

Abstract
A bispecific ligand-directed toxin (BLT), called EGFATFKDEL, consisting of human epidermal growth factor, a fragment of urokinase, and truncated pseudomonas exotoxin (PE38) was assembled in order to target human glioblastoma. Immunogenicity was reduced by mutating seven immunodominant B-cell epitopes on the PE38 molecule to create a new agent, EGFATFKDEL 7mut. In vitro, the drug selectively killed several human glioblastoma cell lines. EGFATFKDEL is our first BLT designed to simultaneously target EGFR on solid tumors and uPAR on the tumor neovasculature. In vitro assays revealed that the agent is effective against glioblastoma cell lines as well as human umbilical vein endothelial cells (HUVEC). Additionally, the bispecific drug displayed enhanced binding to overexpressed epidermal growth factor receptor and urokinase receptor when compared to similar monospecific drugs, EGFKDEL and ATFKDEL. In vivo, an aggressive human glioblastoma cell line was genetically marked with a firefly luciferase reporter gene and administered to the flanks of nude mice. Treatment with intratumoral injections of EGFATFKDEL 7mut eradicated small tumors in over half of the treated mice, which survived with tumor free status at least 100 days post tumor inoculation. ATFKDEL, which primarily targets the tumor neovasculature, prevented tumor growth but did not result in tumor-free mice in most cases. Specificity was shown by treating with an irrelevant BLT control which did not protect mice. Finally, immunization experiments in immunocompetent mice revealed significantly reduced anti-toxin production in EGFATFKDEL 7mut treated groups. Thus, EGFATFKDEL 7mut is an effective drug for glioblastoma therapy in this murine model and warrants further study.
Keywords: Immunotoxin, Pseudomonas exotoxin, Glioblastoma, Xenograft model, EGF
Introduction
Glioblastoma multiforme (GBM) is the most common of all primary intracranial malignancies [1]. While newer chemotherapy agents have shown promise in GBM treatment, the long-term prognosis remains very poor with median survival rates under 2 years [1]. Biological agents, such as targeted toxins are attractive for GBM treatment because glioblastoma rarely metastasizes outside of the cranium. Consequently, targeted toxins may be applicable to GBM through direct administration to tumors via convection enhanced delivery (CED), a method that utilizes catheters and pumps to deliver drugs directly to the tumor site [2–4].
Targeted toxins consist of a catalytic protein toxin directed by a ligand recognizing over-expressed markers on tumor cells [5]. Our group is investigating bispecific ligand directed toxins (BLTs) which link two different tumor selective ligands to a truncated toxin [6–9]. BLTs have been shown to increase targeting capability and drug potency while reducing toxicity [6].
We synthesized a novel BLT, EGFATFKDEL, designed to simultaneously target epidermal growth factor receptors (EGFR) expressed on glioblastoma and urokinase receptors (uPAR) expressed on tumor neovasculature (and glioblastoma cells). The targeting ligands were linked to a truncated pseudomonas exotoxin A (PE38), which has been demonstrated to possess potent anticancer activity [10]. By inhibiting protein synthesis, fewer than 1000 molecules of PE38 delivered to the cytosol are sufficient to bring a complete anti-tumor response [11]. PE38 was modified by adding a Lys-Asp-Glu-Leu (KDEL) C-terminus signal to produce PE38KDEL, which prevents luminal endoplasmic reticulum (ER) protein secretion and provides enhanced anticancer activity [12].
EGFR is overexpressed in 40–50% of GBM cases [1]. Activation of EGFR promotes processes responsible for tumor growth and progression, including proliferation, angiogenesis, invasion, metastasis, and inhibition of apoptosis, making it a popular target for anti-cancer therapy [13]. EGFR-directed therapies have been developed that show promise in preclinical trials [14–17]. Urokinase plasminogen activator (uPA) is associated with a specific urokinase receptor (uPAR) which is over-expressed on numerous human cancer cells. Additionally, uPAR is found on the endothelial neovasculature that sustains tumors’ high metabolic demand [18–22]. Here, we use an amino terminal fragment (ATF) of uPA, which has been shown to bind with high affinity to uPAR [23]. Targeting of both these over-expressed receptors allows us to destroy both solid tumors and the tumor-associated neovasculature using EGFATFKDEL.
The efficacy of ligand-directed toxins and other biologicals in the treatment of solid tumors has been impaired by problems with immunogenicity [24]. Use of targeted toxins requires multiple treatments which results in the generation of antibodies that are mainly produced against the toxin portion of the drug [25, 26]. Onda and Pastan recently mapped seven major immunodominant epitopes in PE38 that can be mutated without loss of catalytic activity [25]. We combined this advancement in toxin “deimmunization” with our observation of enhanced BLT activity to produce a new anti-glioblastoma biological drug called EGFATFKDEL 7mut.
