

Abstract
High salt (HS) intake can change the arterial tone in mice, and the nitric oxide (NO) acts as a mediator to some of the receptors mediated vascular response. The main aim of this study was to explore the mechanism behind adenosine-induced vascular response in HS-fed eNOS+/+ and eNOS−/− mice The modulation of vascular response by HS was examined using aortas from mice (eNOS+/+ and eNOS−/−) fed 4% (HS) or 0.45% (NS) NaCl-diet through acetylcholine (ACh), NECA (adenosine-analog), CGS 21680 (A2A AR-agonist), MS-PPOH (CYP epoxygenase-blocker; 10−5 M), AUDA (sEH-blocker; 10−5 M), and DDMS (CYP4A-blocker; 10−5 M). ACh-response was greater in HS-eNOS+/+ (+59.3 ± 6.3%) versus NS-eNOS+/+ (+33.3 ± 8.0%; P < 0.05). However, there was no response in both HS-eNOS−/− and NS-eNOS−/−. NECA-response was greater in HS-eNOS−/− (+37.4 ± 3.2%) versus NS-eNOS−/− (+7.4.0 ± 3.8%; P < 0.05). CGS 21680-response was also greater in HS-eNOS−/− (+45.4 ± 5.2%) versus NS-eNOS−/−(+5.1 ± 5.0%; P < 0.05). In HS-eNOS−/−, the CGS 21680-response was reduced by MS-PPOH (+7.3 ± 3.2%; P < 0.05). In NS-eNOS−/−, the CGS 21680-response was increased by AUDA (+38.2 ± 3.3%; P < 0.05) and DDMS (+30.1 ± 4.1%; P < 0.05). Compared to NS, HS increased CYP2J2 in eNOS+/+ (35%; P < 0.05) and eNOS−/− (61%; P < 0.05), but decreased sEH in eNOS+/+ (74%; P < 0.05) and eNOS−/− (40%; P < 0.05). Similarly, CYP4A decreased in HS-eNOS+/+ (35%; P < 0.05) and HS-eNOS−/− (34%; P < 0.05). These data suggest that NS causes reduced-vasodilation in both eNOS+/+ and eNOS−/− via sEH and CYP4A. However, HS triggers possible A2AAR-induced relaxation through CYP epoxygenase in both eNOS+/+ and eNOS−/−.
Keywords: Salt, eNOS, CYP2J2, sEH, Relaxation, Adenosine
Introduction
The endothelium plays an important role in the maintenance of vascular tone, and it releases several vasoconstrictors, such as angiotensin II, endothelin-1, reactive oxygen species, and cyclooxygenase-derived metabolites of arachidonic acid [1], as well as several vasodilators like endothelium-derived relaxing factors (EDRF) including nitric oxide (NO), and endothelium-derived hyperpolarizing factor(s) (EDHF) that are the main components of endothelium [2–5]. NO mediates most of the acetylcholine (ACh) regulated vasodilator response in both human and mouse [3–8]. NO also acts as a mediator to some of the receptor mediated agonists and increases shear stress [9, 10].
In recent studies, the authors attempted to block the adenosine-induced vascular relaxation which is mediated by adenosine A2A receptor in mouse aorta with NG-nitro-l-arginine methyl ester (L-NAME), but failed [3–5]. Since a specific inhibitor of endothelial nitric oxide synthase (eNOS) is not available, genetically manipulated mice with targeted deletion of the eNOS (eNOS−/−) have been used by others [11–16] as well as by the authors in this current study to understand the role of eNOS in vascular regulation. Earlier studies with eNOS−/− mice have shown that endothelial NO contributes to the regulation of vascular tone [11–16]. The absence of ACh-induced vasodilation mediated by NO has also been observed in pulmonary and carotid arteries of eNOS−/− mice [9, 13, 17]. In recent studies, the authors have shown ACh-induced vascular response in A2A AR+/+, A2A AR−/−, and high salt (HS)/normal salt (NS)-fed C57BL/6J mice, but adenosine A2A receptor-induced vascular response was not NO or COX dependent [3–5]. It has also been suggested that ACh-dependent vascular relaxation response was abolished in coronary arteries because of the absence of eNOS in eNOS−/− mice [18], and the relaxation induced by bradykinin in coronary circulation of eNOS−/− mice is compensated by arachidonic acid-derived metabolites through CYP450, but not by cyclooxygenase [19].
Salt induces renal CYP epoxygenase, which protects against hypertension [20]. Inhibition of CYP epoxygenase with clotrimazole increases the blood pressure and makes it salt sensitive, while salt itself does not increase blood pressure in rats maintained on a HS-diet [21]. Down-regulation of CYP-epoxygenase activity and inhibition of eicosatrienoic acid (EET) formation are associated with salt-sensitive hypertension [21–23]. Salt induces NO and COX independent vasodilation through A2A AR with upregulation of CYP epoxygenases (CYP2C29 and CYP2J2) in aortas from C57BL/6J mice that is inhibited by MS-PPOH, a CYP450 epoxygenase inhibitor [3, 5]. In the presence of CYP epoxygenases, arachidonic acids generate EETs which are involved in vasodilation, but are very unstable and get converted into diols by soluble epoxide hydrolase (sEH) which causes vasoconstriction. The inhibition of sEH changes vascular contraction to vascular relaxation in NS-fed C57BL/6J mouse aorta [5], and inhibition of CYP4A also changes vascular contraction to vascular relaxation in both A2A AR−/− and NS-fed C57BL/6J mouse aorta [3–5]. Thus, it is not known whether eNOS deficiency plays a role in A2A AR-induced vascular response in HS diet maintained mice, and if so, by what mechanism. Therefore, the main aim of this study was to determine the role of eNOS in A2A AR-mediated vascular response in HS-fed eNOS−/− mice. The proposed hypothesis is that HS enhances NO-independent vascular relaxation through A2A AR and CYP epoxygenase (CYP2J2), while NS limits vascular relaxation through sEH and CYP4A in aorta from eNOS−/− mice.
Materials and methods
Animal use experimental protocols were approved and carried out in accordance with the West Virginia University Institutional Animal Care and Use Committee and were in accordance with the principles and guidelines of the Institute of Laboratory Animal Resources Guide for the Care and Use of Laboratory Animals. Ten-week-old male eNOS+/+ and eNOS−/− mice (Jackson Laboratories, Bar Harbor, ME) were placed on a whole-grain diet containing either NS (0.45% NaCl) or HS (4% NaCl, TD.88311, and TD.92034 diets; Teklad, Madison, WI). All mice were studied for 4–5 weeks after assignment to either the NS or HS group [3].
Organ bath experiments
HS- and NS-fed eNOS+/+ and eNOS−/− mice were killed with pentobarbital sodium (100 mg/kg, i.p.). According to the previous protocol [3, 4], after thoracotomy, the aorta was gently removed, cleaned of fat and connective tissues, and cut transversely into rings of 3–4 mm in length. Extreme care was taken not to damage the endothelium. The rings were mounted vertically between two stainless steel wire hooks. The two rings were suspended in 10 ml organ baths containing modified Krebs-Henseleit buffer (in 118 mM of NaCl, 4.8 mM of KCl, 1.2 mM of MgSO4, 1.2 mM of KH2PO4, 25 mM of NaHCO3, 11 mM of glucose, and 2.5 mM of CaCl2). The pH of the buffer was adjusted and maintained at 7.4 by equilibration with 95% O2 and 5% CO2 at 37°C. The aortic rings were equilibrated for 90 min with a resting force of 1 g, with changes of the bathing solution at 15 min intervals according to the previous protocol [3, 4]. At the end of equilibration period, tissues were contracted with KCl (50 mM) to check the viability. Aortic rings were then constricted with phenylephrine (PE, 10−7 M) and changes in tension were monitored continuously with a fixed range precision force transducer (TSD, 125 C, BIOPAC system) connected to differential amplifier (DA 100B, BIOPAC system). Data were recorded using MP100 WSW, BIOPAC digital acquision system and analyzed using Acknowledge 3.5.7 software (BIOPAC system).
