

Abstract
Background
The enzyme dihydropteroate synthase (DHPS) participates in the de novo synthesis of folate cofactors by catalyzing the formation of 7,8-dihydropteroate from condensation of p-aminobenzoic acid with 6-hydroxymethyl-7,8-dihydropteroate pyrophosphate. DHPS is absent from humans, who acquire folates from diet, and has been validated as an antimicrobial therapeutic target by chemical and genetic means. The bacterium Burkholderia cenocepacia is an opportunistic pathogen and an infective agent of cystic fibrosis patients. The organism is highly resistant to antibiotics and there is a recognized need for the identification of new drugs against Burkholderia and related Gram-negative pathogens. Our characterization of the DHPS active site and interactions with the enzyme product are designed to underpin early stage drug discovery.
Results
An efficient recombinant protein expression system for DHPS from B. cenocepacia (BcDHPS) was prepared, the dimeric enzyme purified in high yield and crystallized. The structure of the apo-enzyme and the complex with the product 7,8-dihydropteroate have been determined to 2.35 Å and 1.95 Å resolution respectively in distinct orthorhombic crystal forms. The latter represents the first crystal structure of the DHPS-pterin product complex, reveals key interactions involved in ligand binding, and reinforces data generated by other structural studies. Comparisons with orthologues identify plasticity near the substrate-binding pocket and in particular a range of loop conformations that contribute to the architecture of the DHPS active site. These structural data provide a foundation for hit discovery. An intriguing observation, an artifact of the analysis, that of a potential sulfenamide bond within the ligand complex structure is mentioned.
Conclusion
Structural similarities between BcDHPS and orthologues from other Gram-negative species are evident as expected on the basis of a high level of sequence identity. The presence of 7,8-dihydropteroate in the binding site provides details about ligand recognition by the enzyme and the different states of the enzyme allow us to visualize distinct conformational states of loops adjacent to the active site. Improved drugs to combat infections by Burkholderia sp. and related Gram-negative bacteria are sought and our study now provides templates to assist that process and allow us to discuss new ways of inhibiting DHPS.
Background
Dihydropteroate synthase (DHPS, EC: 2.5.1.15) catalyses the reaction of 6-hydroxymethyl-7,8-dihydropterin-pyrophosphate with p-aminobenzoic acid (p-ABA) to yield 7,8-dihydropteroate and pyrophosphate (Figure 1). In so doing the enzyme supports the biosynthesis of folate, a key metabolite required to support the synthesis of DNA, and proteins. The provision of folates, either by synthesis in plants and microorganisms or as acquired in the diet by mammals, supports life. The folate biosynthetic pathway is absent from humans and contains several highly valued drug targets for treatments of numerous infections [1,2]. The folate pathway consists of a number of enzymes in addition to DHPS, including: 6-hydroxymethyl-7,8-dihydropterin pyrophosphokinase (HPPK), dihydrofolate synthetase (DHFS) and dihydrofolate reductase (DHFR). Drugs that inhibit DHFR and DHPS are used in the treatment of infections by the apicomplexan parasites Plasmodium sp. and Toxoplasma gondii [2-4]. In these species DHPS is part of a bifunctional enzyme fused to HPPK [5].
Figure 1.

The reaction catalyzed by dihydropteroate synthase. 7,8-dihydropterin pyrophosphate reacts with p-aminobenzoic acid (p-ABA) to yield 7,8-dihydropteroate and pyrophosphate.
The Gram-negative aerobic bacterium Burkholderia cenocepacia is an opportunistic pathogen and a member of the Burkholderia complex, a closely related group of bacteria, which cause particular problems for cystic fibrosis patients [6]. Other members of the genus are also pathogenic; B. pseudomallei is the causal agent for melioidosis [7], a serious infection found primarily in South East Asia, and B. mallei is responsible for glanders, an infection of livestock [8]. A characteristic of Burkholderia species, and one that makes them particularly troublesome pathogens is that they are highly resistant to a wide range of antibiotics [9-12].
DHPS is a validated drug target for the treatment of diseases caused by bacteria and protozoan parasites [1,2]. Sulfonamides in particular are inhibitors of this enzyme and are used as antibiotics [13]. However, increasing levels of resistance to sulfonamides has been observed and there is a need for new drugs to compensate for this [9,14-17]. The value of accurate structural information to support early stage drug discovery is well recognized [18] and characterization of the active site of DHPS from pathogenic organisms has the potential to support the design of new treatments.
Structures of DHPS from Gram-negative and Gram-positive bacteria have been reported [19-23] and also the bifunctional HPPK-DHPS from Saccharomyces cerevisiae and Francisella tularensis [24,25]. The structural studies extend to characterization of the oxidized product analogue, pteroic acid, and a series of pterin derivatives in complex with Bacillus anthracis DHPS (BaDHPS) [21,26] and also a series of pterin derivatives, and a complex of Escherichia coli DHPS (EcDHPS) with sulfanilamide [19]. One structure, that of Thermus thermophilus DHPS complexed with p-ABA has also been solved [Protein Data Bank (PDB) ID: 2DZA]. One observation, reported in several studies [e.g. [20]] and made by our inspection of PDB entries is the pronounced conformational flexibility of loops around the active site. This has resulted in the omission of important residues from the structural models.
Here we report the expression, purification and crystallization of BcDHPS. We describe the crystal structures of the apo-enzyme, the first complex with the actual enzyme product 7,8-dihydropteroate, which produces a more complete view of the active site than most other structures, similarities and differences between DHPS structures, and discuss molecular features that might be exploited to derive novel inhibitors.
