

Abstract
SnoN is an important negative regulator of transforming growth factor-β (TGF-β) signaling that was originally identified as a transforming oncogene in chicken embryonic fibroblasts. Both pro-oncogenic and antioncogenic activities of SnoN have been reported, but its function in normal epithelial cells has not been defined. In the mouse mammary gland, SnoN is expressed at relatively low levels, but it is transiently upregulated at late gestation before being downregulated during lactation and early involution. To assess the effects of elevated levels of SnoN, we generated transgenic mice expressing a SnoN fragment under the control of the mouse mammary tumor virus promoter. In this model system, SnoN elevation increased side-branching and lobular-alveolar proliferation in virgin glands, while accelerating involution in postlactation glands. Increased proliferation stimulated by SnoN was insufficient to induce mammary tumorigenesis. In contrast, elevated levels of SnoN cooperated with polyoma middle T antigen to accelerate the formation of aggressive multifocal adenocarcinomas and to increase the formation of pulmonary metastases. Our studies define functions of SnoN in mammary epithelial cell proliferation and involution, and provide the first in vivo evidence of a pro-oncogenic role for SnoN in mammalian tumorigenesis.
Introduction
The transforming growth factor-β (TGF-β) regulates cell proliferation, apoptosis, motility, differentiation, and extra-cellular matrix production (1). Upon ligand binding, the activated TGF-β receptor kinases phosphorylate Smad2 and Smad3, leading to their heterooligomerization with Smad4 and accumulation in the nucleus. The nuclear Smad complexes interact with coactivators and sequence-specific DNA binding factors to regulate the expression of TGF-β–responsive genes (2–5). SnoN is an important negative regulator of TGF-β signaling (6, 7). It interacts with Smad proteins and represses their transcription activity by disrupting the active heteromeric Smad complex, blocking the interaction of Smad2/3 with transcriptional coactivators and recruiting a transcriptional corepressor complex (6, 8–10).
SnoN was identified as a proto-oncogene that when overexpressed, induces the anchorage-independent growth of quail and chicken embryo fibroblasts (11–14). However, the role of SnoN in human cancer is more complex. Expression of SnoN is elevated in many human carcinomas (15, 16), due to gene amplification, increased protein stability, and transcriptional activation (6, 17–21). Reducing SnoN expression in human breast and lung cancer cells impairs mitogenic transformation in vitro and tumor growth in vivo (22), supporting a pro-oncogenic activity of SnoN. However, SnoN seems to also possess tumor-suppressive functions. Deletion of one copy of the sno gene resulted in increased susceptibility to carcinogen-induced tumor formation (23). More recently, SnoN has been shown to induce premature senescence and inhibit oncogenic transformation and tumorigenesis through upregulating PML and p53 levels (24). Finally, SnoN can also inhibit epithelial-to-mesenchymal transdifferentiation (EMT) and tumor metastasis (22). Thus, the roles of SnoN in mammalian tumorigenesis are complex and maybe context dependent. In addition to cancer cells, SnoN is also expressed ubiquitously in normal cells and tissues, but its function in normal epithelial cells has not been defined (24).
In this study, we used mammary gland as a model system to define the function of SnoN in development and carcinogenesis. The mammary gland consists of a rudimentary branching network of small ducts at birth and undergoes a specific pattern of morphogenesis and differentiation after puberty (25, 26). At puberty, marked growth and branching morphogenesis occur to form the ductal tree that fills the entire mammary fat pad. Extensive branching and alveolar growth occur during pregnancy, with functional differentiation leading to milk production and secretion during lactation. After weaning, the alveolar epithelium undergoes apoptosis and is remodeled back to the state of a mature virgin gland (25, 26). TGF-β signaling is known to play an important role in mammary gland development. All three isoforms, TGF-β1, TGF-β2, and TGF-β3, are expressed in the mammary epithelial cells in mice (27). They inhibit ductal growth, alveolar proliferation, and milk production, and induce apoptosis in the involuting gland (28–31). Consistent with this, mice expressing a dominant-negative type II TGF-β receptor (TβRII) or anti-sense TβRII, or conditional knockout of the TβRII gene in the mammary epithelium exhibit lobular-alveolar hyperplasia (32–35). Although inactivation of TβRII alone is not sufficient for the formation of mammary tumors, in a mouse model of breast cancer induced by the expression of polyoma middle T antigen [mouse mammary tumor virus (MMTV)-PyVmT; refs. 36, 37], it shortens the latency of breast cancer development and increases pulmonary metastasis (32, 38). Conversely, the expression of a constitutively active type I receptor (MMTV-ALK5 T204D or MMTV-TβR1 AAD) in the mammary gland increased the incidence of lung metastases when crossed with the MMTV-c-Neu mice (39, 40), suggesting a tumor-promoting role of TGF-β at late stages of tumorigenesis. TGF-β signaling also regulates postlactational involution, but the underlying mechanism is complex and poorly understood. Although mice expressing the dominant-negative TβRII showed delayed involution, knock out of TβRII displayed increased apoptosis in the developing mammary gland (32, 34). Moreover, overexpression of the constitutively active ALK5 T204D exhibited reduced apoptosis during involution (39). The mechanisms underlying these complex observations remain unclear. The role of the Smad proteins in mammary gland development and tumorigenesis has not been well defined because homozygous deletion of many Smad proteins is embryonically lethal. Smad3-null mice survived to adulthood without apparent defects in mammary gland development. However, these mice showed a delayed involution with a 30% reduction in apoptosis (41).
In normal mammary tissues and untransformed mammary epithelial cell lines, SnoN is expressed at a low level in comparison with that in breast cancer tissues and cells (22) and is predominantly cytoplasmic (42). However, how SnoN expression is regulated during mammary gland development and what roles it plays in this process is not clear, nor is the function of SnoN in breast cancer development in vivo understood. To address these questions, we generated a transgenic mouse expressing a SnoN fragment under the control of the MMTV promoter/enhancer. Using this mouse model, we investigated the functions of SnoN in various stages of mammary gland development and breast cancer progression.
