

Abstract
Background
Investigation of metagenomes provides greater insight into uncultured microbial communities. The improvement in sequencing technology, which yields a large amount of sequence data, has led to major breakthroughs in the field. However, at present, taxonomic binning tools for metagenomes discard 30-40% of Sanger sequencing data due to the stringency of BLAST cut-offs. In an attempt to provide a comprehensive overview of metagenomic data, we re-analyzed the discarded metagenomes by using less stringent cut-offs. Additionally, we introduced a new criterion, namely, the evolutionary conservation of adjacency between neighboring genes. To evaluate the feasibility of our approach, we re-analyzed discarded contigs and singletons from several environments with different levels of complexity. We also compared the consistency between our taxonomic binning and those reported in the original studies.
Results
Among the discarded data, we found that 23.7 ± 3.9% of singletons and 14.1 ± 1.0% of contigs were assigned to taxa. The recovery rates for singletons were higher than those for contigs. The Pearson correlation coefficient revealed a high degree of similarity (0.94 ± 0.03 at the phylum rank and 0.80 ± 0.11 at the family rank) between the proposed taxonomic binning approach and those reported in original studies. In addition, an evaluation using simulated data demonstrated the reliability of the proposed approach.
Conclusions
Our findings suggest that taking account of conserved neighboring gene adjacency improves taxonomic assignment when analyzing metagenomes using Sanger sequencing. In other words, utilizing the conserved gene order as a criterion will reduce the amount of data discarded when analyzing metagenomes.
Background
The investigation of metagenomes, which sequences DNA from mixed environmental samples directly, has provided insights into microbial communities, and is now widely used to study various living microorganisms as a system [1-4]. The major goal of metagenomic studies is to determine the systemic properties of a microbial community, including the genetic, metabolic, ecological, physiological and behavioral aspects of all community members [5-8]. Some high-throughput pipelines have been constructed for high-performance computational analysis of metagenomic data [9,10]. The pipelines facilitate taxonomic binning of huge amounts of sequencing data by referring to databases of known microbial genomes [11-14]. Based on the above approaches, recent investigations have revealed enormous variations among the microbiomes of diverse environments, such as human intestinal and salivary microbiota [15-17], microbial communities growing on sunken whale skeletons [18], and open ocean communities [19,20].
To study genetic materials from natural environmental samples, Sanger sequencing technologies have been used for generating DNA sequences [15,16,20]. Yet, much more metagenomic datasets were conducted using next generation sequencing (NGS) technologies (e.g., Roche GS-FLX, Illumina 1G analyzer, and Applied Biosystems SOLiD) which yield shorter fragments ranging from 30 bp to 350 bp [21]. As huge amount of sequencing data were produced, analysis tools have become a critical player in data interpretation [22]. For example, when scaffolds and contigs are assigned to phylogenetically related groups, GLIMMER [23], GeneMark.hmm [24], and MetaGene [25] are widely used to identify putative coding sequences (CDSs). Subsequently, the taxonomic assignment of CDSs is performed using BLAST [26] or other homology search tools [27] with sequence databases. Recently, some advanced taxonomic assigning tools like MEGAN [28], Phymm [29], PhyloPythia [30] were published. However, the majority of the reads only contain partial coding regions. Thus, they were usually unidentified because of the limited match length. For example, in two distal gut microbiomes, approximately 40% of 139,521 high-quality reads were discarded after sequence assembly. Moreover, approximately 40% of 50,164 CDSs predicted by using the GLIMMER package were excluded from further analysis due to insignificant BLAST scores [15]. In 13 healthy Japanese individuals, 33% of 1,065,392 shotgun reads failed to assemble, and 25% of 662,548 CDSs (identified by MetaGene) were excluded from further analysis [16]. It is estimated that existing analytical methods discard approximately 30-40% of metagenomic data from Sanger approaches [11,15,16,18,19,31]. Considering the drawback, we were motivated to re-analyzed the discarded reads of metagenomes generated using Sanger sequencing.
To overcome the limitations of current binning approaches, which rely heavily on the BLAST hit score, we propose a method for assigning reads discarded by the original studies (Figure 1). The new approach combines the BLAST search scores (two or more CDSs in a read) and the concept of conserved gene adjacency. The rationale is based on the theory that genomes are shuffled, so local gene-order conservation reflects the specificity of microbial organisms [32]. For example, the conservation of the gene order in prokaryotes is known to be an important feature; hence, it has been used in function inference [33,34]. Since gene order conservation is a genomic feature that is extensively conserved between closely related species [35,36], the trend should be universal in prokaryotic genomes [37]. Furthermore, it is known that overlapping gene pairs are frequently observed in microbial chromosomes [38] and conserved across species [39] in all three transcriptional directional classes: unidirectional (→→), convergent (→←), and divergent (←→) [40,41]. Therefore, we argue that, if a genomic fragment contains two or more adjacent CDSs that are identified by BLASTX, it is reasonable to assign the sequence by using the proposed strategy, which combines two BLASTX hit scores and the adjacency of the two genes.
Figure 1.

Overview of the proposed approach.