Results and Discussion
General comments
An efficient recombinant source of BcDHPS was prepared and a purification protocol established that resulted in approximately 20 mg of pure enzyme from one litre of bacterial culture. The purified protein was used in co-crystallization screens with the inhibitor sulphadoxine or the enzyme product 7,8-dihydropteroate. Distinct orthorhombic crystal forms were obtained. However, analysis of the crystals grown in the presence of sulphadoxine revealed only water molecules in the active site and this therefore represents the apo-BcDHPS structure determined to 2.35 Å resolution. The structure of BcDHPS complexed with 7,8-dihydropteroate was refined to 1.95 Å resolution. Initial phases for the product complex were obtained by molecular replacement calculations using the EcDHPS structure [PDB ID: 1AJ2, [19]], which has 44% amino acid sequence identity to BcDHPS, as the search model. The model for molecular replacement calculations to solve the apo-structure of BcDHPS was the product complex. BcDHPS comprises 292 amino acids. The apo-enzyme crystallized in space group C2221 with one molecule in the asymmetric unit. A number of residues could not be modeled in the electron density maps and so were omitted hence the model for this structure consists of 268 residues. The crystals for the complex structure display space group P212121 and there are two molecules, labeled A and B, in the asymmetric unit. Here also disorder was evident and several residues in each subunit are missing from the model. Subunit A consists of 280 residues and subunit B consists of 281. These subunits are similar in structure and superimpose with an r.m.s.d. of 0.34 Å (over 280 aligned Cα atoms). The two structures superimpose with an r.m.s.d. of 0.37 Å (over 268 aligned Cα atoms, superimposing subunit A of the complex structure). Such a close agreement suggests that there are no gross structural changes upon ligand binding but as will be explained, ligand binding promotes greater order in loops that contribute to the active site.
The Overall Structure and comparison with orthologues
BcDHPS displays a triosephosphate isomerase (TIM)-barrel fold, consisting of eight parallel β-strands surrounded by eight α-helices (Figure 2). At the N-terminus there is an antiparallel alignment of two short β-strands, termed Nβ1 and Nβ2, linked by a tight turn. This is a feature common to some other DHPS structures. We assign β1 as the first strand of the β8:α8 barrel. Residues 234-242 form a short α-helix inserted between β7 and α8 and is considered part of loop 7. Size-exclusion gel filtration indicates that BcDHPS is a dimer, of approximate mass 68 kDa, in solution (data not shown). The apo-enzyme structure has a single polypeptide chain in the asymmetric unit whilst the product complex has two. In the latter structure, chains A and B form a dimer stabilized by an interface with a surface area (~1,200 Å2) that is approximately 10% of that of a single subunit (~12,000 Å2). Residues on α9 of one subunit interact with residues on α6, α7 and α8 of the partner subunit. Nearly 40 amino acids from each subunit participate in around 20 direct hydrogen bonds and salt bridge interactions, plus numerous water mediated contacts between the subunits and The same dimer is formed in the apo-structure by the symmetry operation -x, y, -z + ½.
Figure 2.

The structure of BcDHPS and BcDHPS complex. The overall structure of the BcDHPS dimer with 7,8-dihydropteroate bound. α-helices are shown in red and β-sheets in blue, with C atoms of the ligand 7,8-dihydropteroate shown in yellow.
The alignment of the central β-strands results in the localization of the N-terminal segments of these strands at one end of the barrel and the C-terminal segments at the other. On the N-terminal side there are polypeptide segments that form loops connecting the α-helix to the β-strand that follows. These loops are relatively short and well defined in the electron density maps (data not shown). On the C-terminal side of the TIM-barrel, where the active site is located, there are longer, more flexible loops, called the C-terminal loops, that link a β-strand with the following α-helix. The shorter N-terminal loops of TIM-barrel structures are suggested to contribute stability of the protein fold whereas the C-terminal loops contribute to enzyme activity by creation of the active site [27]. The first C-terminal loop, linking β1 to α1 could not be modeled in either structure, due to poorly defined density, suggesting a significant degree of conformational flexibility. Modeling of loop 2, linking β2 to α2 was possible only in the case of the product complex. The increased order observed for the complex is consistent with the stabilizing effects of ligand binding, as loop 2 contributes to the active site, a feature that will be discussed later.
The overall sequence identity of BcDHPS with the enzyme from other Gram-negative species is in the region of 40-60% and the fold of BcDHPS and dimeric quaternary structure is consistent with that observed for DHPS from other species. Structural overlays of the BcDHPS subunit with orthologous structures identified greatest similarity to E. coli DHPS with r.m.s.d. values of 1.35-1.37 Å [PDB IDs: 1AJ0, 1AJZ and 1AJ2, values given for the complex subunit A, over 264, 264 and 263 aligned Cα atoms]. R.m.s.d. values of 2.1 Å (for both subunit A and B, over 242 (A) and 239 (B) aligned Cα atoms) were observed for DHPS structures from Streptococcus pneumoniae [PDB IDs: 2VEF and 2VEG] and 1.5 Å (value for subunit A, over 239 aligned Cα atoms) for the Mycobacterium tuberculosis DHPS structure [PDB ID: 1EYE]. The orientation of DHPS subunits with respect to each other varies [20] but there is no evidence to suggest that this has an influence on activity. The DHPS active site is formed entirely within a single subunit. The difference in subunit orientation may simply reflect sequence variation between species. The residues that occur at the dimer interface (on α6, α7, α8 and α9) show only a low level of sequence conservation (Figure 3).
Figure 3.

Sequence alignment of three dihydropteroate synthases. The amino acid sequences for DHPS from B. cenocepacia, E. coli and S. aureus are aligned. The secondary structure relating to BcDHPS is shown colored as in Figure 2. Those residues shaded in black are conserved in all three sequences and grey marks conservation in two of the three.