Vascular response of aortas from eNOS+/+/eNOS−/− mice fed HS and NS against ACh
The vascular endothelium was tested in vessels from HS/NS-fed eNOS+/+ and eNOS−/− mice through acetylcholine (ACh, 10−7 M) on precontracted (phenylephrine; PE) aortic rings, as described previously [3, 4]. The preparations were washed several times with Krebs-Henseleit solution and allowed to equilibrate for 30 min before the experiment. The contraction and relaxation responses are expressed as % increase and decrease, respectively of PE-induced precontraction.
Vascular response of aortas from eNOS+/+/eNOS−/− mice fed HS and NS with NECA and CGS 21680
The responsiveness of pre-contracted eNOS+/+ and eNOS−/− aortic rings to non-selective adenosine analog, 5′-N-ethylcarboxamidoadenosine (NECA) or the selective A2A receptor agonist CGS 21680 was obtained by cumulative addition of NECA or CGS 21680 to the organ bath in 1 – log increments to obtain a concentration–response curve as previously described [3, 4]. One concentration–response curve for NECA or CGS 21680 was constructed for each eNOS+/+ and eNOS−/− mouse ring, and all concentration–response determinations were run in parallel on pairs of rings from either HS (eNOS+/+ and eNOS−/−) or NS (eNOS+/+ and eNOS−/−) in the same organ bath.
Effect of CYP epoxygenase, sEH, and CYP4A inhibitors on vascular response of aortas from eNOS+/+/eNOS−/− mice fed HS and NS against CGS 21680
A selective CYP epoxygenase inhibitor, Methylsulfonyl-propargyloxyphenylhexanamide (MS-PPOH, 10 μM) or 12-(3-adamantan-1-yl-ureido) dodecanoic acid (AUDA, 10 μM), a selective soluble epoxide hydrolase inhibitor or a selective CYP ω-hydroxylase inhibitor, dibromo-dodecenyl-methylsulfimide (DDMS, 10 μM) was added 30 min before contraction of the tissue (eNOS+/+ and eNOS−/−) with PE and was present throughout the experiment. These experiments were performed in parallel with four rings from either HS- or NS-fed (eNOS+/+/eNOS−/−) mice. Two rings from each aorta were used as control, while the other two were used with either MS-PPOH or AUDA or DDMS. The control was the same as used in the earlier section of CGS 21680.
Western blot
Aortas from both HS (eNOS+/+/eNOS−/−) and NS (eNOS+/+/eNOS−/−) fed mice were isolated and each sample was treated with 1 ml of lysis buffer (50 mM Tris–HCl, pH 7.4, 1% TritonX-100, 150 mM NaCl, 1 mM EGTA, 1 mM PMSF, 0.25% sodium deoxycholate, 1 μg/ml aprotinin, 1 μg/ml pepstatin, 1 μg/ml leupeptin, 1 mM Na3VO4, and 1 mM NaF) and homogenized on ice as described elsewhere [3, 4]. The samples were transferred to dry ice for 5 min and then thawed on wet ice. After thawing, the samples were vortexed and centrifuged for 5 min at 12,000 rpm at 4°C. Supernatants were sonicated and stored at −80°C. Protein was measured using Bio-Rad assay based on the Bradford dye binding procedure with bovine serum albumin as a standard. The protein mixture was divided into aliquots and stored at −80°C. At the time of analysis, samples were thawed and approximately 30 μg of total protein per lane was loaded on a slab gel. Proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) using 10% acrylamide gels (1 mm thick). After electrophoresis, the proteins on the gel were transferred to nitrocellulose membrane (Hybond-ECL) by electroelution. Protein transfer was confirmed by employing pre-stained molecular weight markers (Bio-Rad Laboratories, Hercules, CA). Following blocking with non-fat dry milk, the nitrocellulose membranes were incubated with monoclonal and polyclonal antibodies cross-reacting with primary antibody for CYP2J2 (Dr. Zeldin, NIEHS/NIH), sEH (gift from Dr. Marowsky, Switzerland), CYP4A (polyclonal, Affinity Bio-Reagents), and β-actin antibody (Santa Cruz) was used to detect as housekeeping proteins for proper loading the samples in each lane. The secondary antibody was a horseradish peroxidase conjugated anti-rabbit IgG. The membranes were developed using enhanced chemiluminescence (Amersham BioSciences) and exposed to X-ray film for appropriate time as described by the authors [3, 4].
Chemicals, drugs and antibodies
Phenylephrine and acetylcholine were dissolved in distilled water. NECA and CGS 21680 (Sigma Chemicals, St. Louis, MO) were dissolved in 100% DMSO as 10 mM stock solutions, which were followed by serial dilutions in distilled water. MS-PPOH (Dr. Falck), and DDMS from Cayman Chemical (Ann Arbor, Michigan) were dissolved in 100% ethanol, AUDA (gift from Dr. Morisseau) was dissolved in DMSO. CYP2J2 (polyclonal, Dr. Zeldin, NIEHS/NIH), sEH (gift from Dr. Marowsky, Switzerland), and CYP4A antibody (polyclonal, Affinity BioReagents, Golden, Colorado) were obtained and used for Western Blot experiments.
Statistical Analysis
Data are reported as mean ± SEM. One-way analysis of variance (ANOVA) was used to compare difference among groups, and two-way ANOVA for repeated measure, followed by Tukey post hoc test to compare the vascular responses between HS (eNOS+/+/eNOS−/−) and NS (eNOS+/+/eNOS−/−), and to antagonist (MS-PPOH, AUDA, and DDMS). Differences were considered significant if P < 0.05. Further, densitometry of western blot analysis (CYP2J2, sEH, and CYP4A) was expressed as mean ± SEM in arbitrary units. All the statistical analyses were performed using Graph Pad Prism statistical package.
Results
Vascular response of aortas from eNOS+/+/eNOS−/− mice fed HS and NS against acetylcholine
Relaxation to ACh (10−7 M) was significantly greater in aortas from HS (+59.3 ± 6.3%) than that of NS (+33.3 ± 8.0%; P < 0.05) fed eNOS+/+ mice (Table 1, Fig. 1). In contrast, ACh-dependent response was virtually absent in aorta from both HS- and NS-fed eNOS−/− mice (+0.8 ± 0.1% vs. +1.5 ± 0.3%, Table 1, Fig. 1).
Table 1.