Materials and Methods
Generation of transgenic mice
Exon 1 of human snoN cDNA encoding amino acid residues 1 to 366 was fused to the Flag tag, subcloned into pBKS-MMTV, and injected into the C57BL6/J embryos. For genotyping by Southern blotting, genomic DNA were digested with Pst1 and hybridized to a transgene-specific snoN probe. Alternatively, the snoN transgene was detected by PCR using the following primer pair: forward primer, 5′-CAGGTGATCTGTGAGAATGC-3′; reverse primer, 5′-CAGAACTCACTCAGACTGTG-3′.
Whole mount analysis, histology, and immunohisto-chemistry
For whole mount analysis, the right fourth inguinal mammary gland was fixed in Carnoy’s solution followed by staining with Carmine Alum (43). For anti-SnoN staining, paraffin-embedded sections were permeabilized with 20 μg/mL proteinase K, blocked, and stained with anti-SnoN (42). For proliferating cell nuclear antigen (PCNA) stain, paraffin-embedded sections were performed with anti-PCNA (1:200: PC-10; Lab vision) as previously described (43). To detect the SnoN transgene, 5-μm cryostat sections were fixed in 4% paraformaldehyde and stained with anti-Flag 1:500, M2, Sigma). Terminal deoxynucleotidyl transferase–mediated dUTP nick end labeling (TUNEL) assay was performed on the paraffin-embedded sections using the DeadEnd Fluorometric TUNEL system kit (Promega).
Western blotting
Frozen tissues including the thoracic second and third mammary glands were homogenized in lysis buffer (24) and centrifuged. One percent NP40 was added to the supernatant followed by an additional centrifugation for 15 minutes. Twenty to 50 μg of protein extracts were analyzed by Western blotting using antibodies specific for AKT 1/2 (H-136), Smad2/3 (Santa Cruz Biotechnology), p-AKT, p-Smad, phospo-signal transducers and activators of transcription 3 (p-STAT3), STAT 3 (Cell Signaling Technology), pSmad2 (generous gift from Aris Moustakas), α-Tubulin (Calbiochem), and actin (MP Biomedicals).
Growth inhibition assay
Primary mammary epithelial cells were isolated from virgin females or mammary tumors at 14 weeks of age (44, 45). Cells (7 × 104) were treated with TGF-β1 for 3 days and the growth of the cells was measured by cell counting (42). All experiments were performed in triplicates.
Mammary tumor development and lung metastasis
Mice were palpated twice weekly from 4 to 14 weeks of age for the development of mammary tumors (36). The time of initial tumor appearance was subjected to Kaplan-Meier analysis and plotted. Once the total tumor size reached 120 mm in diameter, mice were euthanized and the total weight of the tumors, spleen, and lung was determined. The length of survival periods for each mouse was recorded and plotted into a modified Kaplan-Meier survival curve. To analyze lung metastasis, the lungs were fixed in Bouin’s solution and metastatic nodules were counted. Significance of data represented by the Kaplan-Meier survival curves was determined using log-rank (Mantel-Cox) tests. All other P values for various quantifications were determined using unpaired Student’s t tests.
Reverse transcription-PCR
Frozen tissues were homogenized in RLT buffer and total RNA extracted (RNeasy Mini kit; QIAGEN). The following primers were used in reverse transcription-PCR (RT-PCR):
-
Total snoN levels:
forward (F): 5′-AGAGACTCTGTTTGCCCCAAGT-3′;
Reverse (R): 5′-CATGCTAAACTTCTCCTTCATTTC-3′.
-
PAI-1: F: 5′-GGGGCCGTGGAACAAGAATGAGAT-3′,
R: 5′-AGATGTTGGTGAGGGCGGAGAGGT-3′.
-
p21CIP1: F: 5′-CAACCCATCTGCATCCGTTTCA-3′,
R: 5′-GAGTGGGGACCATTCCTGTCTTCA-3′.
Results
Expression of SnoN during mammary gland development
We first examined the expression pattern of endogenous SnoN during mammary gland development by immnohisto-chemistry and RT-PCR. Mammary glands were obtained from virgin (8, 12, 14, 16, 18, and 20 wk), pregnancy (13.5 and 18.5 d), lactating (0.5 and 5 d), and involuting (day 1, 3, and 5 postweaning) mice, and immunostaining of the paraffin-embedded sections was performed with an anti-SnoN antibody that recognize the COOH terminus of SnoN (Fig. 1A–C; ref. 42). In parallel, total RNA was prepared and semiquantitative RT-PCR was performed to measure the expression of endogenous snoN (Fig. 1D). In contrast to the high levels of SnoN in the breast cancer tissues (data not shown), the level of SnoN was low in the virgin glands during puberty but slightly elevated at 20 weeks of age (Fig. 1A). During this period of time, SnoN was detected in the cytoplasm of the quiescent luminal epithelial cells of the ducts and in the proliferating epithelial cells of the lobuli and lateral ducts (Fig. 1A). SnoN expression remained similar in early to midpregnancy but increased significantly in late pregnancy and peaked at day 18.5 (Fig. 1B and D). It then decreased dramatically during lactation and became almost undetectable by day 1 of involution (Fig. 1C and D). Interestingly, SnoN levels increased again at day 3 of involution in the collapsing luminal epithelial cells and gradually returned to the basal level observed at puberty (Fig. 1C and D).
Figure 1.

Expression of SnoN during mammary gland development. SnoN expression in nulliparous mammary glands at 8, 12, and 20 wk of age (A); mammary glands at day 13.5 and 18.5 of gestation, and day 5 of lactation (B); glands at day 1, 3, and 5 of postlactational involution (C). Green, SnoN; blue, Hoechst. D, RT-PCR to measure endogenous SnoN transcript levels at different stages of mammary gland development. β-Actin was used as a loading control.
Thus, SnoN expression is highly dynamic during mammary gland development, suggesting that it may play a role in regulating epithelial differentiation, proliferation, and/or apoptosis.
Generation of the MMTV-SnoN transgenic mice
To examine the function of SnoN during mammary gland development, we generated transgenic mice overexpressing a SnoN fragment (amino acid residues 1–366) in the mammary gland. We chose this SnoN fragment because it is more stable than full-length SnoN due to the absence of lysine residues critical for ubiquitin-mediated degradation (20). In addition, SnoN(1-366) fully retains the transforming and differentiation activity of SnoN as well as the ability to bind to important cellular partners including the Smad proteins and PML (24). Thus, it is more potent than full-length SnoN in repressing TGF-β signaling. Transgenic mice expressing the Flag-tagged human SnoN(1-366) from the MMTV promoter were generated (Fig. 2A) and four transgenic founders were obtained. Expression of the Flag-SnoN(1-366) was confirmed by RT-PCR and Western blotting (Fig. 2A). Two of the founder lines, Tg1 and Tg2, expressed the transgene at high levels and displayed similar phenotypes. In subsequent experiments, only results from Tg1 are shown.
