dysfunction
INTRODUCTION
Several systems, including the central and enteric neural nexuses, interstitial cells of Cajal and smooth muscles provide coordinated regulation of gastrointestinal (GI) motility. The GI smooth muscle itself plays an important role; it contributes to general health and wellness when functioning normally but is also associated with morbidity and mortality when dysfunctional[1-4]. Alterations in GI motility with resultant changes in transit can contribute to abdominal pain, intestinal cramping, diarrhea, constipation and urgency to defecate. In overt inflammatory conditions of the bowel, such as infectious colitis, Crohn’s disease and ulcerative colitis (i.e., inflammatory bowel disease, IBD), there have been longstanding observations of altered motility and impaired function of the intestinal smooth muscle[5-8]. Even functional GI disorders including non-erosive gastro-esophageal reflux disease (NERD), functional dyspepsia and idiopathic motility dysfunction (now classified under the panoptic irritable bowel syndrome,) seem to be associated with transformations in the contractile nature of smooth muscle[9-12].
Accumulated evidence suggests that the delicate balance between microbes, particularly commensal flora, and host defensive responses at the mucosal barrier have a pivotal role in the pathogenesis of chronic intestinal inflammation. The motility apparatus of the GI tract can act as an extension of the mucosal immune system, contributing to the evacuation of the luminal contents and to mucosal defense against noxious stimuli[13]. Motility dysfunction can secondarily induce abnormal growth of the intestinal flora, and the resulting disturbance of the flora can further aggravate mucosal inflammation[14]. This, in turn, would exacerbate intestinal dysmotility. Thus, motility disorders that arise in the context of inflammation or immune activation are clinically important as they can lead to systemic disease. Furthermore, defects in smooth muscle function are associated with the development of toxic megacolon. This condition is characterized by marked dilation of the distal colon and can occur with severe ulcerative colitis[15], in Hirschsprung’s disease[16] and with infectious colitis[17].
In summary, GI motor disorders are reflective of a variety of important disease manifestations of varying etiologies. However, a central mechanistic feature of all these conditions is an alteration in the contractile processes that occur at the level of the GI smooth muscle. Therefore, it is very reasonable to target the molecular events underlying smooth muscle impairment. Although several drugs, including antimuscarinic agents, acetylcholine-releasing drugs, 5-HT3 antagonists, 5-HT4 agonists and dopamine D2 antagonists, are currently used in clinical practice for GI motility disorders, antimuscarinic agents are the only ones that directly affect smooth muscle. In this regard, there is pressure for new pharmacologic agents capable of directly targeting GI smooth muscle for the restoration of normal smooth muscle contractility in the treatment of motility disorders. Recently, we have demonstrated that the extracellular signal-regulated kinase 1/2 (ERK1/2) and p38 mitogen-activated protein kinase (p38MAPK) signaling pathways play important roles in the contractile responses of both normal intestinal smooth muscle and inflamed intestinal smooth muscle[18,19]. In this commentary, we discuss the possibility that MAPK signaling pathways represent ideal targets for generation of novel therapeutics for patients with GI motility disorders.
CONVENTIONAL MECHANISM OF SMOOTH MUSCLE CONTRACTION
GI smooth muscle possesses distinct properties that distinguish it from other types of visceral and vascular smooth muscle[20]. Smooth muscle of the proximal stomach and sphincters exhibits sustained tone, whereas smooth muscle of the distal stomach, small intestine and colon exhibits variable (phasic) tone on which are superimposed rhythmic contractions. Cycles (slow-waves) of membrane depolarization and repolarization originate in pacemaker cells (i.e., interstitial cells of Cajal) and are transmitted to the smooth muscle cells (SMCs). The depolarization of SMCs primarily reflects activation of voltage-gated Ca2+ channels, resulting in Ca2+ entry from the extracellular space. Concurrent stimulation of rhythmic smooth muscle by excitatory neurotransmitters elicits further depolarization and Ca2+ entry and activates intracellular signaling cascades that result in Ca2+ release from intracellular stores.
Although increased intracellular Ca2+ concentration ([Ca2+]) is the paramount signal to initiate smooth muscle contraction, the contractile properties of the SMC are primarily governed by the phosphorylation of the regulatory light chain (LC20) of myosin II[21,22] (Figure 1), which is itself driven by the balance between protein kinases responsible for phosphorylation of LC20 and protein phosphatases responsible for its dephosphorylation. To initiate contraction, increases in [Ca2+] activate myosin light chain kinase (MLCK), a Ca2+/calmodulin-dependent enzyme[23]. MLCK phosphorylates LC20 on Ser-19, resulting in contraction of smooth muscle through increases in myosin ATPase activity and cross-bridge cycling. Smooth muscle myosin light chain phosphatase (MLCP) is responsible for the dephosphorylation of LC20, resulting in relaxation of smooth muscle[24]. It is the balance between MLCK and MLCP activities that dictates the contractile activity of smooth muscle.
Figure 1.
