

Abstract

The use of heteroatom-substituted oxyallyl cations in (4 + 3) cycloadditions has had a tremendous impact on the development of cycloaddition chemistry. Extensive efforts have been exerted toward investigating the effect of oxygen-, sulfur-, and halogen-substituents on the reactivity of oxyallyl cations. Most recently, the use of nitrogen-stabilized oxyallyl cations has gained prominence in the area of (4 + 3) cycloadditions. The following article will provide an overview of this concept utilizing nitrogen-stabilized oxyallyl cations.
Keywords: oxyallyl, (4 + 3), cycloaddition, nitrogen atom, heterocyclic compounds, regioselectivity
Introduction
(4 + 3) cycloadditions employing oxyallyl cations 1 and dienes represent a powerful approach for constructing seven-membered carbocycles 2.1 While this process has been known for almost 50 years, the utility of heteroatom-stabilized oxyallyl cations [Figure 1] has become increasingly prominent in the realm of (4 + 3) cycloadditions because of the ability of the heteroatom to provide an electronically-biased oxyallyl cation that can lead to highly regioselective and stereoselective cycloadditions.2,3
Figure 1.

Types of Oxyallyl Cations in (4 + 3) Cycloadditions.
The use of oxygen-4, sulfur-5, and halogen-substituted6 oxyallyl cations in (4 + 3) cycloadditions has been extensively studied and applied in a number of elegant natural product syntheses.7 However, nitrogen-stabilized oxyallyl cations offer distinct advantages over other heteroatom systems. The trivalency of the nitrogen atom allows: (1) Tethering of a chirality-inducing unit [R*] to generate new stereocenters, (2) inclusion of a coordinating unit [W] to provide conformational rigidity, (3) greater flexibility in designing intramolecular reactions, (4) the ability to tune the electron-donating ability toward the oxyallyl cation, and (5) the amino group can serve as the nitrogen atom source for accessing N-heterocycles and complex natural alkaloids. These remarkable features represent attractive advantages for developing highly stereoselective (4 + 3) cycloadditions.
Recent developments making use of nitrogen-stabilized oxyallyl cations have emerged in the past decade sparking a renewed interest in the field of (4 + 3) chemistry. In this concept article, we will focus specifically on (4 + 3) cycloadditions involving nitrogen-stabilized oxyallyl cations and the application of such methodology.
Oxidopyridinium Ions in (4 + 3) Cycloadditions
The use of oxidopyridinium ions in dipolar cycloadditions has been well-established since the early 1970s.8 While commonly used in (5 + 2) cycloadditions9, Katritzky was one of the first to demonstrate that such intermediates could also undergo a (4 + 3) cycloaddition with dienes [Scheme 1].10 Katritzky found substituted oxidopyridinium ions, obtained through the thermal decomposition of its dimer 3, could be used in a (4 + 3) cycloaddition with 2,3-dimethylbutadiene to give cycloadduct 4 in 75% yield. Although oxidiopyridium ions are cyclic aromatic species, they can also be considered stabilized oxyallyl cations.
Scheme 1.

Oxidopyridinium Ions in (4 + 3) Cycloadditions.
At the same time, Mok and Nye reported similar results using the quinolinium species 5 [Scheme 1].11 A variety of dienes could be used, giving cycloadducts with high exo selectivity. However, when using unsymmetrical dienes, mixtures of regioisomers were obtained.
Over 20 years later, Cha and co-workers utilized Katritzky's method as an approach to the core of sarain A [Scheme 2].12 Slow addition of triethylamine to a mixture of 6 and cyclopentadiene provided the desired endo cycloadduct 7a in 58% yield along with a small amount of the exo product 7b and the (5 + 2) product 8. The endo product 7a was then further functionalized to give the core structure of sarain A.
Scheme 2.

Application Toward Sarain A.
Amino-Stabilized Oxyallyl Cations in (4 + 3) Cycloadditions
To the best of our knowledge, there has been only one report of amino-stabilized oxyallyl cations used in a (4 + 3) cycloaddition by Myers and Barbay.13 They showed that oxidation of 10 led to the unstable α-amino-α′-fluoro ketone 11, which decomposed to generate an amino-stabilized oxyallyl cation and underwent a (4 + 3) cycloaddition in the presence of cyclopentadiene affording cycloadduct 12 in 35% yield.
Diastereoselective (4 + 3)
In the same report, Myers and Barbay showed that use of a chiral imidazolidinone auxiliary could provide diastereoselectivity in the cycloaddition [Scheme 4]. Reaction of 13 with cyclopentadiene in hexafluoroisopropanol gave a mixture of cycloadducts favoring endo product 15 with a diastereoselectivity of 6.8:1. The observed selectivity of the major product was proposed to come from the chiral vinyliminium 14, which favors approach of the diene from the top face as the bottom face is blocked by the benzyl group.
Scheme 4.