Figure 2.
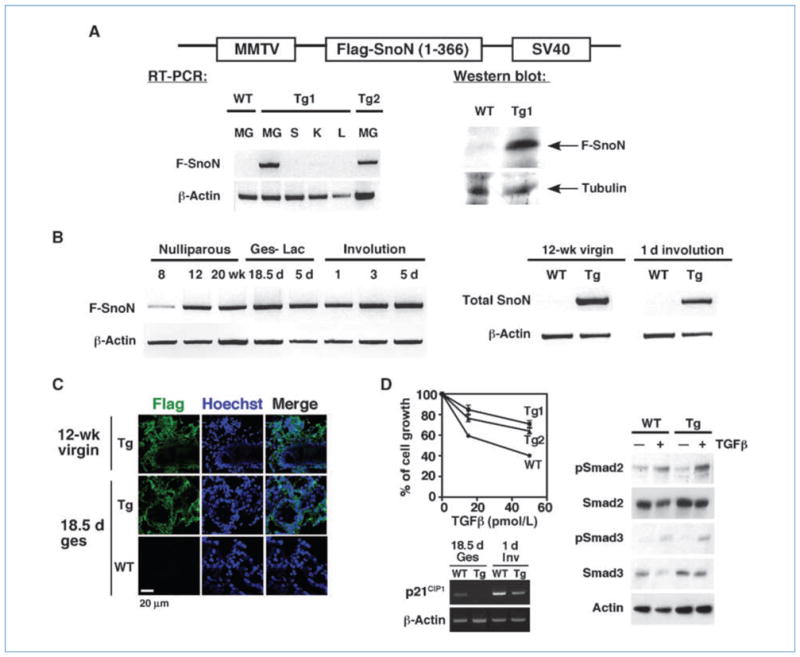
Generation of MMTV-SnoN transgenic Mice. A, top, the transgene construct. Left, RT-PCR measuring the expression of Flag-SnoN(1-366) in the mammary gland (MG), spleen (S), kidney (K), and liver (L) of WT or transgenic line #1 (Tg1) or #2 (Tg2) females. Bottom right, anti-Flag Western blotting of Flag-SnoN(1-366) isolated by immunoprecipitation from gestation day 18.5 mammary glands. Anti-α tubulin was used as a loading control. B, left, RT-PCR showing Flag-SnoN(1-366) levels at various stages of development as indicated. Ges, gestation; Lac, lactation. Right, comparison of total snoN levels between WT and Tg glands at the indicated stages by RT-PCR. C, Flag-SnoN(1-366) showed a similar intracellular localization as endogenous WT SnoN. Immunostaining with anti-Flag (green) was performed on frozen sections. Blue, Hoechst. D, left, growth inhibition assay. Primary mammary epithelial cells isolated from WT, Tg1, and Tg2 mice were incubated with 15 or 50 pmol/L TGF-β1. Top left, the growth of cells was measured and compared with that of unstimulated cells (as 100%). Bottom left, expression of p21CIP1 at 18.5 d of gestation (Ges) and 1 d of involution (Inv) was measured by RT-PCR. Right, Western blotting measuring phospho-Smad2 and phospho-Smad3 levels in total cell lysates prepared from WT or Tg mammary epithelial cells that have been treated with 200 pmol/L TGF-β1 for 30 min. Anti-actin blotting was performed as loading controls.
The expression of the snoN transgene was only detected in the mammary gland but not in spleen, kidney, and liver (Fig. 2A), confirming the tissue-specific expression. It was expressed in the virgin gland at 8 weeks, but this expression was significantly elevated at 12 weeks and remained high throughout pregnancy, lactation, and involution (Fig. 2B, left), consistent with the activity of the MMTV promoter. Using primers that detect both endogenous and transgenic snoN gene, we found that the transgene expression was markedly higher than the endogenous one in most stages of development, especially in the virgin and involuting glands (Fig. 2B, right). Similar to the endogenous SnoN, Flag-SnoN (1-366) was detected in the cytoplasm of mammary epithelial cells and was localized to the ducts and lobular structures during puberty, to the differentiated alveoli during pregnancy and lactation, and to the collapsing secretory epithelial cells during involution (data not shown; Fig. 2C). Thus, the SnoN transgene showed similar pattern and localization of expression as the endogenous protein.
To confirm that overexpression of SnoN antagonizes TGFβ signaling, primary epithelial cells isolated from wild-type (WT) and Tg females were treated with TGF-β1. As expected, although WT epithelial cells responded to TGF-β and were growth arrested, the transgenic cells were significantly less responsive (Fig. 2D, left). Consistent with this, expression of p21CIP1 (Fig. 2D, bottom left) and PAI-1 (data not shown), two TGF-β target genes, was also reduced in the Tg glands. In these cells, the levels of phospho-Smad2 and phospho-Smad3 were indistinguishable from that in the WT cells (Fig. 2D, right), confirming that SnoN affected downstream Smad signaling.
SnoN promotes lobular-alveolar proliferation and side branching
To investigate how SnoN affects branching morphogenesis, mammary glands from nulliparous transgenic and WT littermates were collected at 8, 12, 13, 14, 16, 18, and 20 weeks of age and subjected to whole mount and histologic analyses. The transgenic glands showed a moderate increase in side branching and lobular-alveolar hyperproliferation at 8 and 12 weeks of age (Fig. 3A), and the transgenic ductal branches filled a larger area of mammary fat pad. In addition, the transgenic glands showed a 2-fold increase in the number of primary ducts and side branches (secondary and tertiary branches and alveolar structures) at 8 weeks of age and a 3-fold increase in the number of side branches by 12 weeks (Fig. 3A). These increases were caused by enhanced proliferation as evidenced by an increase in PCNA (Fig. 3B), bromo-deoxyuridine (BrdUrd), and ki67 staining (data not shown), but not by alterations in apoptosis (data not shown). This lobular-alveolar hyperproliferation and increased side branching were maintained until 18 weeks (data not shown). However, by 20 weeks, this difference has disappeared (Fig. 3A, right). Thus, SnoN promotes epithelial proliferation in the virgin glands. Consistent with this, mammary glands from the SnoN knockout mice (−/−; ref. 46) displayed hypoplasia with reduced side branching and lobular-alveolar structures at 12 to 14 weeks than WT littermates (Fig. 3C).
