

Abstract
Vibrio cholerae relies on two main virulence factors, the toxin coregulated pilus (TCP) and cholera toxin, to cause the gastrointestinal disease cholera. TCP is a type IV pilus that mediates bacterial autoagglutination and colonization of the intestine. TCP is encoded by the tcp operon, which also encodes TcpF, a protein of unknown function that is secreted by V. cholerae in a TCP-dependent manner. Although TcpF is not required for TCP biogenesis, a tcpF mutant has a colonization defect in the infant mouse cholera model that is as severe as a pilus mutant. Furthermore, TcpF antisera protects against V. cholerae infection. TcpF has no apparent sequence homology to any known protein. Here we report the de novo x-ray crystal structure of TcpF and the identification of an epitope that is critical for its function as a colonization factor. A monoclonal antibody recognizing this epitope is protective against V. cholerae challenge and adds to the protection provided by an anti-TcpA antibody. These data suggest that TcpF has a novel function in V. cholerae colonization and define a region crucial for this function.
Keywords: bacterial colonization, cholera, diarrheal disease, protein structure, secretion
Introduction
Vibrio cholerae is an aquatic bacterium present in most marine and freshwater environments. The majority of infections caused by V. cholerae can be traced to ingestion of contaminated water. Cholera outbreaks have been reported on every continent and large epidemic outbreaks are relatively common 1. The onset of cholera symptoms occurs 24–48 hours after ingestion of V. cholerae and the disease is characterized by massive secretory diarrhea. Epidemic V. cholerae O1 causes disease by colonizing the intestine and secreting cholera toxin, which initiates a signaling cascade in intestinal epithelial cells that results in elevated Cl− secretion into the intestinal lumen; H2O, HCO3−, and Na+ follow 2; 3 resulting in secretory diarrhea.
In vivo, prior to cholera toxin secretion, the bacteria synthesize another virulence factor: the toxin coregulated pilus (TCP) 4. Both cholera toxin and TCP are regulated by the transcription factor, ToxT 5. TCP is a filamentous structure belonging to the type IVb pilus subclass, assembled by proteins encoded in the tcp operon 6; 7; 8. The TCP filament is comprised of thousands of copies of the pilin subunit, TcpA 10; 11, which is conserved within biotypes and serogroups. The TcpA sequence is almost 100% identical among strains within each biotype, and 81% identical between the El Tor and classical biotypes 9. TCP filaments interact with each other to form bundles and mediate autoagglutination in vitro 11; 12; 13. In the mouse and human intestine there is evidence that TCP mediates microcolony formation at the intestinal surface in vivo 12. TCP is absolutely required for colonization: V. cholerae mutants deficient for TCP production or function are profoundly defective in colonization in both humans and mice 11; 12; 14; 15.
The TCP complex, like all type IV pilus assemblies, resembles a type II secretion system 16, and TCP is necessary for the release of another virulence factor, TcpF, through an undefined mechanism 17. In previous studies, we showed that TcpF is crucial for successful colonization in the infant mouse model of V. cholerae infection 17. In fact, V. cholerae strains lacking TcpF are as deficient in colonization as TCP-negative strains 17; 18; 19. Our initial studies determined that TcpF is not required for TCP-mediated autoagglutination 19 and may therefore have a function in colonization independent from TCP.
Previous studies regarding the mechanism of action of TCP utilized a combination of genetics, immunology and structural biology to characterize the pilus and its mode of action 10; 12; 13; 20; 21. In the present study we utilize this same overall strategy to understand how the structure correlates with function of TcpF. This is especially important since BLAST (http://blast.ncbi.nlm.nih.gov/Blast.cgi) searches with the TcpF sequence returned no protein of significant sequence similarity that might provide clues to its functions. For the initial genetic approach, we used Linker-Scanning Mutagenesis to roughly identify regions of TcpF necessary for in vivo function 22. In the present study we utilize x-ray crystallography in combination with immunological methods with a protective monoclonal antibody to characterize TcpF. Here we report the crystal structure of TcpF and the identification of a functional domain using monoclonal antibodies. The epitope defined by a mAb that decreases V. cholerae colonization was mapped to the surface of TcpF and the functionality of this region was demonstrated by mutational analysis. Our results build on our understanding of this unique protein and its critical role in V. cholerae pathogenesis.
Results
Generation of TcpF mAb13, which protects against V. cholerae challenge
We showed previously that TcpF polyclonal antisera is protective in the infant mouse cholera model 17. In this study we sought to identify monoclonal antibodies and map their epitopes to define functional regions of TcpF. To generate antibodies that recognize TcpF in its native form, we purified TcpF without the use of affinity purification tags. TcpF was purified from the periplasm of V. cholerae strain SJK7, a ΔtcpF strain in which TcpF is expressed exogenously from a plasmid using an arabinose inducible promoter (Table S1). Under these conditions, overexpressed TcpF is secreted via the TCP apparatus but also saturates the extracellular secretion system, which leads to its accumulation in the periplasm 22. Periplasmic TcpF appears at its expected molecular weight, 36 kDa, on a Coomassie-stained gel and an immunoblot, although some degradation products are also present (Supplemental Fig. S1), as seen for endogenously-expressed, secreted TcpF 22.
Purified TcpF was used to immunize mice, after which their spleens were harvested and fused with NS1 myeloma cells to generate hybridomas. Four stable hybridoma clones were selected for analysis. All four clones secrete antibody that recognizes the same epitope of TcpF (see methods below). Data is shown for a single, representative antibody, mAb13. Immunoblot analysis of V. cholerae culture supernatant shows that mAb13 recognizes TcpF with a high degree of specificity (Fig. 1A). mAb13 recognition of TcpF was further confirmed by ELISA using purified TcpF (data not shown).
Figure 1.

mAb13 is protective against V. cholerae infection using the infant mouse cholera model. (A) Western blot of O395 V. cholerae culture supernatants following growth in TCP-inducing conditions, probed with mAb13 (top panel). The same supernatants were probed with an antibody against another secreted protein, GbpA 18, as a loading control to show that cell levels were comparable for each sample. (B) Kaplan-Meier survival curves represent mouse survival after challenge with V. cholerae mixed with purified IgG from polyclonal antiserum (25 mg/ml), mAb13 (2 mg/ml) or PBS. Six mice were inoculated per group. Data are representative of three independent experiments.
To determine if mAb13 could block TcpF functions in colonization, we performed a passive immunization experiment using the infant mouse cholera model. Mice were challenged with wild type O395 bacteria combined with phosphate-buffered saline (PBS), protein G affinity purified IgG from TcpF polyclonal antisera, or mAb13. TcpF antisera was shown previously to be protective in this model 17. The results shown in Fig. 1B demonstrate that protection by mAb13 is comparable to that of the TcpF polyclonal antisera. These data suggest that mAb13 binding disrupts TcpF function in vivo and may thus recognize a region of TcpF that is important for its role as a colonization factor.
mAb13 recognizes TcpF peptides containing residues Q230-K281
To localize the protective epitope recognized by mAb13, a series of TcpF peptides of approximately 80–100 amino acids and overlapping by approximately 50 amino acids, was expressed in E. coli ER2566 (Fig. 2A). The TcpF peptides, named Fpep1-7, were N-terminally His6x tagged to confirm expression and contain a 25 kDa intein tag on the C-terminus to enhance peptide stability 23. The TcpF peptides were evaluated by probing blots of E. coli whole cell extracts expressing each peptide with an anti-His antibody, TcpF polyclonal antisera and mAb13 (Fig. 2B). Both the anti-His antibody and the TcpF antisera bound well to all but one peptide, indicating that the peptides are expressed well, with peptide Fpep5 expressed at reduced levels. TcpF antisera recognizes multiple bands for each peptide, which is likely due to degradation. mAb13 bound to Fpep4, 5, and 7, leading us to conclude that mAb13 recognizes an epitope within the segment common to these peptides, spanning residue Q230 to K281 (Fig. 2C).