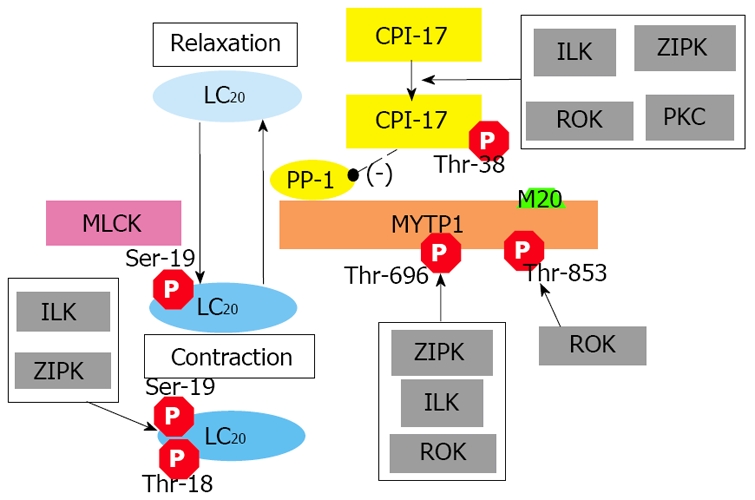
Conventional mechanisms of smooth muscle contraction. Smooth muscle contraction is primarily governed by the phosphorylation of the regulatory light chain (LC20) of myosin II which is in turn driven by the balance between protein kinases responsible for phosphorylation of LC20 and protein phosphatases responsible for its dephosphorylation. Ca2+ sensitization of contractile force can result from the direct phosphorylation of LC20 by Ca2+-independent protein kinases and/or inhibition of myosin phosphatase activity by Ca2+-independent protein kinases. CPI-17: protein kinase C-potentiated inhibitory protein for protein phosphatase 1 of 17 kDa; ILK: integrin-linked kinase; LC20: 20 kDa myosin light chain; MLCK: myosin light chain kinase; MYPT1: myosin targeting subunit 1 of myosin light chain phosphatase; M20: a 20 kDa non-catalytic subunit of myosin light chain phosphatase; PKC: protein kinase C; PP-1: catalytic subunit of protein phosphatase type-1; ROK: Rho-activated protein kinase; ZIPK: zipper-interacting protein kinase.
Although the Ca2+/calmodulin/MLCK pathway plays a crucial role in phosphorylation of LC20, the contraction of many smooth muscle tissues has frequently been observed in the absence of increased [Ca2+] in response to a variety of stimuli, a process commonly referred to as Ca2+ sensitization[22]. Currently, two mechanisms have been proposed to contribute to this phenomenon: (1) the direct phosphorylation of LC20 by Ca2+-independent protein kinases and (2) inhibition of MLCP activity by Ca2+-independent protein kinases. Both integrin-linked kinase (ILK)[25] and zipper-interacting protein kinase (ZIPK)[26,27] can phosphorylate LC20 independently of Ca2+/calmodulin. MLCP functions independently of Ca2+/calmodulin and is regulated by G protein-coupled signaling pathways. Inhibition of MLCP results in greater LC20 phosphorylation and greater force development at given [Ca2+][22]. MLCP activity is regulated directly by phosphorylation of the myosin targeting subunit of MLCP (MYPT1)[24] and/or indirectly via phosphorylation of a protein kinase C (PKC)-potentiated phosphatase inhibitor protein of 17 kDa (CPI-17)[28]. It has been shown that phosphorylation of MYPT1 at Thr-696 (numbering for human sequence) by Rho-associated kinase (ROK)[29], ILK[30] and ZIPK[31] is associated with inhibition of MLCP activity. In contrast, ROK alone is thought to phosphorylate MYPT1 at Thr-853[32], also inhibiting MLCP activity. Alternatively, when CPI-17 is phosphorylated at the regulatory Thr-38 site, it becomes a potent inhibitor of MLCP. Although PKC was the original regulator upstream of CPI-17[28], other protein kinases including ILK[33], ZIPK[34] and ROK[35] have also been demonstrated to phosphorylate CPI-17 at Thr-38.
During intestinal inflammation, it is thought that the smooth muscle undergoes a phenotypic change whereby normal rhythmic contractions are supplanted by sustained Ca2+-independent contractions that persist long after the mucosal response to injury has subsided. It will, therefore, be important to address how different protein kinase networks contribute to Ca2+ sensitization of intestinal smooth muscle contraction. Thus, the study of underlying mechanisms for the regulation of GI smooth muscle contractility will be important for our understanding of the basis for the loss of functional intestinal efficiency that characterizes the inflammatory bowel diseases and other intestinal motility disorders.
CONTRIBUTION OF ERK1/2 AND P38MAPK SIGNALING PATHWAYS TO CONTRACTILE RESPONSE IN NORMAL INTESTINAL SMOOTH MUSCLE
In addition to the protein kinases described above, accumulated evidence has shown that ERK1/2 and p38MAPK can also contribute to smooth muscle contraction[18,36-41]. We have recently examined the relative contributions of ROK, ERK1/2, p38MAPK and PKC to carbachol (CCh)-induced contraction of intestinal smooth muscle. Briefly, the ERK1/2 inhibitor, PD98059, and the p38MAPK inhibitor, SB203580, inhibited CCh-induced contractions of both rat ileal (longitudinal) and colonic (circular) smooth muscles, by 45% and 30% respectively (data not published). Furthermore, GF109203x, a broad PKC inhibitor, had an inhibitory effect (30% inhibition) on CCh-induced contraction in rat colonic smooth muscle, the extent of which was as similar to those observed with PD98059 or SB203580. Interestingly, however, ROK inhibitors Y27632 and H1152 had no effect.
The Ca2+ sensitization process has been examined previously in studies of rat ileal (longitudinal) smooth muscle[18]. When microcystin, a type 1 and type 2A protein phosphatase inhibitor, is applied to permeabilized smooth muscle clamped at pCa 9 (i.e., 1 nmol/L), a sustained contraction is observed that cannot be attributed to MLCK. This Ca2+-independent contraction is thought to result from unmasking of endogenous Ca2+-independent protein kinase activities and induction of the Ca2+ sensitization phenomenon. Pretreatment with either PD98059 or SB203580 inhibited the microcystin-induced contraction of β-escin permeabilized rat ileal smooth muscle strips[18], indicating that both ERK1/2 and p38MAPK were involved. Interestingly, these findings were not observed in rat caudal artery. The microcystininduced contraction at pCa 9 of β-escin permeabilized rat ileal or caudal smooth muscle strips was not affected by pretreatment with Y27632[18,42]. These results indicate that the ERK1/2 and p38MAPK signaling pathways work more extensively than ROK in regulation of smooth muscle contractility, although the effects vary between ileum and colon, and with different agonists[18].