Figure 3.
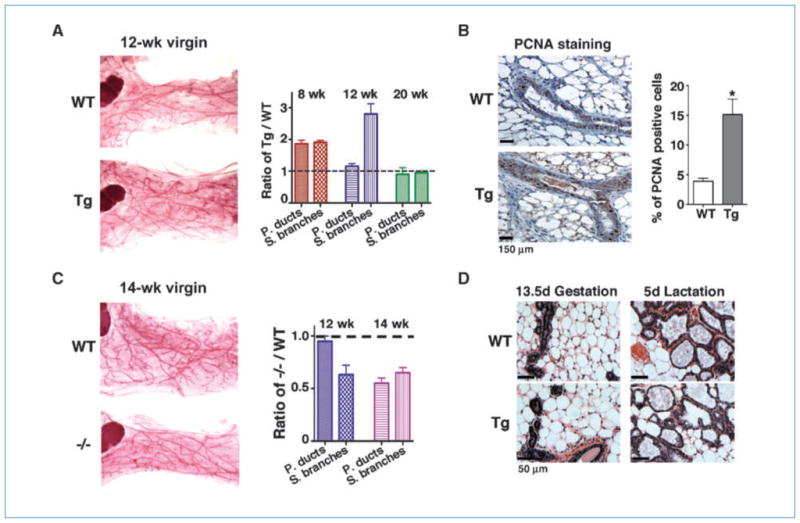
SnoN promotes lobular-alveolar proliferation in the virgin gland. A, left, representative whole mount images of mammary glands from 12-week-old nulliparous WT and Tg littermates. Right, the number of primary ducts and side branches were quantified from H&E sections from 8-wk-old (n = 4 pairs), 12-wk-old (n = 4 pairs), and 20-wk-old (n = 3 pairs) WT and Tg littermates. The data were plotted as the ratio between the Tg and WT glands; dotted line, the WT baseline. B, representative PCNA staining on terminal ducts at 12-wk virgin glands. Right, quantification of PCNA-positive nuclei in the WT and Tg glands (n = 3; P = 0.0140). One thousand nuclei from each mammary gland were counted. C, left, representative whole mount images of WT and SnoN −/− glands. Right, the number of primary ducts and side branches were quantified from H&E sections of 12-wk-old (n = 3) and 14-wk-old (n = 2) WT and −/− glands. Dotted line, WT baseline. D, representative H&E staining of WT and Tg mammary sections at the indicated stages.
During gestation and lactation, no significant difference in the epithelial content and lobular-alveolar structures was detected (Fig. 3D).
Thus, SnoN promotes mammary epithelial cell proliferation in the virgin gland, but does not affect the proliferation and differentiation during gestation and lactation in this transgenic strain.
SnoN accelerates involution
Postweaning involution consists of two distinct phases. The initial phase is triggered by milk accumulation and involves apoptosis of the secretory mammary epithelial cells during the first 48 hours. The second phase is sensitive to the decrease in lactogenic hormones and involves remodeling of alveolar structures, degradation of basement membrane, and replacement of the epithelial cells by adipose tissues (47). To assess the role of SnoN in involution, we performed a histologic analysis of the mammary glands at 1, 3, 5, and 11 days after weaning the pups at 19 days after birth. As shown in Fig. 4, SnoN accelerated involution at day 1 and day 3. The lobular-alveolar structures collapsed much faster in the transgenic mice than those of age- and litter size–matched WT mice (Fig. 4A, left), and roughly seven times more apoptotic bodies were found in the lumen of transgenic alveoli (Fig. 4B). The percentage of epithelial cells in the transgenic glands was 67% compared with 86% in the WT glands at day 1, and 37% compared with 64% at day 3 of involution (Fig. 4A, right). Consistent with this, TUNEL staining showed that more transgenic alveolar epithelial cells underwent apoptosis than WT cells at day 1 and 3 of involution (data not shown; Fig. 4C). However, by day 5 postweaning, the difference between the transgenic and WT glands had largely disappeared (Fig. 4A).
Figure 4.

SnoN accelerates involution. A, representative H&E staining of WT and Tg mammary gland sections at day 1, 3, and 5 post involution (d.p.i) after weaning at day 19 after birth. Right, quantification of the epithelial content in the involuting glands at 1 (P < 0.0001), 3 (P = 0.0026), and 5 (P = 0.4818) d.p.i. B, quantification of the apoptotic bodies in the lumen of collapsing alveoli at 1 d.p.i. Five sections around the lymph node from each gland were analyzed (n = 3 pairs; P = 0.0214). C, TUNEL stain of WT and Tg glands at 1 d.p.i. Right, quantification of apoptotic cells among 2,000 nuclei in each gland was shown to the right (n = 3 pairs, P = 0.0314). D, Western blotting analysis of mammary tissues at 1 d.p.i showed that the transgenic glands exhibited a reduced Akt activation.
Although unlikely, the acceleration of involution in the transgenic mice could be due to the early self-weaning of the pups before day 19. To exclude this possibility, we repeated the above experiment after force-weaning the pups at day 12 after birth. Under this condition, the transgenic glands still involuted faster than WT glands at days 1 and 3 of involution, with a lower epithelial content and more apoptotic bodies in the lumen of the transgenic alveoli (data not shown). Taken together, our results indicate that SnoN promotes postlactational involution. STAT3 and Akt are two pathways known to regulate early stages of involution (39, 48). Although the transgenic mice displayed accelerated involution at day 1 of involution, no significant increase in STAT3 phosphorylation was detected (Fig. 4D). Interestingly, a lower level of phosphorylated Akt was observed in the transgenic glands (Fig. 4D), suggesting reduced survival cues. This is consistent with the accelerated involution of the transgenic glands and also with the previous report that Akt may be activated by TGF-β signaling to regulate involution (39).