In this paper, we study EGFATFKDEL 7mut designed to simultaneously target tumor cells and their neovasculature while reducing the anti-toxin response. The drug shows potent anti-glioblastoma effects in vitro and in vivo in a bioluminescence luciferase reporter gene mouse model in which tumor burden is imaged in real time.
Materials and methods
Construction of EGFATFKDEL and EGFATFKDEL 7mut
Synthesis and assembly of hybrid genes encoding the single-chain EGFATFKDEL was accomplished using DNA-shuffling and DNA cloning techniques. The fully assembled fusion gene (from 5′ end to 3′ end) consisted of an NcoI restriction site, an ATG initiation codon, the genes for human EGF, the downstream 135-amino terminal fragment (ATF) from uPA linked by a 20 amino-acid segment of human muscle aldolase (HMA), the 7 amino-acid EASGGPE linker, the first 362 amino acids of the pseudomonas exotoxin (PE) molecule with KDEL replacing the REDLK at the C terminus, and a NotII restriction site at the 3′ end of the construct. The HMA segment was incorporated into the molecule as a flexible, non-immunogenic linker [27]. The use of the ATF gene fragment was previously described by our laboratory [23, 28]. The resultant 1748 bp NcoI/NotII fragment gene was spliced into the pET28c bacteria expression vector under control of an isopropyl-b-D-thiogalactopyranoside inducible T7 promoter. DNA sequencing analysis (Biomedical Genomics Center, University of Minnesota) was used to verify that the gene was correct in sequence and had been cloned in frame. To create an EGFATFKDEL molecule with decreased immunogenicity (Fig. 1), eight amino acids representing the seven major epitopes on PE38 were mutated using the Quick-Change Site-Directed Mutagenesis Kit (Stratagene. La Jolla CA) [25]. The following amino acids were altered: R490A, R513A, R467A, E548S, K590S, R432G, Q332S, R313A and confirmed by DNA sequencing. Genes for monospecific targeted toxins splicing PE38KDEL to human EGF (EGFKDEL) and mutated uPA fragment (ATFKDEL) were created using the same techniques. CD3CD3KDEL, a bispecific immunotoxin-targeting T-cell surface marker CD3, was made by replacing the DT390 portion of the CD3CD3 (Bic3) molecule described previously with PE38KDEL [29]. 2219ARLKDEL, a BLT which combines VH and VL regions (sFv) for anti-CD22 and anti-CD19, was produced as described previously [7].
Fig. 1.
Construction of EGFATFKDEL 7mut. Sequences for human EGF and the amino terminal fragment of urokinase were linked to a deimmunized PE38KDEL molecule
Isolation of inclusion bodies, refolding and purification
Proteins were produced as described previously with some minor modifications to improve yield and purity [30]. Solubilization of partially purified inclusion bodies was carried out in guanidine hypochloride plus 50 mM dithierythritol in the refolding buffer to decrease protein aggregation. In addition, the purity of protein isolated from the ion exchange column was further enhanced using an FPLC and Supradex 200 size exclusion column (Sigma, Ronconcoma, NY, USA). This modified protocol resulted in a yield of 5–10 mg of protein per liter of culture and a final product with >95% purity.
Cell culture
The human glioblastoma cell lines U87 MG, U118 MG, U373 MG, and T98G were derived from patients with GBM and were obtained from American Type Culture Collection (ATCC, Rockville MD). Human umbilical vein endothelial cells (HUVECs) were obtained from Dr. S. Ramakrishnan (University of Minnesota). Raji cancerous B cells, derived from human Burkitt’s Lymphoma, were used as a control cell line. Cells were grown in either DMEM (U87) or RPMI-1640 media (Raji, U118, U373, and T98) supplemented with 10% fetal bovine serum, 2 mmol/l L-glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin. U87-luc cells, which were stably transfected with vectors containing both the firefly luciferase (luc) and blasticidin resistance genes, were maintained in DMEM with the supplements above and blasticidin. HUVECs were maintained in Medium 199 containing epidermal cell growth media supplement (Invitrogen-Gibco, Carlsbad CA), 15% heat-inactivated fetal bovine serum, and the antibiotics listed above. All carcinoma cells were grown as monolayers and Raji cells in suspension using culture flasks. Cell cultures were incubated in a humidified 37°C atmosphere containing 5% CO2. When adherent cells were 80–90% confluent, they were passaged using trypsin-EDTA for detachment. Only cells with viability >95%, as determined by trypan blue exclusion, were used for experiments.