Diastereoselective (4 + 3) Cycloaddition.
Amido-Stabilized Oxyallyl Cations in (4 + 3) Cycloadditions
By far, amides have been utilized most frequently in cycloadditions involving nitrogen-stabilized oxyallyl cations. The prevalent use of amides in this area is likely due to the stability and ease of preparing such precursors to the oxyallyl cation as well as the ease at which one can control the donating ability of the nitrogen atom toward the oxyallyl cation, thereby affecting its reactivity.
Intermolecular (4 + 3)
Walters was the first to report the use of amido-stabilized oxyallyl cations in a (4 + 3) cycloaddition.14 The reaction of phthalimide-substituted dibromide 16 with furan using Föhlisch's conditions15 afforded cycloadduct 18a in 60% yield via the amido-stabilized oxyallyl cation 17 [Scheme 5]. Some solvolysis product was also observed in the reaction. Interestingly, when using LiClO4, a mixture of two cycloadducts was obtained. Similar results were also found when using cyclopentadiene under varying reaction conditions. The authors proposed that an association of lithium with the oxyallyl species may be responsible for the results.
Scheme 5.

(4 + 3) Cycloaddition of Amido-Stabilized Oxyallyl Cations.
Furthermore, Walters examined the regioselectivity of the cycloaddition when using 2-methyl and 2-methoxyfuran [Table 1].16 In all cases, the anti regiochemistry (19a and 19b) was favored having the phthalimide and furan substituent on opposite sides of the cycloadduct. A small amount of the exo (extended)17 product was obtained in each case. However, using LiClO4 provided the highest overall selectivity for the anti, endo (compact)17 product 19a in reasonable yields, consistent with Föhlisch's results using LiClO4 with an oxygen-stabilized oxyallyl system.18
Table 1.
Regioselectivity in Walters’ (4 + 3) Cycloaddition.
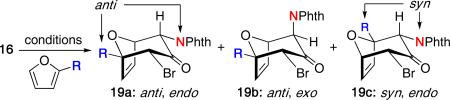 | ||||||
|---|---|---|---|---|---|---|
| entry | conditions | R | anti, endo | anti, exo | syn, endo | yield [%] |
| 1 | CF3CH2OH, Et3N | Me | 3.5 | 1 | 2.7 | 77 |
| 2 | CF3CH2OH, Et3N | OMe | 3 | 3 | 1 | 60 |
| 3 | LiClO4, MeCN, Et3N | Me | 6.5 | 1.4 | 1 | 73 |
| 4 | LiClO4, MeCN, Et3N | OMe | 17 | 1 | 0 | 57 |
Although it is not entirely clear if the nitrogen or bromine provides a stronger electronic bias on the oxyallyl cation leading to the stereochemical outcome, the results are not in stark contrast with recent studies carried out on similar systems (vide infra). Semi-empirical calculations and FMO analysis were unable to provide an adequate mechanistic explanation for these results, but the reaction was predicted to occur through a stepwise process.
We reported using oxazolidinone-substituted allenamides19 as precursors to nitrogen-stabilized oxyallyl cations in order to gain access to (4 + 3) cycloadducts.20 As shown in Scheme 6, epoxidation of allenamide 20 with dimethyldioxirane (DMDO) generates the allene oxide 21, which can open to give the nitrogen-stabilized oxyallyl cation 22. The resulting oxyallyl cation was reacted with cyclopentadiene or furan affording cycloadducts 23 and 24, respectively, in good yields.
Scheme 6.

(4 + 3) Cycloaddition of Allenamides.
Diastereoselective (4 + 3)
Using the same method with various chiral oxazolidinone auxiliaries, we were able to obtain high levels of diastereoselectivity in the intermolecular (4 + 3) cycloaddition [Scheme 7].20 Oxidation of chiral allenamide 25 in the presence of furan led to a mixture of exclusively endo isomers 26a and 26b in 80% yield with a ratio of 75:25 favoring the endo-I isomer. In most cases, the diastereselectivity could be increased to favor essentially one isomer when using ZnCl2.
Scheme 7.

Diastereoselective (4 + 3) Cycloaddition of Chiral Allenamides.
Based on these results, a stereochemical model was proposed in which chelation of the oxygen atoms to zinc would increase the conformational rigidity of the oxyallyl cation, thereby providing greater facial discrimination and enhanced diastereomeric induction. Thus, the resulting stereochemistry of 26a (confirmed by X-ray) would arise via a steric-driven approach of furan to the less hindered face of the oxyallyl cation.
Select examples using different auxiliaries without ZnCl2 are shown in Figure 2. The diastereoselectivities were found to vary greatly depending on the R group of the chiral auxiliary, but high diastereoselectivities were achieved even without the use of ZnCl2 as with 28 and 29. The stereochemistry of the cycloadducts was originally assigned based on the stereochemical assignment made for 26a. However, several of the cycloadducts have since been reassigned based on recent studies (vide infra).
Figure 2.

