

Abstract
In prokaryotic genomes the number of transcriptional regulators is known to be proportional to the square of the total number of protein-coding genes. A toolbox model of evolution was recently proposed to explain this empirical scaling for metabolic enzymes and their regulators. According to its rules, the metabolic network of an organism evolves by horizontal transfer of pathways from other species. These pathways are part of a larger “universal” network formed by the union of all species-specific networks. It remained to be understood, however, how the topological properties of this universal network influence the scaling law of functional content of genomes in the toolbox model. Here we answer this question by first analyzing the scaling properties of the toolbox model on arbitrary tree-like universal networks. We prove that critical branching topology, in which the average number of upstream neighbors of a node is equal to one, is both necessary and sufficient for quadratic scaling. We further generalize the rules of the model to incorporate reactions with multiple substrates/products as well as branched and cyclic metabolic pathways. To achieve its metabolic tasks, the new model employs evolutionary optimized pathways with minimal number of reactions. Numerical simulations of this realistic model on the universal network of all reactions in the KEGG database produced approximately quadratic scaling between the number of regulated pathways and the size of the metabolic network. To quantify the geometrical structure of individual pathways, we investigated the relationship between their number of reactions, byproducts, intermediate, and feedback metabolites. Our results validate and explain the ubiquitous appearance of the quadratic scaling for a broad spectrum of topologies of underlying universal metabolic networks. They also demonstrate why, in spite of “small-world” topology, real-life metabolic networks are characterized by a broad distribution of pathway lengths and sizes of metabolic regulons in regulatory networks.
Author Summary
It has been previously reported that in prokaryotic genomes the number of transcriptional regulators is proportional to the square of the total number of genes. We recently offered a general explanation of this empirical powerlaw scaling in terms of the “toolbox” model in which metabolic and regulatory networks co-evolve together. This evolution is driven by horizontal gene transfer of co-regulated metabolic pathways from other species. These pathways are part of a larger “universal” network formed by the union of all species-specific networks. In the present work we address the question of how topological properties of this universal network influence the powerlaw scaling of regulators in the toolbox model. We also generalize its rules to include reactions with multiple substrates and products, branched and cyclic metabolic pathways, and to account for optimality of metabolic pathways. The main conclusion of our analytical and numerical modeling efforts is that the quadratic scaling is the robust feature of the toolbox model in a broad range of universal network topologies. They also demonstrate why, in spite of “small-world” topology, real-life metabolic networks are characterized by a broad distribution of pathway lengths and sizes of regulons in regulatory networks.
Introduction
In prokaryotic genomes the number of transcriptional regulators is known to
quadratically scale with the total number of protein-coding genes [1]. The
toolbox model of co-evolution of metabolic and regulatory networks was recently
proposed [2] to
explain this scaling in parts of the genome responsible for metabolic functions. In
this model prokaryotes acquire new metabolic capabilities by horizontal transfer of
entire metabolic pathways from other organisms. One can conveniently think of these
new pathways as coming from some “universal network” formed by the union
of metabolic repertoires of all potential donor organisms. The essence of the
toolbox argument [2] can be summarized as follows: as the non-regulatory part
of the genome of an organism (its “toolbox”) grows, it typically needs
to acquire fewer and fewer extra new genes (“tools”) in a pathway
offering it some new metabolic capability (e.g. the ability to utilize a new
nutrient or synthesize a new metabolic product). As a consequence, the number of
pathways and by extension the number of their transcriptional regulators grows
faster than linearly with the number of non-regulatory genes in the genome. While
this qualitative explanation is rather general and therefore does not depend on
specific details such as topology of the universal network, the exact value of the
exponent α connecting the number of transcription factors (equal to
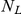 - the number of pathways or leaves of the network) to the
number of metabolites in the metabolic network of an organism
- the number of pathways or leaves of the network) to the
number of metabolites in the metabolic network of an organism
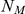 , as
, as 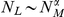 , is in general
model-dependent. In [2] we mathematically derived the quadratic scaling
(
, is in general
model-dependent. In [2] we mathematically derived the quadratic scaling
(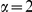 ) for the toolbox model with linear pathways on a fully
connected graph in which any pair of metabolites can in principle be converted to
each other in just one step via a single metabolic reaction. While this situation is
obviously unrealistic from biological standpoint, before present study it remained
the only mathematically treatable variant of the toolbox model. The universality of
the exponent
) for the toolbox model with linear pathways on a fully
connected graph in which any pair of metabolites can in principle be converted to
each other in just one step via a single metabolic reaction. While this situation is
obviously unrealistic from biological standpoint, before present study it remained
the only mathematically treatable variant of the toolbox model. The universality of
the exponent 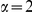 was then corroborated [2] by numerical simulations of the
toolbox model with linearized pathways on the universal network formed by the union
of all metabolic reactions in the KEGG database. While the agreement between the
values of the exponent
was then corroborated [2] by numerical simulations of the
toolbox model with linearized pathways on the universal network formed by the union
of all metabolic reactions in the KEGG database. While the agreement between the
values of the exponent  in these two cases
hinted at underlying general principles at work, the detailed understanding of these
principles remained elusive.
in these two cases
hinted at underlying general principles at work, the detailed understanding of these
principles remained elusive.
The question we address in this study is how the topology of the universal network determines this scaling exponent. To answer this question we first consider and solve a more realistic (yet still mathematically treatable) case in which the universal metabolic network is a directed tree of arbitrary topology. While being closer to reality than previously solved [2] case of fully connected network, the toolbox model on a tree-like universal network still retains a number of simplifications such as strictly linear pathways and one substrate → one product reactions.
To make our approach even more realistic we propose and numerically study a completely new version of the toolbox model incorporating metabolic reactions with multiple substrates and products as well as branched and cyclic metabolic pathways. Furthermore, unlike random linear pathways on a universal network [2] that can be long and therefore suboptimal from an evolutionary standpoint, the new model uses evolutionarily optimized pathways with the smallest number of reactions from the KEGG database sufficient to achieve a given metabolic task.
Results
The toolbox model on a tree-like universal network: General mathematical description
We will first consider the case where the universal metabolic network is a
directed tree. For simplicity in this section we will consider the case of
catabolic pathways, while identical arguments (albeit with opposite direction of
all reactions) apply to anabolic pathways. The root of the tree corresponds to
the central metabolic core of the organism responsible for biomass production.