Flow cytometry analysis of EGFATFKDEL
To measure binding to U87 cells, EGFATFKDEL, EGFKDEL, ATFKDEL, RFB4 (a negative control monoclonal antibody binding to CD22 on B cells), and HD37 (a negative control monoclonal antibody binding to CD19 on B cells) were labeled with fluorescein isothiocyanate (FITC) as described previously [7, 8]. FITC-labeled proteins at 500 nM were incubated with 5 × 105 cells in 200 μl of PBS + 2% FBS on ice for 1 h to allow binding. Following incubation, cells were washed three times and binding was measured using FACSCalibur and CellQuest software (BD Biosciences, San Jose, CA). The percent of positive cells was determined by gating control cells which were not incubated with targeted toxin.
Bioassays to measure cell proliferation
To determine the ability for EGFATFKDEL to inhibit cells in vitro, proliferation assays measuring 3H-thymidine incorporation were used [31]. CD3CD3KDEL, a PE38KDEL-containing molecule which targets CD3 on T cells, was often used as a negative control. Briefly, cells (104/well) were plated out in a 96-well flat-bottomed plate and incubated overnight at 37°C with 5% CO2. The next day, targeted toxins in varying concentrations were added to wells in triplicate. Plates were incubated at 37°C and 5% CO2 for 72 h. [Methyl-3H]-thymidine (GE Healthcare, UK) was added (1 μCi per well) for the final 8 h of incubation. Plates were frozen to detach cells and cells were harvested onto a glass fiber filter, washed, dried, and counted using standard scintillation methods. Background counts in untreated wells ranged from <10 to 500 cpm. Data from proliferation assays are reported as percentage of control counts. Results from bioassays were always reproduced. Protein synthesis assays measuring the incorporation of radioactive leucine were used in some experiments. The assays were similar, except cells were incubated in leucine-free medium. In order to prove that cells were actually killed and not just rendered static, EGFATFKDEL 7mut was tested for its ability to kill U87 cells in vital stain assays using trypan blue staining. These studies confirmed that concentrations of drug that fully inhibited in proliferation assays actually killed the cells (not shown).
Blocking studies were conducted to test the specificity of EGFATFKDEL 7mut. Briefly, EGF or anti-uPA (American Diagnostica, Stamford CT) was added to the media containing 0.1 nM EGFATFKDEL 7mut at a final concentration of 100 nM. Anti-Pseudomonas exotoxin A (Sigma-Aldrich, St. Louis MO) was obtained as whole rabbit serum and added to medium to reach a final dilution of 1:10,000. Resulting mixtures were added to wells containing U87 cells and proliferation was measured as described above. The mouse leukocyte specific antibody anti-Ly5.2 was included as a negative control [32].
Determining immunogenicity of deimmunized EGFATFKDEL 7mut
Mouse immunization studies were used to determine whether mutated EGFATFKDEL 7mut elicited less of an immune response than the unmutated parental EGFATFKDEL molecule. Female BALB/c or C57BL/6 mice (n = 5/group) were injected intraperitoneally once weekly with 0.25 μg of either EGFATFKDEL or EGFATFKDEL 7mut for 62 days. Each week, 5 days after injection, the mice were bled (facial vein collection) to obtain serum. Serum from each mouse was isolated using centrifugation and frozen. The amount of anti-PE38KDEL IgG in each serum sample was measured using indirect ELISA. Briefly, 5 mg of purified recombinant PE38KDEL was added to each well of a 96-well microtiter plate and adhered overnight at 4°C. Unbound protein was washed away with PBS-T and blocking was performed for 1 h with 5% milk/PBS-T. Serum samples were diluted 1:10,000 and 100 μl of each was added to appropriate wells in triplicate. Following 3 h incubation, each well was washed 3 times with PBS-T. Peroxidase-conjugated rabbit anti-mouse IgG (Sigma) was added to each well for a 2 h room temperature incubation. After washing, o-Phenylenediamine dihydrochloride substrate was added to each well. After 30 min, the absorbance at 490 nm was measured using a microplate reader. Quantification of actual anti-PE38KDEL IgG present in each sample was determined by comparing the absorbance values in each well to a standard curve prepared using M40-1 monoclonal anti-PE38KDEL antibody from Dr. Robert Kreitman (NIH, Bethesda, MD).
In vivo efficacy studies of EGFATFKDEL 7mut against U-87 flank tumors
Male nu/nu mice were purchased from the National Cancer Institute, Frederick Cancer Research and Development Center, Animal Production Area and housed in an Association for Assessment and Accreditation of Laboratory Animal Care-accredited specific pathogen-free facility under the care of the Department of Research Animal Resources, University of Minnesota. Animal research protocols were approved by the University of Minnesota Institutional Animal Care and Use Committee. All animals were housed in microisolator cages to minimize the potential of contaminating virus transmission.