Figure 2.

Identification of the epitopic region of TcpF recognized by mAb13. (A) Diagram of peptides. Seven TcpF peptides were constructed, each with an N-terminal His6x tag and a C-terminal intein tag. (B) mAb13 recognizes Fpep4, Fpep5, and Fpep7. Immunoblots of whole cell lysate of V. cholerae or peptide-expressing E. coli strains were first blotted with mAb13 then stripped and re-probed with the TcpF polyclonal antisera and an anti-His antibody to evaluate peptide stability and antibody reactivity. WT indicates culture supernatant from V. cholerae O395 grown under TCP inducing conditions. The 36 kDa TcpF band is recognized by both TcpF antibodies. The calculated molecular weights of the full-length peptides, including the His6x and intein tags, are as follows: Fpep1, 65.2 kDa; Fpep2, 73.2; Fpep3, 66.5; Fpep4, 67.9 Fpep5, 66.0; Fpep6, 83.8; Fpep7, 78.0. (C) Amino acid sequence of the TcpF epitopic region, which corresponds to the sequence shared by Fpep4, Fpep5 and Fpep7. Critical binding residues Glu251 and Glu252 are underlined.
Further delineation of the epitope recognized by mAb13 using phage display
We screened a phage display peptide library with mAb13 to more precisely map its TcpF epitope. Previous researchers have used phage display in a variety of ways, including as a tool to identify epitopes 24. In this study, a phage display peptide library was biopanned with mAb13 for three rounds to select high affinity phage. As a positive control for the selection process, we included the S-20-4 antibody, which binds to V. cholerae Ogawa O-antigen and had been shown previously to bind to phage clone 4P-825. mAb13 selected a number of peptide sequences similar to the TcpF C-terminal segment Y244-E258, which lies within the epitopic region identified in our peptide mapping (Table 1). Notably, two of the phage peptides selected by the monoclonal antibody contain a PXXEE sequence, and several other peptides had a single glutamate, suggesting that the adjacent glutamates present in TcpF at positions 251 and 252 may be important for recognition of TcpF by mAb13.
Table 1.
Phage displayed peptides selected with mAb13
| Unique sequencesa | Antibody binding (A450) | ||||
|---|---|---|---|---|---|
| mAb13 | no Ab | IgG1 | S-20-4 | ||
| TcpF244–258 | VIYDPVYEETVKPYQ | ||||
| 3MD-5 | HDFHDPTLEEQK | 1.830 | 0.138 | 0.175 | 0.218 |
| 3MD-1 | FLPTLEENKGFA | 1.795 | 0.161 | 0.200 | 0.200 |
| 4P-8b | NHNYPPLSLLTF | 0.272 | 0.146 | 0.270 | 1.923 |
underlined amino acids indicate those conserved between phage peptides and TcpF
negative control peptide recognized by anti-LPS antibody S-20-4 identified in Dharamasena et al. 2007.
TcpF residues Glu251 and Glu252 are critical for mAb13 binding
We performed site directed mutagenesis in V. cholerae O395 to alter residues Glu251/252 of TcpF, replacing both amino acids with alanine to determine their importance in mAb13 binding. The altered TcpF protein is expressed and secreted by V. cholerae cells at levels comparable to wild type TcpF (Fig. 3). TcpF:Glu251/252Ala is recognized by polyclonal TcpF antisera but not by mAb13. These data indicate that Glu251 and Glu252 are not essential for TcpF secretion or stability but are critical for its interaction with mAb13.
Figure 3.
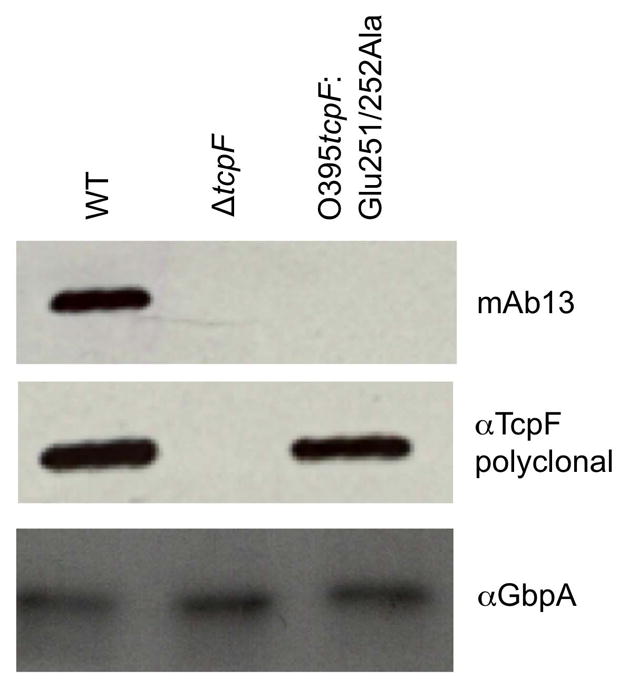
Replacement of Glu251 and Glu252 with alanine results in stable TcpF expression and secretion but disrupts recognition by mAb13. Western blot analysis of secreted TcpF from wild type O395 V. cholerae (WT), O395 ΔtcpF, and O395 tcpF:Glu251/252Ala. A blot of culture supernatant was probed with mAb13, then stripped and reprobed with anti-TcpF polyclonal antibody. The blot was subsequently probed with anti-GbpA as a loading control.
Atomic structure of TcpF
To further our understanding of TcpF and its role in V. cholerae colonization and to understand the involvement of Glu251/252, we determined the x-ray crystal structure of native TcpF to 2.4 Å resolution by dual-wavelength anomalous dispersion methods (Table 2). An interpretable electron density map was obtained for residues 26–318 of the 318-residue mature protein, with the exception of residues 66–71 and 260–261, which lie in surface-exposed loops. The N-terminal 25 residues are not resolved in the electron density map due to disorder or to proteolysis of the N-terminal segment. TcpF has a bilobed structure with an N-terminal domain (NTD, residues 1–185) and a C-terminal domain (CTD, residues 190–318) joined by an extended linker segment (residues 186–189) (Fig. 4A). The overall dimensions of the protein are ~ 73 Å × 36 Å × 24 Å. The globular NTD is comprised of a short twisted β-sheet (β1, β6, β2) encapsulated by 7 short α-helices (α1–α7), with a second twisted β-sheet (β5, β3, β4, β7) forming the floor of this domain (Fig. 4A, B). The central strands of the second β-sheet, β3 and β4, are longer than the outside strands and form a β-hairpin that extends toward the CTD. A disulfide bond between residues Cys34 and Cys47 links the two N-terminal α-helices (α1 and α2).
Table 2.