A recent study showed that the average gene density in prokaryotic genomes is one gene per 1,000 nucleotides [41], which is close to the sequence length yielded by whole genome shotgun sequencing. Thus, we were aware that the read length would be the limitation of this approach. In our study, we only applied the analyses to conventional Sanger reads, which have higher potential to contain adjacent gene information than NGS. We first used simulated metagenomes to estimated the ratio of discarded singletons that may contain at least two neighboring genes [42]. We found that approximately 49% of discarded singletons contained gene pairs in all three transcriptional directional classes. Subsequently, we collected data from conventional metagenome projects that were generated via Sanger sequencing, and re-analyzed the fragments that were discarded from two types of metagenomic data, 13 healthy Japanese individuals [16] and the skeletons of whale carcasses (whale fall) [18]. Two types of genomic fragments, assembled contigs and raw single reads (singletons), were analyzed separately. The results showed that between 12.9% and 31.4% of the discarded data were assigned to taxa. Furthermore, the microbial compositions using discarded data and those reported in previous studies [15,16,18] were highly consistent in the family and phylum ranks. Therefore, we conclude that the proposed metagenomic sequencing approach provide a more comprehensive overview of the functional and taxonomic content of a microbiome.
Results and Discussion
NGS technology facilitates the investigation of microbial communities. Because of the enormous number of short DNA fragments in metagenomic datasets, some bioinformatics tools, such as MEGAN [28], PhymmBL [29] and TACOA [43], have been developed for phylogenetic classification. However, current taxonomic binning methods have to discard a large number of sequences due to low homology scores. To address this problem, we developed a method that assigns discarded genomic fragments by combining the BLAST search scores and the criterion of gene adjacency. First, to assess the feasibility of our approach, we used simulated metagenomes to analyze the distribution of the number of CDSs in discarded singletons. In the simulated data sets, which had different levels of complexity (simLC, simMC and simHC, see Methods), we found that nearly half of the discarded singletons contained two or more partial CDSs (Table 1), suggesting that some of the discarded datasets could still be assigned to taxa.
Table 1.
Number (and ratio) of discarded singletons that did not contain any CDS and those that contain one, two and three or more CDSs in the simulated metagenomes.
| Number of CDS on singleton | simLC | simMC | simHC | |||
|---|---|---|---|---|---|---|
| Singletons | % | Singletons | % | Singletons | % | |
| 0 | 2575 | 6.2 | 2637 | 6.5 | 3986 | 6.0 |
| 1 | 18219 | 44.0 | 18072 | 44.8 | 29926 | 45.0 |
| 2 | 17549 | 42.4 | 16832 | 41.7 | 27874 | 41.9 |
| 3 or more | 3081 | 7.4 | 2838 | 7.0 | 4738 | 7.1 |
| Total singletons | 41424 | 100 | 40379 | 100 | 66524 | 100 |
Binning discarded metagenomic fragments
We used two kinds of metagenomes from whale fall samples (contigs) and healthy Japanese individuals (singletons) respectively (see Methods). Since the singletons were not available in the public domains, we repeated the assembly strategies and obtained similar datasets. As shown in Table 2, between 4,990 and 7,660 discarded contigs were collected from three whale fall microbiomes; and for the Japanese individuals, between 7,078 and 28,244 discarded singletons were collected after the assembly process. Under the proposed approach, between 12.9% and 14.9% of the discarded contigs in the whale fall samples were assigned to taxa. In the group of Japanese individuals, we were able to assign between 16.9% and 31.4% of the discarded singletons (see Table 3) to taxa. Based on the results, we suggest that the proposed binning strategy can be applied for re-analyzing the discarded reads of metagenomic data.
Table 2.
Summary of collected metagenomic fragments.
| Data type I (contigs) | Assigned | Unassigned | ||||
|---|---|---|---|---|---|---|
| Location | Position | Total contigs | CDSsa | Contigsb | Average length (bp) | |
| whale fall sub. 1 | Pacific Ocean, Santa Cruz Basin (N33.30 W 119.22) | section of rib bone | 35975 | 33139 | 7039 | 1167 |
| whale fall sub. 2 | Pacific Ocean, Santa Cruz Basin (N33.30 W 119.22) | bone | 32459 | 32395 | 7660 | 1199 |
| whale fall sub. 3 | West Antarctic Peninsula Shelf (S65.10 W64.47) | bone | 27130 | 26841 | 4990 | 1357 |
| Our duplicationc | ||||||
| Data type II (singletons) | Assigned | Unassigned | ||||
| Sex | Age | Total reads | CDSsd | Singletons | Average length (bp) | |
| Japanese In-A | Male | 45 years | 76434 | 29247 | 13399 | 1057 |
| Japanese In-B | Male | 6 months | 80617 | 14718 | 7078 | 1058 |
| Japanese In-D | Male | 35 years | 84237 | 48033 | 28244 | 1034 |
| Japanese In-E | Male | 3 months | 80852 | 27860 | 10838 | 1124 |
| Japanese In-M | Female | 4 months | 89340 | 26350 | 8456 | 1008 |
| Japanese In-R | Female | 24 years | 85787 | 45438 | 21661 | 998 |
| Japanese F1-S | Male | 30 years | 78452 | 40427 | 15378 | 1005 |
| Japanese F1-T | Female | 28 years | 81348 | 46487 | 21780 | 958 |
| Japanese F1-U | Female | 7 months | 82525 | 27332 | 11791 | 969 |
| Japanese F2-V | Male | 37 years | 80772 | 49411 | 19733 | 1006 |
| Japanese F2-W | Female | 36 years | 79163 | 42750 | 16961 | 1039 |
| Japanese F2-X | Male | 3 years | 80858 | 41337 | 19351 | 1040 |
| Japanese F2-Y | Female | 1.5 years | 79754 | 49315 | 20061 | 990 |
a Genes with best hits at 30% identity or higher in Archaea and Bacteria kingdoms from JGI
b Genes with best hits less than 30% identity in Archaea and Bacteria kingdoms from JGI
c Phred and PCAP assembly package for Japanese samples.
d The number of predicted open-reading frames showing similarity to genes in the "in-house NR database".
Table 3.