For flank tumor studies, mice were injected with 3 × 106 U87-luc cells. Once tumors reached approximately 0.03 cm3 (day 6), mice were divided into groups and treated with EGFATFKDEL 7mut, ATFKDEL, or 2219ARLKDEL. Mice in treated groups were given 2 μg of targeted toxin four times a week for a total of about 5 weeks. All targeted toxins were administered by intratumoral injection in 100 μl volume of sterile saline. Drug was delivered in this volume so that it could be injected in three different directions. Backflow can be a problem, especially when injecting small tumors with high intrastitial pressures. Because of the anti-angiogenic nature of the drug, we reasoned that even if a full dose was not given entirely intratumorally, it would still affect the tissue immediately surrounding the tumor. Tumor size was measured using a digital caliper, and volume was determined as a product of length, width, and height. Treatment-related toxicity was monitored by measuring animal weight.
Mice were imaged in real time and images were captured using Xenogen Ivis imaging system (Xenogen Corporation, Hopkington MA) and analyzed with IGOR Pro 4.09a software (WaveMetrics Inc., Portland OR). Before imaging, mice were anaesthetized using isofluorane gas. All mice received 100 μl of a 30 mg/ml−1 D-luciferin aqueous solution (Gold Biotechnology, St Louis MO) as a substrate for luciferase 10 min before imaging. All images represent a 5 min exposure time and all regions of interest are expressed in units of photons/s/cm2/sr. About 105 photons/s/cm2/sr is the background for luciferase imaging.
Statistical analyses
All statistical analyses were performed using Prism 4 (Graphpad Software, San Diego CA). Groupwise comparisons of single data points were made by Student’s t-tests or one-way ANOVA with Tukey’s multiple comparison tests. P-values < 0.05 were considered significant.
Results
In vitro efficacy of EGFATFKDEL against U87 cells
To determine selective cytotoxicity against human glioblastoma, EGFATFKDEL was tested for its ability to inhibit the protein synthesis of U87 and U87-luc cells in vitro. Figure 2a shows that EGFATFKDEL markedly inhibited U87-luc cells in a dose dependent fashion with an inhibitory concentration 50% (IC50) value of 0.027 nM. The negative control CD3CD3KDEL had minimal effect, confirming the specificity of EGFATFKDEL to EGFR+ uPAR+ GBM cells.
Fig. 2.

Selective activity of EGFATFKDEL against glioblastoma cells in vitro. a EGFATFKDEL was tested against U87-luc cells in a protein synthesis assay. The effects of EGFATFKDEL and T-cell targeting CD3CD3KDEL were determined by analyzing 3H-leucine uptake after a 72-h incubation with targeted toxins. Data are reported as percent control response. Each data point represents an average of triplicate measures ± S.D. b The activities of EGFATFKDEL, EGFATFKDEL 7mut, EGFKDEL, ATFKDEL, and CD3CD3KDEL were tested against HUVEC in 3H-thymidine uptake proliferation assay to determine their ability to target tumor neovasculature. c The specificity of EGFATFKDEL was shown by testing its activity toward EGFR− uPAR− Raji B cells in protein synthesis assays. 2219AR-LKDEL was used as a positive control. d A blocking assay was performed. U87 cells were incubated with 0.1 nM of EGFATFKDEL 7mut and 100 nM of either recombinant anti-urokinase (α-uPA), EGF, recombinant anti-pseudomonas exotoxin (α-PE), or anti-Ly5.2 (α-Ly5.2). The non-specific recombinant α-Ly5.2 was included as a negative control
In vitro efficacy of EGFATFKDEL against endothelial cell lines
EGFATFKDEL is designed to target both solid GBM tumors and their associated endothelial neovasculature. To investigate whether or not the uPAR targeting was effective, we tested our bispecific drug on human umbilical vein endothelial cells (HUVECs). These cells bear similarity to cells in the tumor neovasculature and over-express uPAR but not EGFR. HUVECs were over 166-fold more sensitive to the uPAR-targeting since the IC50 of the monospecific ATFKDEL was 0.092 nM, whereas the IC50 of monospecific EGFKDEL was 15.31 nM (Fig. 2b). However, bispecific EGFATFKDEL had the highest activity with an IC50 of 0.012 nM. Our laboratory has shown that other bispecific targeted toxins are consistently more toxic than their monospecific counterparts [8, 9, 27]. The BLT also had greater activity than an equimolar mixture of monospecific ATFKDEL and EGFKDEL, indicating an advantage of combining both ligands on the same single chain molecule.