CONTRIBUTION OF ERK1/2 AND P38MAPK TO CONTRACTILE RESPONSES IN INFLAMED INTESTINAL SMOOTH MUSCLE
The molecular events underlying the phenotypic responses of the intestine to pathological inflammation are reflected in diverse tissue types, including smooth muscle. We have identified that Ca2+-independent signaling pathways can influence contractile properties of intestinal smooth muscle under inflammatory conditions. Both ERK1/2 and p38MAPK protein kinase pathways are contributors to intestinal hypercontractility under Th2-mediated inflammatory events[19]. Although a hypercontractile response to CCh was observed in Th2 cytokines-related colitis[43,44], it still remains to be determined what types of downstream signaling pathways are involved in generating this response. In our experiments, colitis was induced in BALB/c mice by providing 5% dextran sulfate sodium (DSS) in drinking water for 7 d. Contractile responses of colonic circular smooth muscle strips to 118 mmol/L K+ and carbachol (CCh) were assessed[19]. DSS-induced Th2 colitis in BALB/c mice was indicated by increased IL-4 and IL-6, with no changes in Th1 cytokines. Animals exposed to DSS had increased CCh-induced contraction (3.5-fold) and CCh-induced Ca2+-sensitization (2.2-fold) responses in intact and a-toxin permeabilized colonic smooth muscle, respectively. The contributions of ERK1/2 and p38MAPK to CCh-induced contractions were significantly increased during Th2 cytokines-related colitis. Alternatively Ca2+-independent contraction induced by microcystin was potentiated (1.5-fold) in mice with Th2 cytokines-related colitis. Both ERK1/2 and p38MAPK were found to contribute to this potentiation. Since treatment with Y27632 did not affect either CCh-induced contraction or microcystin-induced, Ca2+-independent contraction in DSS-treated mice, the contribution of ROK to hypercontractility in inflamed colonic circular smooth muscle was determined to be negligible. We also have shown that the ERK1/2- and p38MAPK-associated hypercontractility is accompanied by significant increases in ERK1/2 and p38MAPK expression in the muscularis propria of colonic tissue from DSS-treated mice. Furthermore, we have examined the expression of ERK1/2 and p38MAPK in human colonic smooth muscle from patients with IBD[19] and found that their expression is also altered. Immunohistochemical analysis of total-ERK1/2 and total-p38MAPK was carried out on human colonic sections from non-IBD (normal) and IBD (Crohn’s disease or ulcerative colitis) patients. Interestingly, the positive staining of total-ERK1/2 and p38MAPK in the muscularis propria was increased in sections from patients with ulcerative colitis, compared to Crohn’s disease patients and non-IBD controls. These results are convincing since ulcerative colitis is thought to exhibit a Th2-like cytokine profile[45]. Taken together, these results indicate that murine Th2 colitis resulted in colonic smooth muscle hypercontractility with increased Ca2+-sensitization. Both ERK1/2 and p38MAPK pathways contributed to this contractile dysfunction, and expression of these kinases was altered in patients with ulcerative colitis.
POSSIBLE MECHANISMS BY WHICH ERK1/2 AND P38MAPK CONTRIBUTE TO SMOOTH MUSCLE CONTRACTION
As described above, both the ERK1/2 and the p38MAPK pathways play important roles in contractile response, not only of normal intestinal smooth muscle but also of inflamed intestinal smooth muscle. Although it has yet to be determined precisely how ERK1/2 and p38MAPK signaling pathways contribute to smooth muscle contraction, the following mechanisms, as outlined in Figure 2, can be considered. In one scenario, ERK1/2 and p38MAPK activation can increase the Mg2+-ATPase activity of myosin II. ERK1/2 has been shown to phosphorylate caldesmon, a thin-filament associated protein that prevents the binding of myosin to actin[46]. The phosphorylation of caldesmon by ERK1/2 weakens the affinity of caldesmon toward actin[47], thereby promoting cross-bridge cycling and force development. Alternatively, p38MAPK has been shown to phosphorylate and activate MAPK-activated protein kinase 2 (MAPKAPK-2)[48], which can in turn phosphorylate heat shock protein (HSP) 27[49]. Phosphorylated HSP27 is able to reverse the inhibitory effects of caldesmon on the Mg2+-ATPase activity of myosin II[46,50]. Another possible mechanism by which ERK1/2 and p38MAPK contribute to smooth muscle contraction is through regulation of MLCP activity, although this pathway has yet to be fully established. We have recently shown that both ERK1/2 and p38MAPK are involved in microcystin-induced contraction at pCa 9 in β-escin permeabilized rat ileal smooth muscle strips. Interestingly, increase in microcystin concentration from 1 μmol/L up to 10 μmol/L abolished the inhibitory effects of these ERK1/2 and p38MAPK pathways, suggesting that ERK1/2 and p38MAPK contribute to smooth muscle contraction via inhibition of MLCP activity[18]. We are currently further examining whether ERK1/2 and p38MAPK pathways are involved in MLCP regulation in intact intestinal smooth muscle and determining the underlying mechanisms.
Figure 2.