Peripheral catabolic pathways (branches of the tree) convert external nutrients
(leaves) to this core, while the internal nodes of the tree represent
intermediate metabolites. Each of metabolites is characterized by its distance
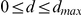 from the root of the network. The universal network has
from the root of the network. The universal network has
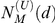 metabolites at distance
metabolites at distance 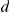 from the root that
included
from the root that
included 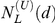 leaves (nutrients used in the first step of catabolic
pathways) and
leaves (nutrients used in the first step of catabolic
pathways) and 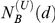 branching points corresponding to intermediate
metabolites generated by more than one metabolic reaction at the next level (see
Figure 1). An
organism-specific network (filled circles and thick edges in Figure 1) at distance
branching points corresponding to intermediate
metabolites generated by more than one metabolic reaction at the next level (see
Figure 1). An
organism-specific network (filled circles and thick edges in Figure 1) at distance
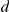 from the root contains
from the root contains 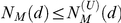 metabolites
composed of
metabolites
composed of 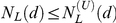 leaves,
leaves, 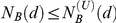 branching points,
and
branching points,
and 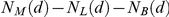 metabolites inside linear branches (“one reaction
in-one reaction out”) . For simplicity we assume that in the universal
network (and thus also in any of its organism-specific subnetworks) no more than
two reaction edges can combine at any node (metabolite), while the most general
case of an arbitrary distribution of branching numbers can be treated in a very
similar fashion.
metabolites inside linear branches (“one reaction
in-one reaction out”) . For simplicity we assume that in the universal
network (and thus also in any of its organism-specific subnetworks) no more than
two reaction edges can combine at any node (metabolite), while the most general
case of an arbitrary distribution of branching numbers can be treated in a very
similar fashion.
Figure 1. An example of organism-specific metabolic network and the corresponding universal network.

The organism-specific metabolic network (filled circles and thick edges)
is always a subset of the universal network (the entire tree). Nodes are
divided into layers based on their distance
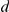 from the
root of the tree. Variables
from the
root of the tree. Variables 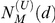 ,
,
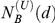 ,
,
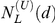 for the
universal network and
for the
universal network and 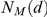 ,
,
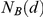 ,
,
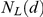 for
species-specific network are illustrated using the layer
for
species-specific network are illustrated using the layer
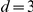 as an
example.
as an
example.
The toolbox model specifies rules by which organism acquires new pathways in the course of its evolution. It consists of the following steps: 1) randomly pick a new nutrient metabolite (a leaf node of the universal network that currently does not belong to the metabolic network of the organism) 2) use the universal network to identify the unique linear pathway which connects the new nutrient to the root of the tree (the metabolic core) and finally 3) add the reactions and intermediate metabolites in the new pathway to the metabolic network of the organism (filled circles and thick edges in Figure 1). One needs to only add those enzymes that are not yet present in the “genome” of the organism. Graphically it means that the new branch of the universal network is extended until it first intersects the existing metabolic network of the organism.
Consider an organism capable of utilizing 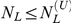 nutrients
represented by leaves in the universal network, where
nutrients
represented by leaves in the universal network, where
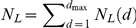 and
and 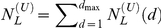 . Since we assume
that each nutrient utilization pathway is controlled by a dedicated
transcriptional regulator sensing its presence or absence in the environment
(e.g. LacR for lactose), the corresponding regulatory network would also have
. Since we assume
that each nutrient utilization pathway is controlled by a dedicated
transcriptional regulator sensing its presence or absence in the environment
(e.g. LacR for lactose), the corresponding regulatory network would also have
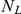 transcription factors (in the model we ignore
transcription factors controlling non-metabolic functions). The non-regulatory
part of the genome consists of
transcription factors (in the model we ignore
transcription factors controlling non-metabolic functions). The non-regulatory
part of the genome consists of 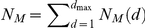 enzymes catalyzing
metabolic reactions (strictly speaking
enzymes catalyzing
metabolic reactions (strictly speaking 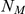 is the number of
metabolites/nodes so that the number of enzymes/edges is
is the number of
metabolites/nodes so that the number of enzymes/edges is
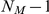 ). Quadratic scaling plots [1] shows the number of
transcriptional regulators
). Quadratic scaling plots [1] shows the number of
transcriptional regulators 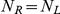 vs. the total
number of genes in the genome (both regulatory and non-regulatory)
vs. the total
number of genes in the genome (both regulatory and non-regulatory)
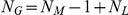 . However, since in all organism-specific networks
NM ≫ NL, the
quadratic scaling between
. However, since in all organism-specific networks
NM ≫ NL, the
quadratic scaling between 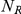 and
and
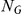 is equivalent to
is equivalent to 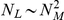 .
.
We further assume that due to random selection 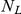 nutrients are
expected to be uniformly distributed among all d levels.
Therefore, the expected number of leaves at a given level is given by
nutrients are
expected to be uniformly distributed among all d levels.
Therefore, the expected number of leaves at a given level is given by
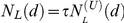 where the fraction
where the fraction 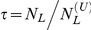 is the same at all
levels. On the other hand the fraction
is the same at all
levels. On the other hand the fraction 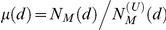 varies from level
to level. It usually tends to increase as one gets closer towards the root of
the tree reaching 1 for d = 0 (the root
node itself). To derive the equation for
varies from level
to level. It usually tends to increase as one gets closer towards the root of
the tree reaching 1 for d = 0 (the root
node itself). To derive the equation for 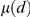 , one first notices
that each of
, one first notices
that each of 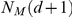 metabolites at level
metabolites at level 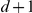 is converted to
another intermediate metabolite at level
is converted to
another intermediate metabolite at level 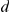 . Due to merging of
pathways at
. Due to merging of
pathways at 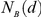 branching points the number of unique intermediate
metabolites at the level
branching points the number of unique intermediate
metabolites at the level 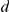 is actually
smaller:
is actually
smaller: 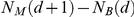 . To calculate
. To calculate 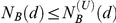 one uses the fact
that each of the two nodes downstream of a branching point in the universal
network is present in the organism-specific network with probability
one uses the fact
that each of the two nodes downstream of a branching point in the universal
network is present in the organism-specific network with probability
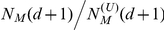 . The probability that they are both present is
. The probability that they are both present is
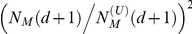 and thus the number of branching points at level
and thus the number of branching points at level
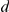 of the organism-specific metabolic network is
of the organism-specific metabolic network is
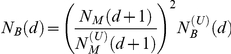 . The intermediate metabolites together with new
nutrients
. The intermediate metabolites together with new
nutrients 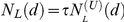 entering at the level
entering at the level 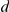 add up to the
total number of metabolites at level
add up to the
total number of metabolites at level 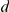 :
:
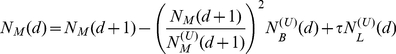 |
(1) |
This equation allows one to iteratively calculate 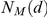 for all
d starting from
for all
d starting from 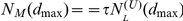 . We will use this
equation to derive the relationship between the number of leaves and the total
number of nodes first for a critical branching tree and then for a supercritical
one.
. We will use this
equation to derive the relationship between the number of leaves and the total
number of nodes first for a critical branching tree and then for a supercritical
one.
The toolbox model on a critical tree
The Galton-Watson branching process [3] is a simple stochastic
process generating random trees, and we will consider its version where a node
can have two, one, or zero neighbors (parents) at the previous level with
probabilities p
2, p
1 and
p
0 correspondingly. If the average number of
parents 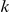 equals one, then the process is referred to as critical,
and if
equals one, then the process is referred to as critical,
and if 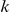 is greater than one then the process is supercritical.
More generally critical and supercritical branching trees can be generated by a
variety of random processes such as e.g. directed percolation [4]. While
for simplicity we used the Galton-Watson branching process in our derivation
below, it can be readily extended to this more general case.
is greater than one then the process is supercritical.
More generally critical and supercritical branching trees can be generated by a
variety of random processes such as e.g. directed percolation [4]. While
for simplicity we used the Galton-Watson branching process in our derivation
below, it can be readily extended to this more general case.