The flexible C-terminal loops are the regions where the most structural variation between DHPS models has been observed. Of particular note are loops 1 and 2. The amino acid residues that comprise these loops are highly conserved in DHPS orthologues (Figure 3) but they are absent from the majority of published crystallographic models. Where the electron density maps have allowed for their inclusion it is noted that the polypeptides display different conformations. This points to a high propensity to disorder due to flexibility and an important role in enzyme function. One role for these loops has been proposed, namely that they close over and shield the active site from bulk solvent during catalysis [20]. To this we would add a potentially important thermodynamic contribution to the enzyme function. Changes in conformational entropy associated with distinct configurations of the loop structures might contribute to the free energy of protein-ligand association [28]. We note the presence of conserved glycine residues at the C-terminal end of β1 and on loop 2 itself (Figure 3) that may contribute to the flexibility in this part of the structure. Loop 2 is closer than loop 1 to the enzyme active site and adjacent to loop 5 (Figure 4). The residues that constitute loop 5 are less well conserved (Figure 3). BcDHPS has 17 residues in this loop, a similar number, 16, are present in the E. coli enzyme, whilst Staphylococcus aureus DHPS has only 9 residues in the loop (Figure 3). This lack of conservation in loop 5 and interactions with the conformationally labile loop 2 likely contributes to the variation observed when different DHPS structures are compared (Figure 4).
Figure 4.

The variations in the positions of active site loops across three DHPS structures. Superposition of the structures of BcDHPS with 7,8-dihydropteroate bound (blue), EcDHPS with 7,8-dihydropterin pyrophosphate bound (green, [PDB ID: 1AJ2]) and SaDHPS with 6-hydroxymethylpterin-diphosphate bound (violet, [PDB ID: 1AD4]).
As an aside we mention the following observation made by a reviewer. At the C-terminal end of β1, leading into loop 1 the amino acid sequence in BcDHPS Asn28-Ala29-Thr30. In EcDHPS and SaDHPS the sequence is Asn-Val-Thr. This is a well-recognized N-glycosylation signal, Asn-X-Ser/Thr [29]. However, in each case DHPS is a cytosolic bacterial enzyme from organisms lacking in glycosides.
Binding of 7,8-dihydropteroate
The active site of DHPS is positioned at the C-terminal end of the central β-barrel. Here, the product of the enzyme reaction, 7,8-dihydropteroate is ordered (Figure 5), placed with the pterin directed down into the barrel with the p-ABA moiety pointing out towards the surface of the protein (Figure 2). Residues located on strands β3, β4, β6, β7 and β8 participate in interactions with the product. The interactions formed between the enzyme and the ligand are very similar in both subunits and therefore only details of subunit A are presented (Figure 6) unless stated otherwise.
Figure 5.

The omit electron density associated with 7,8-dihydropteroate. The blue chicken wire represents the Fo-Fc omit map contoured at 2.5σ. The ligand is colored according to atom type; C yellow, N blue, O red.
Figure 6.

The BcDHPS binding site of 7,8-dihydropteroate. Potential hydrogen bonds formed between N and O atoms are depicted as blue dashed lines. C-H•••O interactions involving C12 as black dashed lines. The ligand is colored as in Figure 5. C atoms of DHPS are colored gray, the S atom of Met144 yellow.
The ligand possesses nine functional groups of which eight participate in direct hydrogen bonds with DHPS. Four hydrogen bond donors, N2, N3, N8 and N14 interact with functional groups on Asn120, Asp191, Asp101 and Thr68 respectively. The N1 atom of the ligand accepts a hydrogen bond donated by Asn120 ND2. On the other side of the pterin, O4 and N5 accept hydrogen bonds donated from Lys233 NZ. This lysine side chain is also held in place by a hydrogen bond donated to the carbonyl oxygen of Gly229. An ordered water molecule interacts with O4 and Asp191 OD1. A carboxylate oxygen, on the p-ABA moiety, accepts hydrogen bonds donated from the main chain amide and side chain hydroxyl groups of Ser234. In subunit A the other oxygen interacts with two water molecules that in turn interact with the carbonyl and amide groups of Gly195 and Met235 respectively (data not shown). In subunit B only the latter interaction is observed. The proximity of C7 to the carbonyl oxygen and OG1 hydroxyl of Thr68, distances range from 3.1 Å to 3.5 Å in subunits A and B, indicates the potential for C-H•••O hydrogen bonds. These are weak but not insignificant stabilizing interactions [30]. The E. coli DHPS structure [PDB ID: 1AJ2] with 7,8-dihydropterin pyrophosphate bound in the active site, shows Thr62 (the equivalent of Thr68 in BcDHPS) interacting with the phosphate groups of the ligand [19]. In our BcDHPS structure Thr68, part of loop 2, interacts with Asp101 and the dihydropterin. This suggests that the conformation of loop 2 alters depending on which ligand is bound in the active site.
The pterin is sandwiched between a cluster of hydrophobic residues (Ile122, Met144 and Phe196) on one side (Figure 6) and the guanidinium group of Arg268 on the other (data not shown). A salt-bridge interaction between Arg268 and Asp62 helps to determine the structure in this part of the active site and Asp62 also interacts with the side chain of Asn120 (data not shown).
The structure of BaDHPS in complex with pteroic acid [21] shares most of the key interactions described above. A notable difference between pteroic acid and 7,8-dihydropteroate is that N8 in the latter is a hydrogen bond donor, in the former a hydrogen bond acceptor. The distances between N8 and the carboxylate of the conserved Asp101 in both structures are close to 3.2 Å. This suggests to us that in the BaDHPS ligand complex Asp101 is likely protonated.