Crystallographic data collection and refinement statistics for TcpF and TcpF-CTD
| Data collection | Native TcpF | SeMet-TcpF | TcpF-CTD | |
|---|---|---|---|---|
| Beamline | ALS 8.2.1 | SSRL 9-2 | SSRL 11-1 | |
| Space group | F432 | F432 | C2221 | |
| Cell dimensions | 224.88, 224.88, 224.88 | 225.30, 225.30, 225.30 | 64.22, 80.3, 56.09 | |
| α, β, γ (°) | 90.0, 90.0, 90.0 | 90.0, 90.0, 90.0 | 90.0, 90.0, 90.0 | |
| Resolution (Å) | 2.40 | 3.00 | 2.10 | |
| Wavelength (Å) | 1.0000 | 0.9116 | 0.9794 | 1.0722 |
| Completeness (%) | 99.4 (97.2) | 99.9 (99.9) | 99.9 (99.9) | 96.1 (91.1) |
| Observed reflections | 220,076 | 181,861 | 181,421 | 50,073 |
| Unique reflections | 19,524 | 10,309 | 10,291 | 8,452 |
| Rsym (%)a, b | 8.2 (84.9) | 11.5 (45.0) | 11.8 (45.0) | 2.5 (7.7) |
| I/σ(I) | 25.5 (2.3) | 5.7 (1.8) | 5.6 (1.8) | 54.9 (22.9) |
| Mosaicity (°) | 0.32 | 0.30 | 0.30 | 1.50 |
|
| ||||
|
Refinement Statistics
| ||||
| Resolution limits (Å) | 19.6–2.4 | 19.4–2.1 | ||
| Molecules/A.U. | 1 | 1 | ||
| Rcryst (%)c | 22.9 | 16.9 | ||
| Rfree (%)d | 25.6 | 23.8 | ||
| No. of reflections used | 18547 | 8029 | ||
| No. of atoms | ||||
| Protein | 2149 | 1060 | ||
| Ligands | 34 | |||
| Water | 132 | 108 | ||
| B-factor (Å2) | ||||
| Average | 58.6 | 21.6 | ||
| Protein | 60.2 | 19.8 | ||
| Ligands | 37.2 | |||
| Water | 33.1 | 19.7 | ||
| RMS deviations | ||||
| Bond lengths (Å) | 0.009 | 0.021 | ||
| Bond angles (°) | 1.13 | 1.82 | ||
| Ramachandran plot | ||||
| Most favoured | 78.9 | 87.0 | ||
| Allowed | 20.2 | 13.0 | ||
| Generously allowed | 0.4 | 0 | ||
| Disallowed | 0.4 | 0 | ||
Values in parentheses correspond to the highest resolution shell
Rsym is the unweighted R value on I between symmetry mates: Σhkl Σj |I(hkl)−<I(hkl)>|/Σhkl ΣI(hkl).
Rcryst = Σhkl ||Fobs(hkl)| − |Fcalc(hkl)||/Σhkl |Fobs(hkl)|.
Rfree is the cross validation R factor for 5% of reflections against which the model was not refined.
Figure 4.

X-ray crystal structure of TcpF. (A) Full-length TcpF is a bilobed structure comprised of an N-terminal domain (NTD, green) and a C-terminal domain (CTD, yellow) joined by a linker (residues 186–189, magenta). (B) Topology diagram of TcpF. (C) Crystal structure of TcpF-CTD. In the absence of the NTD, the linker segment enters the CTD at residue 198. (D) Space-filling representation of TcpF showing the negatively-charged cleft between the NTD and CTD. The protein is colored according to electrostatic potential, calculated using DELPHI 49, where red represents negative charge, blue is positive and white is uncharged (scale −7 kT to +7 kT). (E) TcpF critical binding residues Glu251 and Glu252 for mAb13 recognition. Glu251 and Glu252 (carbon atoms are colored bright green) lie at the lip of the cleft in the C-terminal domain.
The CTD is connected to the NTD by the linker, which is an extension of β7 of the second NTD β-sheet. The CTD consists of two twisted antiparallel β-sheets stacked upon each other in a β-sandwich, with a short α-helix at either end (α8 and α9). The topology of the β-sandwich follows that of the fibronectin type III (FnIII) domain 26, which has an immunoglobulin-like (Ig-like) fold. The “top” β-sheet, which lies closest to the NTD, has 4 anti-parallel strands (β11, β10, β13 and β14) and the bottom β-sheet has 3 anti-parallel strands (β8, β9 and β12). The linker segment connecting the NTD and CTD leads into the first β-strand (β8) of the bottom β-sheet. The curvature of the two β-sheets is such that they together resemble a tight β-barrel, made continuous by contacts between the last few residues of the long terminal strand of the top βsheet (β11) and the short terminal strand (β12′) of the bottom β-sheet. Bulky hydrophobic side chains form the interface between the β-sheets.
Short segments of the CTD main chain as well as some side chain atoms were poorly defined in the electron density map of full length TcpF. To obtain a higher quality electron density map for this region we determined a 2.1 Å resolution crystal structure of a recombinantly expressed CTD, including the linker (TcpF-CTD, residues 186–318) (Fig. 4C). The improved electron density for TcpF-CTD allowed us to fit all backbone and side chain atoms with the exception of distal side chain atoms for Trp227 and Lys318. The TcpF-CTD structure is essentially identical to that of the CTD in the full length TcpF structure for residues 198 onward, with a root mean square deviation of 0.78 Å between Cα carbons over this region. However, the position of the linker and N-terminal segment (residues 186–197) in TcpF-CTD differs substantially from that of the full length protein. Rather than entering this domain from the direction of the NTD and leading into the first β-strand of the bottom β-sheet (β8), this segment is instead extended and forms a β-strand with the corresponding β-sheet in a neighboring symmetry-related molecule within the protein crystal lattice (Supplemental Fig. S2). Two CTD-TcpF molecules, Mol A and Mol B, form a dimer that is aligned along the crystallographic two-fold symmetry axis, and the N-terminal segment of each monomer completes the β-strand for its corresponding monomer partner. This strand-swapping is similar to the arrangement seen in the Type II secretion pseudopilin PulG, in which the C-terminal segment of one subunit completes a β-sheet on the neighboring subunit in the crystallographic dimer 27. The protein arrangement in the TcpF-CTD crystal is clearly an artifact of the N-terminal truncation and crystal packing, and we see no evidence of full length TcpF multimerization but the results suggest that the linker segment is indeed flexible.
In the full length TcpF structure, the NTD and CTD come together to form a curved elongated structure, with the linker located on the convex surface of the protein (Fig. 4D). The interface between the NTD and CTD is minimal, formed mainly from side chain interactions between β7 and its preceding loop in the NTD and the β10–β11 loop of the top β-sheet of the CTD (Fig. 4A, C and D). In addition, Arg147, which lies on the β-hairpin loop (β3–β4) of the NTD, extends across to the CTD β13–β14 loop of the top β-sheet (Fig. 4A, C). These contacts between the NTD and CTD leave a pocket or cleft between the two domains capable of accommodating a spherical volume of roughly 2500 Å3 (Fig. 4A, D) (http://www.ebi.ac.uk/thornton-srv/databases/profunc/). Given the tenuous contacts and flexible linker connecting the two domains, this cleft may open to accommodate an even larger volume. The ceiling of the cleft is formed by α6 and the β-hairpin of the NTD and the floor is formed by the top β-sheet of the CTD. The interior surface of the cleft is lined with a mixture of charged, polar and hydrophobic residues and has an overall negative charge due to the presence of several acidic residues (Fig. 4D). The lower lip of the cleft is lined by negatively charged residues in the CTD, including Glu251.