Overview of experiments and observations, Acetylcholine (10−7 M), NECA (adenosine analog; 10−11–10−5 M), CGS 21680 (selective A2AAR agonist; 10−12–10−6 M), MS-PPOH (CYP-epoxygenase inhibitor; 10−5 M) + CGS 21680, and AUDA (sEH inhibitor; 10−5 M) + CGS 21680 and DDMS (CYP4A inhibitor; 10−5 M) + CGS 21680-dependent concentration response curve of aortas from eNOS+/+ and eNOS−/− mice fed normal salt (NS) and high salt (HS) containing diet
| Group | Ach | NECA | CGS 21680 | MS-PPOH+ CGS 21680 | AUDA+ CGS 21680 | DDMS+ CGS 21680 | CYP2J2 | sEH | CYP4A |
|---|---|---|---|---|---|---|---|---|---|
| NS-eNOS+/+ | + | + | + | − | ++ | ++ | + | + | + |
| Relax | Relax | Relax | Relax | Relax | Relax | ||||
| NS-eNOS−/− | −# | + | + | − | ++ | ++ | + | + | + |
| Relax | Relax | Relax | Relax | Relax | Relax | ||||
| HS-eNOS+/+ | ++* | ++* | ++* | − | ++ | ++ | ++* | −* | −* |
| Relax | Relax | Relax | Relax | Relax | Relax | ||||
| HS-eNOS−/− | −$ | +++# | +++# | − | ++ | ++ | +++# | −# | −# |
| Relax | Relax | Relax | Relax | Relax | Relax |
Representative western blots and densitometric data for CYP2J2 (∼58 kDa), sEH (∼60 kDa), and CYP4A (∼50 kDa) proteins in aortas of eNOS+/+ and eNOS−/− mice fed with HS and NS containing diets. Values are mean ± SEM.
P < 0.05 compared the vascular response of NS (eNOS+/+) with NS (eNOS−/−), HS (eNOS+/+), HS (eNOS−/−), and non-treated groups compared with treated (MS-PPOH, AUDA, DDMS) groups. Also, compared their protein (CYP2J2, sEH, and CYP4A) expression in NS (eNOS+/+) with NS (eNOS−/−), HS (eNOS+/+), and HS (eNOS−/−)
Fig. 1.

Acetylcholine (10−7 M)-dependent response of aortas from eNOS+/+ and eNOS−/− mice fed NS and HS containing diet. Values are mean ± SEM. *$#P < 0.05 compared NS (eNOS+/+) with #NS (eNOS−/−), *HS, (eNOS+/+), and $HS (eNOS−/−), n = 6
Vascular response of aortas from eNOS+/+/eNOS−/− mice fed HS and NS against NECA
Although at lower concentrations (10−11–10−9 M) NECA did not significantly alter the aortic response, increasing NECA concentrations from 10−8 to 10−5 substantially enhanced the relaxation in aortas from both eNOS+/+ and eNOS−/− mice that were fed HS diet (Table 1, Fig. 2). In contrast, increase in the NECA concentration did not produce significant relaxation in aorta from both eNOS+/+ and eNOS−/− mice that were fed NS diet (Table 1, Fig. 2). For example, NECA at 10−6 M produced +30.4 ± 2.4 and +37.4 ± 3.2% relaxation in aortas from HS-fed eNOS+/+ and eNOS−/− mice, respectively, while it produced only +3.2 ± 1.3 and +7.4 ± 3.7% relaxation in aortas from NS-fed eNOS+/+ and eNOS−/− mice, respectively (Table 1, Fig. 2).
Fig. 2.

NECA-induced vascular response in aortic rings of eNOS+/+ and eNOS−/− mice fed with NS and HS containing diet. Values are mean ± SEM. *#P < 0.05 compared NS (eNOS+/+, eNOS−/−) with *HS (eNOS+/+) and #HS (eNOS−/−), n = 6
Vascular response of aortas from eNOS+/+/eNOS−/− mice fed HS and NS against CGS 21680
CGS 21680 produced a concentration-dependent relaxation at concentration from 10−9 to 10−6 M (P < 0.05) in aortas from both eNOS−/− and eNOS+/+) mice that were fed with HS diet (Table 1, Fig. 3). It is to be noted here that the relaxation response at these concentration were significantly greater in aorta from HS-fed eNOS−/− mice (+45.4 ± 5.2% at 10−6 M) than that from NS-fed eNOS−/− mice (+5.1 ± 5.0% at 10−6 M; P < 0.05; Table 1, Fig. 3). Similarly, the relaxation response was more in aorta from HS-fed eNOS+/+ mice (+33.2 ± 3.2% at 10−6 M) than that from NS-fed eNOS−/− mice (+5.9 ± 2.9% at 10−6 M; P < 0.05; Table 1, Fig. 3).
Fig. 3.

CGS 21680-induced vascular response in aortic rings of eNOS+/+ and eNOS−/− mice fed with NS and HS containing diet. Values are mean ± SEM. *#P < 0.05 compared NS (eNOS+/+, eNOS−/−) with*HS (eNOS+/+) and #HS (eNOS−/−), n = 6
Effect of CYP epoxygenase inhibitor on vascular response of aortas from eNOS+/+/eNOS−/− mice fed HS and NS against CGS 21680
CGS 21680 significantly enhanced the relaxation in aorta from HS compared to NS-fed eNOS−/− mice at concentrations ranging from 10−9 to 10−6 M (P < 0.05, Table 1, Fig. 3). As shown in Table 1, Fig. 4, 10 μM MS-PPOH (a highly selective CYP-epoxygenase inhibitor) completely inhibited the CGS 21680-enhanced relaxation at all concentrations (10−9–10−6 M) in aorta from HS-fed eNOS−/− mice. 10 μM MS-PPOH inhibited the 10−6 M CGS 21680 induced relaxation response (from +45.4 ± 5.2 to +7.3 ± 3.2%; P < 0.05) in aorta from HS-fed eNOS−/− mice (Table 1, Fig. 4). MS-PPOH did not change the CGS 21680-induced responses in aorta from NS-fed eNOS−/− mice (Table 1, Fig. 4). The control values presented are adopted from Fig. 3. Use of MS-PPOH in HS-fed eNOS+/+ mouse aorta also caused a reduction in relaxation (to +3.9 ± 3.0%), while the absence of MS-PPOH did not have any effect on relaxation (+30.5 ± 5.2%; P < 0.05, at 10−6 M CGS 21680). No significant effect of MS-PPOH was observed on CGS 21680-induced responses in NS-fed eNOS+/+ mouse aorta (P > 0.05).
Fig. 4.

Effect of MS-PPOH (CYP epoxygenase inhibitor; 10 μM) on CGS 21680 induced vascular response in aortic rings of eNOS−/− mice fed with NS and HS containing diet. The control data adopted from Fig. 3. Values are mean ± SEM. #P < 0.05 compared NS (eNOS−/−), HS (eNOS−/−) + MS-PPOH, and NS (eNOS−/−) + MS-PPOH with #HS (eNOS−/−), n = 6
Effect of sEH inhibitor on vascular response of aortas from eNOS+/+/eNOS−/− mice fed HS and NS against CGS 21680
Increase in the CGS 21680 concentration did not alter response in aorta from NS-fed eNOS−/− mice (data adopted from Fig. 3). In contrast, in the presence of 10 μM AUDA (a sEH inhibitor) with the increase in CGS 21680 concentrations from 10−9 to 10−6 M enhanced the aortic relaxation in a concentration-dependent pattern in NS-fed eNOS−/− mice (Table 1, Fig. 5). In the presence of AUDA, 10−6 M CGS 21680 produced a significantly higher vascular relaxation response in aorta from NS-fed eNOS−/− mice (from +5.1 ± 5.0 to +38.2 ± 3.3%, P < 0.05, Table 1, Fig. 5).The CGS 21680 enhanced aortic relaxation in the presence of AUDA in NS-fed eNOS−/− mice was almost identical to that of aortic relaxation seen in non-AUDA treated HS-fed eNOS−/− mice (Table 1, Fig. 5). In contrast to aorta from NS-fed eNOS−/− mice, AUDA did not alter CGS 21680-mediated relaxation in aorta from HS-fed eNOS−/− mice (Table 1, Fig. 5).The control values are the same as shown in Fig. 3. Also, sEH inhibitor, produced a significantly higher relaxation (+28.9 ± 3.8%, P < 0.05) at 10−6 M of CGS 21680 in NS-fed eNOS+/+ mouse aorta as opposed to control (+5.2 ± 3.2%). In contrast, AUDA had no significant effect on CGS 21680-mediated relaxation in HS-fed eNOS+/+ mouse aorta. There were no significant differences in responses to CGS 21680 among NS-fed eNOS+/+ mouse aorta treated with AUDA, treated HS eNOS+/+ and untreated HS-fed eNOS+/+ mouse aorta (P > 0.05).