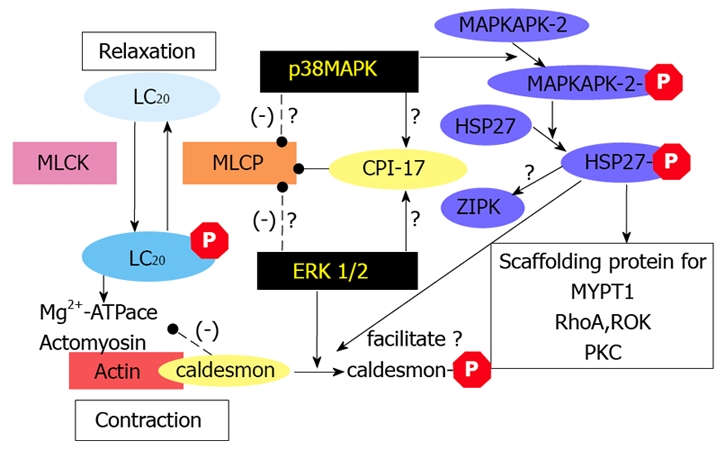
Proposed mechanisms by which ERK1/2 and p38MAPK signaling pathways contribute to smooth muscle contraction. MAPK pathways play important roles in modulating the contractile responses of normal and inflamed intestinal smooth muscles. MAPKs can alter the contractile activity of smooth muscle by (1) increasing the Mg2+-ATPase activity of myosin II, (2) phosphorylating caldesmon, a thin-filament associated protein, and promoting actin-myosin cross-bridges (3) phosphorylating HSP27 to reverse the inhibitory effects of caldesmon, or (4) directly regulating of myosin phosphatase activity. CPI-17: protein kinase C-potentiated inhibitory protein for protein phosphatase 1 of 17 kDa; ERK1/2: extracellular signal-regulated kinase 1/2; HSP27: phosphorylate heat shock protein; MAPKAPK-2: MAPK-activated protein kinase 2; LC20: 20 kDa myosin light chain; MLCK: myosin light chain kinase; MLCP: myosin light chain phosphatase; p38MAPK: p38 mitogen-activated protein kinase; PKC: protein kinase C; ROK: Rho-activated protein kinase; ZIPK: zipper-interacting protein kinase.
MAPK INHIBITORS FOR ERK1/2 AND P38MAPK PATHWAYS AS POTENTIAL THERAPEUTIC TARGETS FOR DISEASES
ERK1/2 and p38MAPK are subfamilies of the MAPKs. In addition to ERK1/2 and p38MAPK, there are two other MAPKs; c-Jun NH2-terminal kinase (JNK1, 2 and 3) and ERK5[51]. ERK1/2 and p38MAPK are activated by a diverse range of stimuli including cytokines, growth factors and matrix proteins that bind to various receptor tyrosine kinases, G-protein coupled receptors, cytokine receptors, and integrins. Signals generated from these cell surface receptors initiate a cascade of signaling events that lead to downstream activation of the MAPKs. ERK1/2 and p38MAPK are activated by phosphorylation of specific Thr and Tyr residues by MAP-kinase kinases (MKK)[51]. MKK1 and MKK2 phosphorylate and activate ERK1 and ERK2, respectively[52], while MKK3 and MKK6 phosphorylate and activate p38MAPK[53]. It has been shown that the ERK1/2 signaling pathway is involved in cell differentiation and proliferation, programmed cell death, cell survival, cell motility and angiogenesis[54]. Alternatively, the p38MAPK signaling pathway plays an especially important role in the production of cytokines such as IL-1, tumor necrosis factor-α (TNF-α), and IL-6[55]. Therefore, the ERK1/2 signaling pathway contributes to various cell cycle-related diseases, including cardiovascular disease[56], cerebral vasospasm[57] and malignancies[58], whereas the p38MAPK signaling pathway is associated with inflammatory diseases including arthritis, psoriasis, IBD and asthma[55]. While PD98059 and SB203580 are classic inhibitors used in many in vitro studies of ERK1/2 and p38MAPK, respectively, several novel kinase inhibitors for ERK1/2 or p38MAPK have already seen use in clinical trials[59]. For example, the benzimidazole derivative, AZD6244, potent second generation inhibitor of MEK1/2, was recently studied in a phase I study to assess its safety, pharmacokinetics and pharmacodynamics in 57 patients with advanced cancer[60]. The 50 % maximal tolerated dose (100 mg BID) was well tolerated with skin rash being the most frequent and dose-limiting side effect. Most other adverse effects were of grade 1 or 2. A strong reduction in ERK1/2 phosphorylation was seen in tumor biopsies and nine patients showed disease stabilization lasting for at least 5 mo. Blocking of the ERK1/2 signaling pathway with MEK1/2 inhibitors (AZD6244, GDC0973, RDEA119, GSK1120212, etc.) has also evaluated in clinical trials for treatment of various malignancies, such as melanoma, breast cancer, colonic cancer, non-small cell lung cancer and a number of leukemias[59]. On the other hand, p38MAPK inhibition has been suggested to be potentially beneficial as a therapeutic strategy in inflammatory disease processes, and several different p38MAPK inhibitors have been tested in animal models of rheumatoid arthritis[61]. In each of these studies, p38MAPK inhibition was shown to reduce disease severity and maintain joint integrity with a reduction in the loss of cartilage and bone. Several p38MAPK inhibitors have advanced into clinical trials on treatment of rheumatoid arthritis in human subjects, although only a few have made it as far as phase II. Unfortunately these compounds have poor safety profiles, including adverse effects in the central nervous system and liver[62] and, as a result, clinical research must move forward cautiously. Clinical trials on MEK1/2 inhibitors and p38MAPK inhibitors are summarized in Table 1.
Table 1.