Identification of structural homologs of TcpF
Since no amino acid sequence homologs have been identified for TcpF, a search of the Protein Structure Database was performed using the DALI server (http://ekhidna.biocenter.helsinki.fi/dali_server/) to identify structural homologs that might provide clues to TcpF functions. No structural homologs were identified for the full length protein, suggesting a unique fold for TcpF, hence the NTD and CTD were used for separate homology searches. Thirteen homologs were identified for the NTD alone (residues 26–185), all having z-scores of 3.1 or lower, indicating low to moderate structural matches. Of these, 12 matches corresponded to the C-terminal domains of the Major Tropism Determinant (Mtd) protein variants, which have a C-type lectin-like domain (CTLD). The CTLD fold is an α-β fold containing two α-helices alternating with two anti-parallel β-sheets 28, an arrangement that is present in the TcpF NTD. Unlike true lectin domains, CTLDs lack motifs for binding calcium and sugar. Superpositions of TcpF-NTD with the CTLDs of Mtd-P1 and domain D5 of the Yersinia pseudotuberculosis invasion protein InvA are shown and discussed in Supplemental Fig. S3 and S4. Despite the protein fold similarity between TcpF-NTD and CTLD-containing proteins, we were unable to infer a functional role for TcpF.
More than 1200 structural homologs were identified for the TcpF CTD using the DALI server, most of which contain Ig-like folds. Members of the Ig superfamily are involved in a diverse array of functions and Ig-like folds are found on many different proteins including antibodies, cell receptors, bacterial chaperones, fibronectin and enzymes 26. Specifically the TcpF CTD has a fibronectin type III family domain (FnIII) fold. The connectivity of the β-strands in the TcpF CTD and other FnIII domains differ from that of the Ig-fold in that the C′ (β11) strand of one β-sheet leads into the second β-sheet instead of continuing in the first β-sheet as it does in the Ig-fold 29 (Supplemental Fig. S5). Some FnIII domains also have an RGD motif that bind integrins 30 but no such sequence is present on TcpF. Thus, while the presence of the FnIII domain in the TcpF CTD is interesting and may suggest a role in protein:protein interactions, it does little to inform us on TcpF functions in V. cholerae colonization. Most likely, the NTD and CTD together form a unique interface that interacts with partner proteins to function in V. cholerae colonization.
Mapping Glu251/252 onto the TcpF crystal structure
Residues Glu251 and Glu252, which are critical for mAb13 recognition of TcpF, are located in the CTD on the lower lip of the cleft (Fig. 4D, E). Both residues lie on β11, the terminal strand of the CTD top β-sheet. The Glu251 side chain is exposed and points into the cleft, whereas the Glu252 side lies on the opposite side of β11, flush with the TcpF surface. Although both residues lie within the FnIII domain, their location is better suited to play a role in interactions involving the TcpF cleft than those involving the ends of the β-sheets, which are typically involved in FnIII domain interactions.
Glu251 and Glu252 are required for function of TcpF in vivo
Since our mAb13 protection data indicated that Glu251 and Glu252 may be important for TcpF function, we tested the ability of the tcpF:Glu251/252Ala V. cholerae mutant (strain CJM073) to colonize the infant mouse. A competitive index study was performed with CJM073 versus a V. cholerae ΔlacZ strain expressing wild type tcpF (strain KSK258). CJM073 was found to have a 4-5-log decrease in colonization efficiency relative to KSK258 (Fig. 5). This colonization defect is similar in magnitude to that of the ΔtcpF strain 17. This result indicates that Glu251 and Glu252 are crucial for TcpF functions in vivo.
Figure 5.

In vivocompetitive index of wild type, TcpF:Glu251/252Ala and ΔtcpF O395 V. cholerae strains competed against the ΔlacZ isogenic reference strain KSK258. The intestines were harvested 24 hours after inoculation and plated on LB containing streptomycin and X-gal. Lactose-positive and -negative colonies were counted. Boxes represent the average counts from each mouse (5 mice per group) and the x represents the combined average within each group. This data is representative of 3 separate experiments with similar results. Student’s T test was used to calculate a P-value of 3.3×10−4 for O395:ΔlacZ vs TcpF:Glu251/252Ala and 1.5×10−4 for O395:ΔlacZ vs ΔtcpF.
mAb13 in combination with anti-TcpA polyclonal antisera provides greater protection than either antibody alone
We were interested in combining low doses of the TcpF monoclonal antibodies with antibodies generated to TcpA as an indication that the function of TcpF is distinct from that of TCP. Anti-TcpA antibodies were previously shown to be protective in the infant mouse cholera model 21. We show here that when mAb13 is added to a sub-protective dose of TcpA antibody there is an increase in survival. Although the differences in overall survival are moderate (60%–80% protective) we consistently see a delay in V. cholerae induced morbidity with the groups co-inoculated with the two antibodies (Fig. 6), supporting the hypothesis that TcpF has a function in colonization that is distinct from the function of TcpA.
Figure 6.

mAb13 and anti-TcpA antibodies confer additional protection to either antibody alone. Kaplan Meier survival curves represent survival after V. cholerae was co-inoculated with mAb13 (1 mg/ml), purified anti-TcpA polyclonal (0.04 mg/ml), or both antibodies. Five mice were inoculated per group. Data are representative of three independent experiments.
Discussion
TcpF is a soluble secreted protein that is critical for V. cholerae colonization. The lack of in vitro functional assays for TcpF and the fact that it shares no apparent amino acid sequence homology to known proteins has made it a challenging protein to study. Here we used an integrated immunologic, genetic and structural approach to advance our understanding of the molecular mechanism by which TcpF mediates colonization by V. cholerae. The architecture of TcpF, with discrete N- and C-terminal domains joined by a linker, suggests that it may use the cleft between the domains to bind to a substrate. The apparent flexibility in the linker segment may allow TcpF to accommodate a fairly large substrate. The TcpF structure appears to be unique, as we uncovered no homologs in our DALI search. Although we identified common folds in the individual domains, a CTLD fold in the NTD and an FnIII fold for the CTD, we failed to discover possible functions for TcpF from the homology searches, consistent with the idea that the TcpF functional domain lies at the interface between the N-and C-terminal domains. This hypothesis is supported by our immunological studies showing that a protective anti-TcpF monoclonal antibody binds to the lower lip of the cleft and blocks TcpF functions, and that residues Glu251 and Glu252 within this region are critical to mAb13 binding and to TcpF function. Most likely, one or both of these residues mediate interactions with an unidentified binding partner to carry out TcpF’s role in V. cholerae colonization. Identifying this TcpF binding partner is a priority in ongoing studies.
Our previous results support an extracellular role for TcpF, possibly as a mediator of bacterial-bacterial or bacterial-epithelial cell interactions 19. Although TcpF appears to operate outside of the bacterial cell, our in vivo competitive index experiments show that secreted TcpF from a wild type strain cannot rescue the colonization defect in strains lacking TcpF (ΔtcpF) 17 or strains that are unable to secrete TcpF (Megli and Taylor, in preparation), indicating that TcpF must act within direct proximity of the cell from which it is secreted. Additionally the synergy in protection from V. cholerae challenge with both the TcpF and TcpA antibodies implies that TcpF has a function distinct from TcpA. Interestingly, the kinetics of clearance of a TcpF mutant are identical to that of a TcpA mutant 19, suggesting that these two proteins act simultaneously during colonization. TcpF may function as a unique mediator of attachment by interacting with both the bacterium and the intestinal epithelium. Studies are currently underway to further define the role of TcpF in colonization by V. cholerae.
We have shown that immunization with TcpF can generate a humoral response and that TcpF-specific antibodies can delay progression of disease in the mouse model of infection in passive immunization studies. These results and our demonstration of synergy between TcpA and TcpF antibodies indicate the potential of a multicomponent subunit vaccine formulation containing both TcpA and TcpF to generate a protective immune response against cholera infection. This principle has been used in the formulation and use of the acellular pertussis vaccine, which is effective and has less toxicity than the previously used killed whole cell vaccine. Antibodies generated against functional determinants of TcpF may directly inhibit TcpF activity or may prevent it from binding its target, thereby preventing V. cholerae colonization. Our ongoing search for TcpF-specific monoclonal antibodies may uncover additional functional sites on TcpF that could contribute to a multicomponent subunit vaccine for V. cholerae.