Summary of reassignments using discarded metagenomic data.
| Data type I (contigs) | Re-assigned | |||||
|---|---|---|---|---|---|---|
| Contigs | Contigs | Average length (bp) | Rate (%) | r (phylum) | r (family) | |
| whale fall sub. 1 | 7039 | 1050 | 1388 | 14.9 | 0.98 | 0.92 |
| whale fall sub. 2 | 7660 | 995 | 1295 | 12.9 | 0.98 | 0.77 |
| whale fall sub. 3 | 4990 | 720 | 1400 | 14.4 | 0.97 | 0.79 |
| Data type II (singletons) | Re-assigned | |||||
| Singletons | Singletons | Average length (bp) | Rate (%) | r (phylum) | r (family) | |
| Japanese In-A | 13399 | 3542 | 1074 | 26.4 | 0.95 | 0.85 |
| Japanese In-B | 7078 | 2050 | 1073 | 28.9 | 0.99 | 0.90 |
| Japanese In-D | 28244 | 5542 | 1061 | 16.9 | 0.89 | 0.72 |
| Japanese In-E | 10838 | 2888 | 1129 | 26.6 | 0.99 | 0.95 |
| Japanese In-M | 8546 | 2159 | 1057 | 25.2 | 0.93 | 0.86 |
| Japanese In-R | 21661 | 3993 | 1020 | 18.4 | 0.96 | 0.80 |
| Japanese F1-S | 15378 | 3216 | 1018 | 20.9 | 0.95 | 0.82 |
| Japanese F1-T | 21780 | 4395 | 971 | 20.1 | 0.93 | 0.59 |
| Japanese F1-U | 11791 | 3711 | 983 | 31.4 | 0.99 | 0.99 |
| Japanese F2-V | 19733 | 4007 | 1020 | 20.3 | 0.90 | 0.61 |
| Japanese F2-W | 16961 | 4011 | 1052 | 23.6 | 0.89 | 0.77 |
| Japanese F2-X | 19351 | 4402 | 1054 | 22.7 | 0.92 | 0.66 |
| Japanese F2-Y | 20061 | 4766 | 1002 | 23.7 | 0.96 | 0.82 |
The consistency between binning with discarded fragments and that in the original studies was tested by the Pearson correlation coefficient (r).
The consistency of binning with discarded fragments compared to the strategies in previous studies
To validate our approach, we compared the proposed taxonomic binning strategy using discarded datasets with the strategies in previous studies [15,16,18]. We used Pearson correlation coefficient to evaluate the similarity of the two groups. For taxonomic assignments using homology search tools, reads were assigned down to the class, order, family, and genus ranks [3,5,11,14,16,29,43]. Therefore, we separated the comparison into phylum and family ranks to describe the similarity between the original results and our binning results. We found that the results derived by our taxonomic binning strategy and those reported in previous studies were consistent. The correlation coefficients were 0.94 ± 0.03 in the phylum rank and 0.80 ± 0.11 in the family rank (Table 3). For example, the compositional view of Japanese individual F1-U showed a high degree of similarity between the two binnings (Figure 2). The correlation coefficient was 0.99 in both the phylum rank and the family rank. The consistency between the two datasets indicates that taxonomic binning using discarded data is as representative as the binning strategies used in previous studies.
Figure 2.
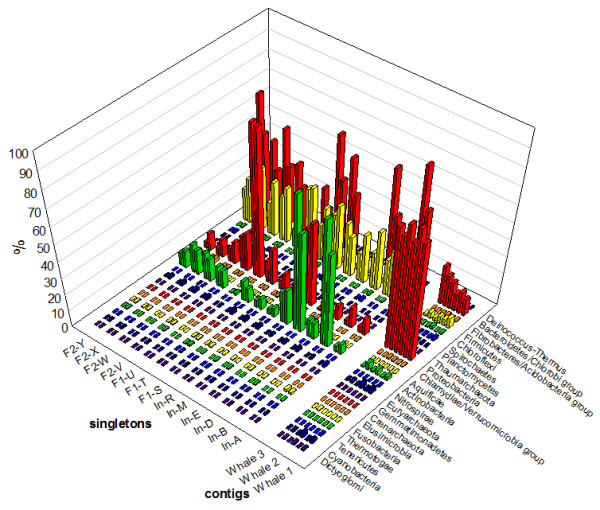
Compositional View of 16 microbiomes in the phylum rank. The bars depict the detailed contribution of microbiomes with 22 phyla represented on two types of genomic fragments, contigs and singletons. For each microbiome, the similarity of binning between our re-assignments and that of the original studies was compared. The consistency of the two datasets is represented by the Pearson correlation coefficient (Table 3).
To further evaluate our approach, we used 10,000 simulated singletons (simMC) for taxonomic binning to quantify the performance of our analysis. As shown in Table 4, the discarded singletons with the length of ~1 kb (Table 2) were correctly assigned with sensitivity between 36.8-25.9% and specificity between 93.3-79.0% between phylum and genus (using E-value 10-2, hits numbers 250). The hit number is positively correlated with the sensitivity but is negatively correlated with specificity, while the E-values do not seem to affect accuracy. In comparison with same method but without considering the gene adjacency, our approach showed a slight decrease in specificity but increased in sensitivity. For example, in family and genus ranking, the sensitivity is approximately four times higher than the method that does not consider gene adjacency (Table 4). Furthermore, because of the lack of similar analysis for discarded reads, here, we referred to previous studies using whole metagenomic data. For example, in TACOA [43], which reportedly performed better than PhyloPythia [44], the average sensitivity for binning 1 kb singletons ranged from 71% in the superkingdom rank to 22% in the class rank; and the average specificity ranged from 73% in the superkingdom rank to 64% in the class rank. Although our dataset sources (discarded dataset) were different from TACOA (whole dataset), the results indicate that with suitable filters and criteria, reliable information in the discarded data can be retrieved.