MEK1/2 or p38MAPK inhibitors in ongoing clinical trials
| Inhibitor | Sponsor | Phase | Study title | Status | |
| MEK1/2 inhibiors | |||||
| AZD6244 | AstraZeneca | Phase II | Randomised study to compare the efficacy of AZD6244 vs TMZ | In progress | |
| National Cancer Centre, Singapore | Phase I/II | AZD6244 and sorafenib in advanced hepatocellular carcinoma | In progress | ||
| University of Oxford | Phase II | Docetaxel with or Without AZD6244 in melanoma (DOC-MEK) | In progress | ||
| Massachusetts General Hospital | Phase II | AZD6244 in cancers with BRAF mutations | In progress | ||
| Other than listed above, almost 20 clinical trials are in progress. | |||||
| GDC0973 | Genentech | Phase I | A study of relative bioavailability and food effect study of GDC-0973 in healthy subjects | Completed | |
| Genentech | Phase I | Study of GDC-0973/XL518 in patients with solid tumors | In progress | ||
| RDEA119 | Ardea Biosciences, Inc | Phase I/II | RDEA119 and sorafenib combination dose escalation study | In progress | |
| GSK1120212 | GlaxoSmithKline | Phase I | Investigate safty, pharmacokinetics and pharmacodynamics of GSK2118436 & GSK1120212 | In progress | |
| GlaxoSmithKline | Phase I | A study of the GSK MEK inhibitor GSK1120212 and everolimus in cancer subjects | In progress | ||
| Other than listed above, 10 clinical trials are in progress. | |||||
| p38MAPK inhibitors | |||||
| VX-702 | Vertex Pharmaceuticals Incorporated | Phase II | A study in rheumatoid arthritis with an investigational oral p38MAP kinase inhibitor VX-702 | Completed | |
| Vertex Pharmaceuticals Incorporated | Phase II | Phase 2 clinical study in RA with an investigational oral p38 MAP kinase inhibitor VX-702 | Completed | ||
Information was obtained from ClinicalTrials.gov (http://clinicaltrials.gov/ct2/home)
ERK1/2 AND P38MAPK PATHWAYS ARE NEW POTENTIAL THERAPEUTIC TARGETS FOR PATIENTS WITH GI MOTILITY DISORDERS
As the ERK1/2 and p38MAPK signaling pathways are already being exploited for therapeutics development in a broad range of diseases (discussed above), they may also be possible new therapeutic targets for GI motility disorders that accompany gastrointestinal smooth muscle dysfunction. Patients suffering from symptoms associated with altered gastrointestinal motility experience decreased quality of life. Although several medications including anti-muscarinic agents, acetylcholine-releasing drugs, 5-HT3 antagonists, 5-HT4 agonists and dopamine D2 antagonists are currently available in clinical practice for GI motility disorders, there are still cases where their therapeutic efficacy is not satisfactory. The ERK1/2 and p38MAPK signaling pathways play an important role in the contractile response not only of normal intestinal smooth muscle but also of inflamed intestinal smooth muscle. These pathways represent ideal targets for generation of novel therapeutics for patients with GI motility disorders. Since several kinase inhibitors for ERK1/2 or p38MAPK are already available and used in clinical trials as described above, blockade of the ERK1/2 or p38MAPK signaling pathway with selective kinase inhibitors may be a good approach for developing new therapeutics for GI motility disorders.However, the potential toxicity of systemic ERK1/2 or p38MAPK inhibition, which may affect a multitude of growth factor signaling pathways that regulate cell proliferation and tissue homeostasis, will need to be addressed before new therapeutics can be developed. MEK1/2 inhibitors can be used in clinical trials only in several advanced, life-threatening cancers where there are no better therapeutic options. In these cases, the benefits of using MEK inhibitors for treatment could outweigh their side effects and toxicity. On risk-benefit considerations[59], currently available MEK1/2 inhibitors are not sufficiently beneficial and safe to be used in clinical trials in humans with GI motility disorders. Therefore, we eagerly await the next generations of ERK1/2 and p38MAPK signaling pathway inhibitors. These compounds, which may avoid systemic adverse effects because of greater specificity with reduced toxicity or smooth muscle tissue-selective delivery, could become a new therapeutic option for patients with GI motility disorders.
Footnotes
Supported by the Research Grant from the Canadian Institutes for Health Research, and Partly by an Alberta Innovates–Health Solutions Senior Scholar Award and Canada Research Chair in Smooth Muscle Pathophysiology
Peer reviewers: Gregory M Holmes, Associate Professor, Department of Neuroscience, Pennington Biomedical Research Center, 6400 Perkins Road, Baton Rouge, LA 70808, United States
S- Editor Zhang HN L- Editor Hughes D E- Editor Liu N
References
- 1.Barbara G, De Giorgio R, Stanghellini V, Cremon C, Corinaldesi R. A role for inflammation in irritable bowel syndrome? Gut. 2002;51 Suppl 1:i41–i44. doi: 10.1136/gut.51.suppl_1.i41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barbara G, De Giorgio R, Stanghellini V, Cremon C, Salvioli B, Corinaldesi R. New pathophysiological mechanisms in irritable bowel syndrome. Aliment Pharmacol Ther. 2004;20 Suppl 2:1–9. doi: 10.1111/j.1365-2036.2004.02036.x. [DOI] [PubMed] [Google Scholar]