In vitro specificity of EGFATFKDEL
To further demonstrate the specificity of EGFATFKDEL, EGFATFKDEL was incubated with EGFR− uPAR− Raji B cell cells (Fig. 2c). Raji cells were not affected by EGFATFKDEL, EGFKDEL, or ATFKDEL. In contrast, 2219ARLKDEL, a bispecific toxin targeting both CD22 and CD19 expressed on Raji cells [7], killed the cells as anticipated (IC50 = 0.015 nM).
To determine that both high affinity cytokines, EGF and ATF, on the EGFATFKDEL 7mut molecule bound to their appropriate receptors, anti-urokinase antibody (α-uPA), EGF, and anti-pseudomonas exotoxin antibody (α-PE) were incubated with a known cytotoxic concentration of EGFATFKDEL 7mut in attempt to block killing (Fig. 2d). All of these blocking agents inhibited the ability of EGFATFKDEL 7mut to kill U87 cells. In contrast, anti-Ly5.2 (α-Ly5.2), an irrelevant control antibody, did not block EGFATFKDEL 7mut activity. These results confirm that both EGF and ATF ligands are active on the EGFATFKDEL 7mut molecule.
Binding of EGFATFKDEL to U87 Cells
The efficacy of all targeted toxins is dependent on cell surface binding and internalization. Thus, we measured ligand-mediated cell binding of EGFATFKDEL, EGFKDEL, ATFKDEL, and the negative controls, HD37 and RFB4, on U87 cells in Fig. 3. All of the targeted toxins were FITC-labeled, incubated with U87 cells at 500 nM, and analyzed using flow cytometry. The percentage of positive cells was 22.8 and 34.8% with monospecific EGFKDEL-FITC and ATFKDEL-FITC, respectively. In contrast, the same cells reacted with bispecific EGFATFKDEL-FITC were 58% positive, indicating superior binding of the BLT. The mean fluorescent intensity (MFI) values for EGFATFKDEL, EGFKDEL and ATFKDEL were 30.5, 18.4 and 19.6, respectively. Cells stained with anti-B cell negative control HD37-FITC or RFB4-FITC showed little binding ability.
Fig. 3.
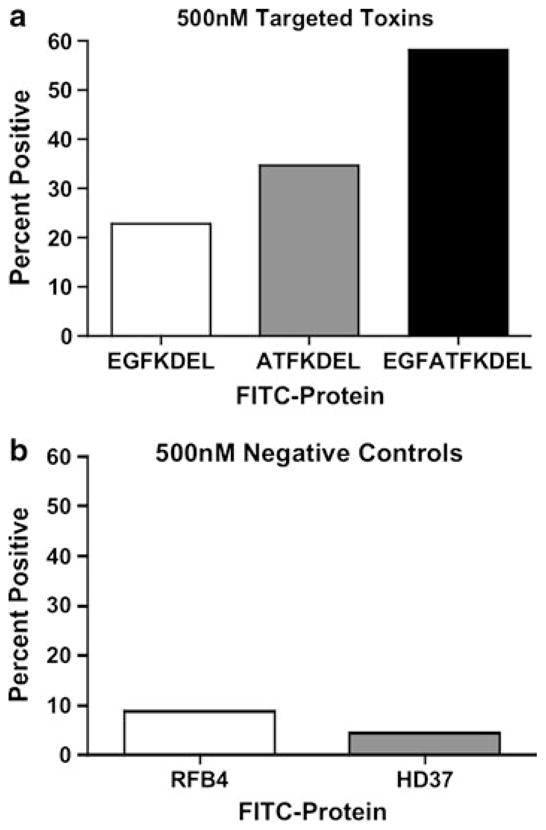
The ability for EGFKDEL, ATFKDEL, and EGFATFKDEL to bind to U87 cells. Percent positive values from flow cytometry analysis are graphed for U87 cells incubated 500 nM of EGFKDEL (a), ATFKDEL (a), EGFATFKDEL (a), control RFB4 (b), or control HD37 (b)
Reducing the immunogenicity of EGFATFKDEL
To determine whether EGFATFKDEL had been successfully deimmunized, groups of immunocompetent BALB/c mice were immunized weekly with 0.25 μg of either non-mutated EGFATFKDEL or mutated EGFATFKDEL 7mut. Mice were used because the same seven major epitopes are recognized in mice and humans [33]. Animals were immunized i.p. over the period of 62 days. Serum samples were obtained weekly and analyzed using ELISA to detect anti-PE38KDEL IgG. The results of the immunization experiment are summarized in Fig. 4a and show statistical differences between the anti-toxin responses of the two groups. After nine injections (day 62), the EGFATFKDEL 7mut group showed no immune response, while the EGFATFKDEL group had an average anti-PE38KDEL response of 5,664 μg/ml. A similar evaluation of C57BL/6 mice showed the same results (not shown).