The principal geometric difference between supercritical and critical trees is
that in the former case the number of nodes in a layer
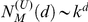 exponentially grows with
exponentially grows with 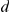 [3], while in
a critical tree it grows at most algebraically (for the Galton-Watson critical
process
[3], while in
a critical tree it grows at most algebraically (for the Galton-Watson critical
process 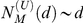 [3]). The
other difference is that while the critical branching process always stops on
its own at a certain finite height
[3]). The
other difference is that while the critical branching process always stops on
its own at a certain finite height 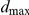 , a supercritical
process will go on forever so that to generate a tree one has to manually
terminate it at a predefined layer
, a supercritical
process will go on forever so that to generate a tree one has to manually
terminate it at a predefined layer 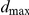 . The most
significant feature of a critical tree is that it has much longer branches than
a supercritical one of the same size. Indeed, the diameter (the maximal height)
of a random critical tree with
. The most
significant feature of a critical tree is that it has much longer branches than
a supercritical one of the same size. Indeed, the diameter (the maximal height)
of a random critical tree with 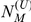 nodes is
nodes is
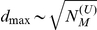 while in a supercritical tree it is much shorter:
while in a supercritical tree it is much shorter:
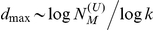 . Thus supercritical trees (unlike their critical
counterparts) have the small world property.
. Thus supercritical trees (unlike their critical
counterparts) have the small world property.
A random critical network where each node has at most has two parents in the
previous layer is defined by 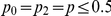 . Indeed, in this
case
. Indeed, in this
case 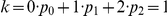 . In such network
. In such network 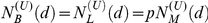 and hence the Eq.
(1) can be rewritten as
and hence the Eq.
(1) can be rewritten as
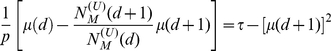 |
(2) |
A critical branching process that has not terminated by level d
satisfies 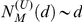 or
or 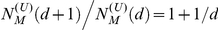 . More generally if
. More generally if
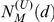 algebraically increases with
algebraically increases with
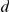 ,
, 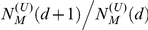 asymptotically
approaches 1 as
asymptotically
approaches 1 as
| (3) |
Here 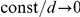 as
as 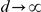 , thus for
, thus for
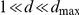
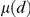 remains approximately constant and according to Eq. (2)
this constant ratio
remains approximately constant and according to Eq. (2)
this constant ratio 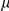 is defined by
is defined by
| (4) |
This quadratic relation is exact in a critical branching tree where each node can
branch out into at most two nodes at the next layer, and it is still correct to
a leading order in 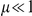 for a critical
branching tree with arbitrary branching ratios (see “Quadratic relation
between
for a critical
branching tree with arbitrary branching ratios (see “Quadratic relation
between 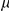 and
and  for general
critical branching processes” of Text S1). Furthermore, one can show (see
“Calculation of the average
for general
critical branching processes” of Text S1). Furthermore, one can show (see
“Calculation of the average 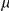 in the toolbox
model on a critical tree” of Text S1) that in large critical networks the
overall fraction of metabolites present in organism-specific metabolic network
is very close to this stationary limit of
in the toolbox
model on a critical tree” of Text S1) that in large critical networks the
overall fraction of metabolites present in organism-specific metabolic network
is very close to this stationary limit of 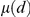 :
:
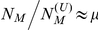 .
.
As was explained in the previous section the ratio
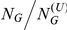 between the total number
between the total number 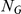 of
metabolic-related genes in the genome of an organism and its theoretical maximal
value
of
metabolic-related genes in the genome of an organism and its theoretical maximal
value 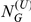 for a genome containing the entire universal network is
also given by
for a genome containing the entire universal network is
also given by 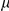 . Furthermore, in our model the number of leaves is equal
to the number of nutrient-utilizing pathways or, alternatively, their
transcriptional regulators
. Furthermore, in our model the number of leaves is equal
to the number of nutrient-utilizing pathways or, alternatively, their
transcriptional regulators 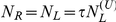 . Thus like in a
much simpler model of Ref. [2] the toolbox model on any critical tree-like universal
network gives rise to quadratic scaling of the number of transcription factors
with the total number of genes:
. Thus like in a
much simpler model of Ref. [2] the toolbox model on any critical tree-like universal
network gives rise to quadratic scaling of the number of transcription factors
with the total number of genes:
| (5) |
The geometrical properties of the universal network such as its total number of
nodes/edges 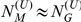 and number of leaves/branches
and number of leaves/branches
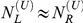 determine the prefactor of this scaling law. Simulation
of the toolbox model on the critical tree (Figure 2) verified our mathematical
predictions with the best fit to binned datapoints in Figure 2 giving the exponent
α = 1.9±0.1.
determine the prefactor of this scaling law. Simulation
of the toolbox model on the critical tree (Figure 2) verified our mathematical
predictions with the best fit to binned datapoints in Figure 2 giving the exponent
α = 1.9±0.1.
Figure 2.
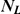 vs.
vs. 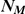 .
.

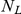 is the number of leaves in an organism-specific
metabolic network and equal to the number of transcriptional regulators
of corresponding nutrient-utilizing pathways, while
is the number of leaves in an organism-specific
metabolic network and equal to the number of transcriptional regulators
of corresponding nutrient-utilizing pathways, while
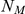 is the
total number of nodes/metabolites in this netowrk. The data are
generated by the toolbox model on critical universal network with sizes
around 2000. Solid line
is the
total number of nodes/metabolites in this netowrk. The data are
generated by the toolbox model on critical universal network with sizes
around 2000. Solid line 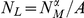 , where the
exponent
, where the
exponent 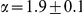 and the
prefactor
and the
prefactor 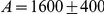 , are the
best fits to the binned data.
, are the
best fits to the binned data.
The toolbox model on a supercritical tree
For a supercritical branching process 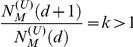 and according to
Eq. (1) (See SI for the derivation) the steady state value
and according to
Eq. (1) (See SI for the derivation) the steady state value
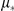 of
of 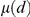 satisfies
satisfies
| (6) |
Here 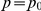 and
and 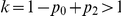 . Notice that for
. Notice that for
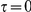 one has two solutions for
one has two solutions for
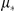 :
: 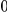 and
and
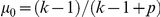 . This indicates transition in which for
. This indicates transition in which for
 exactly at zero one has
exactly at zero one has 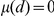 , while for an
arbitrary small yet positive
, while for an
arbitrary small yet positive  the value of
the value of
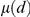 asymptotically converges to
asymptotically converges to
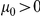 for
for 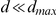 . This transition
resembles the first order phase transition, e.g., liquid-gas transition, where
right at the transition point very small variation of the external parameter
such as temperature (analogous to
. This transition
resembles the first order phase transition, e.g., liquid-gas transition, where
right at the transition point very small variation of the external parameter
such as temperature (analogous to  in this model)
results in a large jump of the order parameter such as density (analogous to our
in this model)
results in a large jump of the order parameter such as density (analogous to our
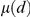 ). (See [5] for details), The number of layers over which this
conversion is taking place is itself a function of
). (See [5] for details), The number of layers over which this
conversion is taking place is itself a function of
 and for small
and for small  it is large. For
exponentially growing supercritical networks and for small
it is large. For
exponentially growing supercritical networks and for small
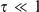 , the network average value of
, the network average value of
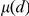 defined as
defined as 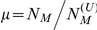 satisfies
satisfies
| (7) |
Note that this equation connecting 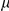 and
and
 (see SI for detailed derivation) is markedly different
from Eq. (6) for steady state value
(see SI for detailed derivation) is markedly different
from Eq. (6) for steady state value 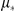 in middle
layers.
in middle
layers.