- 3.Pfeiffer RF. Intestinal Dysfunction. Pfeiffer RF, Bodis-Wollner I, editors. Current Clinical Neurology Parkinson's Disease and Non Motor dysfunction. Human Press. 2005. pp. 115–127. [Google Scholar]
- 4.Vonderohe MR, Camilleri M, Kvols LK, Thomforde GM. Motor dysfunction of the small bowel and colon in patients with the carcinoid syndrome and diarrhea. N Engl J Med. 1993;329:1073–1078. doi: 10.1056/NEJM199310073291503. [DOI] [PubMed] [Google Scholar]
- 5.Akiho H, Deng Y, Blennerhassett P, Kanbayashi H, Collins SM. Mechanisms underlying the maintenance of muscle hypercontractility in a model of postinfective gut dysfunction. Gastroenterology. 2005;129:131–141. doi: 10.1053/j.gastro.2005.03.049. [DOI] [PubMed] [Google Scholar]
- 6.Khan WI, Collins SM. Gut motor function: immunological control in enteric infection and inflammation. Clin Exp Immunol. 2006;143:389–397. doi: 10.1111/j.1365-2249.2005.02979.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vermillion DL, Huizinga JD, Riddell RH, Collins SM. Altered small intestinal smooth muscle function in Crohn's disease. Gastroenterology. 1993;104:1692–1699. doi: 10.1016/0016-5085(93)90647-u. [DOI] [PubMed] [Google Scholar]
- 8.Vrees MD, Pricolo VE, Potenti FM, Cao W. Abnormal motility in patients with ulcerative colitis: the role of inflammatory cytokines. Arch Surg. 2002;137:439–445. doi: 10.1001/archsurg.137.4.439. [DOI] [PubMed] [Google Scholar]
- 9.Iwakiri K, Hayashi Y, Kotoyori M, Tanaka Y, Kawami N, Sano H, Takubo K, Sakamoto C, Holloway RH. Defective triggering of secondary peristalsis in patients with non-erosive reflux disease. J Gastroenterol Hepatol. 2007;22:2208–2211. doi: 10.1111/j.1440-1746.2006.04817.x. [DOI] [PubMed] [Google Scholar]
- 10.Colecchia A, Sandri L, Staniscia T, Vestito A, Capodicasa S, Portincasa P, Mazzella G, Roda E, Festi D. Gallbladder motility and functional gastrointestinal disorders. Dig Liver Dis. 2003;35 Suppl 3:S30–S34. doi: 10.1016/s1590-8658(03)00091-4. [DOI] [PubMed] [Google Scholar]
- 11.Saito YA, Strege PR, Tester DJ, Locke GR 3rd, Talley NJ, Bernard CE, Rae JL, Makielski JC, Ackerman MJ, Farrugia G. Sodium channel mutation in irritable bowel syndrome: evidence for an ion channelopathy. Am J Physiol Gastrointest Liver Physiol. 2009;296:G211–G218. doi: 10.1152/ajpgi.90571.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang M, Leung FP, Huang Y, Bian ZX. Increased colonic motility in a rat model of irritable bowel syndrome is associated with up-regulation of L-type calcium channels in colonic smooth muscle cells. Neurogastroenterol Motil. 2010;22:e162–e170. doi: 10.1111/j.1365-2982.2009.01467.x. [DOI] [PubMed] [Google Scholar]
- 13.Collins SM. The immunomodulation of enteric neuromuscular function: implications for motility and inflammatory disorders. Gastroenterology. 1996;111:1683–1699. doi: 10.1016/s0016-5085(96)70034-3. [DOI] [PubMed] [Google Scholar]
- 14.Ohama T, Hori M, Ozaki H. Mechanism of abnormal intestinal motility in inflammatory bowel disease: how smooth muscle contraction is reduced? J Smooth Muscle Res. 2007;43:43–54. doi: 10.1540/jsmr.43.43. [DOI] [PubMed] [Google Scholar]
- 15.Gan SI, Beck PL. A new look at toxic megacolon: an update and review of incidence, etiology, pathogenesis, and management. Am J Gastroenterol. 2003;98:2363–2371. doi: 10.1111/j.1572-0241.2003.07696.x. [DOI] [PubMed] [Google Scholar]
- 16.Siegman MJ, Butler TM, Mooers SU, Trinkle-Mulcahy L, Narayan S, Adam L, Chacko S, Haase H, Morano I. Hypertrophy of colonic smooth muscle: contractile proteins, shortening velocity, and regulation. Am J Physiol. 1997;272:G1571–G1580. doi: 10.1152/ajpgi.1997.272.6.G1571. [DOI] [PubMed] [Google Scholar]
- 17.Gilbert RJ, Triadafilopoulos G, Pothoulakis C, Giampaolo C, LaMont JT. Effect of purified Clostridium difficile toxins on intestinal smooth muscle. I. Toxin A. Am J Physiol. 1989;256:G759–G766. doi: 10.1152/ajpgi.1989.256.4.G759. [DOI] [PubMed] [Google Scholar]
- 18.Ihara E, Moffat LD, Ostrander JM, Walsh MP, Macdonald JA. Characterization of protein kinase pathways responsible for Ca2+ sensitization in rat ileal longitudinal smooth muscle. Am J Physiol Gastrointest Liver Physiol. 2007;293:G699–G710. doi: 10.1152/ajpgi.00214.2007. [DOI] [PubMed] [Google Scholar]
- 19.Ihara E, Beck PL, Chappellaz M, Wong J, Medlicott SA, MacDonald JA. Mitogen-activated protein kinase pathways contribute to hypercontractility and increased Ca2+ sensitization in murine experimental colitis. Mol Pharmacol. 2009;75:1031–1041. doi: 10.1124/mol.108.049858. [DOI] [PubMed] [Google Scholar]
- 20.Murthy KS. Signaling for contraction and relaxation in smooth muscle of the gut. Annu Rev Physiol. 2006;68:345–374. doi: 10.1146/annurev.physiol.68.040504.094707. [DOI] [PubMed] [Google Scholar]
- 21.Somlyo AP, Somlyo AV. Signal transduction and regulation in smooth muscle. Nature. 1994;372:231–236. doi: 10.1038/372231a0. [DOI] [PubMed] [Google Scholar]
- 22.Somlyo AP, Somlyo AV. Ca2+ sensitivity of smooth muscle and nonmuscle myosin II: modulated by G proteins, kinases, and myosin phosphatase. Physiol Rev. 2003;83:1325–1358. doi: 10.1152/physrev.00023.2003. [DOI] [PubMed] [Google Scholar]