Fig. 5.

Effect of AUDA (sEH inhibitor; 10 μM) on CGS 21680 induced vascular response in aortic rings of eNOS−/− mice fed with NS and HS containing diet. The control data adopted form Fig. 3. Values are mean ± SEM. *$#P < 0.05 compared NS (eNOS−/−) with *HS (eNOS−/−), #HS + AUDA (eNOS−/−) and $NS + AUDA (eNOS−/−), n = 6
Effect of CYP4A inhibitor on vascular response of aortas from eNOS+/+/eNOS−/− mice fed HS and NS against CGS 21680
Increase in the CGS 21680 concentration did not alter the basal response present in aorta from NS-fed eNOS−/− mice (data adopted from Fig. 3). In the presence of 10 μM DDMS (a CYP4A inhibitor), increase in the concentration (from 10−9 to 10−6 M) of CGS 21680 produced significantly higher relaxation (from +5.1 ± 5.0 to +30.1 ± 4.1% at 10−6 M; P < 0.05, Table 1, Fig. 6) in aorta from NS-fed eNOS−/− mice. In contrast, DDMS did not alter the CGS 21680 enhanced relaxation in aorta from HS eNOS−/− mice (Table 1, Fig. 6). The CGS 21680 enhanced vascular relaxation in presence of DDMS in aorta from NS-fed eNOS−/− mice were almost similar to that of aorta from HS-fed eNOS−/− mice (Table 1, Fig. 6). The control values are the same as shown in Fig. 3. Also, CYP4A inhibitor, produced significantly higher relaxation (+29.1 ± 4.1%; P < 0.05) at 10−6 M of CGS 21680 in NS-fed eNOS+/+ mouse aorta as opposed to control (+5.2 ± 3.2%). In contrast, DDMS had no significant effect on CGS 21680-mediated relaxation in HS-fed eNOS+/+ mouse aorta. There were no significant differences in responses to CGS 21680 among NS-fed eNOS+/+ mouse aorta treated with DDMS, treated HS-fed eNOS+/+ mouse aorta and untreated HS-fed eNOS+/+ mouse aorta (P > 0.05).
Fig. 6.

Effect of DDMS (CYP4A inhibitor; 10 μM) on CGS 21680 induced vascular response in aortic rings of eNOS−/− mice fed with NS and HS containing diet. The control data adopted from Fig. 3. Values are mean ± SEM. *$#P < 0.05 compared NS (eNOS−/−) with. *HS (eNOS−/−), #HS + DDMS (eNOS−/−) and $NS + DDMS (eNOS−/−), n = 6
Expression of CYP2J2, sEH and CYP4A proteins in aortas from eNOS+/+/eNOS−/− mice fed HS and NS
Western blot analyses indicate that the expression of CYP2J2 protein (∼58 kDa) was significantly (35%) enhanced in aorta from HS-fed eNOS+/+ mice than that in NS-fed eNOS+/+ mice (Table 1, Fig. 7). Also, there was a significant increase in CYP2J2 protein expression (61%) in aorta from HS compared to NS-fed eNOS−/− mice (Table 1, Fig. 7). As shown in Table 1 and Fig. 8, the sEH protein (∼60 kDa) expression was reduced by 75% in aorta from HS-fed eNOS+/+ mice than that of NS-fed eNOS+/+ mice (Table 1, Fig. 8). Similarly, the sEH protein expression was also significantly (39%) reduced in aorta from HS compared to NS-fed eNOS−/− mice (Table 1, Fig. 8). Figure 9 shows the Western blot analyses for CYP4A (∼50 kDa) protein. The expression of CYP4A protein level was significantly (35%) reduced in aorta from HS-fed eNOS+/+ mice compared to NS-fed eNOS+/+ mice (Table 1, Fig. 9). The CYP4A protein expression was also significantly reduced in aorta from HS fed compared to NS-fed eNOS−/− mice (Table 1, Fig. 9).
Fig. 7.
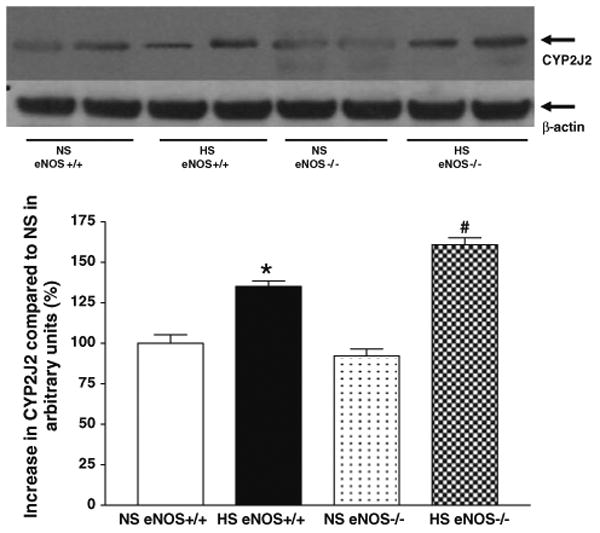
Representative western blots and densitometric data for CYP2J2 protein in aortas of eNOS+/+ and eNOS−/− mice fed with HS and NS containing diets. Values are mean ± SEM, *#P < 0.05 compared NS (eNOS+/+, eNOS−/−) aortas with *HS (eNOS+/+) and #HS (eNOS−/−), n = 4
Fig. 8.
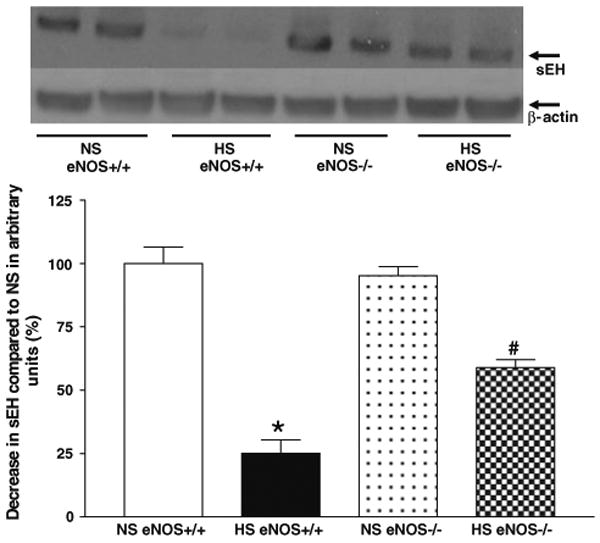
Representative western blots and densitometric data for sEH protein in aortas of eNOS+/+ and eNOS−/− mice fed with HS and NS containing diets. Values are mean ± SEM, *#P < 0.05 compared NS (eNOS+/+, eNOS−/−) aortas with *HS (eNOS+/+) and #HS (eNOS−/−), n = 5
Fig. 9.