(4 + 3) Cycloadducts Derived from Chiral Allenamides.
This methodology was subsequently applied by Kozlowski and Hsung toward the synthesis of new chiral ligands used for the asymmetric alkylation of aldehydes.21 As shown in Scheme 8, the chiral ligand 33 was prepared from cycloadducts 31 through a 4 step sequence involving hydrogenation, reduction of the ketone, cleavage of the auxiliary to give the amino alcohol 32 and finally alkylation of the primary amine. Reaction of benzaldehyde with ZnEt2 and 5 mol% of the chiral ligands provided the desired alcohol 34 in excellent yields and enantiomeric excess. Predictive 3D-QSSR calculations were also performed which correlated closely with the experimental results.
Scheme 8.

Synthesis of Chiral Ligands from (4 + 3) Cycloadducts.
We also explored the use of chiral enamides as an alternative approach toward chiral nitrogen-stabilized oxyallyl cations [Scheme 9].22 Epoxidation of chiral enamide 35 followed by acidic methanolysis led to a mixture of aminals 36a and 36b. Oxidation and subsequent enol ether formation of the major isomer led to the cycloaddition precursor 37 in 64% yield. Upon treatment with catalytic TMSOTf and 10 equivalents of furan, 37 underwent the (4 + 3) cycloaddition affording 38 in a modest 30% yield but with good diastereoselectivity favoring the endo-I isomer. An equal amount of product with only single bond formation to furan was also obtained. No other examples using this methodology have since been reported.
Scheme 9.

Diastereoselective (4 + 3) Cycloaddition Derived from Enamides.
MaGee and Walters also reported a diastereoselective (4 + 3) cycloaddition using their methodology with chiral oxazolidinone auxiliaries.23 As shown in Table 2, reaction of chiral bromoketone 39 with furan or cyclopentadiene under the standard conditions gave cycloadducts 40a and 40b in approximately a 2:1 ratio favoring the endo-I product.
Table 2.
Diastereoselective (4 + 3) Cycloaddition of α-Bromoketones.
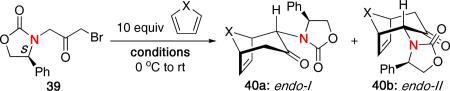 | |||||
|---|---|---|---|---|---|
| entry | conditions | X | endo / exo | endo 1:11 | yield [%] |
| 1 | CF3CH2OH, Et3N | 0 | 93:7 | 66:34 | 70 |
| 2 | CF3CH2OH, Et3N | CH2 | 93:7 | 66:34 | 65 |
| 3 | MgBr2, MeCN, Et3N | 0 | 98:2 | 98:2 | 40 |
| 4 | MgBr2, MeCN, Et3N | CH2 | 86:14 | 86:14 | 45 |
When using MgBr2, they observed a remarkable improvement in diastereoselectivity, obtaining almost exclusively one endo product with furan, although the yield was somewhat diminished. Similar results were also found when using cyclopentadiene under the Lewis acid conditions.
The resulting stereochemistry of the major product was proposed to arise from oxyallyl cation 41, in which the solvent or a Lewis acid can chelate [Figure 3]. An endo approach of furan away from the phenyl substituent on the oxazolidinone would then lead to the observed product. It is worth noting that these findings agreed closely with the results reported by us.19
Figure 3.

Proposed Model for Diastereoselective (4 + 3) Cycloaddition.
Interestingly, when employing a bulkier oxazolidinone derived from camphanic acid, Walters and MaGee observed a complete reversal in stereochemistry of the cycloadduct [Scheme 10]. Reaction of 42 under the reaction conditions led to a 90% yield of the endo-II cycloadduct 43, consistent with a non-chelated transition state. Attempts to reverse the stereochemical outcome through a chelation pathway using Lewis acids were unsuccessful.
Scheme 10.

Reversal in Stereochemistry for (4 + 3).
A similarly unexpected reversal in stereochemistry was also reported by us in the (4 + 3) cycloaddition of allenamides with substituted furans [Scheme 11].24 Oxidation of allenamide 25 in the presence of 2-methylfuran and ZnCl2 led to the predicted cycloadduct 44, consistent with the endo-I pathway previously observed. However, when the reaction was run in the presence of 2-methyl furoate, a complete reversal in selectivity was observed in favor of the endo-II product 45. The use of ZnCl2 had little effect on the diastereoselectivity with 2-methyl furoate.
Scheme 11.

Reversal in Stereochemistry with Substituted Furans.
These intriguing results prompted Houk and Hsung to examine the (4 + 3) cycloaddition of several chiral nitrogen-stabilized oxyallyl cations in hopes of providing an improved rationale for the observed outcomes.25 In the originally proposed model (Scheme 7), it was thought that the oxyallyl cation possessed a Z configuration with the two oxygen atoms on the same side of the Cα-N double bond. Thus, the stereoselectivity was believed to be controlled by steric repulsion between furan and the phenyl group, leading to the major product observed experimentally. Moreover, the Z-oxyallyl cation had a suitable geometry for forming a chelate, thus explaining the increased stereoselectivity in the presence of ZnCl2.
However, DFT calculations revealed that the E-oxyallyl cation was actually the preferred configuration [Figure 4]. Transition state structures of the E-oxyallyl cation with furan were also calculated to be lower in energy than the Z-oxyallyl cation. Furthermore, while the presence of ZnCl2 was found to lower the activation barrier for the ensuing cycloaddition, there was no change in the preferred configuration of the oxyallyl species and thus, no chelation would be involved.
Figure 4.