- 23.Taylor DA, Stull JT. Calcium dependence of myosin light chain phosphorylation in smooth muscle cells. J Biol Chem. 1988;263:14456–14462. [PubMed] [Google Scholar]
- 24.Hartshorne DJ, Ito M. and Erdodi F. Role of protein phosphatase type 1 in contractile functions: myosin phosphatase. J Biol Chem. 2004;279:37211–37214. doi: 10.1074/jbc.R400018200. [DOI] [PubMed] [Google Scholar]
- 25.Deng JT, Van Lierop JE, Sutherland C, Walsh MP. Ca2+-independent smooth muscle contraction. a novel function for integrin-linked kinase. J Biol Chem. 2001;276:16365–16373. doi: 10.1074/jbc.M011634200. [DOI] [PubMed] [Google Scholar]
- 26.Borman MA, MacDonald JA, Haystead TA. Staurosporine inhibition of zipper-interacting protein kinase contractile effects in gastrointestinal smooth muscle. Biochem Cell Biol. 2007;85:111–120. doi: 10.1139/o06-209. [DOI] [PubMed] [Google Scholar]
- 27.Niiro N, Ikebe M. Zipper-interacting protein kinase induces Ca2+-free smooth muscle contraction via myosin light chain phosphorylation. J Biol Chem. 2001;276:29567–29574. doi: 10.1074/jbc.M102753200. [DOI] [PubMed] [Google Scholar]
- 28.Eto M, Ohmori T, Suzuki M, Furuya K, Morita F. A novel protein phosphatase-1 inhibitory protein potentiated by protein kinase C. Isolation from porcine aorta media and characterization. J Biochem. 1995;118:1104–1107. doi: 10.1093/oxfordjournals.jbchem.a124993. [DOI] [PubMed] [Google Scholar]
- 29.Kimura K, Ito M, Amano M, Chihara K, Fukata Y, Nakafuku M, Yamamori B, Feng J, Nakano T, Okawa K, et al. Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho-kinase) Science. 1996;273:245–248. doi: 10.1126/science.273.5272.245. [DOI] [PubMed] [Google Scholar]
- 30.Kiss E, Muranyi A, Csortos C, Gergely P, Ito M, Hartshorne DJ, Erdodi F. Integrin-linked kinase phosphorylates the myosin phosphatase target subunit at the inhibitory site in platelet cytoskeleton. Biochem J. 2002;365:79–87. doi: 10.1042/BJ20011295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Borman MA, MacDonald JA, Muranyi A, Hartshorne DJ, Haystead TA. Smooth muscle myosin phosphatase-associated kinase induces Ca2+ sensitization via myosin phosphatase inhibition. J Biol Chem. 2002;277:23441–23446. doi: 10.1074/jbc.M201597200. [DOI] [PubMed] [Google Scholar]
- 32.Wilson DP, Susnjar M, Kiss E, Sutherland C, Walsh MP. Thromboxane A2-induced contraction of rat caudal arterial smooth muscle involves activation of Ca2+ entry and Ca2+ sensitization: Rho-associated kinase-mediated phosphorylation of MYPT1 at Thr-855, but not Thr-697. Biochem J. 2005;389:763–774. doi: 10.1042/BJ20050237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Deng JT, Sutherland C, Brautigan DL, Eto M, Walsh MP. Phosphorylation of the myosin phosphatase inhibitors, CPI-17 and PHI-1, by integrin-linked kinase. Biochem J. 2002;367:517–524. doi: 10.1042/BJ20020522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.MacDonald JA, Borman MA, Muranyi A, Somlyo AV, Hartshorne DJ, Haystead TA. Identification of the endogenous smooth muscle myosin phosphatase-associated kinase. Proc Natl Acad Sci USA. 2001;98:2419–2424. doi: 10.1073/pnas.041331498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Koyama M, Ito M, Feng J, Seko T, Shiraki K, Takase K, Hartshorne DJ, Nakano T. Phosphorylation of CPI-17, an inhibitory phosphoprotein of smooth muscle myosin phosphatase, by Rho-kinase. FEBS Lett. 2000;475:197–200. doi: 10.1016/s0014-5793(00)01654-9. [DOI] [PubMed] [Google Scholar]
- 36.Lee DW, Park SY, Ryu JS, Kim SH, Im CU, Choi SH, Lee SE, Ko SK, Sohn UD. Relaxation effect of synthetic ceramide analogues in cat esophageal smooth muscle cells. Korean J Physiol Pharmacol. 2008;12:137–142. doi: 10.4196/kjpp.2008.12.4.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee HM, Won KJ, Kim J, Park HJ, Kim HJ, Roh HY, Lee SH, Lee CK, Kim B. Endothelin-1 induces contraction via a Syk-mediated p38 mitogen-activated protein kinase pathway in rat aortic smooth muscle. J Pharmacol Sci. 2007;103:427–433. doi: 10.1254/jphs.fp0070039. [DOI] [PubMed] [Google Scholar]
- 38.Kim J, Lee YR, Lee CH, Choi WH, Lee CK, Bae YM, Cho S, Kim B. Mitogen-activated protein kinase contributes to elevated basal tone in aortic smooth muscle from hypertensive rats. Eur J Pharmacol. 2005;514:209–215. doi: 10.1016/j.ejphar.2005.03.030. [DOI] [PubMed] [Google Scholar]
- 39.Kwon S, Fang LH, Kim B, Ha TS, Lee SJ, Ahn HY. p38 Mitogen-activated protein kinase regulates vasoconstriction in spontaneously hypertensive rats. J Pharmacol Sci. 2004;95:267–272. doi: 10.1254/jphs.fpj03091x. [DOI] [PubMed] [Google Scholar]
- 40.Pearce WJ, Williams JM, Chang MM, Gerthoffer WT. ERK inhibition attenuates 5-HT-induced contractions in fetal and adult ovine carotid arteries. Arch Physiol Biochem. 2003;111:36–44. doi: 10.1076/apab.111.1.36.15143. [DOI] [PubMed] [Google Scholar]
- 41.Puri RN, Fan YP, Rattan S. Role of pp60c-src and p44/42 MAPK in ANG II-induced contraction of rat tonic gastrointestinal smooth muscles. Am J Physiol Gastrointest Liver Physiol. 2002;283:G390–G399. doi: 10.1152/ajpgi.00025.2002. [DOI] [PubMed] [Google Scholar]