Sulfonamides, which are used to treat infections by Burkholderia [31], inhibit DHPS by binding in the p-ABA binding site, as shown through competition, resistance and structural data [19,20,22]. The complex of EcDHPS with sulfanilamide showed the ligand positioned between Lys221 and Arg63. This is similar to the placement of the p-ABA groups in the pteroic acid and 7,8-dihydropteroate complexes, which are near to loop 2. Mutations in residues that contribute to loop 2 have been noted to correlate with resistance to sulfonamide and dapsone; in particular the equivalent residues to Thr68 in BcDHPS [19,20]. The alignment of three bacterial DHPS sequences (Figure 3) shows that positions equivalent to 234 in our structure are occupied either with serine or arginine. Given that Ser234 interacts with the p-ABA moiety the difference between serine and arginine has interesting implications regarding differences in ligand binding sites across species.
The availability of high-resolution crystal structures offers opportunities to exploit structure-based methods to derive information on DHPS inhibitors that would support early stage drug discovery. The pterin binding pocket of DHPS is highly conserved across species. In the enzyme:product complex for BcDHPS we described the contributions of 14 amino acids above; ten of these residues are strictly conserved (Figure 3) and two, Gly229 and Met235, only use main chain functional groups to bind the 7,8-dihydropteroate. The structures clearly reveal the necessity for a planar entity and the arrangement of hydrogen bond donors and acceptors required to optimize interactions in the pterin binding pocket. The presence of an ordered water molecule mediating interactions between the ligand the enzyme suggests a position where a new hydrogen bonding interaction might be sought. Unfortunately the disorder (flexibility) of loop 2 means that many of the crystal structures have limitations with respect to drug design. We were fortunate that the product complex of BcDHPS allowed us to model loop 2 and the complete structure we obtained provides an improved template suited for in silico screening. We recognize that there is flexibility around the edge of the active site part of the binding site and that this flexibility might make an important thermodynamic contribution to DHPS function. This observation suggests that ligands able to interact with and reduce conformational freedom of loop 2 have the potential to increase the free energy of ligand binding, as might substrate, and these could provide suitable chemical matter for future development.
A Potential Cys-Arg Sulfenamide Bond
During refinement of the BcDHPS:dihydropteroate complex we noted an unusual feature in, first the electron density surrounding the side chains of Cys258 and Arg262, and then in omit difference maps that is worthy of mention. The density suggested the possibility of a covalent linkage, possibly a sulfenamide bond between Cys258 SG and Arg262 NH2 (Figure 7). The crystallographic analysis of the apo-enzyme clearly showed the normal side chains not interacting with each other (Figure 8). Our first assessment was that conformational flexibility had produced a mixture of rotamers that were overlapping. However, there is no evidence of an alternative rotamer for either residue. We were surprised that the feature was conserved in each subunit of the asymmetric unit and sought data to identify if an unusual covalent linkage was present. The analysis of freshly purified protein solution using Fourier Transform Ion Cyclotron Resonance mass spectrometry and Top-Down fragmentation failed to find any evidence for an unusual S-N covalent linkage. Similarly analysis of DHPS crystals, from the same batch of protein that gave the complex structure, by trypsin cleavage followed by mass spectrometry did not find any evidence to substantiate S-N bond formation.
Figure 7.
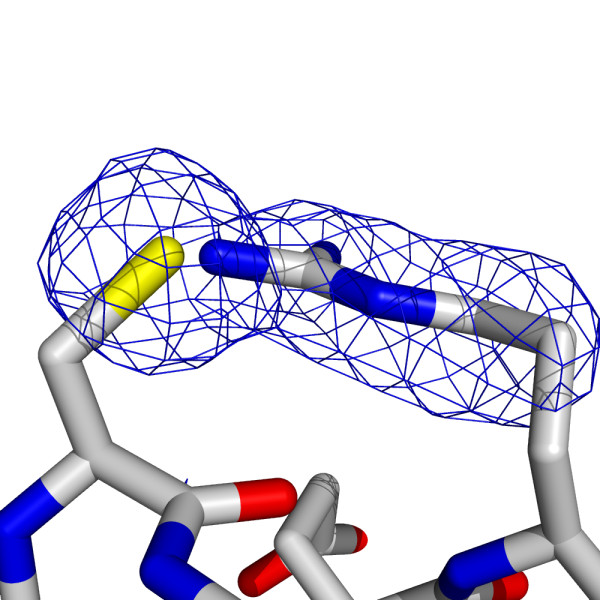
The positions of Cys258 and Arg262 in the product complex. The blue chicken wire represents the Fo-Fc omit map contoured at 3.5σ.
Figure 8.
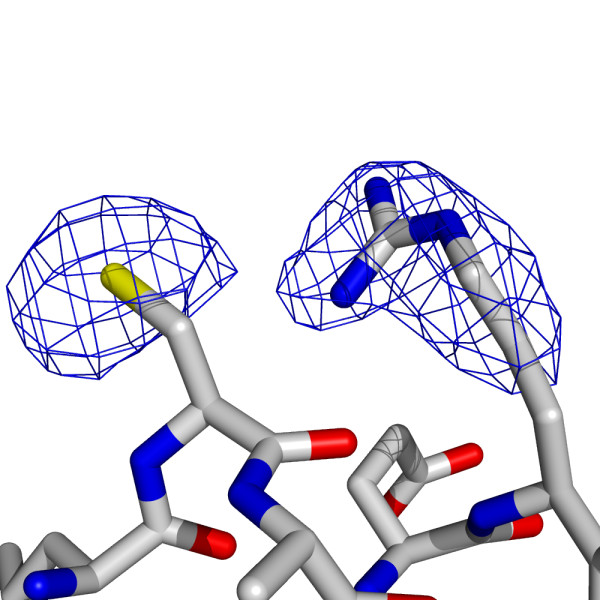
The positions of Cys258 and Arg262 in apo-BcDHPS. The blue chicken wire represents the Fo-Fc omit map contoured at 3.5σ.