In conclusion, while the toolbox model on a critical universal network is
characterized by a quadratic scaling between  and
and
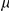 (see Eq. (4)), the same model on a supercritical,
exponentially expanding universal network gives rise to a linear scaling of
(see Eq. (4)), the same model on a supercritical,
exponentially expanding universal network gives rise to a linear scaling of
 vs.
vs. 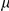 albeit with
logarithmic corrections (see Eq. (7)). This difference in exponent equally
applies to the scaling of the number of regulators
albeit with
logarithmic corrections (see Eq. (7)). This difference in exponent equally
applies to the scaling of the number of regulators
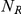 vs. the total number of genes
vs. the total number of genes
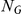 in the toolbox model on critical and supercritical
universal network.
in the toolbox model on critical and supercritical
universal network.
Simulation of the toolbox model on the KEGG network with linearized pathways
To test our mathematical results for a more realistic version of the universal
tree we linearized pathways and reactions in the network formed by the union of
all reactions in the KEGG database [6]. To this end we generated
a random spanning tree on the KEGG network by the following algorithm: the
metabolite pyruvate was selected as the root of the tree. We then randomly
picked a metabolite located upstream of it and generated a linear pathway (tree
branch) as a self-avoiding random walk on the KEGG network extended until it
either merges with another pathway or reaches the root of the tree. This step
was repeated until all upstream metabolites were covered. The resulting spanning
tree was then used as the universal network on which we simulated the toolbox
model by gradually increasing the number of pathways
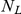 and recording the total number of metabolites
and recording the total number of metabolites
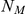 in organism-specific metabolic networks. Our numerical
simulations generated approximately quadratic scaling
in organism-specific metabolic networks. Our numerical
simulations generated approximately quadratic scaling
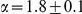 (see Ref. [2]).
(see Ref. [2]).
To better understand the origins of this scaling we investigated the topology of
the underlying universal tree. The criticality of a tree is defined by the
asymptotic value of the ratio 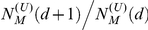 for large
for large
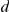 : for supercritical trees it reaches a limit
: for supercritical trees it reaches a limit
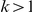 , while for critical ones it converges to 1 as described
in Eq. (3). Figure 3 showing
, while for critical ones it converges to 1 as described
in Eq. (3). Figure 3 showing
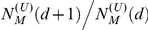 vs.
vs. 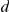 in the linearized
KEGG network convincingly demonstrates its criticality. Thus the quadratic
scaling between the number of transcriptional regulators and the number of
metabolites in the toolbox model simulated on the linearized KEGG network is
explained by the mathematical formalism described in previous sections.
in the linearized
KEGG network convincingly demonstrates its criticality. Thus the quadratic
scaling between the number of transcriptional regulators and the number of
metabolites in the toolbox model simulated on the linearized KEGG network is
explained by the mathematical formalism described in previous sections.
Figure 3.
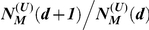 vs.
vs. 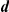 for
KEGG-based universal network with linearized pathways.
for
KEGG-based universal network with linearized pathways.

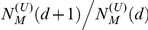 (the ratio of the number of metabolites at two
consecutive layers) plotted as a function of
(the ratio of the number of metabolites at two
consecutive layers) plotted as a function of
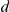 (the layer
number) for KEGG-based universal network with linearized pathways. Solid
line: measurement, dotted line: its expected profile,
(the layer
number) for KEGG-based universal network with linearized pathways. Solid
line: measurement, dotted line: its expected profile,
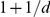 , in a
critical branching tree. The error bars reflect standard deviation in
different spanning trees used to linearize the KEGG network.
, in a
critical branching tree. The error bars reflect standard deviation in
different spanning trees used to linearize the KEGG network.
In addition to using a random spanning tree to linearize the KEGG network we also
tried a version using minimal paths. In this version the universal network is
generated by randomly picking a metabolite and connecting it to the root of the
tree (pyruvate) by the shortest path. At a first glance such “minimal
path” selection appears to be reasonable from evolutionary standpoint.
Indeed, evolution would favor simpler and shorter pathways in order to minimize
the expenditure of resources to achieve a given metabolic goal [7] .
However, the minimal paths version of linearization of the KEGG resulted in a
supercritical universal network with logarithmically short branches
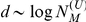 . As predicted for supercritical trees (Eq. (7)) the
toolbox model in this case had an approximately linear scaling of the number of
transcriptional regulators (leaves of branches on the network) with the total
number of metabolites: the measured best fit exponent was only
. As predicted for supercritical trees (Eq. (7)) the
toolbox model in this case had an approximately linear scaling of the number of
transcriptional regulators (leaves of branches on the network) with the total
number of metabolites: the measured best fit exponent was only
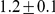 .
.
How do we reconcile the evolutionary pressure apparently selecting for minimal
pathways with dramatically wrong scaling properties of this model? We believe
that most of the ultra-short “small world” pathways generated by
minimal paths on the KEGG network are unrealistic from biochemical standpoint.
Indeed, highly connected co-factors often position metabolites with very
different chemical formulas in close proximity to each other. For example, the
KEGG reaction R00134: 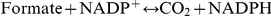 would appear as a
miraculous “one-step” conversion of carbon dioxide into formate,
while the reaction R03546:
would appear as a
miraculous “one-step” conversion of carbon dioxide into formate,
while the reaction R03546: 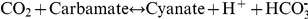 would artificially
link carbon dioxide and cyanate. The combination of these two reactions gives
rise to equally impossible two-step path: formate → CO2 →
cyanate. As a consequence of such artificial shortcuts branches of the universal
network linearized by minimal paths are much shorter than they are in reality.
.This problem is at least partially alleviated by 1) removing unusually
high-degree nodes corresponding to common co-factors such as H2O,
ATP, NAD in the metabolic network so that some unrealistic paths are eliminated,
and also 2) using random spanning tree instead of the shortest paths. In Ref.
[2] we
followed both of these recipes to successfully reproduce the quadratic scaling
in real-life data. Still no linearization procedure could completely avoid
biochemically meaningless shortcuts. In the next section we introduce and study
a new considerably more realistic version of the toolbox model operating on
branched and interconnected universal networks. Pathways in this version of the
toolbox model satisfy the evolutionary requirements for minimal size. Proper
treatment of metabolic reactions with multiple substrates prevents biochemically
meaningless shortcuts and as a consequence restores the quadratic scaling.
would artificially
link carbon dioxide and cyanate. The combination of these two reactions gives
rise to equally impossible two-step path: formate → CO2 →
cyanate. As a consequence of such artificial shortcuts branches of the universal
network linearized by minimal paths are much shorter than they are in reality.
.This problem is at least partially alleviated by 1) removing unusually
high-degree nodes corresponding to common co-factors such as H2O,
ATP, NAD in the metabolic network so that some unrealistic paths are eliminated,
and also 2) using random spanning tree instead of the shortest paths. In Ref.