SnoN accelerates tumor development
Although SnoN promotes branching morphogenesis and lobular-alveolar hyperproliferation, overexpression of SnoN alone was insufficient to drive spontaneous tumor development for up to 24 months (data not shown). To determine whether high levels of SnoN accelerate breast cancer progression, we crossed the Tg1 mice with the MMTV-PyVmT mice that develop spontaneous breast cancer (36, 37). Development of breast cancer in the double PyVmT/SnoN mice was monitored and compared with that in PyVmT mice. Under our experimental conditions, the PyVmT mice developed tumors at a median age of 46 days with a median survival age of 109 days (Fig. 5A and B). Interestingly, the PyVmT/SnoN mice developed tumors at a median age of 38 days with a median survival period of 93 days (Fig. 5A and B). Not only did the PyVmT/SnoN mice develop tumors more rapidly, these tumors were also bigger and more aggressive than those in the PyVmT mice (Fig. 5C and D). Whole mount analysis showed that the PyVmT/SnoN glands at 8 weeks had more aggressive primary tumors and were filled with tumor nodules, in contrast to the PyVmT glands at the same age (Fig. 5C). Histologic analysis showed that both mice developed highly invasive mammary adenocarcinomas with no apparent differences in the primary tumors (data not shown).
Figure 5.

SnoN accelerates tumor development. A, Kaplan-Meier curve showed that mice expressing PyVmT/SnoN (n = 21) developed tumors with a median latency of 38 d, whereas those expressing PyVmT (n = 15) developed tumors with a medium latency of 46 d (P = 0.0002). B, a modified Kaplan-Meier survival curve showed that the PyVmT/SnoN females (n = 11) die at a median age of 93 d, whereas the PyVmT mice (n = 10) died at 109 d (P = 0.0002). C, whole mount image of 8-wk PyVmT and PyVmT/SnoN glands. D, total tumor weight of PyVmT/SnoN (n = 18) and PyVmT (n = 10) mice at 14 wk of age (P = 0.0218).
The PyVmT mice also developed pulmonary metastasis by 14 to 16 weeks of age. To determine the effects of SnoN on metastasis, we measured the lung and spleen weights in the PyVmT/SnoN mice. When sacrificed at the same time, the PyVmT/SnoN mice had a significantly higher wet lung weight than their PyVmT littermates, whereas no difference was observed in the mean wet spleen weight (data not shown). Ninety percent of the PyVmT/SnoN mice had pulmonary metastasis versus only 30% of the PyVmT mice at 13 to 14 weeks (Fig. 6A). The level of metastatic lesions in the PyVmT/SnoN mice was also increased significantly compared with that in the PyVmT mice, which had very few surface metastases if any at 13 to 14 weeks (Fig. 6B).
Figure 6.
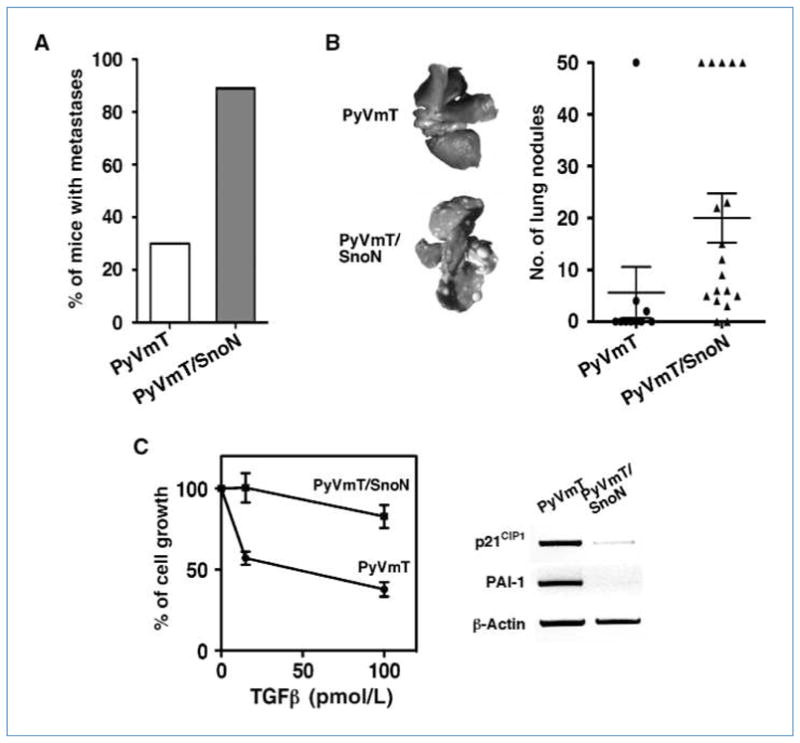
SnoN promotes pulmonary metastases. A, detection of lung metastasis at 14 wk of age in the PyVmT/SnoN (n = 18) and PyVmT females (n = 10). B, metastatic tumor nodules on the surface of the lungs were counted and showed in the graph. C, left, primary tumor cells isolated from PyVmT and PyVmT/SnoN mice were treated with 15 pmol/L or 100 pmol/L TGF-β1. The growth of cells was measured and compared with nontreated cells. Right, RT-PCR measuring PAI-1 transcript levels in the PyVmT and PyVmT/SnoN tumors at 14 wk.
Because high levels of SnoN can antagonize TGF-β signaling, we wondered whether the PyVmT/SnoN tumor cells displayed a reduced TGF-β signaling. Primary mammary tumor cells were isolated from either the PyVmT or PyVmT/SnoN mice and treated with TGF-β. As expected, TGF-β–induced phosphorylation of Smad2 or Smad3 was not significantly different between the two mice (data not shown). Although the PyVmT tumor cells were still sensitive to the antiproliferative effect of TGF-β, the PyVmT/SnoN cells were significantly more resistant (Fig. 6C, left). Moreover, expression of p21CIP1 and PAI-1 was also lower in the PyVmT/SnoN tumors, consistent with the reduced TGF-β responses in cells with high levels of SnoN (Fig. 6C, right).
Taken together, our results showed that high levels of SnoN accelerate mammary tumor growth in vivo and promote pulmonary metastases, possibly through repressing TGF-β signaling, resulting in increased proliferation and tumor growth.