These residues are located towards the C-terminal end of α8, at the surface of the protein and contribute to the dimer interface. The residues are not conserved in orthologues, and any interaction is unlikely to be physiologically relevant. A covalent modification may have occurred due to the experimental conditions in this particular instance and would likely have followed from oxidation of the cysteine.
Conclusion
The structure of B. cenocepacia DHPS has been solved in the presence and absence of a product from the reaction that it catalyses, 7,8-dihydropteroate. These structures closely resemble published DHPS structures from other species. Loop 2 has been modeled in the BcDHPS product complex. This loop, which is highly conserved in terms of amino acid sequence and inherently flexible, makes important contributions to the creation of the active site. The structure of the BcDHPS:7,8-dihydropteroate complex, with loop 2 modeled provides a useful template to aid the design of new inhibitors of this enzyme.
Methods
Protein expression and purification
The gene encoding BcDHPS [UniProtKB Q1BXC8] was amplified from genomic DNA (Belgian Co-ordinated Collections of Micro-organisms, bacteria collection (BCCM/LMG)) using 5'-CATATGTCCCCGTTCCTTCCCGC-3' and 5'-GGATCCTTATCGTTGCCGCGCGGCT-3' as the forward and reverse primers respectively (Thermo Scientific). The restriction sites for NdeI and BamHI are in bold. The PCR product was ligated into pCR2.1-TOPO using the TOPO® TA Cloning® Kit (invitrogen) and cloned into a modified pET15b (Novagen) expression vector. This vector produces the protein with a N-terminal hexa-His tag with a Tobacco Etch Virus (TEV) protease cleavage site. DNA sequencing confirmed the identity of the construct and the vector was heat-shock transformed into E. coli BL21 (DE3) cells for expression.
Cells were grown at 37°C in 1 L of Luria-Bertani media supplemented with carbenicillin (50 μgL-1). Gene expression was induced, once an OD600 of 0.6 was reached, by addition of 1 mM isopropyl-β-D-thiogalactopyranoside. The culture was maintained for a further 16 hours at 22°C. Cells were harvested by centrifugation at 3,500 g for 30 min at 4°C.
The cells were resuspended into a lysis buffer (20 mL, 50 mM Tris-HCl, pH 7.5, 250 mM NaCl, 25 mM imidazole) containing DNAse I (0.2 mg) and two tablets of a cocktail of EDTA-free protease inhibitors (Roche) and lysed using a French press at 16,000 psi. The resultant lysate was centrifuged at 35,000 g for 30 min at 4°C and the supernatant loaded onto a pre-equilibrated HisTrap HP 5 mL column (GE Healthcare) pre-charged with Ni2+. A linear gradient of imidazole, 25 mM to 1 M was applied to elute BcDHPS and the fractions were analyzed by sodium dodecyl sulfate polyacrylamide gel electrophoresis. The BcDHPS containing samples were pooled and incubated with His-tagged TEV protease (2 mg) at 30°C for 3 hours and then dialyzed into 50 mM Tris-HCl, pH 7.5, 250 mM NaCl. The protein was loaded onto a HisTrap HP 5 ml column and eluted as described above, untagged BcDHPS did not bind to the column. A Superdex 200 26/60 size exclusion column (GE Healthcare) equilibrated with 50 mM Tris-HCl, 250 mM NaCl pH 7.5 was used to further purify the protein. This column had previously been calibrated with molecular weight standards, blue dextran (>2,000 kDa), thyroglobulin (669 kDa), ferritin (440 kDa), aldolase (158 kDa), conalbumin (75 kDa), ovalbumin (43 kDa), carbonic anhydrase (29.5 kDa), ribonuclease A (13.7 kDa) and aprotinin (6.5 kDa); (GE Healthcare; data not shown).
The protein eluted with one peak of a mass of approximately 68 kDa corresponding to a dimer. Fractions containing the protein were pooled and concentrated using Amicon Ultra devices (Millipore) to 30 mg L-1 for subsequent use. The purity and identity of the protein was further confirmed by matrix-assisted laser desorption/ionization mass spectrometry, with a yield of approximately 20 mg L-1 of cell culture.
Crystallization and data collection
Crystallization conditions were screened using the sitting drop vapor diffusion method in 96-well plates. This was achieved with a Phoenix liquid handling system (Rigaku, Art Robins Instruments) and the commercially available screens Classics (Qiagen) and JCSG+ (Molecular Dimensions). Optimized conditions were achieved using hanging drop vapor diffusion methods with drops consisting of 1 μL protein solution and 1 μL reservoir solution.
Crystals of apo-BcDHPS were obtained with reservoir conditions of 0.3 M Tris-HCl, pH8 and 10% polyethylene glycol (PEG) 8000 and protein solution with BcDHPS at 7.5 mg mL-1 and sulphadoxine (2 mM; Sigma-Aldrich). Rectangular crystals with approximate dimensions 0.2 mm × 0.2 mm × 0.15 mm grew in one week. A specimen was transferred to a mixture of reservoir solution and 40% PEG 400, for approximately 15 seconds, then transferred into a stream of cold nitrogen (-170°C) and a data set collected.