Fig. 4.

The immunogenicity of EGFATFKDEL 7mut a The immune response to EGFATFKDEL and EGFATFKDEL 7mut was determined by measuring anti-PE38KDEL serum IgG on weekly samples of mice treated with 0.25 μg of EGFATFKDEL (n = 5) or EGFATFKDEL 7mut (n = 5). Measurements were made using an indirect ELISA and quantification of antibodies was determined using a standard curve generated with M40-1 anti-PE38KDEL antibody. b Mutated EGFATFKDEL 7mut was compared to unmutated parental EGFATFKDEL against U87 cells in a proliferation assay. 3H-thymidine incorporation was measured after 72-h incubation with targeted toxins. Data are reported as percentage of control response and measurements were made in triplicates ± S.D. CD3CD3KDEL was the negative control
In vitro comparison of EGFATFKDEL and EGFATFKDEL 7mut
To verify that EGFATFKDEL retained activity after the mutation, the mutated and non-mutated drugs were compared in vitro against the U87 cell line. Mutated EGFATFKDEL 7mut consistently had greater activity than parental EGFATFKDEL. Figure 4b shows the IC50 of EGFATFKDEL 7mut as 0.007 nM and the IC50 of EGFATFKDEL as 0.044 nM. A slight increase in toxicity was also observed in HUVEC cells where the IC50 of EGFATFKDEL 7mut was 0.0017 nM compared to 0.012 nM in the parental EGFATFKDEL (Fig. 2b). Similar increases in activity have been observed in other mutant PE38KDEL-containing drug studies but the cause is unknown [34]. Control CD3CD3KDEL was not toxic.
The effect of EGFATFKDEL 7mut is shown against other glioblastoma cell lines including U118, U373, and T98 in Table 1. The bispecific drug was effective against all of the glioblastoma cell lines tested and one head and neck cancer cell line, UMSCC-11B, but not against Raji (Fig. 2c).
Table 1.
The effect of EGFATFKDEL on the activity of various glioblastoma cell lines
| Cell line | Cancer type | EGFATFKDEL 7mut IC50 (nM) |
|---|---|---|
| U87 | Glioblastoma | 6.5 × 10−3 |
| U87-luc | Glioblastoma | 7.9 × 10−3 |
| U118 | Glioblastoma | 4.8 × 10−8 |
| U373 | Glioblastoma | <1.0 × 10−8 |
| T98 | Glioblastoma | 9.17 × 10−8 |
| UMSCC-11B | Head & neck squamous cell carcinoma | 3.2 × 10−5 |
| Raji | B cell lymphoma | >100 |
These human cell lines were cultured in vitro and treated with drug. Thymidine or leucine incorporation was measured by standard scintillation counting methods as described in the methods. IC50 concentrations (or the amount of drug inhibiting activity 50%) were determined
In vivo efficacy of EGFATFKDEL 7mut against U87-luc flank tumors
To study the in vivo activity of EGFATFKDEL 7mut, we used a bioluminescence luciferase reporter gene mouse model with U87-luc cells in athymic mice. U87-luc cells stably express firefly luciferase without affecting cell growth. This model enables real-time imaging of flank tumors. U87-luc tumors were grown on each mouse’s flank. Mice were treated with 2 μg of EGFATFKDEL 7mut (n = 7), or 2219ARLKDEL (n = 3) four consecutive days each week. This was called one course of treatment and 5 courses were given over a 5 week period (day 6 through 40). A third group of mice was untreated (n = 5). Our treatment regimen was designed from our other previously published flank tumor models with BLT [8, 9]. Treatment was continued in the hopes of maintaining tumor-free mice and to potentially yield long-term survivors. Also, 21 treatments were given to show that mice survive despite continued treatment. An image of luciferase activity for each mouse was obtained weekly. Figure 5a, b, and c show the images for representative mice in different treatment groups, and demonstrates that EGFATFKDEL 7mut decreased tumor-associated bioluminescence compared to untreated mice or mice treated with control 2219ARLKDEL.
Fig. 5.