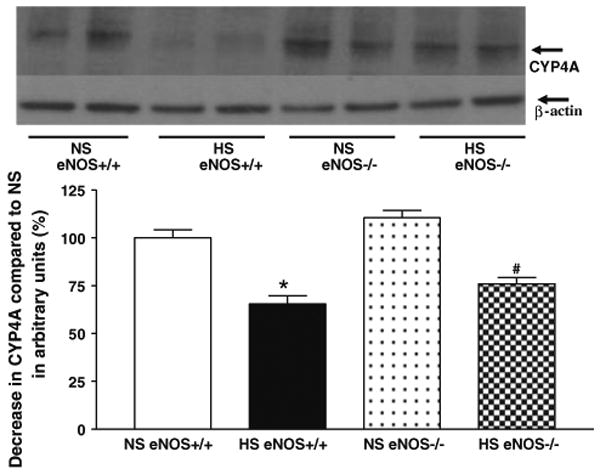
Representative western blots and densitometric data for CYP4A protein in aortas of eNOS+/+ and eNOS−/− mice fed with HS and NS containing diets. Values are mean ± SEM, *#P < 0.05 compared NS (eNOS+/+, eNOS−/−) aortas with *HS (eNOS+/+) and # HS (eNOS−/−) n = 5
Discussion
This study utilizes eNOS+/+ and eNOS−/− mice to assess the functional and biochemical consequences of HS and NS feeding on vascular response. This study demonstrates that ACh-induced relaxation is present in both HS- and NS-fed eNOS+/+, but not in eNOS−/− mouse aortas. This study also shows that: (1) adenosine A2A receptor regulates the eNOS-independent vascular relaxation through CYP epoxygenase in aorta of HS-fed eNOS−/− mice; (2) the expression of CYP epoxygenase (CYP2J2) protein was enhanced in aortas of both HS-fed eNOS+/+ and eNOS−/− mice; (3) both sEH and ω-hydroxylase (CYP4A) inhibitor reversed the reduced vasodilation into enhanced relaxation in aortas of both NS-fed eNOS+/+ and eNOS−/− mice; (4) HS down regulated both sEH and CYP4A protein expression in aortas of both eNOS+/+ and eNOS−/− mice; while (5) NS up regulated both sEH and CYP4A protein expression in aortas of both eNOS+/+ and eNOS−/− mice.
The observation that ACh induced vascular relaxation in aorta of HS/NS-fed eNOS+/+, but not in eNOS−/− mice suggests that NO generated by eNOS is essential for ACh-induced vascular relaxation (Fig. 1). Similarly, the absence of ACh-induced response in aorta [13] and carotid arteries [9] has been shown in regular-diet fed-eNOS-null mice. In humans, NOS inhibition causes a significant reduction in exercise hyperemia in the forearm [24, 25]; while in rats NOS inhibition reduces hind limb blood flow during treadmill exercise [26]. In contrast, other studies have shown that NOS inhibition does not affect the increase of blood flow evoked in forearm or leg exercise in humans, rats, and rabbits [27–30]. Also, Ray and Marshall [31] reported that during exercise, adenosine originates from skeletal muscle fibers independently of NO and acts directly on adenosine A2A AR on the vascular smooth muscle to evoke vasodilatation. As far as the absence of eNOS in eNOS−/− mice, the coronary flow is also not altered [14], and it has been suggested that either up-regulation of different NOS isoform (s) or enzyme(s) that produce other vasodilators as an alternate mechanism proposed for the unaltered coronary flow in eNOS null mice [14]. However, this is not the case with adenosine A2A receptor-induced vasodilation, where L-NAME and indomethacin (COX inhibitor) are unable to block aortic relaxation in A2A AR+/+ and high salt fed C57BL/6J mice [3–5]. Similar results have been shown in bradykinin-induced coronary flow in eNOS−/− mice, indicating that alternate NOS isoform (s) are not involved in eNOS deficient mice [19]. Elevated PGI2 production in bradykinin-induced vasodilation is also not responsible for compensation of eNOS deficiency in eNOS−/− mice [19]. This finding is that the HS-diet leads to a greater NECA-induced vasodilation in aortas of eNOS−/− mice, suggests that eNOS deficiency enhances adenosine-induced vascular relaxation. The results from this study show that NECA (a non-selective adenosine analog)-induced vascular response in aorta of HS-fed eNOS+/+ and NS-fed eNOS+/+ mice, respectively, is similar to that of the earlier reports with HS- and NS-fed C57BL/6J mice [3, 5]. Further, similar to the earlier report with HS-fed C57BL/6J mice, the selective A2A receptor agonist CGS 21680 also caused vasodilation of aorta from HS-fed, but not in NS-fed eNOS+/+ mice [3, 5]. CGS 21680 (A2A AR agonist) also caused vasodilation in aorta of HS-fed eNOS−/− similar to eNOS+/+ mice, but a degree of higher relaxation in aorta of HS-fed eNOS−/− compared to HS-fed eNOS+/+ mice was observed. These findings strongly suggest that HS-induced vascular relaxation in eNOS−/− mice is A2A AR-dependent, but is NO independent as it was in HS-fed C57BL/6J mice aorta [3, 5]. Also, this study demonstrates that CYP epoxygenase is involved in the regulation of A2A AR-induced vasodilation in HS-fed eNOS−/− and eNOS+/+ mice. The epoxygenation of arachidonic acid by CYP epoxygenases may result in the formation of four different EET regioisomers (5,6-, 8,9-, 11,12- and 14,15-EET) that may be involved in vasodilation as reported [4, 32]. Since the CYP epoxygenase-derived metabolites are capable of hyperpolarizing and relaxing smooth muscle cells, the CYP epoxygenase derivatives are recognized as endothelium-derived hyperpolarization factors [4, 33–41]. In this study, MS-PPOH (a CYP epoxygenase inhibitor) is also able to block the CGS 21680-induced vasodilation in aorta of HS-fed eNOS−/− mice. However, the vascular response in aorta of NS-fed eNOS−/− mice is not changed. These observations suggest that NO-independent CGS 21680-induced vascular relaxation is mediated by endothelial CYP epoxygenases, including CYP2J2 in aorta from both HS-fed eNOS+/+ and eNOS−/− mice.
This data also demonstrates that AUDA (a sEH inhibitor) alters the vascular response by converting the lack of response against CGS 21680 to an enhanced vasodilation in aorta of NS-fed eNOS−/− mice. The absence of significant difference between AUDA treated and untreated control aorta of HS-fed eNOS−/− mice suggests that the sEH enzyme is involved in the inhibition of vascular relaxation in NS-fed eNOS−/− mice. Usually, the conversion of EETs into diols by sEH diminishes the EETs mediated vascular relaxation [42]. Thus, the inhibition of sEH results in EETs accumulation and prolongs the period of vascular relaxation [42], as the authors have seen in this study where lack of response against CGS 21680 to an enhanced vasodilation in aorta of NS-fed eNOS−/− mice. the authors have also observed the down-regulation of sEH protein expression in aorta from HS-fed eNOS+/+ and eNOS−/− mice, whereas increased expression of sEH has been seen in aorta from NS-fed eNOS+/+ and eNOS−/− mice in this study.