[2] we
followed both of these recipes to successfully reproduce the quadratic scaling
in real-life data. Still no linearization procedure could completely avoid
biochemically meaningless shortcuts. In the next section we introduce and study
a new considerably more realistic version of the toolbox model operating on
branched and interconnected universal networks. Pathways in this version of the
toolbox model satisfy the evolutionary requirements for minimal size. Proper
treatment of metabolic reactions with multiple substrates prevents biochemically
meaningless shortcuts and as a consequence restores the quadratic scaling.
The toolbox model on KEGG network with branched pathways and multi-substrate reactions
Real metabolic reactions routinely include multiple inputs (substrates) and multiple outputs (products) (see Table 1 and Table 2 for statistics in the KEGG database). Furthermore, metabolic networks often have two or more alternative pathways generating the same set of end-products from the same set of nutrients. Both these factors result in metabolic networks that are branched and interconnected. Here we propose and simulate a more realistic version of the toolbox model. The most prominent feature of the new model of pathways is the “AND” function acting on inputs of multi-substrate reactions. It reflects the constraint that a reaction cannot be carried out unless all its substrates are present.
Table 1. The distribution of irreversible reactions classified by their numbers of substrates and products.
| The number of products of an irreversible reaction | ||||||
| The number of substrates of an irreversible reaction | 1 | 2 | 3 | 4 | 5 | |
| 1 | 157 | 141 | 4 | |||
| 2 | 82 | 491 | 95 | 7 | ||
| 3 | 1 | 123 | 170 | 31 | 1 | |
| 4 | 10 | 73 | 15 | |||
| 5 | 1 | |||||
Table 2. The distribution of reversible reactions classified by their numbers substrates/products.
| The number of substrates/products at one end of a reversible reaction | |||||
| The number of substrates/products at the opposite end of a reversible reaction | 1 | 2 | 3 | 4 | 5 |
| 1 | 143 | 231 | 6 | ||
| 2 | 553 | 284 | 15 | ||
| 3 | 106 | 69 | 1 | ||
| 4 | 6 | 3 | |||
The new version of the toolbox model simulates addition of anabolic pathways aimed at production of new metabolites from those the model organism can currently synthesize (its current metabolic core). The new pathways are optimal in the sense that they contain the smallest number of reactions necessary to synthesize the desired end-product. As for previous versions of the toolbox model, one can modify the rules of this model to apply to catabolic pathways but for simplicity we will limit the following discussion to anabolic pathways. The rules of the new model are:
At the beginning of the simulation, the model organism starts with a “seed” metabolic network consisting of 40 metabolites classified by the KEGG as parts of central carbohydrate metabolism, plus a number of “currency” metabolites such as water, ATP and NAD (see the section “Seed metabolites of the scope expansion” of Text S1 for additional details). It is assumed that our organism is able to generate all of these metabolites by some unspecified catabolic pathways.
At each step a new metabolite that cannot yet be synthesized by the organism is randomly selected from the “scope” [8] of our seed metabolites. This scope consists of all metabolites that in principle could be synthesized from the seed metabolites using all reactions listed in the KEGG database (see Ref. [8] for details).
To search for the minimal pathway that converts core metabolites to this target we first perform the “scope expansion” [8] of the core until it first reaches the target. In the course of this expansion reactions and metabolites are added step by step (or layer by layer). Each layer consists of all KEGG reactions that have all their substrates among the metabolites in the current metabolic core of the organism (light blue area in Figure 4) and those generated by reactions in all the previous layers. (See Figure 4 for an illustration).
Next we trace back added reactions starting from the target and progressively moving to lower levels. One starts by finding the reaction responsible for fabrication of the target metabolite and adding it to the new pathway (if several such reactions exist in the last layer we randomly choose one of them). In case of multi-layer expansion process some substrates of this reaction are not among the core metabolites (otherwise this reaction would be in the first layer). One then goes down one layer and adds the reactions fabricating these missing substrates. This is repeated all the way down to the first level of the original expansion. The resulting pathway includes the minimal (or nearly minimal) set of reactions needed to generate the target metabolite from the current metabolic core of the organism. Starting from the next step of the model the target and all intermediate metabolites become part of the metabolic core. Genes for enzymes catalyzing these new reactions are assumed to be horizontally transferred to the genome of the organism. The newly added metabolic pathway is assumed to have a dedicated transcriptional regulator so that the number of transcription factors in our model is always equal to the number of pathways or their target metabolites.
Steps 1–5 are repeated until metabolic network of the organism reaches its maximal size. At this stage it includes the entire scope [8] of the starting set of metabolites in step 1.
Figure 4. Diagram of a new pathway added to the metabolic network of the organism.

The diagram explains different types of metabolites and reactions. Reactions (squares) in the added pathway use base substrates (yellow circles with horizontal shading) from the metabolic core of the organism (light blue area) to produce the target metabolite (the red circle). Added pathway generates intermediate products (green circles) as well as byproducts that are not further converted to the target (blue circles). Products of some reactions feed back into the metabolic core (yellow circles with vertical shading). Reactions are labeled with expansion steps at which they were added to the pathway.
Numerical simulation of this model shows that the number of transcriptional
regulators scales with the number of metabolites with power
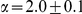 (Figure
5). This is consistent with quadratic scaling we observed and
mathematically derived for a simpler model with linearized pathways composed of
single-substrate reactions.
(Figure
5). This is consistent with quadratic scaling we observed and
mathematically derived for a simpler model with linearized pathways composed of
single-substrate reactions.
Figure 5.
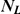 vs.
vs. 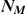 of toolbox
model with branched pathways and multi-substrate reactions.
of toolbox
model with branched pathways and multi-substrate reactions.

The scaling between the number of regulated pathways (leaves),
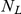 and the
number of metabolites,
and the
number of metabolites, 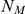 , in
metabolic networks generated by the toolbox model with branched pathways
and multi-substrate reactions. Solid line with slope
2.0+/−0.1 is the best fit to the data. Error bars reflect the
standard deviation of
, in
metabolic networks generated by the toolbox model with branched pathways
and multi-substrate reactions. Solid line with slope
2.0+/−0.1 is the best fit to the data. Error bars reflect the
standard deviation of 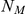 at a given
value of
at a given
value of 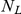 in 9
realizations of the model (see he section “Error analysis of the
toolbox model” of Text S1 for our estimation methods
and error analysis).
in 9
realizations of the model (see he section “Error analysis of the
toolbox model” of Text S1 for our estimation methods
and error analysis).
The mathematical formalism derived in the previous sections is limited to
tree-like universal networks and thus does not directly apply to the new model.