Discussion
Although SnoN is expressed in all normal tissues and cells, its function has not been defined. Here, we show that SnoN expression is dynamically regulated during mammary gland development. It is expressed predominantly in the cytoplasm at a relatively low level in the virgin mammary gland and throughout all stages of development but is significantly up-regulated at late gestation/early lactation stages. This suggests that SnoN expression may be sensitive to hormonal signaling and may play important roles in mammary gland development. Indeed, using transgenic mice overexpressing a SnoN fragment in the mammary gland, we found that SnoN promotes mammary epithelial cell proliferation and accelerates postlactational involution. In the MMTV-PyVmT background, SnoN further accelerates tumor growth and pulmonary metastases. Thus, our study suggests that SnoN contains pro-oncogenic activity in vivo.
The observation that SnoN promotes mammary epithelial cell hyperproliferation is consistent with the phenotypes of mice lacking a functional TβRII in the mammary gland that showed increased proliferation and even hyperplasia (34, 35). Because SnoN can repress the Smad proteins, an obvious question is whether the effects of SnoN on proliferation and involution require antagonism of TGF-β/Smad signaling. The phenotypes of our transgenic mice as well as the SnoN knockout mice that displayed mammary hypoplasia are consistent with the Smad-dependent model of SnoN action. Although this is not sufficient to exclude the possibility that SnoN can also promote cell proliferation in a Smad-independent manner, it lends a strong support to the notion that the ability of SnoN to repress Smad signaling contributes significantly to the hyperproliferation phenotypes.
Our study revealed a novel function of SnoN in accelerating involution. Specifically, SnoN seems to affect the initial phase of involution that involves apoptosis of secretory epithelial cells in response to milk accumulation. Mutations in other TGF-β signaling components have also been shown to affect involution. In particular, deletion of TβRII results in an increase in apoptosis (34) and the constitutively active TβRI T204D delayed involution, due probably to an increase in phosphoinositide 3-kinase (PI3)/Akt–mediated survival signaling (39). These results suggested a prosurvival function of TGF-β during involution and are consistent with the accelerated apoptosis observed in our transgenic mice. Interestingly, our transgenic mice also displayed a decreased Akt activation during involution, suggesting that SnoN may reduce PI3K/Akt-mediated survival signaling. How SnoN affects Akt activation awaits future investigation. Interestingly, Akt activity was not altered in the transgenic virgin glands and the PyVmT/SnoN tumors (data not shown), indicating that SnoN may regulate AKT activity differently in the involuting glands versus virgin glands and tumors.
Both pro-oncogenic and antioncogenic roles of SnoN in mammalian tumorigenesis have been proposed. The pro-oncogenic activity of SnoN depends on its ability to repress TGF-β signaling, whereas its antioncogenic activity relies on its ability to activate p53 (24). Our result that SnoN accelerates PyVmT breast tumor growth and metastasis supports a pro-oncogenic activity of SnoN in vivo. It is worth noting that in this PyVmT breast cancer model, the p53 pathway is inactivated by the middle T antigen (49, 50). Thus, the antioncogenic branch of SnoN may have been silenced, allowing the pro-oncogenic activity of SnoN to be fully manifested. Supporting this idea, we found that although the PyVmT tumor cells are still responsive to TGF-β–induced growth inhibition, the PyVmT/SnoN tumor cells are not, suggesting that SnoN may promote the growth of these tumors through repressing TGF-β signaling.
Previously, we have reported that SnoN may inhibit epithelial-to-mesenchymal transdifferentiation and breast cancer metastasis using the MB-MDA-231 model (22). We did not observe any inhibition of metastasis in this transgenic mouse model. This could be due to differences in the mechanisms underlying metastasis between the MMTV-PyVmT mice and MB-MDA-231 model. In the MMTV-PyVmT model, tumor cells may metastasize “en masse” in a process dependent on the recruitment of bone marrow–derived proinflammatory cells to the tumor invasion front by the tumor-produced chemokines. TGF-β suppresses the production of these chemokines and thus inhibits metastasis of MMTV-PyVmT tumors (38). In contrast in the MB-MDA-231 cell–initiated metastasis model, the metastasis relies on epithelial-to-mesenchymal transdifferentiation and mobility of the breast cancer cells, a process enhanced by TGF-β (22). Thus, the complex roles of SnoN in breast cancer metastasis may depend on the metastasis models used and reflects the complexity of TGF-β signaling on tumorigenesis.
In summary, our study has provided the first direct evidence for the pro-oncogenic function of SnoN in vivo and revealed important functions of SnoN in mammary gland development. Future work will further determine the signaling mechanisms underlying these processes.
Acknowledgments
We thank Dr. Mina Bissell and Rana Mroue for their advices and support; Dr. Lin He and Margaux Bennett for helping with the PCNA staining; Drs. Steve Ruzin and Denise Schichnes for assistance with microscopy; Fangjiu Zhang, Christopher Hendrick, and Angeline Protacio for technical assistance; and Stanford transgenic facility for generating the transgenic mice.
Grant Support
NIH RO1 CA101891, Philip Morris External Research Program grant 019016 (K. Luo), and DOD BCRP predoctoral fellowship (N. Jahchan).