Table 4.
Sensitivity and specificity of taxonomic binning at different taxonomic ranks using discarded dataset of simMC.
| Accuracy | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Criteria | P | O | F | G | ||||||
| adjacency | E-value | hits | Sn | Sp | Sn | Sp | Sn | Sp | Sn | Sp |
| with | 1e-2 | 50 | 31.8 | 97.2 | 28.3 | 90.8 | 27.6 | 84.8 | 24.1 | 88.8 |
| 150 | 34.9 | 94.5 | 30.8 | 86.2 | 30.0 | 79.3 | 25.3 | 82.0 | ||
| 250 | 36.8 | 93.3 | 31.7 | 84.0 | 30.7 | 77.0 | 25.9 | 79.0 | ||
| 350 | 37.9 | 92.7 | 32.0 | 83.0 | 30.7 | 75.8 | 25.7 | 77.6 | ||
| 1e-4 | 50 | 29.7 | 97.4 | 27.8 | 91.1 | 27.4 | 85.1 | 23.7 | 89.2 | |
| 150 | 34.1 | 94.9 | 30.3 | 87.1 | 29.5 | 80.2 | 25.5 | 83.1 | ||
| 250 | 36.0 | 93.7 | 31.4 | 84.9 | 30.3 | 77.8 | 25.7 | 80.2 | ||
| 350 | 37.1 | 93.2 | 31.7 | 83.9 | 30.5 | 76.7 | 25.8 | 78.7 | ||
| 1e-6 | 50 | 29.1 | 97.5 | 27.3 | 91.2 | 26.8 | 85.1 | 23.4 | 89.2 | |
| 150 | 33.1 | 94.9 | 29.7 | 87.1 | 28.8 | 80.2 | 24.7 | 83.1 | ||
| 250 | 35.0 | 93.7 | 30.8 | 84.9 | 29.8 | 77.8 | 25.5 | 80.2 | ||
| 350 | 36.2 | 93.2 | 31.3 | 83.9 | 30.2 | 76.7 | 25.8 | 78.7 | ||
| without | 1e-2 | 50 | 31.3 | 99.6 | 9.6 | 97.8 | 5.9 | 89.1 | 4.1 | 93.3 |
| 150 | 20.0 | 99.7 | 7.3 | 94.0 | 5.2 | 88.1 | 4.5 | 92.0 | ||
| 250 | 17.5 | 99.6 | 7.3 | 94.0 | 5.2 | 88.1 | 4.5 | 92.3 | ||
| 350 | 16.3 | 99.5 | 7.3 | 94.0 | 5.2 | 88.1 | 4.5 | 92.3 | ||
Criteria considering gene adjacency and without considering gene adjacency were tested separately. P: phylum, O: order, F: family, G: genus. Sn and Sp denote sensitivity (%) and Specificity (%).
It has been observed that HGT (horizontal gene transfer) occurs frequently in prokaryotes [45]. Such a mechanism of genetic variability within a species may create bias in taxonomic binning based on a traditional homology search method. However, not all genes are equally itinerant, and they do not exhibit the same HGT behavior [46,47]. Preferential HGT correlates strongly with the functions of different types of genes. For example, informational genes (those involved in transcription, translation, and related processes) are far less likely to be transferred horizontally than operational genes (e.g. housekeeping functions) because they are complex systems [46]. In genome wide studies using 116 prokaryotes [48], the authors reported 46,759 HGT events in a total of 3,245,653 ORFs, but the horizontal transfer clusters (more than one gene) were relatively low (only 1,357 cases). Our approach considers the BLAST search scores and the criterion of conserved gene adjacency. Hence, the bias resulting from HGT should be relatively low compared to that of other approaches using a single hit.
Conclusions
Since a large amount of metagenomic data generated using Sanger sequencing fails to satisfy the cut-off for taxonomic binning, we introduce a criterion based on a genomic feature, namely, the conservation of gene adjacency between prokaryotes. Our analysis suggests that considering the conserved neighboring gene adjacency reduces the amount of data discarded by current methods. In fact, a latest update of MEGAN software has incorporated similar analysis for pair reads, and the assignment for LCA-gene has been improved considering the conserved adjacency [49]. In addition, we are aware that the vast majority of recent metagenomic datasets were produced by NGS technologies (e.g., Roche GS-FLX, Illumina 1G analyzer, and Applied Biosystems SOLiD), and our analysis can only be applied to datasets with longer reads, such as Sanger. Yet, Roche's first-generation instrument, 454 GS 20 (released in 2005), yielded 100-bp reads, the latest version GS Junior System (released in 2009, Roche) already yielded demonstrably higher read lengths, exceeding 500 bp. Hopefully, the limitations of sequence length will be resolved in the near future, and our study will provide a basis for analyzing metagenomic data.
Methods
Collection of metagenomes and microbial genome sequence
Figure 1 shows an overview of our methodology. We used two kinds of metagenome samples: sunken whale skeletons (whale fall) and human distal guts. Three independent whale fall samples were collected in 2005 [18]. The assembled sequence data was downloaded from NCBI ftp://ftp.ncbi.nih.gov/genbank/wgs/ with accession numbers AAFY01000001-AAFY01028151 (whale fall 1), AAFZ01000001-AAFZ01029934 (whale fall 2), and AAGA01000001-AAGA01026232 (whale fall 3). The microbiomes of distal guts were collected from 13 healthy Japanese individuals (six individuals and members of two unrelated families) [16]. The data was downloaded from the Human Metagenome Consortium, Japan (HMGJ, http://www.metagenome.jp/). Table 2 summarizes the metagenomic fragments that we collected.