DDMS (a CYP4A inhibitor) has also been identified to cause alteration to vascular response (enhanced dilation) in aortas from NS-fed eNOS−/− mice. A significant difference was not observed between DDMS treated and non-treated control in aorta of HS-fed eNOS−/− mice. At this point, the authors do not know how the regulation of vascular tone is being balanced between CYP2J2 and CYP4A in DDMS treated and non-treated control in aorta of HS-fed eNOS−/− mice. However, these observations suggest that the CYP4A enzyme also plays a critical role in controlling the vascular relaxation in aorta of NS-fed eNOS−/− mice. These findings also suggest that the increased expression of CYP2J2 epoxygenase, and the decreased expression of sEH and CYP4A in aortas from both HS-fed eNOS−/− and eNOS+/+ mice are important contributors of vascular relaxation. Further, the decrease in CYP2J2 expression, and the increased expression of both sEH and CYP4A are the major contributors of the vessels to limit relaxation response in aorta from both NS-fed eNOS+/+ and eNOS−/− mice. Also, these findings indicate that A2A AR causes concentration-dependent vasorelaxation in aorta from both HS-fed eNOS−/− and eNOS+/+ mice with 61% (P < 0.05) and 35% (P < 0.05) up-regulation of CYP2J2 protein, respectively compared to their respective NS-fed eNOS−/− and eNOS+/+ mice. HS-diet enhanced the responses to NECA and CGS 21680 to a greater extent in eNOS−/− than in eNOS+/+ mice. Because, in HS-fed eNOS−/− mouse aorta, the CYP2J2 protein expression was 26% (P < 0.05) more than the HS-fed eNOS+/+, and more CYP2J2 enzymes in HS-fed eNOS−/− mouse aorta helps in the process of epoxgenation to generate more EETs from arachidonic acids leading to more vasodilation compared to HS-fed eNOS+/+ mice. This observation suggests that in the event of NO deficiency, the increased CYP epoxygenases activity including CYP2J2 might possibly serve as alternate or enhanced mechanism for vasodilation as reported by others, the role of EDHF in regular diet fed mice [43, 44]. As the authors discussed earlier, HS feeding enhances vascular relaxation in C57BL/6J mouse aortas through upregulation of A2A AR and CYP epoxygenases (CYP2C29 and CYP2J2) [3, 5], and EETs are generated through CYP epoxygenases (CYP2C29 and CYP2J2), which mediates vasodilator response in HS-fed rat preglomerular microvessels to A2A AR agonists [45]. Activation of A2A AR is coupled to EETs release, and stimulates adenylyl cyclase in HS-fed rat preglomerular microvessels [45]. In this study, there is vascular relaxation through A2A AR and CYP2J2 in aortas from both HS-fed eNOS−/− and eNOS+/+ mice compared to NS-fed eNOS−/− and eNOS+/+ mice. Therefore, the data conclude that there is a possibility of A2A AR coupled with CYP2J2 activity, and that may be involved in cAMP-dependent PKA-mediated pathway through activation of adenylyl cyclase lead to NO-independent vascular relaxation in aortas from both HS-fed eNOS−/− and eNOS+/+ mice. Similarly, others have shown that EET regioisomers are involved in cAMP-dependent PKA-mediated pathway via activation of adenylyl cyclase in vitro cell culture models [46–48] as well as in HS-fed rat preglomerular microvessels [45].
In summary, this study demonstrates that ACh induced relaxation in both HS- and NS-fed aorta is dependent on NO produced by eNOS, whereas adenosine A2A receptor-modulated vasodilation is dependent on CYP epoxygenases including CYP2J2 in aorta of HS-fed eNOS−/− and eNOS+/+ mice. Recently, the authors have also reported that adenosine A2A receptor-induced vascular relaxation is independent of NO and COX, but dependent on CYP epoxygenases including CYP2C29 and CYP2J2 via generation of EETs in both HS-fed C57BL/6J and A2A AR+/+ mouse aorta [3–5]. Further, this study also demonstrates the role of sEH and CYP4A in regulating the vascular response in aortas of NS-fed eNOS−/− and eNOS+/+ mice. These findings suggest that there is a relationship between the availability of A2A ARs, functional capacity of endothelium to increase CYP2J2 enzyme, down-regulate sEH and CYP4A lead to NO-independent vascular relaxation in aorta from HS-fed eNOS−/− and eNOS+/+ mice, while less availability of A2A ARs, less functional capacity of endothelium to increase CYP2J2 enzyme, up-regulation of sEH and CYP4A lead to limited vascular relaxation in aorta from NS-fed eNOS−/− and eNOS+/+ mice. Additional studies with gene manipulation technology is necessary to unravel the mechanism(s) by which adenosine receptors regulate controlling vascular tone with A2A AR coupled with CYP2J2 in cAMP-dependent PKA-mediated pathway via activation of adenylyl cyclase in HS- and NS-fed mice.
Acknowledgments
The authors would like to thank Dr. Mustafa for his support (HL 027339 and MAN is Co-Investigator in HL 094447), AHA 2250298 (MAB), GM 31278 (JRF), and the Intramural Research Program of the NIEHS/NIH-Z01 ES025034 (DCZ).
Abbreviations
- EDRF
Endothelium-derived relaxing factors
- EDHF
Endothelium-derived hyperpolarizing factor
- L-NAME
NG-nitro-l-arginine methyl ester
- sEH
Soluble epoxide hydrolase
- eNOS
Endothelial nitric oxide synthase
- EET
Eicosatrienoic acid
- NECA
5′-N-ethylcarboxamidoadenosine
- MS-PPOH
Methylsulfonyl-propargyloxyphenylhexanamide
- AUDA
12-(3-adamantan-1-yl-ureido)dodecanoic acid
- DDMS
Dibromo-dodecenyl-methylsulfimide
Contributor Information
Mohammed A. Nayeem, Email: mnayeem@hsc.wvu.edu, Department of Physiology and Pharmacology, Center for Cardiovascular and Respiratory Sciences, West Virginia University, 3051 Robert C. Byrd Health Science Center-North, 1 Medical Center Drive, P. O. Box 9229, Morgantown, WV 26506, USA.