Preferred Geometries of Nitrogen-Stabilized Oxyallyl Cations.
Perhaps most surprising was the preference found for the reaction of furan from the more hindered face of the oxyallyl cation when calculated for the phenyl-substituted oxazolidinone [46, Figure 5]. This was attributed to a stabilizing CH−π interaction between the C3 hydrogen on furan and the phenyl substituent. Thus, the stereoselectivity leading to 26a was controlled not by steric repulsion, but by “steric attraction”. This is also in agreement with work done by Harmata and Houk involving alkoxy-substituted siloxyallyl cations.26
Figure 5.

Revised Models for Oxyallyl-Furan (4 + 3) Cycloadditions.
Further investigation into some of the other previously reported cycloadducts also led to a revision of their initially assigned stereochemistry. As the benzyl group in 47 is unable to participate in a CH−π interaction, a steric-driven approach would be expected to control the stereochemistry, thereby leading to the endo-II cycloadduct 27. This was confirmed through X-ray analysis of the major isomer.
Furthermore, in the case of the diphenyl isopropyl oxazolidinone, the stereochemistry of the major product 29 (confirmed by X-ray) can be attributed to both steric and CH−π interactions. The favored transition state 48 benefits from minimization of repulsive interactions with the isopropyl group as well as a stabilizing CH−π interaction with one of the C5-phenyl substituents leading to excellent selectivity for one isomer.
It is worth noting that with the isopropyl-substituted oxazolidinone, the diastereoselectivity was only 45:55 (vide supra), thereby accentuating the impact that non-bonding CH−π interactions can have on the stereochemical outcome of such cycloadditions. This rationale using only an E-oxyallyl cation with steric versus CH−π interactions would also explain the reversal in selectivity observed by Walters and MaGee in their system.22
We also reported the diastereoselective (4 + 3) cycloaddition with pyrroles as an approach to tropinone alkaloids.27 As shown in Scheme 12, reaction of allenamide 25 with the Boc-protected pyrrole under DMDO oxidation led to the desired cycloadduct 49 in 76% and a ratio of 82:18 favoring the endo-I product. Slow addition of DMDO via a syringe pump was required for the chemoselective oxidation of the allenamide over the pyrrole. Similar diastereoselectivities were obtained with pyrrole using various chiral oxazolidinones as was reported with furan.20 Cycloadduct 50 was derivatized from 51 in order to confirm the stereochemistry by X-ray analysis.
Scheme 12.

(4 + 3) Cycloaddition with Pyrroles
In light of the new mechanistic insights involving CH−π interactions,25 some of the structures of the tropinone adducts have been revised. Presumably, the stereochemistry of 49 and 51 resulted from CH−π interactions, 52 and 54 resulted from steric repulsions, and the excellent selectivity obtained for 53 and 55 would be due to an integration of both interactions as observed with furan.
The cycloaddition with pyrroles was also applied in an approach toward the core of parvineostemonine [Scheme 13].27 Starting from 51, hydrogenation and Boc deprotection gave amine 56 in 73% yield. N-allylation and subsequent allylation at the α-substituted position afforded diene 58 which was finally subjected to ring-closing metathesis to give the core of parvineostemonine 59 in 36% yield.
Scheme 13.

An Approach Toward Parvineostemonine.
Intramolecular (4 + 3)
Nitrogen-Tethered
Intramolecular (4 + 3) cycloadditions allow for an increased level of complexity in the resulting product with the ability to make polycyclic ring systems in a single step.1,2 We were the first to demonstrate the use of nitrogen-stabilized oxyallyl cations in an intramolecular (4 + 3) cycloaddition.28 The initial report focused on the use of N-tethered allenamides in which the diene is tethered through the nitrogen atom. As shown in Scheme 14, DMDO oxidation of allenamides 60 and 61 afforded the desired cycloadducts 62 and 63, respectively, as single diastereomers. Slow addition of DMDO via syringe pump was critical to prevent competing oxidation of furan. A small amount of the epoxidized cycloadduct 64 was also obtained from the reaction of 60.
Scheme 14.

N-Tethered Intramolecular (4 + 3) Cycloaddition.
A mechanistic model was presented based on the preferred conformations of substituted oxyallyl cations [Figure 6]. While two possible approaches, 65-endo and 65-exo, would both lead to the observed stereochemistry, it was proposed that 65-exo should dominate because it possesses a preferred W-conformation, whereas 65-endo would experience more A1,3 strain.1,2
Figure 6.