Footnotes
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Siegel PM, Massague J. Cytostatic and apoptotic actions of TGF-β in homeostasis and cancer. Nat Rev Cancer. 2003;3:807–21. doi: 10.1038/nrc1208. [DOI] [PubMed] [Google Scholar]
- 2.Attisano L, Wrana JL. Signal transduction by the TGF-β superfamily. Science. 2002;296:1646–7. doi: 10.1126/science.1071809. [DOI] [PubMed] [Google Scholar]
- 3.Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-β family signalling. Nature. 2003;425:577–84. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- 4.Massague J, Wotton D. Transcriptional control by the TGF-β/Smad signaling system. EMBO J. 2000;19:1745–54. doi: 10.1093/emboj/19.8.1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moustakas A, Pardali K, Gaal A, Heldin CH. Mechanisms of TGF-β signaling in regulation of cell growth and differentiation. Immunol Lett. 2002;82:85–91. doi: 10.1016/s0165-2478(02)00023-8. [DOI] [PubMed] [Google Scholar]
- 6.Stroschein SL, Wang W, Zhou S, Zhou Q, Luo K. Negative feedback regulation of TGF-β signaling by the SnoN oncoprotein. Science. 1999;286:771–4. doi: 10.1126/science.286.5440.771. [DOI] [PubMed] [Google Scholar]
- 7.Sun Y, Liu X, Ng-Eaton E, Lodish HF, Weinberg RA. SnoN and Ski protooncoproteins are rapidly degraded in response to transforming growth factor β signaling. Proc Natl Acad Sci U S A. 1999;96:12442–7. doi: 10.1073/pnas.96.22.12442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Akiyoshi S, Inoue H, Hanai J, et al. c-Ski acts as a transcriptional co-repressor in transforming growth factor-β signaling through interaction with smads. J Biol Chem. 1999;274:35269–77. doi: 10.1074/jbc.274.49.35269. [DOI] [PubMed] [Google Scholar]
- 9.Luo K, Stroschein SL, Wang W, et al. The Ski oncoprotein interacts with the Smad proteins to repress TGFβ signaling. Genes Dev. 1999;13:2196–206. doi: 10.1101/gad.13.17.2196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu JW, Krawitz AR, Chai J, et al. Structural mechanism of Smad4 recognition by the nuclear oncoprotein Ski: insights on Ski-mediated repression of TGF-β signaling. Cell. 2002;111:357–67. doi: 10.1016/s0092-8674(02)01006-1. [DOI] [PubMed] [Google Scholar]
- 11.Boyer PL, Colmenares C, Stavnezer E, Hughes SH. Sequence and biological activity of chicken snoN cDNA clones. Oncogene. 1993;8:457–66. [PubMed] [Google Scholar]
- 12.Nomura N, Sasamoto S, Ishii S, Date T, Matsui M, Ishizaki R. Isolation of human cDNA clones of ski and the ski-related gene, sno. Nucleic Acids Res. 1989;17:5489–500. doi: 10.1093/nar/17.14.5489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pearson-White S. SnoI, a novel alternatively spliced isoform of the ski protooncogene homolog, sno. Nucleic Acids Res. 1993;21:4632–8. doi: 10.1093/nar/21.19.4632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pearson-White S, Crittenden R. Proto-oncogene Sno expression, alternative isoforms and immediate early serum response. Nucleic Acids Res. 1997;25:2930–7. doi: 10.1093/nar/25.14.2930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang F, Lundin M, Ristimaki A, et al. Ski-related novel protein N (SnoN), a negative controller of transforming growth factor-β signaling, is a prognostic marker in estrogen receptor-positive breast carcinomas. Cancer Res. 2003;63:5005–10. [PubMed] [Google Scholar]
- 16.Medrano EE. Repression of TGF-β signaling by the oncogenic protein SKI in human melanomas: consequences for proliferation, survival, and metastasis. Oncogene. 2003;22:3123–9. doi: 10.1038/sj.onc.1206452. [DOI] [PubMed] [Google Scholar]
- 17.Bonni S, Wang HR, Causing CG, et al. TGF-β induces assembly of a Smad2-2 ubiquitin ligase complex that targets SnoN for degradation. Nat Cell Biol. 2001;3:587–95. doi: 10.1038/35078562. [DOI] [PubMed] [Google Scholar]
- 18.Edmiston JS, Yeudall WA, Chung TD, Lebman DA. Inability of transforming growth factor-β to cause SnoN degradation leads to resistance to transforming growth factor-β-induced growth arrest in esophageal cancer cells. Cancer Res. 2005;65:4782–8. doi: 10.1158/0008-5472.CAN-04-4354. [DOI] [PubMed] [Google Scholar]
- 19.Imoto I, Pimkhaokham A, Fukuda Y, et al. SNO is a probable target for gene amplification at 3q26 in squamous-cell carcinomas of the esophagus. Biochem Biophys Res Commun. 2001;286:559–65. doi: 10.1006/bbrc.2001.5428. [DOI] [PubMed] [Google Scholar]
- 20.Stroschein SL, Bonni S, Wrana JL, Luo K. Smad3 recruits the anaphase-promoting complex for ubiquitination and degradation of SnoN. Genes Dev. 2001;15:2822–36. doi: 10.1101/gad.912901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhu Q, Pearson-White S, Luo K. Requirement for the SnoN oncoprotein in transforming growth factor β-induced oncogenic transformation of fibroblast cells. Mol Cell Biol. 2005;25:10731–44. doi: 10.1128/MCB.25.24.10731-10744.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhu Q, Krakowski AR, Dunham EE, et al. Dual role of SnoN in mammalian tumorigenesis. Mol Cell Biol. 2007;27:324–39. doi: 10.1128/MCB.01394-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shinagawa T, Dong HD, Xu M, Maekawa T, Ishii S. The sno gene, which encodes a component of the histone deacetylase complex, acts as a tumor suppressor in mice. EMBO J. 2000;19:2280–91. doi: 10.1093/emboj/19.10.2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pan D, Zhu Q, Luo K. SnoN functions as a tumour suppressor by inducing premature senescence. EMBO J. 2009;28:3500–13. doi: 10.1038/emboj.2009.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hennighausen L, Robinson GW. Information networks in the mammary gland. Nat Rev Mol Cell Biol. 2005;6:715–25. doi: 10.1038/nrm1714. [DOI] [PubMed] [Google Scholar]
- 26.Sternlicht MD. Key stages in mammary gland development: the cues that regulate ductal branching morphogenesis. Breast Cancer Res. 2006;8:201. doi: 10.1186/bcr1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Robinson SD, Silberstein GB, Roberts AB, Flanders KC, Daniel CW. Regulated expression and growth inhibitory effects of transforming growth factor-β isoforms in mouse mammary gland development. Development. 1991;113:867–78. doi: 10.1242/dev.113.3.867. [DOI] [PubMed] [Google Scholar]