To obtain information about gene adjacency, we downloaded microbial genomes from the NCBI ENTREZ Genome Project database http://www.ncbi.nlm.nih.gov/genomes/lproks.cgi. A total of 3,072,893 protein sequences were obtained from 939 complete microbial genomes and 576 plasmids in August 2009. The sequences had to be processed by formatdb before they could be used by the BLAST program.
Collection of discarded genomic fragments
We analyzed two types of discarded genomic fragments: contigs that failed to meet the criteria in the original studies and singletons that were left for analysis. The discarded contigs, which were obtained from the DOE Joint Genome Institute (JGI, http://www.jgi.doe.gov/), contained genes that failed to pass the 30% BLAST identity cut-off, or they had no hits in the Archaea and Bacteria kingdoms of each microbiome.
To collect the discarded singletons, we followed the assembly strategy described in Kurokawa K et al. [16]. For the 13 Japanese samples, the original trace archives (chromatogram files) were downloaded from the DNA Data Bank of Japan (DDBJ, http://www.ddbj.nig.ac.jp/). To read the DNA sequence chromatogram files, we adopted the Phred program [50,51], which is widely used for base-calling and characterizing the quality of DNA sequences. Finally, the shotgun reads from the 13 samples were assembled using the PCAP software [52] with the default parameters. The number and average length of the remaining singletons from the Japanese individuals are shown in Table 2. The slight differences between our statistics and those reported in previous studies may be due to different parameter settings.
Collection of simulated datasets
To estimate the proportion of discarded singletons that contain at least two genes from real metagenomes, we downloaded three simulated metagenomic data sets of varying complexity as benchmarks and calculated the number of CDSs in each singleton. The three simulated datasets, a low-complexity community (simLC), a moderate-complexity community (simMC) and a high-complexity community (simHC), were compiled by combining sequencing reads randomly selected from 113 genomes [42]. After assembling the simulated datasets using Phrap (v3.57), all remaining singletons were published by the Department of Energy (DOE) Joint Genome Institute. They are available through the Integrated Microbial Genome (IMG) system. In addition, we also used simMC to evaluate the performance of our taxonomic assignment method. In total, there are 15,197 contigs and 40,379 singletons that Phrap assembler failed to assemble. We randomly selected 10,000 non-redundant singletons from simMC for analysis.
Taxonomic assignment of discarded genomic fragments
To incorporate the conservation of gene order into the taxonomic classification, each discarded genomic fragment was screened for protein encoding genes via a BLASTX search against the NCBI ENTREZ Genome Project database. An expected cut-off value (E) of 10-5 was used to select the top 250 potential coding elements as the default settings. (We discuss the selection criteria in Accuracy evaluation using simulated datasets).
Normally, the best hits are selected from BLAST results, but best hits do not provide information on adjacent genes. Therefore, the top 250 hits were selected instead. In our strategies, adjacent gene pair is a pair of genes that are directly next to each other in a given chromosome. Thus, each hit was grouped with its corresponding species. These hits were then compared in a pair-wise fashion in order to identify adjacent CDSs. The transcriptional direction (unidirectional (→→), convergent (→←), and divergent (←→)) of all identified adjacent CDSs should be consistent with the genomic arrangement of reference genomes. Next, the pairs with inconsistent genomic arrangement were removed. Subsequently, among the remaining pairs, we ran the lowest common ancestor (LCA) algorithm used in MEGAN [28] to analyze the data. The source code is provided in the supplementary material (see [Additional file 1]). It requires perl and Basic Local Alignment Search Tool (BLAST) on the work station. The program has been tested by using several resources listed in Collection of discarded genomic fragments and in Collection of simulated datasets.
Comparison of binning discarded fragments in the proposed approach and the original studies
To assess the consistency of binning results using the discarded dataset and the binning results reported in original studies, we compared the quantitative contribution of microorganisms in discarded data set and original data set. Contigs and singletons were performed separately. Because the phylogenetic taxonomies constructed by the NR database (used in previous studies) and the NCBI ENTREZ Genome Project database (used in our study) were not consistent, we selected 22 phyla and 166 families that were consistent in both databases to estimate the similarity of the binning results (see [Additional file 2]). To quantify the similarity, we calculated Pearson correlation coefficient. We found that, in each environment, the taxonomic binning was dominated by a limited number of phylotypes; and the remaining phylotypes only made a small contribution. To avoid over-estimation resulting from the latter, all phylotypes less than five were combined before calculating Pearson correlation coefficient in both datasets.
Accuracy evaluation using simulated datasets
The selection of appropriate criteria may have a critical effect on our system's performance. In Table 4, the relationships between the criteria (E-value threshold (10-2, 10-4, and 10-6) and BLAST hits numbers (50, 150, 250 and 350)) and the accuracy of our system were evaluated using a simulated discarded dataset. Twelve combinations (3 E-values * 4 BLAST hits numbers) were tested for the performance evaluation.
Taxonomic reassignment for simulated data was evaluated by comparing the assignments made by our method to those of the real corresponding taxa in different taxonomic ranks (i.e., species, genus, family, order, class, phylum and superkingdom). In this study, we employed the adapted definition of sensitivity and specificity [43,53]. The accuracy was evaluated for each taxonomic class. Let the i-th taxonomic class of taxonomic rank r be denoted as class i. The true positives (TPi) are defined as the number of genomic fragments correctly assigned to class i; the false positives (FPi) are defined as the number of fragments from any class j ≠ i that is wrongly assigned as i. The false negatives (FNi) are defined as the number of fragments from class i that is erroneously assigned to any other class j ≠ i. For a genomic fragment whose taxonomic class cannot be inferred, the algorithm classifies it as "unclassified". The unclassified (Ui) are the numbers of fragments from class i that cannot be assigned to a taxonomic class.