In vivo efficacy of EGFATFKDEL 7mut against U87-luc tumors. Weekly images show the luciferase activity of U87-luc tumors in individual mice during and after intratumoral treatment. Here, three representative untreated mice (a), three mice treated with control 2219ARLKDEL (b), and three mice treated with EGFATFKDEL 7mut (c) are shown. Mice were treated on days 6, 7, 8, 9, 12, 13, 14, 15, 19, 20, 21, 22, 26, 27, 28, 29, 33, 34, 35, 36, and 40. Red crosses indicate mouse death. Mice given control 2219ARLKDEL received drug on the same schedule as EGFATFKDEL 7mut. On the right of the images, we graphed the total bioluminescence activity for each of the individual mice in the entire treatment group over time. The images are shown in grayscale
In addition to bioluminescence, tumor volumes in these mice were measured two times per week with calipers. The tumor volumes of EGFATFKDEL 7mut-treated mice were markedly reduced compared to the controls. These tumor volumes were significantly different when compared with the untreated group at days 26, 29, 33, and 36 (P < 0.05, Fig. 6a). In the long-term, even after 110 days, four out of the eight EGFATFKDEL 7mut treated mice were tumor-free. One of the tumor-free mice died on day 55 from rapid weight loss and potentially toxicity. Another tumor-free mouse died on day 99 from unknown causes. In a second experiment, EGFATFKDEL 7mut again had potent anti-tumor activity (Fig. 6b). However, differences were not significant in this experiment due to variances in tumor sizes (n = 6/group).
Fig. 6.

a In addition to measuring bioluminescence of U87 tumors, we measured tumor growth with calipers. Thus, tumor volume in three dimensions was measured for each animal in Fig. 5 and the average tumor volume was plotted versus time. The stars indicate the points on the curve in which EGFATFKDEL treatment differed significantly from the untreated controls (P < 0.05 as determined by one-way ANOVA with Tukey’s test for multiple comparison). b Tumor growth was measured in a second similar experiment (n = 6/group) and plotted similarly
In order to determine whether similar anti-tumor responses would be observed with monospecific drugs, groups of mice in the same experiment as Fig. 6 were treated with EGFKDEL and ATFKDEL at similar drug concentrations as EGFATFKDEL. Figure 7 shows the animals treated with ATFKDEL and 4 of 5 of these animals did not elicit an anti-tumor response after 40 days, but bioluminescence measurements remained static. All EGFKDEL treated animals died. These data indicate that the monospecific forms are not as effective at comparable drug concentrations.
Fig. 7.

In the same experiment as Fig. 5 an extra group of mice (n = 5/group) was treated with monospecific ATFKDEL. As in Fig. 5 we measured total bioluminescence activity for each of the individual mice in the treatment group and graphed it over time
Discussion
The original contribution of this research is the development of EGFATFKDEL 7mut, a promising new anti-glioblastoma agent with potential for clinical development. Our laboratory has designed and published BLTs which simultaneously bind to dual independent receptors on the surface of tumor cells [6, 8, 9, 27, 29, 30, 35]. However, EGFATFKDEL is the first targeted toxin that simultaneously attacks tumor cells and the blood supply that feeds them. Angiogenesis, or generation of neovasculature, is a complex process which involves several elements including endothelial cells, stromal cells, and factors from the extracellular matrix. There has been tremendous interest in drugs which target the neovasculature [36]. For example, bevacizumab and other anti-angiogenic drugs currently in clinical trials have demonstrated that the vasculature can be successfully targeted with antibodies and other neovasculature targeting strategies [36]. Although angiogenesis inhibitors possess impressive potential, their success has been limited mostly by the fact that tumor regressions are often only partial [36]. Since EGFATFKDEL directly attacks the most prominent cells in the tumor (vascular cells and tumor cells), we believe that effects will be magnified because the toxic effect of PE is catalytic and irreversible. Our in vitro data with HUVECs shows that our drug can potentially impact tumor neovasculature. While we acknowledge that HUVECs are not an optimal model of tumor neovasculature, our studies show that the ATF portion of our drug effectively targeted uPAR on endothelial cells. Our in vivo data supports this because we have demonstrated that EGFATFKDEL 7mut is highly effective in a mouse model in which flank tumors were induced by the injection of the human glioblastoma line U87. Tumor-free survivors were observed beyond day 100, despite the fact that treatment ended on day 40, indicating that drug responses were durable.
Other properties that set EGFATFKDEL 7mut apart from other biological targeted toxins are its toxin modifications. First, the KDEL sequence was added to the c-terminus to enhance potency. The KDEL modification does not necessarily result in an increased therapeutic window and it is possible that the advantage of increased potency may be negated by enhanced toxicity. To address this issue KDEL-modified EGFATF 7mut could be compared to non-KDEL modified EGFATF 7mut in future studies.