- 42.Wilson DP, Sutherland C, Borman MA, Deng JT, Macdonald JA, Walsh MP. Integrin-linked kinase is responsible for Ca2+-independent myosin diphosphorylation and contraction of vascular smooth muscle. Biochem J. 2005;392:641–648. doi: 10.1042/BJ20051173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Akiho H, Blennerhassett P, Deng Y, Collins SM. Role of IL-4, IL-13, and STAT6 in inflammation-induced hypercontractility of murine smooth muscle cells. Am J Physiol Gastrointest Liver Physiol. 2002;282:G226–G232. doi: 10.1152/ajpgi.2002.282.2.G226. [DOI] [PubMed] [Google Scholar]
- 44.Akiho H, Lovato P, Deng Y, Ceponis PJ, Blennerhassett P, Collins SM. Interleukin-4- and -13-induced hypercontractility of human intestinal muscle cells-implication for motility changes in Crohn's disease. Am J Physiol Gastrointest Liver Physiol. 2005;288:G609–G615. doi: 10.1152/ajpgi.00273.2004. [DOI] [PubMed] [Google Scholar]
- 45.Heller F, Florian P, Bojarski C, Richter J, Christ M, Hillenbrand B, Mankertz J, Gitter AH, Burgel N, Fromm M, et al. Interleukin-13 is the key effector Th2 cytokine in ulcerative colitis that affects epithelial tight junctions, apoptosis, and cell restitution. Gastroenterology. 2005;129:550–564. doi: 10.1016/j.gastro.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 46.Sobue K, Sellers JR. Caldesmon, a novel regulatory protein in smooth muscle and nonmuscle actomyosin systems. J Biol Chem. 1991;266:12115–12118. [PubMed] [Google Scholar]
- 47.Huang R, Li L, Guo H, Wang CL. Caldesmon binding to actin is regulated by calmodulin and phosphorylation via different mechanisms. Biochemistry. 2003;42:2513–2523. doi: 10.1021/bi0268605. [DOI] [PubMed] [Google Scholar]
- 48.Cohen P. The search for physiological substrates of MAP and SAP kinases in mammalian cells. Trends Cell Biol. 1997;7:353–361. doi: 10.1016/S0962-8924(97)01105-7. [DOI] [PubMed] [Google Scholar]
- 49.Rouse J, Cohen P, Trigon S, Morange M, AlonsoLlamazares A, Zamanillo D, Hunt T, Nebreda AR. A novel kinase cascade triggered by stress and heat shock that stimulates MAPKAP kinase-2 and phosphorylation of the small heat shock proteins. Cell. 1994;78:1027–1037. doi: 10.1016/0092-8674(94)90277-1. [DOI] [PubMed] [Google Scholar]
- 50.Somara S, Bitar KN. Phosphorylated HSP27 modulates the association of phosphorylated caldesmon with tropomyosin in colonic smooth muscle. Am J Physiol Gastrointest Liver Physiol. 2006;291:G630–G639. doi: 10.1152/ajpgi.00350.2005. [DOI] [PubMed] [Google Scholar]
- 51.Avruch J. MAP kinase pathways: the first twenty years. Biochim Biophys Acta. 2007;1773:1150–1160. doi: 10.1016/j.bbamcr.2006.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pearson G, Robinson F, Beers Gibson T, Xu BE, Karandikar M, Berman K, Cobb MH. Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr Rev. 2001;22:153–183. doi: 10.1210/edrv.22.2.0428. [DOI] [PubMed] [Google Scholar]
- 53.Brancho D, Tanaka N, Jaeschke A, Ventura JJ, Kelkar N, Tanaka Y, Kyuuma M, Takeshita T, Flavell RA, Davis RJ. Mechanism of p38 MAP kinase activation in vivo. Genes Dev. 2003;17:1969–1978. doi: 10.1101/gad.1107303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yoon S, Seger R. The extracellular signal-regulated kinase: multiple substrates regulate diverse cellular functions. Growth Factors. 2006;24:21–44. doi: 10.1080/02699050500284218. [DOI] [PubMed] [Google Scholar]
- 55.Sweeney SE, Firestein GS. Mitogen activated protein kinase inhibitors: where are we now and where are we going? Ann Rheum Dis. 2006;65 Suppl 3:iii83–iii88. doi: 10.1136/ard.2006.058388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang Y. Mitogen-activated protein kinases in heart development and diseases. Circulation. 2007;116:1413–1423. doi: 10.1161/CIRCULATIONAHA.106.679589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Satoh M, Parent AD, Zhang JH. Inhibitory effect with antisense mitogen-activated protein kinase oligodeoxynucleotide against cerebral vasospasm in rats. Stroke. 2002;33:775–781. doi: 10.1161/hs0302.103734. [DOI] [PubMed] [Google Scholar]
- 58.Friday BB, Adjei AA. Advances in targeting the Ras/Raf/MEK/Erk mitogen-activated protein kinase cascade with MEK inhibitors for cancer therapy. Clin Cancer Res. 2008;14:342–346. doi: 10.1158/1078-0432.CCR-07-4790. [DOI] [PubMed] [Google Scholar]
- 59.Fremin C, Meloche S. From basic research to clinical development of MEK1/2 inhibitors for cancer therapy. J Hematol Oncol. 2010;3:8. doi: 10.1186/1756-8722-3-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Adjei AA, Cohen RB, Franklin W, Morris C, Wilson D, Molina JR, Hanson LJ, Gore L, Chow L, Leong S, et al. Phase I pharmacokinetic and pharmacodynamic study of the oral, small-molecule mitogen-activated protein kinase kinase 1/2 inhibitor AZD6244 (ARRY-142886) in patients with advanced cancers. J Clin Oncol. 2008;26:139–146. doi: 10.1200/JCO.2007.14.4956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Loeser RF, Erickson EA, Long DL. Mitogen-activated protein kinases as therapeutic targets in osteoarthritis. Curr Opin Rheumatol. 2008;20:581–586. doi: 10.1097/BOR.0b013e3283090463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang J, Shen B, Lin A. Novel strategies for inhibition of the p38 MAPK pathway. Trends Pharmacol Sci. 2007;28:286–295. doi: 10.1016/j.tips.2007.04.008. [DOI] [PubMed] [Google Scholar]