Nevertheless, one generally expects the quadratic scaling to be limited only to
critical, “large world” networks in which organisms with small
genomes initially tend to acquire sufficiently long pathways. As noted before,
from purely topological standpoint the KEGG network has a “small
world” property making long pathways unlikely. It is important to check if
the realistic treatment of multi-substrate reactions did in fact restore the
“large world” property and criticality to the KEGG universal network
by increasing the minimal number of steps required for connecting target
metabolites to the metabolic core. To quantify the criticality of the expansion
process as before we use the ratio 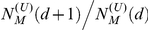 where
where
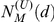 denotes the number of metabolites reached at step
denotes the number of metabolites reached at step
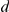 of the scope expansion starting from the initial seed
subset of metabolites. As in the case of critical branching trees this ratio
asymptomatically converges to 1 thus confirming the criticality of the scope
expansion process. The mere existence of ∼40 steps in this process (the
x-axis in Figure 6) can
serve as evidence in favor of “large world” character of the KEGG
universal network characterized by the existence of long pathways.
of the scope expansion starting from the initial seed
subset of metabolites. As in the case of critical branching trees this ratio
asymptomatically converges to 1 thus confirming the criticality of the scope
expansion process. The mere existence of ∼40 steps in this process (the
x-axis in Figure 6) can
serve as evidence in favor of “large world” character of the KEGG
universal network characterized by the existence of long pathways.
Figure 6.
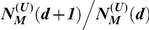 vs.
vs. 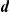 for the
universal network consisting of all KEGG reactions.
for the
universal network consisting of all KEGG reactions.

The ratio 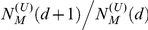 of the
number of metabolites at two consecutive layers of the scope expansion
process plotted versus the layer number
of the
number of metabolites at two consecutive layers of the scope expansion
process plotted versus the layer number 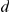 . Scope
expansion was performed for the universal network consisting of all KEGG
reactions. The dashed line is the mathematical expectation of the same
curve in a critical branching process.
. Scope
expansion was performed for the universal network consisting of all KEGG
reactions. The dashed line is the mathematical expectation of the same
curve in a critical branching process.
Geometrical properties of branched pathways in the model
Unlike linearized pathways in the original version of the toolbox model [2], branched
pathways in the more realistic model from previous section are interesting
objects in their own right. We identified several geometrical properties of
these pathways (see Figure 4
for illustration) quantifying their position relative to the core network to
which they were added: 1) n
border rxn–the
number of added reactions that are connected (as a substrate or a product) with
at least one metabolite in the core, 2)
n
base–the number of metabolites in the core
that serve as substrates to reactions in the added pathway, 3)
n
feedback–the number of core metabolites
that are products of reactions in the new pathway, 4)
n
byproduct–the number of final metabolic
products of the added pathway that are neither core metabolites nor the target,
5) length-the number of steps (layers of the scope expansion process) it takes
to transform core metabolites into the target product. 4 illustrates the
definition of these parameters while Figure 7 and Figure 8 plot these parameters as a function
of 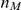 (the number of metabolites in the added pathway) or
(the number of metabolites in the added pathway) or
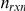 (the number of reactions in the added pathway).
(the number of reactions in the added pathway).
Figure 7. nbyproduct vs.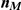 .
.

Faster-than-linear scaling of the number of byproducts,
nbyproduct, and the total number of metabolites,
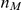 , in
individual branched pathways illustrated in Figure 4. Data for individual
pathways were logarithmically binned along the x-axis. Hence y-axis can
be and are below 1 due to pathways with 0 byproducts. The solid line
with exponent 1.7+/−0.1 is the best fit to the
logarithmically-binned data shown in this plot. Readers can refer to the
section “Analysis of number of by-product of the pathways of the
toolbox model on the metabolic network with branched pathways and
multi-substrates reactions” of Text
S1 for our estimation methods and error analysis.
, in
individual branched pathways illustrated in Figure 4. Data for individual
pathways were logarithmically binned along the x-axis. Hence y-axis can
be and are below 1 due to pathways with 0 byproducts. The solid line
with exponent 1.7+/−0.1 is the best fit to the
logarithmically-binned data shown in this plot. Readers can refer to the
section “Analysis of number of by-product of the pathways of the
toolbox model on the metabolic network with branched pathways and
multi-substrates reactions” of Text
S1 for our estimation methods and error analysis.
Figure 8. Various linear relationships on the individual pathways.

Approximately linear relationship between a) pathway's length and
its number of reactions 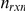 , ) b) the
number of border reactions, nborder rxn, and the total number
of reactions,
, ) b) the
number of border reactions, nborder rxn, and the total number
of reactions, 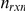 , c) the
number of base metabolites, nbase, and the total number of
metabolites,
, c) the
number of base metabolites, nbase, and the total number of
metabolites, 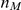 , d) the
number of metabolites receiving feedback, nfeedback, and the
total number of metabolites,
, d) the
number of metabolites receiving feedback, nfeedback, and the
total number of metabolites, 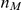 . These
different geometrical properties of individual pathways are illustrated
in Figure 4. Sizes
of circles are proportional to the logarithm of the number of discrete
(x, y) pairs contributing to this point.
. These
different geometrical properties of individual pathways are illustrated
in Figure 4. Sizes
of circles are proportional to the logarithm of the number of discrete
(x, y) pairs contributing to this point.
Approximately linear relationship between n
border rxn
vs. 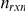 (Figure
8a) suggests that added pathways tend to be located at or near the
surface of the core metabolic network of the organism. Most of reactions in
these pathways use metabolites from this core network either as substrates
(n
base) or as products
(n
feedback). Further analysis indicates that
“currency metabolites” (common co-factors that serve as substrates
or products of many reactions) constitute a significant fraction
(∼25%) of all core metabolites involved in border reactions (see the
section “Analysis of the currency metabolites in the toolbox model”
of Text
S1 for details). On the other hand, the fact that the number of steps
in a pathway (its length) constitutes a good fraction of its overall number of
reactions
(Figure
8a) suggests that added pathways tend to be located at or near the
surface of the core metabolic network of the organism. Most of reactions in
these pathways use metabolites from this core network either as substrates
(n
base) or as products
(n
feedback). Further analysis indicates that
“currency metabolites” (common co-factors that serve as substrates
or products of many reactions) constitute a significant fraction
(∼25%) of all core metabolites involved in border reactions (see the
section “Analysis of the currency metabolites in the toolbox model”
of Text
S1 for details). On the other hand, the fact that the number of steps
in a pathway (its length) constitutes a good fraction of its overall number of
reactions 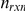 implies that, in spite of these numerous
“shortcut” connections to the core, added pathways remain very thin
and essentially linear. That is to say, these pathways tend to work as a single
“conveyor belt” sequentially converting intermediate products into
each other instead of having two or more parallel “processing lines”
and assembling final products of these lines only at final stages of the
pathway. This finding provides an intuitive reason why models with branched and
linearized pathways have similar scaling properties. One can argue that this is
because pathways in both models are essentially linear. Yet, in spite of their
linearity and optimality (each has the smallest number of reactions to generate
the target from the core) added pathways in the new version of the model are
very different from shortest paths on the universal network. As illustrated in
Figure 9 the average
pathway length is several times longer than the geometrically shortest path
between the target and the core.
implies that, in spite of these numerous
“shortcut” connections to the core, added pathways remain very thin
and essentially linear. That is to say, these pathways tend to work as a single
“conveyor belt” sequentially converting intermediate products into
each other instead of having two or more parallel “processing lines”
and assembling final products of these lines only at final stages of the
pathway. This finding provides an intuitive reason why models with branched and
linearized pathways have similar scaling properties. One can argue that this is
because pathways in both models are essentially linear. Yet, in spite of their
linearity and optimality (each has the smallest number of reactions to generate
the target from the core) added pathways in the new version of the model are
very different from shortest paths on the universal network. As illustrated in
Figure 9 the average
pathway length is several times longer than the geometrically shortest path
between the target and the core.