We have previously described proteins involved in attachment 18, microcolony formation 8; 11 and motility 31 and proposed a model in which all of these components (motility, attachment, microcolony formation and secretion of TcpF) comprise a series of steps leading to successful colonization of the mammalian host 19. The exact function of TcpF in colonization has yet to be determined, but this report provides new genetic and immunologic details and further illustrates the requirement of TcpF in colonization. We further present the unique atomic structure of TcpF and map a functional region on this structure, thus paving the way for structure-based mutagenesis to delineate TcpF functions. Finally, our data suggest that targeting of each discrete colonization step may prove to be a successful strategy in the design of a subunit vaccine to V. cholerae.
Materials and Methods
Bacterial strains and growth conditions
The strains and plasmids used in this study are listed in Supplemental Table S1. Briefly, bacteria were grown in Luria-Bertani (LB) broth pH 7.0 at 37°C for 16 hr or in LB broth pH 6.5 at 30°C for 16 hr for TCP inducing conditions.
Bacterial constructs
TcpF constructs and derivatives were generated using the primers listed in Supplemental Table S2. PCR products encoding TcpF peptides were digested then ligated into pTXB1 (NEB). The constructs were then transformed into the protease deficient E. coli cell line ER2566 (NEB). V. cholerae CJM073 (TcpF:Glu251/252Ala) was constructed by amplifying DNA 500 base pairs upstream and downstream of the mutations using primer pairs F290A and cmF.alaboth and F290D and cmF.epiD1. PCR products were digested by restriction enzymes then ligated into pKAS32 and electroporated into S17λpir 32. Allelic exchange was performed with CJM035 (O395 pMIN1) 33 as previously described 32. The expression vector for TcpF-CTD, pAY04, was constructed by cloning the tcpF gene fragment encoding residues 186–318 into the Nde1 and BamH1 sites of the pET15b vector, which encodes an N-terminal His6x-tag (Novagen). All constructs were verified by DNA sequencing.
Purification of TcpF for monoclonal antibody production and crystal structure determination
V. cholerae strain SJK7 was grown in 200 ml LB broth, pH 6.5, 100 μg/ml ampicillin at 30°C for 16 hours. TcpF expression was induced with 0.01% arabinose. The following day, cultures were centrifuged at 3,500 × g for 20 min and the cells pellet was resuspended in 20 ml phosphate-buffered saline (PBS) containing 10 mM EDTA, 10 mM EGTA and Complete Protease Inhibitor Cocktail tablets (Roche). Polymyxin B was added at 8.1 × 104 U/ml to lyse the outer membrane and the mixture was incubated on ice for 10 min. Cellular debris was removed by centrifugation at 10,000 × g for 15 min, and TcpF was precipitated from the periplasmic fraction using 50% ammonium sulfate (W/V). The solution was placed on a rocker at 4°C for 2 hrs then centrifuged at 10,000 × g for 30 min. The protein pellet was resuspended in 5 ml of buffer (50 mM HEPES pH 7.0, 150 mM NaCl, 10 mM EDTA, 10 mM EGTA) and applied to a Sepharose size exclusion column (GE Healthcare). TcpF-containing fractions were identified by sodium-dodecyl sulfate polyacrylamide gel electrophoresis (SDS -PAGE) and Coomassie Blue staining, then pooled and concentrated to 7.0 mg/ml using a Centriplus centrifugal filter device (Millipore) for mouse injections or to 15 mg/ml using a stirred cell concentrator (Amicon) for crystallization. TcpF used for immunization was filtered through a Pierce Detoxi Gel Column to remove lipopolysaccharide (LPS).
Selenomethionine substituted TcpF (SeMet-TcpF) for crystallization was expressed in M9 minimal media containing appropriate amino acids 34. Briefly, V. cholerae SJK7 cells from overnight culture were diluted 1/100 in 1 L minimal media and grown to an OD600 ~ 0.5–0.6 at 37°C, then amino acids (100 mg/L of lysine, threonine, phenylalanine, 50 mg/L of leucine, isoleucine, valine, 60 mg/L of L-selenomethionine) were added to each liter of M9 culture. After 15 minutes, arabinose was added to 0.01% and cells were grown for 12–16 hrs at 30°C. Cells were harvested and lysed and TcpF was purified as described above, with 1 mM dithiothreitol included in the buffers. SeMet-TcpF was concentrated to 15 mg/ml using the Amicon stirred cell concentrator (Millipore) and flash-frozen in liquid nitrogen and stored at −80°C. The monodispersity of the protein was confirmed using Dynamic Light Scattering (Wyatt Technologies).
For the purification of the CTD, E. coli BL21 cells (Novagen) carrying pAY04 were grown in LB at 37°C with 100 μg/ml ampicillin. Once cells reached an OD600 of ~0.4, they were induced with 1.0 M IPTG for 6 hours at 30°C. Cells were harvested by centrifugation and lysed by sonication. TcpF-CTD was purified by metal affinity chromatography using a nickel-nitrilo-triacetate column (GE Healthcare) and dialyzed with buffer (20 mM HEPES, 50 mM NaCl, 1 mM EDTA, 1 mM EGTA) with the addition of thrombin protease at 4°C for 2 days to remove the His-tag. His-tag-free TcpF-CTD was concentrated by dialysis in a 6,000 MWCO membrane and further purified by size exclusion chromatography using a Sephacryl S-100 column (GE Healthcare). Peak fractions were analyzed for protein concentration and purity using SDS-PAGE and fractions > 95% purity were concentrated to 14 mg/ml and frozen at −80°C.
Antibody production
Female BALB/c mice of 6 weeks of age (Harlan Laboratories) were immunized with 50 μg of antigen prepared in Titermax adjuvant (Sigma), given intraperitoneally in two sites. Splenocytes were harvested five days after the last boost. Hybridomas were prepared by fusing NS1 myeloma cells (ATCC) with splenocytes using 50% polyethylene glycol. Hybrids were selected by reactivity in ELISA using adsorbed antigen. Selected hybrids were subcloned by limiting dilution before use in experiments. Monoclonal antibody production was performed at the Dartmouth Monoclonal Core Facility. Monoclonal and polyclonal IgG were purified using a Montage antibody purification kit (Invitrogen) and resuspended in PBS.
Expression of TcpF peptides and immunoblot analysis
Gene fragments encoding TcpF peptides were inserted into plasmid pTXB1 and expressed in E. coli ER2566. E. coli strain ER2566 was grown at 37°C for 16 hr and V. cholerae was grown overnight in TCP-inducing conditions. TcpF peptide expression was induced with 1 mM IPTG. Cells were pelleted by centrifugation and culture supernatants were run through a 0.22 μm filter and mixed with SDS-PAGE lysis buffer. Protein content was quantified using BCA protein quantification kit (Pierce). Whole cells and cellular fractions were suspended in SDS-PAGE lysis buffer with 2-mercaptoethanol. Samples were boiled and 20 μg of protein or 20 μl of supernatant was loaded on a polyacrylamide gel and subjected to electrophoresis. Proteins were then transferred to a nitrocellulose membrane and blocked in 3% bovine serum albumin (BSA) in Tris-buffered saline (TBS) with 0.05% Tween (TBST) solution (blocking buffer). The membrane was washed in TBST and incubated with primary antibodies in blocking buffer. Membranes were washed again then incubated with the corresponding horse-radish peroxidase (HRP)-conjugated secondary antibody in blocking buffer. Antibody binding was visualized using Amersham ECL Western Blotting Detection Reagents (GE) followed by autoradiography.