Crystals of the complex were obtained with reservoir conditions of 0.1 M Tris-HCl pH 8, 10% PEG 8000, 0.3 M MgCl2. The protein solution (7.5 mg mL-1) was incubated at 4°C for 4 hours with 7,8-dihydropteroate (2 mM; Schircks Laboratories). The crystals consisted of multiple plates with dimensions 0.2 mm × 0.2 mm × 0.10 mm. A fragment of a crystal, of dimensions 0.1 mm × 0.1 mm × 0.05 mm. was transferred to a mixture of the reservoir solution and 25% ethylene glycol prior to cooling. The samples diffracted well and a data set was collected.
Data for both crystals were collected in-house with a Micromax 007 rotating anode generator (copper Kα, λ = 1.5418 Å) and R-AXIS IV++ dual image plate detector (Rigaku). Processing and scaling of the data was carried out using MOSFLM [32] and SCALA [33]. The apo-BcDHPS crystal belonged to space group C2221, with unit cell parameters a = 73.9 Å, b = 89.43 Å, c = 87.60 Å, α = β = γ = 90 °. There is one subunit in the asymmetric unit with VM values of 2.3 Å3 Da-1 and a solvent content of approximately 50%.
The crystal of the BcDHPS:7,8-dihydropteroate complex displayed space group P212121, with unit cell parameters a = 73.66 Å, b = 86.92 Å, c = 88.79Å, α = β = γ = 90 °. Two subunits in the asymmetric unit gives VM of 2.3 Å3 Da-1 and solvent content of approximately 50%. The complex structure was solved in PHASER [34] using molecular replacement with the E. coli DHPS structure [PDB ID: 1AJ2]. The structure of apo-BcDHPS was solved using the complex structure for molecular replacement. Alteration of the model, including addition of solvent molecules was achieved in COOT [35]. The model was refined using REFMAC5 [36] with non-crystallographic symmetry (NCS) restraints in the initial rounds of refinement. Translation, libration, screw analysis (TLS) was also applied [37]. Crystallographic statistics are presented in Table 1. R.m.s.d. values comparing the previously published structures were achieved using DALI [38]. The figures were prepared with PyMOL [39]. Amino-acid sequence alignments were carried out using the program MUSCLE [40]. The analysis of the dimer interface used the PISA server [41].
Table 1.
Crystallographic statistics
| Apo-BcDHPS | BcDHPS complex | |
|---|---|---|
| Data Collection | C2221 | P212121 |
| Unit Cell (Å) | 73.9 89.43 87.60 | 73.66 86.92 88.79 |
| Resolution Range (Å) | 57.25-2.35 (2.48-2.35) | 29.60-1.95 (2.001-1.95) |
| Number of Reflections | 84141 | 183983 |
| Number of Unique Reflections | 12564 | 41750 |
| Completeness (%) | 99.5 (97.1) | 98.9 (93.6) |
| Multiplicity | 6.7 (4.2) | 4.4 (3.7) |
| Rmerge (%)† | 11.7 (36.4) | 5.4 (36.9) |
| <I/s(I)> | 10.0 (2.4) | 14.4 (3.1) |
| Refinement | ||
| Rwork‡ | 0.208 | 0.176 |
| Rfree§ | 0.274 | 0.211 |
| r.m.s.d. from ideal values, bond lengths (Å) | 0.0083 | 0.0109 |
| r.m.s.d. from ideal values, bond angles (°) | 2.1597 | 1.2320 |
| DPI* (Å) | 0.3917 | 0.1496 |
| Ramachandran analysis | ||
| Residues in favored regions (%) | 95.4 | 96.7 |
| Residues in allowed regions (%) | 0.4 | 1.3 |
| Model Statistics | ||
| Wilson B-factor (Å2) | 47.7 | 27.4 |
| Protein residues: | ||
| No. in subunit A & B | 268 | 280 281 |
| B-factor A & B (Å2) | 39.5 | 27.8 30.4 |
| Additional groups: | ||
| Ligand B-factor in subunit A & B | NA | 31.4 33.1 |
| Water No./B-factor | 89/35.9 | 324/21.9 |
| Ethylene glycol No./B-factor | 4/35.5 | 4/38.7 |
| Chloride No./B-factor | NA | 1/34.3 |
Values in parentheses refer to the highest resolution shell. †Rmerge = ∑hkl∑i|Ii(hkl) - <I(hkl)>|/ ∑hkl∑i Ii(hkl); where Ii(hkl) is the intensity of the ith measurement of reflection hkl and <I(hkl)> is the mean value of Ii(hkl) for all i measurements. ‡Rwork = ∑hkl||Fo|-|Fc||/∑|Fo|, where Fo is the observed structure factor and Fc is the calculated structure factor. §Rfree is the same as Rcryst except calculated with a subset, 5%, of data that are excluded from refinement calculations. *The diffraction-component precision index as defined by Cruickshank [42].
Protein Data Bank accession numbers
Coordinates and structure factors have been deposited with accession code 2Y5J (apo-enzyme) and 2Y5S (product complex).
Abbreviations
E.C.: Enzyme Commission; HPPK: 6-hydroxymethyl-7,8-dihydropterin pyrophosphokinase; PEG: polyethylene glycol; PDB: Protein Data Bank; r.m.s.d.: root mean square deviation; DHPS: dihydropteroate synthase TEV: Tobacco Etch Virus; TLS: Translation/Libration/Screw.
Authors' contributions
REM collected data, solved and refined the structure of the complex, completed refinement of the apo-structure. Investigated the Cys-Arg bond and drafted the manuscript. GOB cloned, expressed, purified and crystallized the DHPS complex. JMD crystallized apo-DHPS, collected data and contributed to the refinement. VAR prepared the expression system. TCE aided the refinement of the complex structure. WNH conceived of the study, contributed to data analysis and checking, and manuscript completion. All authors read and approved the final manuscript.
Contributor Information
Rachel E Morgan, Email: r.e.morgan@dundee.ac.uk.