Figure 9. Comparison of lengths of the pathways and shortest distances of the targets from the core.

The lengths of the pathways are represented by circles and solid line, while the shortest distances of the targets from the core are represented by crosses and dotted line.
As can be seen from Figure 7, most of added pathways (around 97%) do not generate any byproducts. They only produce the intended target and n feedback metabolites in the core network of the organism to which they were added. The relative scarcity of byproducts suggests that pathways in our model satisfy the evolutionary constrains imposed on real-life organisms. Indeed, as previously proposed in Ref [9] it makes sense to assume that evolution favors pathways that achieve a given metabolic goal using the smallest number of enzymes and at the same time striving to generate the maximal possible yield. Unnecessary byproducts not only reduce the yield of the desired metabolic target, they also might become toxic in high concentrations and thus would require extra transporter proteins to pump them out.
Discussion
The small world property of complex biomolecular networks has been extensively discussed in the literature during the last decade (see [10]–[12] for earliest reports in metabolic and protein interaction networks correspondingly). It was often assumed that the small world effect positively contributes to the robustness of the network by providing multiple redundant pathways for target production in metabolic networks or for propagation of signals along regulatory and protein interaction networks. In addition to its positive aspects the small world property in biomolecular networks also has a potentially negative side by facilitating system-wide propagation of undesirable cross-talk [13]. In the course of evolution different strategies appeared allowing organism to limit and attenuate these unwelcome side effects of global connectivity.
The extent of small world topology in metabolic networks has been recently disputed
in [14]. There it
was argued that many shortcuts in simple graph representations of metabolic networks
are meaningless from biochemical standpoint. By taking into account additional
structural information about metabolites Arita [14] dramatically increased the
diameter of the metabolic network in E. coli. In our simulations of
the toolbox model we also encountered limitations of the simple graph representation
giving rise to small world topology of metabolic networks. Small world by definition
implies very short pathways (or equivalently supercritical network branching with
exponentially growing lists of neighbors at distance 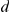 ), which in its turn
prevents the appearance of quadratic scaling in the linear toolbox model.
), which in its turn
prevents the appearance of quadratic scaling in the linear toolbox model.
How to reconcile this apparent contradiction? The answer known from pioneering studies of R. Heinrich and collaborators (see e.g. [8], [15], [16] ) is to altogether abandon the simple graph representation in favor of realistic treatment of multi-substrate reactions. A metabolic reaction with two or more substrates will not proceed at any rate until all these metabolites are present in the cell. This implicit “AND” function operating on inputs of multi-substrate metabolic reactions makes reaching a given metabolic target much harder task and ultimately leads to dramatically longer pathways (Figure 9 quantifies this effect). These longer pathways in turn reinstate the quadratic scaling in the version of the toolbox model that was introduced in the previous section. This leads to the novel conclusion of our study that, when multi-substrate reactions are properly taken into account, the small world (supercritical) topology of the metabolic universe disappears in favor of the “large world” topology characteristic of critical branching networks. The increase in the effective diameter of the network due to this effect is dramatic. One goes from 3–4 steps diameter typical of a small world network of [12], [11] to ∼8 steps of [14] and finally to 30–40 layers in the scope expansion process shown in Figure 6 (see also Figure 6 of [8]).
These arguments lead us to adapt the “scope expansion” algorithm by Heinrich et al [8] to pathway acquisition in the toolbox model. Not only did it restore the “large world” properties such as quadratic scaling to the model, it also made the added pathways plausible from evolutionary standpoint. Unlike linear random walk pathways on KEGG network used in [2], pathways in the new version of the toolbox model have the smallest number of KEGG reactions to achieve their metabolic task (production of the target metabolite from the set of metabolites already present in organism's network). As can be seen in Figure 7 a large fraction of these pathways also does not generate any byproducts. Accumulation of such byproducts inside a cell is potentially dangerous and would require specialized proteins to excrete them to the environment. The lack of byproducts also means that the useful yield of an added pathway is at or near its theoretical maximum. This is consistent with the fact that real biological pathways are optimized in the course of evolution to increase their yield while simultaneously reducing the number of reaction steps [7], [17], [18].
Optimality of metabolic pathways in central carbon metabolism was recently discussed
in Ref. [17]. There
it was shown that some (but not all) of these pathways coincide with the shortest
walks in the space of possible metabolic transformations. This study also estimated
a typical metabolic substrate can in principle be converted into any of the 20
different products in just one step. This quickly adds up to a very large number of
biochemically feasible paths connecting metabolites to each other. However, this
exponential growth does not necessarily result in a small world universal metabolic
network. Indeed, evolutionary optimization leaves just a tiny fraction of these
biochemically feasible reactions to be realized in any organism. The universal
metabolic network formed by the union of all organism-specific metabolic networks is
thus dramatically sparser than the set of all reactions allowed by the basic rules
of biochemistry. Thus, as demonstrated in Ref. [8] and the present study, the
number of metabolites one could generate in N steps starting from a small core
network and using KEGG-listed metabolic reactions instead of expanding as
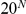 grows with N much more slowly (algebraically). The overall
picture consistent with both our observations and those of Ref. [17] is that
exponentially large, supercritical tree of all possible biochemical transformations
is first pruned to an evolutionary optimized critical universal network out of which
individual organisms select a subset of reactions necessary to accomplish their
metabolic goals: that is to utilize nutrients in their environment and generate
metabolic targets necessary for their operation.
grows with N much more slowly (algebraically). The overall
picture consistent with both our observations and those of Ref. [17] is that
exponentially large, supercritical tree of all possible biochemical transformations
is first pruned to an evolutionary optimized critical universal network out of which
individual organisms select a subset of reactions necessary to accomplish their
metabolic goals: that is to utilize nutrients in their environment and generate
metabolic targets necessary for their operation.
Simplified “toy” models based on artificial chemistry reactions have a long history of being used to reveal fundamental organizational principles of metabolic networks:
The recent model of Riehl et al [18] uses the simplest possible metabolites distinguished from each other only by the number of atoms of one element (e.g. carbon). All reactions in this case are of ligation/cleavage type (e.g.
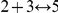 ) constrained
only by mass conservation. In spite of utmost simplicity of this artificial
chemistry, the optimal pathways in this model display a surprisingly rich
set of properties and bear some similarity to real-life metabolic
pathways.
) constrained
only by mass conservation. In spite of utmost simplicity of this artificial
chemistry, the optimal pathways in this model display a surprisingly rich
set of properties and bear some similarity to real-life metabolic
pathways.The study of Pfeiffer et el [19] emphasizes the role of different chemical groups forming metabolites. They consider another artificial chemistry where metabolites are defined by binary strings indicating presence or absence of each of
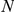 different
chemical groups, and reactions transferring one such chemical group from one
substrate which has it to another substrate which initially does not.