Screening phage displayed peptide libraries
Phage display libraries, in which randomized 12-amino acid linear peptides (X12) or 7-amino acid constrained peptides (CX7C) displayed on the minor coat protein (pIII) of M13 phage (NEB), were screened as previously described 25. Fifty μl of protein A/G agarose beads (Pierce Biotechnology) were blocked for one hour with blocking buffer (0.1 M NaHCO3, 5 mg/ml BSA, pH 8.6) and washed with TBST. One ml of mAb13 hybridoma supernatant was incubated with blocked A/G agarose beads for 20 min at room temperature, followed by washing with TBST. Phage virions from the phage display library (2×1011 particles) were diluted with TBST to a final volume of 200 μl and incubated with antibodies captured on A/G agarose beads at room temperature for 20 minutes. The resin was washed with TBST to remove the unbound phage. Bound phage were eluted with 1 ml of glycine-HCl (pH 2.2) and immediately neutralized with 150 μl of 1 M Tris-HCl (pH 9.1). Eluted phage were titered to determine the number of phage captured by the antibody coated A/G agarose beads and then amplified by infecting E. coli strain ER2738. Amplified phage were purified by precipitation with PEG/NaCl (20% polyethelene glycol, 2.5 M NaCl). Three rounds of biopanning were carried out to enrich phage, as indicated by a large increase in phage titer from one round to the next. After the enrichment, phage plaques were selected and the gene III region was sequenced by automated dideoxyoligonucleotide sequencing as described below.
DNA sequencing of phage gene III peptide-encoding regions
To determine the nucleotide sequence encoding the pIII-displayed peptides, phage clones were propagated in 2 ml cultures of logarithmically growing E. coli ER2738 for 4 to 5 hours at 37°C. To recover the phage, the bacteria were removed from the culture suspension by a brief centrifugation (10,000 × g for 10 sec). A 1-ml aliquot of the culture supernatant was mixed with 400 μl of 20% polyethylene glycol and 2.5 M NaCl solution, and the mixture was centrifuged (10,000 × g) for 10 min. The phage pellet was resuspended with 100 μl of iodide buffer (10 mM Tris-HCl pH 8.0, 1 mM EDTA, 4 M NaI) and 250 μl of absolute ethanol and centrifuged for 10 min. The phage DNA was washed with 70% ethanol, dried, and resuspended in 30 μl of TE buffer (10 mM Tris-HCl, 1 mM EDTA). One hundred ng of the phage DNA was sequenced using the −96 gIII sequencing primer (NEB).
Phage Binding Assays
ELISA was used to assess the binding of phage peptides to mAb13. mAb13 was coated onto microtiter plates at 8 μg/ml. An ELISA was carried out as previously described 25. Briefly, microtiter plates were treated with blocking buffer then purified phage particles (1010) in 100 μl of TBST were added and incubated at room temperature for 1 hr. Plates were washed 6 times with 100 μl TBST then 100 μl of a 1:3300 dilution of HRP-conjugated anti-M13 antibody (Amersham Biosciences) was added and plates were incubated for 1 hr. Next, the substrate 3,3′,5,5′-tetramethylbenzidine (Sigma) was added and reactions were stopped with 100 μl of 3 M HCl once color appeared. Absorbance was measured at 450 nm with a kinetic microplate reader (Molecular Devices). The X12 library was also screened with monoclonal antibody S-20-4 to identify phage clone 4P-8 as previously described 25 to ensure that the screening conditions were optimal.
Infant mouse cholera model
V. cholerae strains were grown overnight in LB pH 7.0 at 30°C. For antibody protection experiments, overnight cultures were diluted 1:10 and then mixed 1:1 with purified antibody in PBS to give the following concentrations: 25 mg/ml for the TcpF polyclonal antibody; 1 mg/ml for the mAb13; and 1 mg/ml of mAb13 with 0.04 mg/ml TcpA polyclonal antibody. Fifty μl of the V. cholerae-antibody mixture was inoculated intragastrically into 5 day old CD1 mice and survival was recorded periodically after challenge.
For competitive index analysis the strains were grown overnight then mixed with the ΔlacZ reference strain KSK258 at a ratio of 1:1 and diluted 1:100. The competitions were carried out by inoculating 50 μl of the V. cholerae mixture intragastrically into 4–5 day old CD1 mice. At 24 hrs, the intestines were removed, suspended in 4 ml LB with 10% glycerol and homogenized in a tissue homogenizer. A dilution series was plated on LB-agar with streptomycin and 5-bromo-4-chloro-3-indolyl-β-D-galactoside (X-gal) and after 24 hr incubation, lactose-positive and -negative colonies were counted. The competitive index was reported as the ratio of the output and input of the experimental strain in comparison to the reference strain as previously described 19.
TcpF crystallization and x-ray diffraction data collection and processing
TcpF crystals were grown at 20°C using the hanging drop vapor-diffusion method. Full length native TcpF crystals were grown by mixing equal volumes (2 μl) of protein solution (15 mg/ml in 25 mM HEPES pH 7.0, 150 mM NaCl, 10 mM EDTA and 10 mM EGTA) and reservoir solution (1.0 M (NH4)2HPO4, 100 mM imidazole pH 8.0, 240 mM NaCl). Crystals formed after 8 months of growth. SeMet-TcpF crystals grew in similar conditions after 6–7 months. TcpF-CTD crystals were grown by mixing 2 μl of protein solution (15 mg/ml in 20 mM HEPES, 50 mM NaCl, 1 mM EDTA and 1 mM EGTA) and 2 μl of reservoir solution (1.6 M (NH4)2SO4, 100 mM MES pH 6.5, 10% v/v dioxane). Crystals appeared after 1 month. All crystals were frozen and stored in liquid nitrogen in mother liquor with 20% glycerol. A native full length TcpF data set was collected at the Advanced Light Source (ALS) Beamline 8.2.1. Raw data were processed and scaled using the XDS suite 35. For SeMet TcpF crystals, an x-ray fluorescence scan was performed and datasets were collected at the inflection point and high energy remote wavelengths at the Stanford Synchrotron Radiation Laboratory (SSRL) Beamline 9–2 using the Blu-Ice package 36; 37. Individual SeMet-TcpF data sets were processed with MOSFLM 38 and scaled with SCALA 39. Diffraction data for the TcpF-CTD crystals were collected at SSRL Beamline 11-1 and processed and scaled using XDS suite 35. The statistics for data collection for native TcpF, SeMet-TcpF and TcpF-CTD are provided in Table 2. The solvent contents for native TcpF and TcpF-CTD crystals were 63% and 46%, respectively, as determined from their respective Matthew’s coefficients 40.
TcpF structure determination and refinement
Experimental phases were obtained for SeMet-TcpF by multiple-wavelength anomalous-dispersion (MAD) method. Four selenium atoms were located and initial phases were calculated using SOLVE 41. Phases were improved by density modification procedures such as solvent flattening and histogram matching using RESOLVE 42. The first model was built in COOT 43 and the clear density for the majority of the N-terminal domain allowed the trace of the main chain and most of the side chains. However the density for the carboxy terminus was weak and the main chain of the core structure could not be built. A rigid-body refinement of the first model with the native data set followed by density modification and phase extension by DM 39 yielded a more interpretable map which facilitated building of more side chains and some parts of the C-terminal domain. By iterative cycles of restrained refinement using REFMAC544 and model building in COOT, we arrived at the final electron density map. The model was further improved by employing TLS refinement in REFMAC5. Water oxygens were located using ARP/wARP 45. TcpF-CTD data were processed and scaled using the XDS suite 35. The TcpF-CTD structure was solved by molecular replacement using C-terminal domain of the full length TcpF as model. PHASER 46 gave a good solution with high Z-scores (RFZ=7.6, TFZ=16.3). The electron density map after density modification by DM was sufficient to trace the backbone and model many side chains that were disordered in the full length structure. Restrained refinement with atomic isotropic B-factor brought the Rfree to 28.9%. The model was further improved by TLS refinement in REFMAC5. ARP/wARP 45 was used to locate water oxygens. Co-ordinates for dioxane, glycerol and sulfates were obtained through HIC-UP (http://xray.bmc.uu.se/hicup/). Both TcpF and TcpF-CTD models were validated using PROCHECK 47 and MolProbity 48. Refinement statistics are shown in Table 2 for both the native and the TcpF-CTD structures.