Model for N-Tethered Intramolecular (4 + 3) Cycloaddition.
Longer tethered allenamides were also tolerated under the reaction conditions, although diminished stereoselectivity was observed. However, when cyclic-tethered allenamides 66 were employed, excellent diastereoselectivities were achieved regardless of tether length [Scheme 15]. The observed stereochemistry was rationalized based on a similar 67-exo approach. The proposed model also agrees with the recent studies which suggest a preferred E-oxyallyl cation (vide supra).25
Scheme 15.

(4 + 3) Cycloaddition of Cyclic-Tethered Allenamides.
Most recently, we examined a new class of N-sulfonyl substituted allenamides and their improved reactivity in the intramolecular (4 + 3) cycloaddition.29 As shown in Scheme 16, allenamide 69 was prepared in six steps starting from furfural. Reaction with DMDO at -78 °C led to the desired cycloadduct 70 with four stereocenters in 94% yield and a diastereomeric ratio of ≥95:5 for the major isomer shown. The use of N-sulfonyl allenamides allowed a more practical and expedient approach for obtaining diastereoselectivity in the cycloaddition.
Scheme 16.

(4 + 3) Cycloaddition of N-Sulfonyl Allenamides.
Carbon-Tethered
The (4 + 3) cycloaddition has also been reported with dienes tethered onto the allene itself and could serve as a potential route to the aromadendrane alkaloids [Scheme 17].30 We demonstrated that DMDO oxidation of α-tethered allenamide 71 afforded the cycloadduct 73, containing a quaternary stereocenter, in 75% yield and with excellent disastereoselectivity. An E-oxyallyl cation 72 and endo approach of furan was proposed to lead to the major isomer, although it is unclear as to whether or not CH−π interactions play a role.
Scheme 17.

α-Tethered Intramolecular (4 + 3) Cycloaddition
In the same report, we also explored the use of γ-tethered allenamides in the (4 + 3) cycloaddition [Scheme 18]. Treatment of γ-tethered allenamide 71 with DMDO led to cycloadduct 72a as the major isomer when n was equal to 1 or 2 carbons. While the stereochemistry of the protected alcohol seemed to have an effect on the resulting stereochemistry of the products, the axial chirality of the allene did not play a role since its stereochemistry is scrambled upon formation of the oxyallyl cation.
Scheme 18.

γ-Tethered Intramolecular (4 + 3) Cycloadditions.
Interestingly, when the tether length was increased to n = 3, the stereochemical outcome was reversed in favor of 72b. Other examples using butadiene also displayed a reversal in stereochemistry. These intriguing results suggested an overall change in the preferred conformation of the oxyallyl cation depending on the tether length or the diene used. While models were proposed to rationalize the stereochemical outcome, further work will need to be done to provide a better mechanistic understanding. Computational studies have not yet been reported on these intramolecular systems.
Catalytic Asymmetric (4 + 3) Cycloadditions
Surprisingly, there have only been two reports of catalytic asymmetric (4 + 3) cycloadditions31 using nitrogen-stabilized oxyallyl cations, but the potential for future development in this area is promising and merits further exploration.
Harmata was the first to report the catalytic asymmetric (4 + 3) cycloaddition utilizing a chiral organocatalyst with pentadienals.32 As shown in Scheme 19, reaction of pentadienal 73 with 2,5-disubstituted furans in the presence of 20 mol% chiral amine catalyst and trifluoroacetic acid afforded the cycloadducts 75 with good yields and up to 89% enantiomeric excess. The reaction presumably proceeds through the vinylogous nitrogen-stabilized oxyallyl cation 74 in which the top face is blocked by the benzyl substituent of the catalyst thereby leading to an endo approach of furan from the bottom face.
Scheme 19.

Organocatalytic Asymmetric (4 + 3) Cycloaddition.
The only other catalytic asymmetric (4 + 3) cycloaddition was reported by Hsung using chiral Lewis acid catalysts [Scheme 20].33 Reacting achiral allenamide 20 with DMDO and various dienes in the presence of catalytic CuOTf and the chiral bisoxazoline ligand shown afforded the desired cycloadducts 76a/b in good yields and up to 99% enantiomeric excess. The use of molecular sieves and AgSbF6 was found to increase the overall yield and selectivities.
Scheme 20.