Gaëlle O Batot, Email: g.batot@gmail.com.
Jennifer M Dement, Email: j.m.dement@dundee.ac.uk.
Vincenzo A Rao, Email: v.a.rao@dundee.ac.uk.
Thomas C Eadsforth, Email: t.eadsforth@dundee.ac.uk.
William N Hunter, Email: w.n.hunter@dundee.ac.uk.
Acknowledgements
This work was funded by The Wellcome Trust [grants 082596 and 083481] and the European Commission Seventh Framework Programme (FP7/2007-2013), the Aeropath project. We thank Dr D. J. Clarke at The University of Edinburgh and Dr D. J. Lamont and K. Beattie at The University of Dundee, for mass spectrometry investigations into the potential cysteine-arginine bond.
References
- Bermingham A, Derrick JP. The folic acid biosynthesis pathway in bacteria: evaluation of potential for antibacterial drug discovery. Bioessays. 2002;24:637–648. doi: 10.1002/bies.10114. [DOI] [PubMed] [Google Scholar]
- Nzila A. Inhibitors of de novo folate enzymes in Plasmodium falciparum. Drug Discov Today. 2006;11:939–44. doi: 10.1016/j.drudis.2006.08.003. [DOI] [PubMed] [Google Scholar]
- Montoya JG, Liesenfeld O. Toxoplasmosis. Lancet. 2004;363:1965–1976. doi: 10.1016/S0140-6736(04)16412-X. [DOI] [PubMed] [Google Scholar]
- Aspinall TV, Joynson DH, Guy E, Hyde JE, Sims PF. The molecular basis of sulfonamide resistance in Toxoplasma gondii and implications for the clinical management of toxoplasmosis. J Infect Dis. 2002;185:1637–1643. doi: 10.1086/340577. [DOI] [PubMed] [Google Scholar]
- Triglia T, Cowman AF. Primary structure and expression of the dihydropteroate synthetase gene of Plasmodium falciparum. Proc Natl Acad Sci USA. 1994;91:7149–7153. doi: 10.1073/pnas.91.15.7149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George AM, Jones PM, Middleton PG. Cystic fibrosis infections: treatment strategies and prospects. FEMS Microbiol Lett. 2009;300:153–164. doi: 10.1111/j.1574-6968.2009.01704.x. [DOI] [PubMed] [Google Scholar]
- Wiersinga WJ, van der Poll T, White NJ, Day NP, Peacock SJ. Melioidosis: insights into the pathogenicity of Burkholderia pseudomallei. Nat Rev Microbiol. 2006;4:272–282. doi: 10.1038/nrmicro1385. [DOI] [PubMed] [Google Scholar]
- Whitlock GC, Estes DM, Torres AG. Glanders: off to the races with Burkholderia mallei. FEMS Microbiol Lett. 2007;277:115–122. doi: 10.1111/j.1574-6968.2007.00949.x. [DOI] [PubMed] [Google Scholar]
- Livermore DM. Has the era of untreatable infections arrived? J Antimicrob Chemother. 2009;64(Suppl 1):i29–36. doi: 10.1093/jac/dkp255. [DOI] [PubMed] [Google Scholar]
- Pitt TL, Kaufmann ME, Patel PS, Benge LC, Gaskin S, Livermore DM. Type characterization and antibiotic susceptibility of Burkholderia (Pseudomonas) cepacia isolates from patients with cystic fibrosis in the United Kingdom and the Republic of Ireland. J Med Microbiol. 1996;44:203–210. doi: 10.1099/00222615-44-3-203. [DOI] [PubMed] [Google Scholar]
- Golini G, Cazzola G, Fontana R. Molecular epidemiology and antibiotic susceptibility of Burkholderia cepacia-complex isolates from an Italian cystic fibrosis centre. Eur J Clin Microbiol Infect Dis. 2006;25:175–180. doi: 10.1007/s10096-006-0099-x. [DOI] [PubMed] [Google Scholar]
- Drevinek P, Mahenthiralingam E. Burkholderia cenocepacia in cystic fibrosis: epidemiology and molecular mechanisms of virulence. Clin Microbiol Infect. 2010;16:821–830. doi: 10.1111/j.1469-0691.2010.03237.x. [DOI] [PubMed] [Google Scholar]
- McCullough JL, Maren TH. Inhibition of dihydropteroate synthetase from Escherichia coli by sulfones and sulfonamides. Antimicrob Agents Chemother. 1973;3:665–669. doi: 10.1128/aac.3.6.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sköld O. Sulfonamide resistance: mechanisms and trends. Drug Resist Updat. 2000;3:155–160. doi: 10.1054/drup.2000.0146. [DOI] [PubMed] [Google Scholar]
- Triglia T, Menting JG, Wilson C, Cowman AF. Mutations in dihydropteroate synthase are responsible for sulfone and sulfonamide resistance in Plasmodium falciparum. Proc Natl Acad Sci USA. 1997;94:13944–13949. doi: 10.1073/pnas.94.25.13944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iliades P, Meshnick SR, Macreadie IG. Dihydropteroate synthase mutations in Pneumocystis jiroveci can affect sulfamethoxazole resistance in a Saccharomyces cerevisiae model. Antimicrob Agents Chemother. 2004;48:2617–2623. doi: 10.1128/AAC.48.7.2617-2623.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iliades P, Meshnick SR, Macreadie IG. Mutations in the Pneumocystis jirovecii DHPS gene confer cross-resistance to sulfa drugs. Antimicrob Agents Chemother. 2005;49:741–748. doi: 10.1128/AAC.49.2.741-748.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter WN. Structure-based ligand design and the promise held for antiprotozoan drug discovery. J Biol Chem. 2009;284:11749–11753. doi: 10.1074/jbc.R800072200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Achari A, Somers DO, Champness JN, Bryant PK, Rosemond J, Stammers DK. Crystal structure of the anti-bacterial sulfonamide drug target dihydropteroate synthase. Nat Struct Biol. 1997;4:490–497. doi: 10.1038/nsb0697-490. [DOI] [PubMed] [Google Scholar]