Supplementary Material
Acknowledgments
We thank the beamline staff at ALS and SSRL and Virginia Rath (Reciprocal Space Consulting) for their assistance in data collection. Funding was provided by National Institute of Health (NIH) grant AI 025096 to RKT and Canadian Institutes of Health Research (CIHR) grant RGPIN312152 to LC. LC is a recipient of a Michael Smith Foundation for Health Research Scholar Award and a CIHR New Investigator Award. CJM was funded by NIH T32 training grant AI007519. SJK, MRR and ASWY designed the purification protocol for TcpF. Phage library screening was done by MND. Protein purification and structure determination were performed by ASWY, SK and LC. All other molecular experiments and data compilation were completed by CJM with some assistance from MRR under the guidance of RKT and LC.
Abbreviations
- BSA
bovine serum albumin
- Cm
chloramphenicol
- CTD
C-terminal domain
- CTLD
C-type lectin-like domain
- EDTA
ethylenediamine tetraacetic acid
- EGTA
ethylene glycol tetraacetic acid
- ELISA
enzyme-linked immunosorbent assay
- FnIII
fibronectin type III
- Gm
gentamicin
- HRP
horse-radish peroxidase
- Ig
immunoglobulin
- LB
Luria-Bertani
- MAD
multiple-wavelength anomalous-dispersion
- NTD
N-terminal domain
- PBS
phosphate-buffered saline
- PEG
polyethylene glycol
- SDS-PAGE
sodium-dodecyl sulfate polyacrylamide gel electrophoresis
- SeMet
selenomethionine
- Sm
streptomycin
- TBS
Tris-buffered saline
- TBST
TBS with 0.05% Tween
- TCP
toxin coregulated pilus/pili
- X-gal
5-bromo-4-chloro-3-indolyl-β-D-galactoside
Footnotes
Accession numbers
The atomic coordinates for the full length TcpF and TcpF-CTD have been deposited in the Protein Data Bank under accession numbers 3OC5 and 3OC8, respectively.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mekalanos JJ, Rubin EJ, Waldor MK. Cholera: molecular basis for emergence and pathogenesis. FEMS Immunol Med Microbiol. 1997;18:241–248. doi: 10.1111/j.1574-695X.1997.tb01052.x. [DOI] [PubMed] [Google Scholar]
- 2.Broeck DV, Horvath D, De Wolf MJS. Vibrio cholerae: Cholera toxin. Int J Biochem Cell Biol. 2007;39:1771–1775. doi: 10.1016/j.biocel.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 3.Sanchez J, Holmgren J. Cholera toxin structure, gene regulation and pathophysiological and immunological aspects. Cell Mol Life Sci. 2008;65:1347–1360. doi: 10.1007/s00018-008-7496-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee SH, Hava DL, Waldor MK, Camilli A. Regulation and temporal expression patterns of Vibrio cholerae virulence genes during infection. Cell. 1999;99:625–634. doi: 10.1016/s0092-8674(00)81551-2. [DOI] [PubMed] [Google Scholar]
- 5.Brown RC, Taylor RK. Organization of tcp, acf, and toxT genes within a ToxT-dependent operon. Mol Microbiol. 1995;16:425–439. doi: 10.1111/j.1365-2958.1995.tb02408.x. [DOI] [PubMed] [Google Scholar]
- 6.Jude BA, Taylor RK. Genetics of Vibrio cholerae Colonization and Motility. In: Faruque SM, Nair GB, editors. Vibrio cholerae: Genomics and Molecular Biology. Horizon Scientific Press; 2008. pp. 67–99. [Google Scholar]
- 7.Manning PA. The tcp gene cluster of Vibrio cholerae. Gene. 1997;192:63–70. doi: 10.1016/s0378-1119(97)00036-x. [DOI] [PubMed] [Google Scholar]
- 8.Tripathi SA, Taylor RK. Membrane association and multimerization of TcpT, the cognate ATPase ortholog of the Vibrio cholerae toxin-coregulated-pilus biogenesis apparatus. J Bacteriol. 2007;189:4401–4409. doi: 10.1128/JB.00008-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rhine JA, Taylor RK. TcpA pilin sequences and colonization requirements for O1 and O139 Vibrio cholerae. Mol Microbiol. 1994;13:1013–1020. doi: 10.1111/j.1365-2958.1994.tb00492.x. [DOI] [PubMed] [Google Scholar]
- 10.Craig L, Taylor RK, Pique ME, Adair BD, Arvai AS, Singh M, Lloyd SJ, Shin DS, Getzoff ED, Yeager M, Forest KT, Tainer JA. Type IV pilin structure and assembly: X-ray and EM analyses of Vibrio cholerae toxin-coregulated pilus and Pseudomonas aeruginosa PAK pilin. Mol Cell. 2003;11:1139–1150. doi: 10.1016/s1097-2765(03)00170-9. [DOI] [PubMed] [Google Scholar]
- 11.Taylor RK, Miller VL, Furlong DB, Mekalanos JJ. Use of phoA gene fusions to identify a pilus colonization factor coordinately regulated with cholera toxin. Proc Natl Acad Sci USA. 1987;84:2833–2837. doi: 10.1073/pnas.84.9.2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kirn TJ, Lafferty MJ, Sandoe CM, Taylor RK. Delineation of pilin domains required for bacterial association into microcolonies and intestinal colonization by Vibrio cholerae. Mol Microbiol. 2000;35:896–910. doi: 10.1046/j.1365-2958.2000.01764.x. [DOI] [PubMed] [Google Scholar]
- 13.Lim MS, Ng D, Zong S, Arvai AS, Taylor RK, Tainer JA, Craig L. Vibrio cholerae El Tor TcpA crystal structure and mechanism for pilus-mediated microcolony formation. Mol Microbiol. 2010;77:755–770. doi: 10.1111/j.1365-2958.2010.07244.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Herrington DA, Hall RH, Losonsky G, Mekalanos JJ, Taylor RK, Levine MM. Toxin, toxin-coregulated pili, and the toxR regulon are essential for Vibrio cholerae pathogenesis in humans. J Exp Med. 1988;168:1487–1492. doi: 10.1084/jem.168.4.1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tacket CO, Taylor RK, Losonsky G, Lim Y, Nataro JP, Kaper JB, Levine MM. Investigation of the roles of toxin-coregulated pili and mannose-sensitive hemagglutinin pili in the pathogenesis of Vibrio cholerae O139 infection. Infect Immun. 1998;66:692–695. doi: 10.1128/iai.66.2.692-695.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sandkvist M. Biology of type II secretion. Mol Microbiol. 2001;40:271–283. doi: 10.1046/j.1365-2958.2001.02403.x. [DOI] [PubMed] [Google Scholar]
- 17.Kirn TJ, Bose N, Taylor RK. Secretion of a soluble colonization factor by the TCP type 4 pilus biogenesis pathway in Vibrio cholerae. Mol Microbiol. 2003;49:81–92. doi: 10.1046/j.1365-2958.2003.03546.x. [DOI] [PubMed] [Google Scholar]
- 18.Kirn TJ, Jude BA, Taylor RK. A colonization factor links Vibrio cholerae environmental survival and human infection. Nature. 2005;438:863–866. doi: 10.1038/nature04249. [DOI] [PubMed] [Google Scholar]