Catalytic Asymmetric (4 + 3) Cycloaddtion of Allenamides
A working model was proposed based on previous studies for asymmetric catalysis using C2-symmetric ligands.34 Chelation of the oxyallyl cation to the copper catalyst would create four quadrants. Out of the two productive front quadrants, the top endo approach would be favored as the bottom approach would have unfavorable steric interactions with the phenyl substituent. While the model correctly predicts the observed enantioselectivity, its validity is unclear in light of our recent computational work suggesting the preference for the E-oxyallyl cation. Further mechanistic studies would prove useful in this regard.
The reaction also displayed interesting regioselectivity with unsymmetrical furans. Depending on the position of the substituents on furan, regioselectivities were found to favor either the syn or the anti products 76a and 76b, respectively. Regioselectivities were high in most cases, although certain furans were found to give diminished enantioselectivities. While an ade quate explanation for the regioselectivity was unclear at the time, a mechanistic study on the observed regiochemistry has since been reported (vide infra).35
First Regiochemical Models In Oxyallyl Cation (4 + 3) Cycloadditions
The use of nitrogen-stabilized oxyallyl cations with unsymmetrical dienes in (4 + 3) cycloadditions has been reported to provide high levels of regioselectivity in many cases.16,23,24 However, there have been few systematic studies concerning the regioselectivity of oxyallyls and the influence of substituents on the diene.3e,23 Furthermore, there had not been any coherent let alone predictive model reported for the regioselectivities in (4 + 3) cycloadditions.
Houk and Hsung reported the first systematic study on the regioselectivity between furans and nitrogen-stabilized oxyallyl cations using the oxazolidinone-substituted system.35 As shown in Scheme 21, reaction of allenamide 20 with 3-methylfuran or ethyl 3-furoate gave predominantly the anti product 77 having the oxazolidinone and furan substituent on opposite sides of the cycloadduct. However, when using 2-methylfuran or methyl 2-furoate, the regiochemistry reversed giving predominantly the syn cycloadduct 78. Inclusion of a Lewis acid generally increased the yield, but did not alter the regioselectivity.
Scheme 21.

Regioselective (4 + 3) Cycloaddition.
The reaction was calculated to be a concerted process, although the transition states showed slightly more bonding at the ω enolate terminus. Furthermore, only the E-oxyallyl cation was involved in the reaction, consistent with our previous computational work.25 DFT calculations on the regioselectivities predicted the correct major product for each furan and a predictive set of regiochemical models were proposed as shown in Figure 7. With electron-withdrawing ester groups, the regiochemical preference was consistent with electronic effects as a conjugate addition type of approach. In contrast, steric effects likely governed the regioselectivities for methylfurans in which the oxyallyl attacks the less substituted position with 2-methylfuran or avoids steric hindrance with the oxazolidinone for 3-methylfuran.
Figure 7.

Summary of Regioselectivity with Unsymmetrical Furans.
Other donor-substituted oxyallyl cations have often been calculated to undergo (4 + 3) cycloadditions in a stepwise process, with the oxyallyl cation exhibiting electrophilic behaviour [Figure 8].4a,16,26,36. In contrast, nitrogen-substituted oxyallyl cations are a distinct class of oxyallyl displaying ambiphilic properties and thus, are truly stabilized oxyallyl cations. These features provide oxazolidinone-substituted oxyallyls with well-defined and unique regiochemical properties, allowing a coherent and predictive set of models to be established. These models can also serve as useful guidelines for future studies in other (4 + 3) cycloadditions.
Figure 8.

Heteroatom- vs. Nitrogen-Stabilized Oxyallyl Cations.
Summary and Outlook
The chemistry of heteroatom-stabilized oxyallyl cations remains a fertile ground for continued exploration. Within this realm, nitrogen-stabilized oxyallyl cations represent of unique class of allylic cations that have allowed for the development of highly stereoselective (4 + 3) cycloadditions.
The use of nitrogen-stabilized oxyallyl cations in (4 + 3) cycloadditions offers distinct adva ntages for the construction of nitrogen heterocycles and complex natural alkaloids. From the development of new reaction methodology to its application in asymmetric catalysis and natural product synthesis, the utility of (4 + 3) cycloadditions with nitrogen-stabilized oxyallyl cations has grown tremendously. It is our hope that this concept article will stimulate further growth and development in this field.
Scheme 3.