- Baca AM, Sirawaraporn R, Turley S, Sirawaraporn W, Hol WGJ. Crystal structure of Mycobacterium tuberculosis 6-hydroxymethyl-7,8-dihydropteroate synthase in complex with pterin monophosphate: new insight into the enzymatic mechanism and sulfa-drug action. J Mol Biol. 2000;302:1193–1212. doi: 10.1006/jmbi.2000.4094. [DOI] [PubMed] [Google Scholar]
- Babaoglu K, Qi J, Lee RE, White SW. Crystal structure of 7,8-dihydropteroate synthase from Bacillus anthracis: mechanism and novel inhibitor design. Structure. 2004;12:1705–1717. doi: 10.1016/j.str.2004.07.011. [DOI] [PubMed] [Google Scholar]
- Levy C, Minnis D, Derrick JP. Dihydropteroate synthase from Streptococcus pneumoniae: structure, ligand recognition and mechanism of sulfonamide resistance. Biochem J. 2008;412:379–388. doi: 10.1042/BJ20071598. [DOI] [PubMed] [Google Scholar]
- Hampele IC, D'Arcy A, Dale GE, Kostrewa D, Nielsen J, Oefner C, Page MG, Schönfeld HJ, Stüber D, Then RL. Structure and function of the dihydropteroate synthase from Staphylococcus aureus. J Mol Biol. 1997;268:21–30. doi: 10.1006/jmbi.1997.0944. [DOI] [PubMed] [Google Scholar]
- Lawrence MC, Iliades P, Fernley RT, Berglez J, Pilling PA, Macreadie IG. The three-dimensional structure of the bifunctional 6-hydroxymethyl-7,8 dihydropterin pyrophosphokinase/dihydropteroate synthase of Saccharomyces cerevisiae. J Mol Biol. 2005;348:655–670. doi: 10.1016/j.jmb.2005.03.021. [DOI] [PubMed] [Google Scholar]
- Pemble CW, Mehta PK, Mehra S, Li Z, Nourse A, Lee RE, White SW. Crystal structure of the 6-hydroxymethyl-7,8-dihydropterin pyrophosphokinase- dihydropteroate synthase bifunctional enzyme from Francisella tularensis. PLoS One. 2010;5:e14165. doi: 10.1371/journal.pone.0014165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hevener KE, Yun MK, Qi J, Kerr ID, Babaoglu K, Hurdle JG, Balakrishna K, White SW, Lee RE. Structural studies of pterin-based inhibitors of dihydropteroate synthase. J Med Chem. 2010;53:166–177. doi: 10.1021/jm900861d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wierenga RK. The TIM-barrel fold: a versatile framework for efficient enzymes. FEBS Letters. 2001;492:193–198. doi: 10.1016/S0014-5793(01)02236-0. [DOI] [PubMed] [Google Scholar]
- Frederick KK, Marlow MS, Valentine KG, Wand AJ. Conformational entropy in molecular recognition by proteins. Nature. 2007;448:325–329. doi: 10.1038/nature05959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavel Y, von Heijne G. Sequence differences between glycosylated and non-glycosylated Asn-X-Thr/Ser acceptor sites: implications for protein engineering. Protein Eng. 1990;3:433–442. doi: 10.1093/protein/3.5.433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard GA, McAuley-Hecht K, Brown T, Hunter WN. Do C-H•••O hydrogen bonds contribute to the stability of nucleic acid base pairs? Acta Crystallogr D Biol Crystallogr. 1995;51:136–139. doi: 10.1107/S0907444994004713. [DOI] [PubMed] [Google Scholar]
- Avgeri SG, Matthaiou DK, Dimopoulos G, Grammatikos AP, Falagas ME. Therapeutic options for Burkholderia cepacia infections beyond co-trimoxazole: a systematic review of the clinical evidence. Int J Antimicrob Agents. 2009;33:394–404. doi: 10.1016/j.ijantimicag.2008.09.010. [DOI] [PubMed] [Google Scholar]
- Leslie AGW. The integration of macromolecular diffraction data. Acta Crystallogr D Biol Crystallogr. 2006;62:48–57. doi: 10.1107/S0907444905039107. [DOI] [PubMed] [Google Scholar]
- Evans P. Scaling and assessment of data quality. Acta Crystallogr D Biol Crystallogr. 2006;62:72–82. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Lebedev A, Wilson KS, Dodson EJ. Efficient anisotropic refinement of macromolecular structures using FFT. Acta Crystallogr D Biol Crystallogr. 1999;55:247–255. doi: 10.1107/S090744499801405X. [DOI] [PubMed] [Google Scholar]
- Painter J, Merritt EA. TLSMD web server for the generation of multi-group TLS models. J Appl Cryst. 2006;39:109–111. doi: 10.1107/S0021889805038987. [DOI] [Google Scholar]
- Holm L, Rosenström P. Dali server: conservation mapping in 3D. Nucl Acids Res. 2010;38:W545–549. doi: 10.1093/nar/gkq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLano WL. The PyMOL Molecular Viewer. 2002. http://www.pymol.org
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. (2007) [DOI] [PubMed] [Google Scholar]
- Cruickshank DWJ. Remarks about protein structure precision. Acta Cryst. 1999;D55:583–601. doi: 10.1107/s0907444998012645. [DOI] [PubMed] [Google Scholar]