Darryl C. Zeldin, Division of Intramural Research, NIEHS/NIH, RTP, Durham, NC 27709, USA
Matthew A. Boegehold, Department of Physiology and Pharmacology, Center for Cardiovascular and Respiratory Sciences, West Virginia University, 3051 Robert C. Byrd Health Science Center-North, 1 Medical Center Drive, P. O. Box 9229, Morgantown, WV 26506, USA
John R. Falck, Department of Biochemistry, UTSWMC, Dallas, TX 75390, USA
References
- 1.Wong WT, Wong SL, Tian XY, Huang Y. Endothelial dysfunction: the common consequence in diabetes and hypertension. J Cardiovasc Pharmacol. 2010;55:300–307. doi: 10.1097/fjc.0b013e3181d7671c. [DOI] [PubMed] [Google Scholar]
- 2.Brandes RP, Schmitz-Winnenthal FH, Feletou M, Godecke A, Huang PL, Vanhoutte PM, Fleming I, Busse R. An endothelium-derived hyperpolarizing factor distinct from NO and prostacyclin is a major endothelium-dependent vasodilator in resistance vessels of wild-type and endothelial NO synthase knockout mice. Proc Natl Acad Sci USA. 2000;97:9747–9752. doi: 10.1073/pnas.97.17.9747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nayeem MA, Ponnoth DS, Boegehold MA, Zeldin DC, Falck JR, Mustafa SJ. High-salt diet enhances mouse aortic relaxation through adenosine A2A receptor via CYP epoxygenases. Am J Physiol Regul Integr Comp Physiol. 2009;296:R567–R574. doi: 10.1152/ajpregu.90798.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nayeem MA, Poloyac SM, Falck JR, Zeldin DC, Ledent C, Ponnoth DS, Ansari HR, Mustafa SJ. Role of CYP epoxygenases in A2A AR-mediated relaxation using A2A AR-null and wild-type mice. Am J Physiol Heart Circ Physiol. 2008;295:H2068–H2078. doi: 10.1152/ajpheart.01333.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nayeem MA, Zeldin DC, Boegehold MA, Morisseau C, Marowsky A, Ponnoth DS, Roush KP, Falck JR. Modulation by salt intake of the vascular response mediated through adenosine A2A receptor: role of CYP epoxygenase and soluble epoxide hydrolase. Am J Physiol Regul Integr Comp Physiol. 2010;299(1):R325–R333. doi: 10.1152/ajpregu.00823.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Elhusseiny A, Hamel E. Muscarinic—but not nicotinic—acetylcholine receptors mediate a nitric oxide-dependent dilation in brain cortical arterioles: a possible role for the M5 receptor subtype. J Cereb Blood Flow Metab. 2000;20:298–305. doi: 10.1097/00004647-200002000-00011. [DOI] [PubMed] [Google Scholar]
- 7.Rosenblum WI, Nishimura H, Nelson GH. Endothelium-dependent l-Arg- and L-NMMA-sensitive mechanisms regulate tone of brain microvessels. Am J Physiol. 1990;259:H1396–H1401. doi: 10.1152/ajpheart.1990.259.5.H1396. [DOI] [PubMed] [Google Scholar]
- 8.Sobey CG, Faraci FM. Effects of a novel inhibitor of guanylyl cyclase on dilator responses of mouse cerebral arterioles. Stroke. 1997;28:837–842. doi: 10.1161/01.str.28.4.837. discussion 842-833. [DOI] [PubMed] [Google Scholar]
- 9.Faraci FM, Heistad DD. Regulation of the cerebral circulation: role of endothelium and potassium channels. Physiol Rev. 1998;78:53–97. doi: 10.1152/physrev.1998.78.1.53. [DOI] [PubMed] [Google Scholar]
- 10.Faraci FM, Sobey CG. Role of soluble guanylate cyclase in dilator responses of the cerebral microcirculation. Brain Res. 1999;821:368–373. doi: 10.1016/s0006-8993(99)01110-5. [DOI] [PubMed] [Google Scholar]
- 11.Huang PL. Mouse models of nitric oxide synthase deficiency. J Am Soc Nephrol. 2000;11(Suppl 16):S120–S123. [PubMed] [Google Scholar]
- 12.Shesely EG, Maeda N, Kim HS, Desai KM, Krege JH, Laubach VE, Sherman PA, Sessa WC, Smithies O. Elevated blood pressures in mice lacking endothelial nitric oxide synthase. Proc Natl Acad Sci USA. 1996;93:13176–13181. doi: 10.1073/pnas.93.23.13176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang PL, Huang Z, Mashimo H, Bloch KD, Moskowitz MA, Bevan JA, Fishman MC. Hypertension in mice lacking the gene for endothelial nitric oxide synthase. Nature. 1995;377:239–242. doi: 10.1038/377239a0. [DOI] [PubMed] [Google Scholar]
- 14.Godecke A, Decking UK, Ding Z, Hirchenhain J, Bidmon HJ, Godecke S, Schrader J. Coronary hemodynamics in endothelial NO synthase knockout mice. Circ Res. 1998;82:186–194. doi: 10.1161/01.res.82.2.186. [DOI] [PubMed] [Google Scholar]
- 15.Fagan KA, McMurtry I, Rodman DM. Nitric oxide synthase in pulmonary hypertension: lessons from knockout mice. Physiol Res. 2000;49:539–548. [PubMed] [Google Scholar]
- 16.Fagan KA, Fouty BW, Tyler RC, Morris KG, Jr, Hepler LK, Sato K, LeCras TD, Abman SH, Weinberger HD, Huang PL, McMurtry IF, Rodman DM. The pulmonary circulation of homozygous or heterozygous eNOS-null mice is hyperresponsive to mild hypoxia. J Clin Invest. 1999;103:291–299. doi: 10.1172/JCI3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Steudel W, Scherrer-Crosbie M, Bloch KD, Weimann J, Huang PL, Jones RC, Picard MH, Zapol WM. Sustained pulmonary hypertension and right ventricular hypertrophy after chronic hypoxia in mice with congenital deficiency of nitric oxide synthase 3. J Clin Invest. 1998;101:2468–2477. doi: 10.1172/JCI2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chataigneau T, Feletou M, Huang PL, Fishman MC, Duhault J, Vanhoutte PM. Acetylcholine-induced relaxation in blood vessels from endothelial nitric oxide synthase knockout mice. Br J Pharmacol. 1999;126:219–226. doi: 10.1038/sj.bjp.0702300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ding Z, Godecke A, Schrader J. Contribution of cytochrome P450 metabolites to bradykinin-induced vasodilation in endothelial NO synthase deficient mouse hearts. Br J Pharmacol. 2002;135:631–638. doi: 10.1038/sj.bjp.0704472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Capdevila JH, Wei S, Yan J, Karara A, Jacobson HR, Falck JR, Guengerich FP, DuBois RN. Cytochrome P-450 arachidonic acid epoxygenase. Regulatory control of the renal epoxygenase by dietary salt loading. J Biol Chem. 1992;267:21720–21726. [PubMed] [Google Scholar]
- 21.Makita K, Takahashi K, Karara A, Jacobson HR, Falck JR, Capdevila JH. Experimental and/or genetically controlled alterations of the renal microsomal cytochrome P450 epoxygenase induce hypertension in rats fed a high salt diet. J Clin Invest. 1994;94:2414–2420. doi: 10.1172/JCI117608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hao CM, Breyer MD. Physiologic and pathophysiologic roles of lipid mediators in the kidney. Kidney Int. 2007;71:1105–1115. doi: 10.1038/sj.ki.5002192. [DOI] [PubMed] [Google Scholar]
- 23.Zhao X, Pollock DM, Inscho EW, Zeldin DC, Imig JD. Decreased renal cytochrome P450 2C enzymes and impaired vasodilation are associated with angiotensin salt-sensitive hypertension. Hypertension. 2003;41:709–714. doi: 10.1161/01.HYP.0000047877.36743.FA. [DOI] [PubMed] [Google Scholar]