- 19.Kirn TJ, Taylor RK. TcpF is a soluble colonization factor and protective antigen secreted by El Tor and classical O1 and O139 Vibrio cholerae serogroups. Infect Immun. 2005;73:4461–4470. doi: 10.1128/IAI.73.8.4461-4470.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li J, Lim MS, Li S, Brock M, Pique ME, Woods VL, Jr, Craig L. Vibrio cholerae toxin-coregulated pilus structure analyzed by hydrogen/deuterium exchange mass spectrometry. Structure. 2008;16:137–148. doi: 10.1016/j.str.2007.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sun D, Seyer JM, Kovari I, Sumrada RA, Taylor RK. Localization of protective epitopes within the pilin subunit of the Vibrio cholerae toxin-coregulated pilus. Infect Immun. 1991;59:114–118. doi: 10.1128/iai.59.1.114-118.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Krebs SJ, Kirn TJ, Taylor RK. Genetic mapping of secretion and functional determinants of the Vibrio cholerae TcpF colonization factor. J Bacteriol. 2009;191:3665–3676. doi: 10.1128/JB.01724-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sharma SS, Chong S, Harcum SW. Intein-mediated protein purification of fusion proteins expressed under high-cell density conditions in E. coli. J Biotechnol. 2006;125:48–56. doi: 10.1016/j.jbiotec.2006.01.018. [DOI] [PubMed] [Google Scholar]
- 24.Sidhu SS, Koide S. Phage display for engineering and analyzing protein interaction interfaces. Curr Opin Struct Biol. 2007;17:481–487. doi: 10.1016/j.sbi.2007.08.007. [DOI] [PubMed] [Google Scholar]
- 25.Dharmasena MN, Jewell DA, Taylor RK. Development of peptide mimics of a protective epitope of Vibrio cholerae Ogawa O-antigen and investigation of the structural basis of peptide mimicry. J Biol Chem. 2007;282:33805–33816. doi: 10.1074/jbc.M707314200. [DOI] [PubMed] [Google Scholar]
- 26.Bork P, Holm L, Sander C. The immunoglobulin fold. Structural classification, sequence patterns and common core. J Mol Biol. 1994;242:309–320. doi: 10.1006/jmbi.1994.1582. [DOI] [PubMed] [Google Scholar]
- 27.Kohler R, Schafer K, Muller S, Vignon G, Deiderichs K, Philippsen A, Ringler P, Pugsley AP, Engel A, Welte W. Structure and assembly of the pseudopilin PulG. Mol Microbiol. 2004;54:647–664. doi: 10.1111/j.1365-2958.2004.04307.x. [DOI] [PubMed] [Google Scholar]
- 28.Weis WI, Taylor ME, Drickamer K. The C-type lectin superfamily in the immune system. Immunol Rev. 1998;163:19–34. doi: 10.1111/j.1600-065x.1998.tb01185.x. [DOI] [PubMed] [Google Scholar]
- 29.Leahy DJ, Hendrickson WA, Aukhil I, Erickson HP. Structure of a fibronectin type III domain from tenascin phased by MAD analysis of the selenomethionyl protein. Science. 1992;258:987–9891. doi: 10.1126/science.1279805. [DOI] [PubMed] [Google Scholar]
- 30.Leiss M, Beckmann K, Giros A, Costell M, Fassler R. The role of integrin binding sites in fibronectin matrix assembly in vivo. Curr Opin Cell Biol. 2008;20:502–507. doi: 10.1016/j.ceb.2008.06.001. [DOI] [PubMed] [Google Scholar]
- 31.Martinez RM, Dharmasena MN, Kirn TJ, Taylor RK. Characterization of two outer membrane proteins, FlgO and FlgP, that influence Vibrio cholerae motility. J Bacteriol. 2009;191:5669–5679. doi: 10.1128/JB.00632-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Skorupski K, Taylor RK. Positive selection vectors for allelic exchange. Gene. 1996;169:47–52. doi: 10.1016/0378-1119(95)00793-8. [DOI] [PubMed] [Google Scholar]
- 33.Nye MB, Pfau JD, Skorupski K, Taylor RK. Vibrio cholerae H-NS silences virulence gene expression at multiple steps in the ToxR regulatory cascade. J Bacteriol. 2000;182:4295–4303. doi: 10.1128/jb.182.15.4295-4303.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fan QR, Mosyak L, Winter CC, Wagtmann N, Long EO, Wiley DC. Structure of the inhibitory receptor for human natural killer cells resembles haematopoietic receptors. Nature. 1997;389:96–100. doi: 10.1038/38028. [DOI] [PubMed] [Google Scholar]
- 35.Kabsch H. Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J App Crystallogr. 1993;26:795–800. [Google Scholar]
- 36.Gonzales A, Moorhead P, McPhillips SE, Song J, Sharp K, Taylor JR, Adams PD, Sauter NK, Soltis SM. Web-Ice: integrated data collection and analysis for macromolecular crystallography. J App Crystallogr. 2008;41:176–184. [Google Scholar]
- 37.McPhillips TM, McPhillips SE, Chiu HJ, Cohen AE, Deacon AM, Ellis PJ, Garman E, González A, Sauter NK, Phizackerley RP, Soltis SM, Kuhn P. Blu-Ice and the Distributed Control System:Software for Data Acquisition and Instrument Control at MacromolecularCrystallography Beamlines. J Synchrotron Rad. 2002;9:401–406. doi: 10.1107/s0909049502015170. [DOI] [PubMed] [Google Scholar]
- 38.Leslie AGW. Recent changes to the MOSFLM package for processing film and image plate data Joint CCP4 + ESF-EAMCB Newsletter on Prot. Crystallogr. 1992:26. [Google Scholar]
- 39.Collaborative Computational Project, Number 4, 1994: The CCP4 Suite: Programs for Protein Crystallography. Acta Crystallogr. 1994;D50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 40.Matthews BW. Solvent content of protein crystals. J Mol Biol. 1968;33:491–7. doi: 10.1016/0022-2836(68)90205-2. [DOI] [PubMed] [Google Scholar]
- 41.Terwilliger TC, Berendzen J. Automated MAD and MIR structure solution. Acta Crystallogr. 1999;D55(Pt 4):849–861. doi: 10.1107/S0907444999000839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Terwilliger TC. Maximum-likelihood density modification. Acta Crystallogr D Biol Crystallogr. 2000;56:965–972. doi: 10.1107/S0907444900005072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 44.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 45.Perrakis A, Morris R, Lamzin VS. Automated protein model building combined with iterative structure refinement. Nat Struct Biol. 1999;6:458–463. doi: 10.1038/8263. [DOI] [PubMed] [Google Scholar]
- 46.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Laskowski RA, McArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- 48.Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB, 3rd, Snoeyink J, Richardson JS, Richardson DC. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nuc Acids Res. 2007;35:W375–383. doi: 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Oron A, Wolfson H, Gunasekaran K, Nussinov R. Using DelPhi to compute electrostatic potentials and assess their contribution to interactions. Curr Protoc Bioinformatics. 2003;Chapter 8(Unit 8):4. doi: 10.1002/0471250953.bi0804s02. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.