- 28.Bierie B, Moses HL. Tumour microenvironment: TGFβ: the molecular Jekyll and Hyde of cancer. Nat Rev Cancer. 2006;6:506–20. doi: 10.1038/nrc1926. [DOI] [PubMed] [Google Scholar]
- 29.Wakefield LM, Piek E, Bottinger EP. TGF-β signaling in mammary gland development and tumorigenesis. J Mammary Gland Biol Neoplasia. 2001;6:67–82. doi: 10.1023/a:1009568532177. [DOI] [PubMed] [Google Scholar]
- 30.Pierce DF, Jr, Johnson MD, Matsui Y, et al. Inhibition of mammary duct development but not alveolar outgrowth during pregnancy in transgenic mice expressing active TGF-β 1. Genes Dev. 1993;7:2308–17. doi: 10.1101/gad.7.12a.2308. [DOI] [PubMed] [Google Scholar]
- 31.Pierce DF, Jr, Gorska AE, Chytil A, et al. Mammary tumor suppression by transforming growth factor β 1 transgene expression. Proc Natl Acad Sci U S A. 1995;92:4254–8. doi: 10.1073/pnas.92.10.4254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gorska AE, Jensen RA, Shyr Y, Aakre ME, Bhowmick NA, Moses HL. Transgenic mice expressing a dominant-negative mutant type II transforming growth factor-β receptor exhibit impaired mammary development and enhanced mammary tumor formation. Am J Pathol. 2003;163:1539–49. doi: 10.1016/s0002-9440(10)63510-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lenferink AE, Magoon J, Pepin MC, Guimond A, O’Connor-McCourt MD. Expression of TGF-β type II receptor antisense RNA impairs TGF-β signaling in vitro and promotes mammary gland differentiation in vivo. Int J Cancer. 2003;107:919–28. doi: 10.1002/ijc.11494. [DOI] [PubMed] [Google Scholar]
- 34.Forrester E, Chytil A, Bierie B, et al. Effect of conditional knockout of the type II TGF-β receptor gene in mammary epithelia on mammary gland development and polyomavirus middle T antigen induced tumor formation and metastasis. Cancer Res. 2005;65:2296–302. doi: 10.1158/0008-5472.CAN-04-3272. [DOI] [PubMed] [Google Scholar]
- 35.Gorska AE, Joseph H, Derynck R, Moses HL, Serra R. Dominant-negative interference of the transforming growth factor β type II receptor in mammary gland epithelium results in alveolar hyperplasia and differentiation in virgin mice. Cell Growth Differ. 1998;9:229–38. [PubMed] [Google Scholar]
- 36.Guy CT, Cardiff RD, Muller WJ. Induction of mammary tumors by expression of polyomavirus middle T oncogene: a transgenic mouse model for metastatic disease. Mol Cell Biol. 1992;12:954–61. doi: 10.1128/mcb.12.3.954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guy CT, Muthuswamy SK, Cardiff RD, Soriano P, Muller WJ. Activation of the c-Src tyrosine kinase is required for the induction of mammary tumors in transgenic mice. Genes Dev. 1994;8:23–32. doi: 10.1101/gad.8.1.23. [DOI] [PubMed] [Google Scholar]
- 38.Bierie B, Stover DG, Abel TW, et al. Transforming growth factor-β regulates mammary carcinoma cell survival and interaction with the adjacent microenvironment. Cancer Res. 2008;68:1809–19. doi: 10.1158/0008-5472.CAN-07-5597. [DOI] [PubMed] [Google Scholar]
- 39.Muraoka-Cook RS, Shin I, Yi JY, et al. Activated type I TGFβ receptor kinase enhances the survival of mammary epithelial cells and accelerates tumor progression. Oncogene. 2006;25:3408–23. doi: 10.1038/sj.onc.1208964. [DOI] [PubMed] [Google Scholar]
- 40.Siegel PM, Shu W, Cardiff RD, Muller WJ, Massague J. Transforming growth factor β signaling impairs Neu-induced mammary tumorigenesis while promoting pulmonary metastasis. Proc Natl Acad Sci U S A. 2003;100:8430–5. doi: 10.1073/pnas.0932636100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang YA, Tang B, Robinson G, et al. Smad3 in the mammary epithelium has a nonredundant role in the induction of apoptosis, but not in the regulation of proliferation or differentiation by transforming growth factor-β. Cell Growth Differ. 2002;13:123–30. [PubMed] [Google Scholar]
- 42.Krakowski AR, Laboureau J, Mauviel A, Bissell MJ, Luo K. Cytoplasmic SnoN in normal tissues and nonmalignant cells antagonizes TGF-β signaling by sequestration of the Smad proteins. Proc Natl Acad Sci U S A. 2005;102:12437–42. doi: 10.1073/pnas.0504107102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bascom JL, Fata JE, Hirai Y, Sternlicht MD, Bissell MJ. Epimorphin overexpression in the mouse mammary gland promotes alveolar hyperplasia and mammary adenocarcinoma. Cancer Res. 2005;65:8617–21. doi: 10.1158/0008-5472.CAN-05-1985. [DOI] [PubMed] [Google Scholar]
- 44.Novaro V, Roskelley CD, Bissell MJ. Collagen-IV and laminin-1 regulate estrogen receptor α expression and function in mouse mammary epithelial cells. J Cell Sci. 2003;116:2975–86. doi: 10.1242/jcs.00523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fata JE, Mori H, Ewald AJ, et al. The MAPK(ERK-1,2) pathway integrates distinct and antagonistic signals from TGFα and FGF7 in morphogenesis of mouse mammary epithelium. Dev Biol. 2007;306:193–207. doi: 10.1016/j.ydbio.2007.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pearson-White S, McDuffie M. Defective T-cell activation is associated with augmented transforming growth factor β sensitivity in mice with mutations in the Sno gene. Mol Cell Biol. 2003;23:5446–59. doi: 10.1128/MCB.23.15.5446-5459.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Watson CJ. Involution: apoptosis and tissue remodelling that convert the mammary gland from milk factory to a quiescent organ. Breast Cancer Res. 2006;8:203. doi: 10.1186/bcr1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chapman RS, Lourenco PC, Tonner E, et al. Suppression of epithelial apoptosis and delayed mammary gland involution in mice with a conditional knockout of Stat3. Genes Dev. 1999;13:2604–16. doi: 10.1101/gad.13.19.2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Qian W, Kashuba E, Magnusson KP, et al. Role of p53 mutation in polyomavirus-induced tumorigenesis. J Gen Virol. 1997;78:893–903. doi: 10.1099/0022-1317-78-4-893. [DOI] [PubMed] [Google Scholar]
- 50.Qian W, Wiman KG. Polyoma virus middle T and small t antigens cooperate to antagonize p53-induced cell cycle arrest and apoptosis. Cell Growth Differ. 2000;11:31–9. [PubMed] [Google Scholar]