Generation of an Amino-Stabilized Oxyallyl Cation .
Acknowledgements
Financial support from NIH [GM066055] is greatly appreciated.
References
- 1.For reviews, see: Harmata M. Chem. Commun. 2010;46:8886–8903. doi: 10.1039/c0cc03620j.Battiste MA, Pelphrey PM, Wright DL. Chem. Eur. J. 2006;12:3438–3447. doi: 10.1002/chem.200501083.Antoline JE, Hsung RP. ChemTracts. 2005;18:207–214.Hartung IV, Hoffmann HMR. Angew. Chem. Int. Ed. 2004;43:1934–1949. doi: 10.1002/anie.200300622.Harmata M, Rashatasakhon P. Tetrahedron. 2003;59:2371–2395.Harmata M. Acc. Chem. Res. 2001;34:595–605. doi: 10.1021/ar000064e. Also see; Davies HML. In: Advances in Cycloaddition. Harmata M, editor. Vol. 5. JAI Press; 1998. pp. 119–164.West FG. In: Advances in Cycloaddition. Lautens M, editor. Vol. 4. JAI; Greenwich: 1997. pp. 1–40.Rigby JH, Pigge FC. Org. React. 1997;51:351–478.Harmata M. Tetrahedron. 1997;53:6235–6280.Fort AW. J. Am. Chem. Soc. 1962;82:4979–4981. l The designation (m + n) is used here in accordance with Woodward–Hoffmann/IUPAC conventions for describing cycloadditions based on the number of atoms, as opposed to the bracketed [m + n] designation which indicates the number of electrons involved.
- 2.For reviews on heteroatom-substituted oxyallyl cations, see: Harmata M. Chem. Commun. 2010;46:8904–8922. doi: 10.1039/c0cc03621h.Harmata M. Adv. Synth. Catal. 2006;348:2297–2306.Harmata M. Recent Res. Devel. In Organic Chem. 1997;1:523–535.
- 3.For diastereoselective (4 + 3) cycloadditions, see: Davies HML, Dai X. J. Am. Chem. Soc. 2004;126:2692–2693. doi: 10.1021/ja039908q.Prié G, Prévost N, Twin H, Fernandes SA, Hayes JF, Shipman, M M. Angew. Chem. Int. Ed. 2004;43:6517–6519. doi: 10.1002/anie.200461084.Grainger RS, Owoare RB, Tisselli P, Steed JW. J. Org. Chem. 2003;68:7899–7902. doi: 10.1021/jo034356f.Montanã AM, Grima PM. Tetrahedron. 2002;58:4769–4786.Beck H, Stark CBW, Hoffman HMR. Org. Lett. 2000;2:883–886. doi: 10.1021/ol991386p., and reference 11 cited within. Harmata M, Rashatasakhon P. Synlett. 2000:1419–1422.Cho SY, Lee JC, Cha JK. J. Org. Chem. 1999;64:3394–3395. doi: 10.1021/jo990262n.Harmata M, Jones DE, Kahraman M, Sharma U, Barnes CL. Tetrahedron Lett. 1999;40:1831–1834.Kende AS, Huang H. Tetrahedron Lett. 1997;38:3353–3356.Harmata M, Jones DE. J. Org. Chem. 1997;62:1578–1579.
- 4.For leading examples of oxygen-substituted oxyallyl cations, see: Sáez JA, Arnó M, Domingo LR. Tetrahedron. 2005;61:7538–7545.Harmata M, Kahraman M, Adenu G, Barnes CL. Heterocycles. 2004;62:583–618.Sáez JA, Arnó M, Domingo LR. Org. Lett. 2003;5:4117–4120. doi: 10.1021/ol035652h.Funk RL, Aungst RA. Org. Lett. 2001;3:3553–3555. doi: 10.1021/ol016668f.Harmata M, Sharma U. Org. Lett. 2000;2:2703–2705. doi: 10.1021/ol006281x.Masuya K, Domon K, Tanino K, Kuwajima I. J. Am. Chem. Soc. 1998;120:1724–1731.Harmata M, Elomari S, Barnes CJ. J. Am. Chem. Soc. 1996;118:2860–2871., and references cited within. For a leading example on pseudo-oxygen-stabilized oxyallyl cations, see: Chung WK, Lam SK, Lo B, Liu LL, Wong W-T, Chiu P. J. Am. Chem. Soc. 2009;131:4556–4557. doi: 10.1021/ja807566t.
- 5.For examples of sulfur-substituted oxyallyl cations, see: Hardinger SA, Bayne C, Kantorowski E, McClellan LL, Nuesse M-A. J. Org. Chem. 1995;60:1104–1105.Harmata M, Gamlath CB. J. Org. Chem. 1988;53:6154–6156.
- 6.For examples of halogen-substituted oxyallyl cations, see: Harmata M, Wacharasindhu S. Org. Lett. 2005;7:2563–2565. doi: 10.1021/ol050598l.Lee K, Cha JK. Org. Lett. 1999;1:523–526. doi: 10.1021/ol990709e.
- 7.Lee JC, Jin S-J, Cha JK. J. Org. Chem. 1998;63:2804–2805.Lee JC, Cha JK. Tetrahedron. 2000;56:10175–10184.Cha JK, Lee JC. J. Am. Chem. Soc. 2001;123:3243–3246. doi: 10.1021/ja0101072. and reference 6.
- 8.Katritzky AR, Takeuchi Y. J. Am. Chem. Soc. 1970;92:4134–4136. For a review on oxidopyridium ions, see: Katritzky AR, Dennis N. Chem. Rev. 1989;89:827–861.