The sensitivity (Sni) for a taxonomic class i is defined as the percentage of fragments from class i correctly classified. It is computed by:
The reliability (expressed in percentage) of the predictions made by the classifier for class i is denoted as specificity (Spi). It is measured using the following equation:
To select appropriate E-value threshold, the data in Table 4 were examined. Since the results indicated that the E-values do not affect the performance of taxonomic binning, we selected a loose criterion (E-value 10-5) as default. The hit number is positively correlated with the sensitivity but is negatively correlated with specificity, (Table 4), the hit number 250 was selected as default considering the sensitivity, specificity and also the run-time required.
Authors' contributions
DW and HKT formulated the studies, participated in the experiment design, and drafted the manuscript. FCHW participated in the program design of the study and helped draft the manuscript. CHS, MTH and TYW collected the data and performed the statistical analysis. All the authors read and approved the final manuscript.
Supplementary Material
Reassignment_using_gene_adjacency.pl. Perl script for reassignment using gene adjacency.
Supplemental Table S1. The phylotypes used to estimate the binning similarity.
Contributor Information
Francis C Weng, Email: manpower@gate.sinica.edu.tw.
Chien-Hao Su, Email: d95922033@ntu.edu.tw.
Ming-Tsung Hsu, Email: mingtsung@iis.sinica.edu.tw.
Tse-Yi Wang, Email: tseyiwang@gmail.com.
Huai-Kuang Tsai, Email: hktsai@iis.sinica.edu.tw.
Daryi Wang, Email: dywang@gate.sinica.edu.tw.
Acknowledgements
We wish to thank the reviewers for their valuable comments and suggestions, which helped us to improve our analysis. This work was supported by the National Science Council of Taiwan under grants NSC99-2621-B-001-005-MY2, NSC99-2627-B-001-005-MY3 to DW, and grant NSC98-2221-E-001-015, NSC98-2627-B-001-003 to HKT.
References
- Vieites JM, Guazzaroni ME, Beloqui A, Golyshin PN, Ferrer M. Metagenomics approaches in systems microbiology. FEMS Microbiol Rev. 2009;33(1):236–255. doi: 10.1111/j.1574-6976.2008.00152.x. [DOI] [PubMed] [Google Scholar]
- Hugenholtz P, Tyson GW. Microbiology: Metagenomics. Nature. 2008;455(7212):481–483. doi: 10.1038/455481a. [DOI] [PubMed] [Google Scholar]
- Pignatelli M, Aparicio G, Blanquer I, Hernandez V, Moya A, Tamames J. Metagenomics reveals our incomplete knowledge of global diversity. Bioinformatics. 2008;24(18):2124–2125. doi: 10.1093/bioinformatics/btn355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tringe SG, Rubin EM. Metagenomics: DNA sequencing of environmental samples. Nat Rev Genet. 2005;6(11):805–814. doi: 10.1038/nrg1709. [DOI] [PubMed] [Google Scholar]
- Biddle JF, Fitz-Gibbon S, Schuster SC, Brenchley JE, House CH. Metagenomic signatures of the Peru Margin subseafloor biosphere show a genetically distinct environment. Proceedings of the National Academy of Sciences. 2008;105(30):10583–10588. doi: 10.1073/pnas.0709942105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooper SD, Raes J, Foerstner KU, Harrington ED, Dalevi D, Bork P. A Molecular Study of Microbe Transfer between Distant Environments. PLoS ONE. 2008;3(7):e2607. doi: 10.1371/journal.pone.0002607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbaugh PJ, Ley RE, Hamady M, Fraser-Liggett CM, Knight R, Gordon JI. The Human Microbiome Project. Nature. 2007;449(7164):804–810. doi: 10.1038/nature06244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser C, Alm EJ, Polz MF, Spratt BG, Hanage WP. The Bacterial Species Challenge: Making Sense of Genetic and Ecological Diversity. Science. 2009;323(5915):741–746. doi: 10.1126/science.1159388. [DOI] [PubMed] [Google Scholar]
- Meyer F, Paarmann D, D'Souza M, Olson R, Glass E, Kubal M, Paczian T, Rodriguez A, Stevens R, Wilke A, Wilkening J, Edwards R. The metagenomics RAST server - a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinformatics. 2008;9(1):386. doi: 10.1186/1471-2105-9-386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdivia-Granda W. The next meta-challenge for Bioinformatics. Bioinformation. 2008;2(8):358–362. doi: 10.6026/97320630002358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause L, Diaz NN, Goesmann A, Kelley S, Nattkemper TW, Rohwer F, Edwards RA, Stoye J. Phylogenetic classification of short environmental DNA fragments. Nucl Acids Res. 2008;36(7):2230–2239. doi: 10.1093/nar/gkn038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Mering C, Hugenholtz P, Raes J, Tringe SG, Doerks T, Jensen LJ, Ward N, Bork P. Quantitative Phylogenetic Assessment of Microbial Communities in Diverse Environments. Science. 2007;315(5815):1126–1130. doi: 10.1126/science.1133420. [DOI] [PubMed] [Google Scholar]