- 24.Dyke CK, Proctor DN, Dietz NM, Joyner MJ. Role of nitric oxide in exercise hyperaemia during prolonged rhythmic handgripping in humans. J Physiol. 1995;488(Pt 1):259–265. doi: 10.1113/jphysiol.1995.sp020964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Duffy SJ, New G, Tran BT, Harper RW, Meredith IT. Relative contribution of vasodilator prostanoids and NO to metabolic vasodilation in the human forearm. Am J Physiol. 1999;276:H663–H670. doi: 10.1152/ajpheart.1999.276.2.H663. [DOI] [PubMed] [Google Scholar]
- 26.Hirai T, Visneski MD, Kearns KJ, Zelis R, Musch TI. Effects of NO synthase inhibition on the muscular blood flow response to treadmill exercise in rats. J Appl Physiol. 1994;77:1288–1293. doi: 10.1152/jappl.1994.77.3.1288. [DOI] [PubMed] [Google Scholar]
- 27.Shoemaker JK, Halliwill JR, Hughson RL, Joyner MJ. Contributions of acetylcholine and nitric oxide to forearm blood flow at exercise onset and recovery. Am J Physiol. 1997;273:H2388–H2395. doi: 10.1152/ajpheart.1997.273.5.H2388. [DOI] [PubMed] [Google Scholar]
- 28.Radegran G, Saltin B. Nitric oxide in the regulation of vasomotor tone in human skeletal muscle. Am J Physiol. 1999;276:H1951–H1960. doi: 10.1152/ajpheart.1999.276.6.H1951. [DOI] [PubMed] [Google Scholar]
- 29.Persson MG, Gustafsson LE, Wiklund NP, Hedqvist P, Moncada S. Endogenous nitric oxide as a modulator of rabbit skeletal muscle microcirculation in vivo. Br J Pharmacol. 1990;100:463–466. doi: 10.1111/j.1476-5381.1990.tb15829.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yamada M, Ishikawa T, Fujimori A, Goto K. Local neurogenic regulation of rat hindlimb circulation: role of calcitonin gene-related peptide in vasodilatation after skeletal muscle contraction. Br J Pharmacol. 1997;122:703–709. doi: 10.1038/sj.bjp.0701422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ray CJ, Marshall JM. Nitric oxide (NO) does not contribute to the generation or action of adenosine during exercise hyperaemia in rat hindlimb. J Physiol. 2009;587:1579–1591. doi: 10.1113/jphysiol.2008.163691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McGiff JC, Carroll MA. Cytochrome P450-dependent arachidonate metabolites, renal function and blood pressure regulation. Adv Prostaglandin Thromboxane Leukot Res. 1991;21B:675–682. [PubMed] [Google Scholar]
- 33.Bauersachs J, Hecker M, Busse R. Display of the characteristics of endothelium-derived hyperpolarizing factor by a cytochrome P450-derived arachidonic acid metabolite in the coronary microcirculation. Br J Pharmacol. 1994;113:1548–1553. doi: 10.1111/j.1476-5381.1994.tb17172.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Campbell WB, Gebremedhin D, Pratt PF, Harder DR. Identification of epoxyeicosatrienoic acids as endothelium-derived hyperpolarizing factors. Circ Res. 1996;78:415–423. doi: 10.1161/01.res.78.3.415. [DOI] [PubMed] [Google Scholar]
- 35.Fisslthaler B, Popp R, Kiss L, Potente M, Harder DR, Fleming I, Busse R. Cytochrome P450 2C is an EDHF synthase in coronary arteries. Nature. 1999;401:493–497. doi: 10.1038/46816. [DOI] [PubMed] [Google Scholar]
- 36.Gebremedhin D, Harder DR, Pratt PF, Campbell WB. Bioassay of an endothelium-derived hyperpolarizing factor from bovine coronary arteries: role of a cytochrome P450 metabolite. J Vasc Res. 1998;35:274–284. doi: 10.1159/000025594. [DOI] [PubMed] [Google Scholar]
- 37.Miura H, Gutterman DD. Human coronary arteriolar dilation to arachidonic acid depends on cytochrome P-450 monooxygenase and Ca2+-activated K+ channels. Circ Res. 1998;83:501–507. doi: 10.1161/01.res.83.5.501. [DOI] [PubMed] [Google Scholar]
- 38.Mombouli JV, Vanhoutte PM. Endothelium-derived hyperpolarizing factor(s): updating the unknown. Trends Pharmacol Sci. 1997;18:252–256. [PubMed] [Google Scholar]
- 39.Oltman CL, Weintraub NL, VanRollins M, Dellsperger KC. Epoxyeicosatrienoic acids and dihydroxyeicosatrienoic acids are potent vasodilators in the canine coronary microcirculation. Circ Res. 1998;83:932–939. doi: 10.1161/01.res.83.9.932. [DOI] [PubMed] [Google Scholar]
- 40.Popp R, Bauersachs J, Hecker M, Fleming I, Busse R. A transferable, beta-naphthoflavone-inducible, hyperpolarizing factor is synthesized by native and cultured porcine coronary endothelial cells. J Physiol. 1996;497(Pt 3):699–709. doi: 10.1113/jphysiol.1996.sp021801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Quilley J, McGiff JC. Is EDHF an epoxyeicosatrienoic acid? Trends Pharmacol Sci. 2000;21:121–124. doi: 10.1016/s0165-6147(00)01445-0. [DOI] [PubMed] [Google Scholar]
- 42.Fang X, Kaduce TL, Weintraub NL, Harmon S, Teesch LM, Morisseau C, Thompson DA, Hammock BD, Spector AA. Pathways of epoxyeicosatrienoic acid metabolism in endothelial cells. Implications for the vascular effects of soluble epoxide hydrolase inhibition. J Biol Chem. 2001;276:14867–14874. doi: 10.1074/jbc.M011761200. [DOI] [PubMed] [Google Scholar]
- 43.Bauersachs J, Popp R, Hecker M, Sauer E, Fleming I, Busse R. Nitric oxide attenuates the release of endothelium-derived hyperpolarizing factor. Circulation. 1996;94:3341–3347. doi: 10.1161/01.cir.94.12.3341. [DOI] [PubMed] [Google Scholar]
- 44.Nishikawa Y, Stepp DW, Chilian WM. Nitric oxide exerts feedback inhibition on EDHF-induced coronary arteriolar dilation in vivo. Am J Physiol Heart Circ Physiol. 2000;279:H459–H465. doi: 10.1152/ajpheart.2000.279.2.H459. [DOI] [PubMed] [Google Scholar]
- 45.Cheng MK, Doumad AB, Jiang H, Falck JR, McGiff JC, Carroll MA. Epoxyeicosatrienoic acids mediate adenosine-induced vasodilation in rat preglomerular microvessels (PGMV) via A2A receptors. Br J Pharmacol. 2004;141:441–448. doi: 10.1038/sj.bjp.0705640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Node K, Ruan XL, Dai J, Yang SX, Graham L, Zeldin DC, Liao JK. Activation of Galpha s mediates induction of tissue-type plasminogen activator gene transcription by epoxyeicosatrienoic acids. J Biol Chem. 2001;276:15983–15989. doi: 10.1074/jbc.M100439200. [DOI] [PubMed] [Google Scholar]
- 47.Popp R, Brandes RP, Ott G, Busse R, Fleming I. Dynamic modulation of interendothelial gap junctional communication by 11,12-epoxyeicosatrienoic acid. Circ Res. 2002;90:800–806. doi: 10.1161/01.res.0000015328.20581.d6. [DOI] [PubMed] [Google Scholar]
- 48.Li PL, Chen CL, Bortell R, Campbell WB. 11,12-Epoxyeicosatrienoic acid stimulates endogenous mono-ADP-ribosylation in bovine coronary arterial smooth muscle. Circ Res. 1999;85:349–356. doi: 10.1161/01.res.85.4.349. [DOI] [PubMed] [Google Scholar]