- 9.Peese KM, Gin DY. J. Am. Chem. Soc. 2006;128:8734–8735. doi: 10.1021/ja0625430.Peese KM, Gin DY. Org. Lett. 2005;7:3323–3325. doi: 10.1021/ol051184v., and references cited within.
- 10.a Dennis N, Ibrahim B, Katritzky AR. J. Chem. Soc., Chem. Commun. 1974:500–501. [Google Scholar]; b Dennis N, Ibrahim B, Katritzky AR. J. Chem. Soc., Perkin Trans. 1. 1976;21:2296–2307. [Google Scholar]
- 11.Mok K-L, Nye MJ. J. Chem. Soc., Chem. Commun. 1974:608–610. [Google Scholar]
- 12.Sung MJ, Lee HI, Chong Y, Cha JK. Org. Lett. 1999;1:2017–2019. doi: 10.1021/ol9911932.; Lee I, Sung MJ, Lee HB, Cha JK. Heterocycles. 2004;62:407–422. For isolation of sarain A, see: Cimino G, Mattia CA, Mazzarella L, Puliti R, Scognamiglio G, Spinella A, Trivellone E. Tetrahedron. 1989;45:3863–3872.
- 13.Myers AG, Barbay JK. Org. Lett. 2001;3:425–428. doi: 10.1021/ol006931x. [DOI] [PubMed] [Google Scholar]
- 14.Walters MA, Arcand HR, Lawrie DJ. Tetrahedron Lett. 1995;36:23–26. [Google Scholar]
- 15.a Föhlisch B, Gottstein W, Herter R, Wanner I. J. Chem. Res., Synop. 1981:246–247. [Google Scholar]; b Föhlisch B, Wolf E. J. Chem. Res., Synop. 1983:166. [Google Scholar]
- 16.Walters MA, Arcand HR. J. Org. Chem. 1996;61:1478–1486. [Google Scholar]
- 17.For a review on Hoffmann's notations, see: Hoffmann HMR. Angew. Chem. 1973;85:877–894.; Angew. Chem., Int. Ed. Engl. 1973;12:819–835.; see also: Hoffmann HMR, Joy DR. J. Chem. Soc. B. 1968:1182–1186.
- 18.Föhlisch B, Krimmer D, Gehrlach E, Kaeshammer D. Chem. Ber. 1988;121:1585–1594. [Google Scholar]
- 19.For reviews on the chemistry and synthesis of allenamides, see: Hsung RP, Wei L-L, Xiong H. Acc. Chem. Res. 2003;36:773–782. doi: 10.1021/ar030029i.
- 20.Xiong H, Hsung RP, Berry CR, Rameshkumar C. J. Am. Chem. Soc. 2001;123:7174–7175. doi: 10.1021/ja0108638. [DOI] [PubMed] [Google Scholar]
- 21.Huang J, Ianni JC, Antoline JE, Hsung RP, Kozlowski MC. Org. Lett. 2006;8:1565–1568. doi: 10.1021/ol0600640. [DOI] [PubMed] [Google Scholar]
- 22.Xiong H, Hsung RP, Shen L, Hahn JM. Tetrahedron Lett. 2002;43:4449–4453. [Google Scholar]
- 23.MaGee DI, Godineau E, Thornton PD, Walters MA, Sponholtz DJ. Eur. J. Org. Chem. 2006:3667–3680. [Google Scholar]
- 24.Antoline JE, Hsung RP. Synlett. 2008:739–744. [Google Scholar]
- 25.Krenske EH, Houk KN, Lohse AG, Antoline JE, Hsung RP. Chem. Sci. 2010;1:387–392. doi: 10.1039/C0SC00280A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Krenske EH, Houk KN, Harmata M. Org. Lett. 2010;12:444–447. doi: 10.1021/ol902591k. [DOI] [PubMed] [Google Scholar]
- 27.Antoline JE, Hsung RP, Huang J, Song Z, Li G. Org. Lett. 2007;9:1275–1278. doi: 10.1021/ol070103n.. For isolation of parvineostemonine, see: Ke CQ, He ZS, Yang YP, Ye Y. Chin. Chem. Lett. 2003;14:173–175.
- 28.Xiong H, Huang J, Ghosh SK, Hsung RP. J. Am. Chem. Soc. 2003;125:12694–12695. doi: 10.1021/ja030416n. [DOI] [PubMed] [Google Scholar]
- 29.Lohse AG, Hsung RP, Leider MD, Ghosh SK. J. Org. Chem. 2011 doi: 10.1021/jo200147h. Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rameshkumar C, Hsung RP. Angew. Chem., Int. Ed. 2004;43:615–618. doi: 10.1002/anie.200352632. For isolation of aromadendrane alkaloids, see: Braekman J-C, Daloze D, Deneubourg F, Huysecom J, Vandevyver G. Bull. Soc. Chim. 1989;98:869–875.
-
31.For an enantioselective formal (4 + 3) cycloaddition, see: Dai X, Davies HML. Adv. Synth. Cat. 2006;348:2449–2456.
- 32.Harmata M, Ghosh SK, Hong X, Wacharasindhu S, Kirchhoefer P. J. Am. Chem. Soc. 2003;125:2058–2059. doi: 10.1021/ja029058z. [DOI] [PubMed] [Google Scholar]
- 33.Huang J, Hsung RP. J. Am. Chem. Soc. 2005;127:50–51. doi: 10.1021/ja044760b. [DOI] [PubMed] [Google Scholar]
- 34.For a recent review on C2-symmetric ligands in asymmetric catalysis, see: Desimoni G, Faita G, Jørgensen KA. Chem. Rev. 2006;106:3561–3651. doi: 10.1021/cr0505324.
- 35.Lohse AG, Krenske EH, Antoline JE, Houk KN, Hsung RP. Org. Lett. 2010;12:5506–5509. doi: 10.1021/ol1023745. [DOI] [PMC free article] [PubMed] [Google Scholar]