- Wilhelm L, Tripp HJ, Givan S, Smith D, Giovannoni S. Natural variation in SAR11 marine bacterioplankton genomes inferred from metagenomic data. Biology Direct. 2007;2(1):27. doi: 10.1186/1745-6150-2-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monier A, Claverie J-M, Ogata H. Taxonomic distribution of large DNA viruses in the sea. Genome Biology. 2008;9(7):R106. doi: 10.1186/gb-2008-9-7-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill SR, Pop M, DeBoy RT, Eckburg PB, Turnbaugh PJ, Samuel BS, Gordon JI, Relman DA, Fraser-Liggett CM, Nelson KE. Metagenomic Analysis of the Human Distal Gut Microbiome. Science. 2006;312(5778):1355–1359. doi: 10.1126/science.1124234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurokawa K, Itoh T, Kuwahara T, Oshima K, Toh H, Toyoda A, Takami H, Morita H, Sharma VK, Srivastava TP, Taylor TD, Noguchi H, Mori H, Ogura Y, Ehrlich DS, Itoh K, Takagi T, Sakaki Y, Hayashi T, Hattori M. Comparative Metagenomics Revealed Commonly Enriched Gene Sets in Human Gut Microbiomes. DNA Res. 2007;14(4):169–181. doi: 10.1093/dnares/dsm018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasidze I, Li J, Quinque D, Tang K, Stoneking M. Global diversity in the human salivary microbiome. Genome Res. 2009;19(4):636–643. doi: 10.1101/gr.084616.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tringe SG, von Mering C, Kobayashi A, Salamov AA, Chen K, Chang HW, Podar M, Short JM, Mathur EJ, Detter JC, Bork P, Hugenholtz P, Rubin EM. Comparative Metagenomics of Microbial Communities. Science. 2005;308(5721):554–557. doi: 10.1126/science.1107851. [DOI] [PubMed] [Google Scholar]
- Venter JC, Remington K, Heidelberg JF, Halpern AL, Rusch D, Eisen JA, Wu D, Paulsen I, Nelson KE, Nelson W, Fouts DE, Levy S, Knap AH, Lomas MW, Nealson K, White O, Peterson J, Hoffman J, Parsons R, Baden-Tillson H, Pfannkoch C, Rogers Y-H, Smith HO. Environmental Genome Shotgun Sequencing of the Sargasso Sea. Science. 2004;304(5667):66–74. doi: 10.1126/science.1093857. [DOI] [PubMed] [Google Scholar]
- Yooseph S, Sutton G, Rusch DB, Halpern AL, Williamson SJ, Remington K, Eisen JA, Heidelberg KB, Manning G, Li W, Jaroszewski L, Cieplak P, Miller CS, Li H, Mashiyama ST, Joachimiak MP, van Belle C, Chandonia J-M, Soergel DA, Zhai Y, Natarajan K, Lee S, Raphael BJ, Bafna V, Friedman R, Brenner SE, Godzik A, Eisenberg D, Dixon JE, Taylor SS. et al. The Sorcerer II Global Ocean Sampling Expedition: Expanding the Universe of Protein Families. PLoS Biol. 2007;5(3):e16. doi: 10.1371/journal.pbio.0050016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrosino JF, Highlander S, Luna RA, Gibbs RA, Versalovic J. Metagenomic pyrosequencing and microbial identification. Clin Chem. 2009;55(5):856–866. doi: 10.1373/clinchem.2008.107565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shendure J, Ji H. Next-generation DNA sequencing. Nat Biotechnol. 2008;26(10):1135–1145. doi: 10.1038/nbt1486. [DOI] [PubMed] [Google Scholar]
- Delcher AL, Harmon D, Kasif S, White O, Salzberg SL. Improved microbial gene identification with GLIMMER. Nucleic Acids Research. 1999;27(23):4636–4641. doi: 10.1093/nar/27.23.4636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukashin AV, Borodovsky M. GeneMark.hmm: new solutions for gene finding. Nucleic Acids Res. 1998;26(4):1107–1115. doi: 10.1093/nar/26.4.1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noguchi H, Park J, Takagi T. MetaGene: prokaryotic gene finding from environmental genome shotgun sequences. Nucleic Acids Res. 2006;34(19):5623–5630. doi: 10.1093/nar/gkl723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215(3):403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Bork P, Koonin EV. Predicting functions from protein sequences--where are the bottlenecks? Nat Genet. 1998;18(4):313–318. doi: 10.1038/ng0498-313. [DOI] [PubMed] [Google Scholar]
- Huson DH, Auch AF, Qi J, Schuster SC. MEGAN analysis of metagenomic data. Genome Res. 2007;17(3):377–386. doi: 10.1101/gr.5969107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady A, Salzberg SL. Phymm and PhymmBL: metagenomic phylogenetic classification with interpolated Markov models. Nat Meth. 2009;6(9):673–676. doi: 10.1038/nmeth.1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHardy AC, Martin HG, Tsirigos A, Hugenholtz P, Rigoutsos I. Accurate phylogenetic classification of variable-length DNA fragments. Nat Methods. 2007;4(1):63–72. doi: 10.1038/nmeth976. [DOI] [PubMed] [Google Scholar]
- Martin Garcia H, Ivanova N, Kunin V, Warnecke F, Barry KW, McHardy AC, Yeates C, He S, Salamov AA, Szeto E, Dalin E, Putnam NH, Shapiro HJ, Pangilinan JL, Rigoutsos I, Kyrpides NC, Blackall LL, McMahon KD, Hugenholtz P. Metagenomic analysis of two enhanced biological phosphorus removal (EBPR) sludge communities. Nat Biotechnol. 2006;24(10):1263–1269. doi: 10.1038/nbt1247. [DOI] [PubMed] [Google Scholar]