Plausible evolutionary rules of their model give rise to complex scale-free
metabolic networks emerging from the simple initial condition of
different
chemical groups, and reactions transferring one such chemical group from one
substrate which has it to another substrate which initially does not.
Plausible evolutionary rules of their model give rise to complex scale-free
metabolic networks emerging from the simple initial condition of
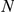 completely non-specific transferase enzymes.
completely non-specific transferase enzymes.Finally the artificial chemistry studied by Hintze et al [20] has molecules composed of three different types of atoms with different valences. Metabolites are linear molecules in which every atom is connected to others by as many bonds as specified by its valence. This model with rather complicated rules of evolution is then used to shed light on topics such as robustness and modularity of metabolic networks.
In our study we used the real-life (even if incomplete and sometimes noisy) metabolic universe of all reactions in the KEGG database. The only simplifying approximations remaining in the new most realistic version of the toolbox model is random selection of metabolic targets in the course of evolution and easy availability of any subset of KEGG reactions for horizontal transfer. Both these approximations can be relaxed in later versions of the model. Another promising direction is to extend the toolbox model to artificial chemistry universal networks of Refs. [18], [19], [20]. While taking away from the realism of the model such extensions might help to broaden our intuition about what topological properties of the universal network determine the scaling properties of its species-specific subnetworks.
Materials and Methods
The universal network used in our study consists of the union of all reactions listed in the KEGG database. The directionality of reactions and connected pairs of metabolites were inferred from the map version of the reaction formula: ftp.genome.jp/pub/kegg/ligand/reaction/reaction?mapformula.lst. The universal network with linearized pathways used to construct Figure 2 and Figure 3 consists of 1813 metabolites upstream of pyruvate and 2745 reaction edges out of which 1782 are irreversible and 963 are reversible. The metabolic network with branched and cyclic pathways used to construct Figure 5– 9 consists of 1861metabolites located downstream from the central metabolism and reachable from it by the scope expansion algorithm of Ref. [8]. It has 2819 reactions out of which 1402 are irreversible and the remaining 1417 are reversible. Table 1 and Table 2 shows the statistics for the number of substrates and products of these reactions. The list of core metabolites is obtained from KEGG Pathways Modules in the category “central carbohydrate metabolism” and extended with “currency” metabolites including water, ATP and NAD. Simulations were done in Matlab and Octave.
Supporting Information
Supplementary information.
(0.37 MB DOC)
Acknowledgments
Additional support of this work was provided by the DOE Systems Biology Knowledgebase project “Tools and Models for Integrating Multiple Cellular Networks”.
Footnotes
The authors have declared that no competing interests exist.
Work at Brookhaven National Laboratory was carried out under Contract No. DE-AC02-98CH10886, Division of Material Science, US Department of Energy. Part of this study was supported by the program “Computational Biology and Bioinformatic Methods to Enable a Systems Biology Knowledgebase” ( DE-FOA-0000143) of the Office of Biological and Environmental Research, US Department of Energy. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.van Nimwegen E. Scaling laws in the functional content of genomes. Trends Genet. 2003;19:479–484. doi: 10.1016/S0168-9525(03)00203-8. [DOI] [PubMed] [Google Scholar]
- 2.Maslov S, Krishna S, Pang TY, Sneppen K. Toolbox model of evolution of prokaryotic metabolic networks and their regulation. Proc Natl Acad Sci U S A. 2009;106:9743–9748. doi: 10.1073/pnas.0903206106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Athreya KB, Ney PE. New York: Dover Publications; 2004. Branching Processes. [Google Scholar]
- 4.Broadbent SR, Hammersley JM. Percolation Processes. Math. Proc. Camb. Phil. Soc. 1957;53:629–641. [Google Scholar]
- 5.Huang K. Statistical mechanics. New York: Wiley; 1987. 17.5 THE VAN DER WAALS EQUATION OF STATE.426 [Google Scholar]
- 6.Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Meléndez-Hevia E, Torres NV. Economy of design in metabolic pathways: further remarks on the game of the pentose phosphate cycle. J. Theor. Biol. 1988;132:97–111. doi: 10.1016/s0022-5193(88)80193-0. [DOI] [PubMed] [Google Scholar]
- 8.Handorf T, Ebenhöh O, Heinrich R. Expanding metabolic networks: scopes of compounds, robustness, and evolution. J. Mol. Evol. 2005;61:498–512. doi: 10.1007/s00239-005-0027-1. [DOI] [PubMed] [Google Scholar]
- 9.Meléndez-Hevia E, Torres NV. Economy of design in metabolic pathways: further remarks on the game of the pentose phosphate cycle. J. Theor. Biol. 1988;132:97–111. doi: 10.1016/s0022-5193(88)80193-0. [DOI] [PubMed] [Google Scholar]
- 10.Wagner A. The Yeast Protein Interaction Network Evolves Rapidly and Contains Few Redundant Duplicate Genes. Mol Biol Evol. 2001;18:1283–1292. doi: 10.1093/oxfordjournals.molbev.a003913. [DOI] [PubMed] [Google Scholar]
- 11.Wagner A, Fell DA. The small world inside large metabolic networks. Proc. Biol. Sci. 2001;268:1803–1810. doi: 10.1098/rspb.2001.1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jeong H, Tombor B, Albert R, Oltvai ZN, Barabasi A. The large-scale organization of metabolic networks. Nature. 2000;407:651–654. doi: 10.1038/35036627. [DOI] [PubMed] [Google Scholar]
- 13.Maslov S, Ispolatov I. Propagation of large concentration changes in reversible protein-binding networks. Proc Natl Acad Sci U S A. 2007;104:13655–13660. doi: 10.1073/pnas.0702905104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Arita M. The metabolic world of Escherichia coli is not small. Proc Natl Acad Sci U S A. 2004;101:1543–1547. doi: 10.1073/pnas.0306458101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Handorf T, Christian N, Ebenhöh O, Kahn D. An environmental perspective on metabolism. J. Theor. Biol. 2008;252:530–537. doi: 10.1016/j.jtbi.2007.10.036. [DOI] [PubMed] [Google Scholar]
- 16.Ebenhöh O, Handorf T, Heinrich R. A cross species comparison of metabolic network functions. Genome Inform. 2005;16:203–213. [PubMed] [Google Scholar]
- 17.Noor E, Eden E, Milo R, Alon U. Central Carbon Metabolism as a Minimal Biochemical Walk between Precursors for Biomass and Energy. Mol. Cell. 2010;39:809–820. doi: 10.1016/j.molcel.2010.08.031. [DOI] [PubMed] [Google Scholar]
- 18.Riehl WJ, Krapivsky PL, Redner S, Segrè D. Signatures of Arithmetic Simplicity in Metabolic Network Architecture. PLoS Comput Biol. 2010;6:e1000725. doi: 10.1371/journal.pcbi.1000725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pfeiffer T, Soyer OS, Bonhoeffer S. The evolution of connectivity in metabolic networks. PLoS Biol. 2005;3:e228. doi: 10.1371/journal.pbio.0030228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hintze A, Adami C. Evolution of Complex Modular Biological Networks. PLoS Comput Biol. 2008;4:e23. doi: 10.1371/journal.pcbi.0040023. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary information.
(0.37 MB DOC)