- Huynen MA, Bork P. Measuring genome evolution. Proc Natl Acad Sci USA. 1998;95(11):5849–5856. doi: 10.1073/pnas.95.11.5849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overbeek R, Fonstein M, D'Souza M, Pusch GD, Maltsev N. The use of gene clusters to infer functional coupling. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(6):2896–2901. doi: 10.1073/pnas.96.6.2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dandekar T, Snel B, Huynen M, Bork P. Conservation of gene order: a fingerprint of proteins that physically interact. Trends in Biochemical Sciences. 1998;23(9):324–328. doi: 10.1016/S0968-0004(98)01274-2. [DOI] [PubMed] [Google Scholar]
- Tamames J, Casari G, Ouzounis C, Valencia A. Conserved clusters of functionally related genes in two bacterial genomes. J Mol Evol. 1997;44(1):66–73. doi: 10.1007/PL00006122. [DOI] [PubMed] [Google Scholar]
- Tamames J. Evolution of gene order conservation in prokaryotes. Genome Biology. 2001;2(6):research0020.0021–research0020.0011. doi: 10.1186/gb-2001-2-6-research0020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mushegian AR, Koonin EV. Gene order is not conserved in bacterial evolution. Trends in Genetics. 1996;12(8):289–290. doi: 10.1016/0168-9525(96)20006-X. [DOI] [PubMed] [Google Scholar]
- Palleja A, Harrington E, Bork P. Large gene overlaps in prokaryotic genomes: result of functional constraints or mispredictions? BMC Genomics. 2008;9(1):335. doi: 10.1186/1471-2164-9-335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda Y, Nakayama Y, Tomita M. On dynamics of overlapping genes in bacterial genomes. Gene. 2003;323:181–187. doi: 10.1016/j.gene.2003.09.021. [DOI] [PubMed] [Google Scholar]
- Normark S, Bergstrom S, Edlund T, Grundstrom T, Jaurin B, Lindberg FP, Olsson O. Overlapping genes. Annu Rev Genet. 1983;17:499–525. doi: 10.1146/annurev.ge.17.120183.002435. [DOI] [PubMed] [Google Scholar]
- Rogozin IB, Makarova KS, Natale DA, Spiridonov AN, Tatusov RL, Wolf YI, Yin J, Koonin EV. Congruent evolution of different classes of non-coding DNA in prokaryotic genomes. Nucleic Acids Res. 2002;30(19):4264–4271. doi: 10.1093/nar/gkf549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mavromatis K, Ivanova N, Barry K, Shapiro H, Goltsman E, McHardy AC, Rigoutsos I, Salamov A, Korzeniewski F, Land M, Lapidus A, Grigoriev I, Richardson P, Hugenholtz P, Kyrpides NC. Use of simulated data sets to evaluate the fidelity of metagenomic processing methods. Nat Meth. 2007;4(6):495–500. doi: 10.1038/nmeth1043. [DOI] [PubMed] [Google Scholar]
- Diaz N, Krause L, Goesmann A, Niehaus K, Nattkemper T. TACOA - Taxonomic classification of environmental genomic fragments using a kernelized nearest neighbor approach. BMC Bioinformatics. 2009;10(1):56. doi: 10.1186/1471-2105-10-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHardy AC, Martin HG, Tsirigos A, Hugenholtz P, Rigoutsos I. Accurate phylogenetic classification of variable-length DNA fragments. Nat Meth. 2007;4(1):63–72. doi: 10.1038/nmeth976. [DOI] [PubMed] [Google Scholar]
- Gogarten JP, Townsend JP. Horizontal gene transfer, genome innovation and evolution. Nat Rev Microbiol. 2005;3(9):679–687. doi: 10.1038/nrmicro1204. [DOI] [PubMed] [Google Scholar]
- Jain R, Rivera MC, Lake JA. Horizontal gene transfer among genomes: the complexity hypothesis. Proc Natl Acad Sci USA. 1999;96(7):3801–3806. doi: 10.1073/pnas.96.7.3801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhaxybayeva O, Lapierre P, Gogarten JP. Genome mosaicism and organismal lineages. Trends Genet. 2004;20(5):254–260. doi: 10.1016/j.tig.2004.03.009. [DOI] [PubMed] [Google Scholar]
- Nakamura Y, Itoh T, Matsuda H, Gojobori T. Biased biological functions of horizontally transferred genes in prokaryotic genomes. Nat Genet. 2004;36(7):760–766. doi: 10.1038/ng1381. [DOI] [PubMed] [Google Scholar]
- Mitra S, Schubach M, Huson DH. Short clones or long clones? A simulation study on the use of paired reads in metagenomics. BMC Bioinformatics. 2010;11(Suppl 1):S12. doi: 10.1186/1471-2105-11-S1-S12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewing B, Green P. Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 1998;8(3):186–194. [PubMed] [Google Scholar]
- Ewing B, Hillier L, Wendl MC, Green P. Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 1998;8(3):175–185. doi: 10.1101/gr.8.3.175. [DOI] [PubMed] [Google Scholar]
- Huang X, Wang J, Aluru S, Yang SP, Hillier L. PCAP: a whole-genome assembly program. Genome Res. 2003;13(9):2164–2170. doi: 10.1101/gr.1390403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldi P, Brunak S, Chauvin Y, Andersen CA, Nielsen H. Assessing the accuracy of prediction algorithms for classification: an overview. Bioinformatics. 2000;16(5):412–424. doi: 10.1093/bioinformatics/16.5.412. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Reassignment_using_gene_adjacency.pl. Perl script for reassignment using gene adjacency.
Supplemental Table S1. The phylotypes used to estimate the binning similarity.