Another, important toxin modification was the “deimmunization” of EGFATFKDEL 7mut. Kreitman et al. showed that the clinical efficacy of treatment with targeted toxins against solid tumors hinges on the ability to give multiple treatments or sustained treatment which enables the drug to penetrate a solid tumor [37]. Toxins may be administered locally to treat tumors in sensitive organs, but targeted toxins must still be used repeatedly or via sustained delivery to achieve positive results. The major problem with this is that neutralizing antibodies will be generated that significantly reduce efficacy over time. To address this issue, investigators used an expansive library of anti-PE monoclonal antibodies to epitope map prominent molecular regions which elicited the strongest antibody response. Fortunately, the immunogenic regions of PE were mapped in seven distinct epitotic areas, and not distributed throughout the molecule. We constructed our PE-based BLT and mutated key amino acids in each of the 7 regions without compromising toxin activity. The immune response to the resultant second-generation drug was reduced by 80–90% in a validated mouse model [34].
Our immunization experiments are designed to evoke gradual immune responses by administering low concentrations of drug on a weekly basis. Initial responses are mild, but they grow exponentially, as seen in Fig. 4. Responses likely could be expedited by using a more aggressive immunization regimen employing greater dosages and more immunizations. However, from other studies we know that neutralizing antibodies are present when antitoxin levels reach about 500 μg/ml. Consequently, analysis of early and low antibody production is important. In our experiments, anti-toxin levels in some mice treated with the parental drug exceeded this threshold after only four injections (day 26). After eight injections, none of the mice treated with the mutant drug had reached 20% of this threshold. However, it is possible that extending the experiment beyond day 62 could reveal that the response to the mutant is delayed and not eliminated.
Another concern of our mouse immunogenicity studies is that major histocompatibility complex (MHC) haplotypes differ in their presentation of peptide fragments in the MHC groove and a different haplotype might present different peptides. In other words, basic immunology dictates that a danger of mutating B cell recognizing regions of the toxin is that peptides regarded as immunogenic by one MHC polymorphism, may not be regarded as immunogenic by a different polymorphism. Thus, we used two mice strains, BALB/c and C57BL/6, with entirely different MHC haplotypes. We observed significant anti-toxin reductions in both strains, indicating that the strength of mutating all 7 regions was enough to overcome differences in MHC polymorphisms. However, if the remaining anti-toxin response cannot be eliminated by future mutation of other B cell recognizing amino acids, we may need to pursue mutation of the T cell recognizing toxin regions.
A final concern of the immunogenicity studies is whether mice are useful for studying human anti-toxin responses. Kreitman and Pastan have treated over 300 patients with non-mutated PE targeted toxins [33]. Nagata studied the antibodies in patients with high titers of anti-PE antibodies and found they bind to the same seven epitopes which regulate toxin B cell recognition in mice indicating that mice are a useful model for human anti-PE responses [33].
Of course, there are other potential issues when applying findings from mouse studies to humans. In humans, we do not know whether treatments will be separated by days or weeks or how the disruption of the blood-brain barrier (BBB) by tumor and surgery may expedite immunologic recognition. We do know that antitoxin antibodies developed in a surprisingly high proportion of 73% of CED patients in phase 1 clinical studies where patients received intracranial IL-13 spliced to the same pseudomonas exotoxin used in our studies [26]. Also, now that we know PE can be deimmunized, some will argue that any additional clinical trials should be performed with deimmunized PE. (The same way that most clinical trials are no longer performed without humanizing antibodies.) Knowing that we can now reduce the anti-toxin response at least 80%, we believe that deimmunization will be necessary in future trials.
In this paper, we showed that in a mouse flank tumor model, EGFATFKDEL 7mut was impressively effective against GBM tumors. Although flank tumor models are useful for determining drug efficacy against vascularized tumors, they are not ideal. For our study, drug was injected directly into small, palpable, and established tumors because targeted toxins have been vigorously pursued for IC therapy in which they are delivered directly into the tumor. A more sophisticated model would use controlled IC delivery via CED in which drug is pumped through a catheter directly into the brain tumor [2–4]. This is an established model in our laboratory and these studies are underway [38]. Another delivery option that will require further exploration is systemic delivery. Due to its vascular reactivity, the drug may be highly effective systemically because of its ability to disrupt the BBB. Additionally, treatment in conjunction with hyperosmolar mannitol may enhance the drug’s BBB disruptive capabilities.
We have noticed that toxin mutation (deimmunization) increases the activity of EGFATFKDEL 7mut, but not other BLT [6]. Onda et al. noted that certain toxin mutations enhanced the activity of one targeted toxin as well [34]. In these instances, different toxin mutation combinations were compared, but only one ligand was explored. Several explanations are possible, but we favor binding change as a possible reason. Binding has a major impact on the activity of targeted toxins [39]. Any mutation can affect positions of alpha helices, beta strands, and turns. This consequentially impacts configuration and these shifts can improve binding. Sequence variances can also affect refolding quality which can also affect binding. This could explain why mutation of EGFATFKDEL enhances activity, but mutation of EGF4KDEL (a PE38KDEL, EGF and interleukin-4 containing targeted toxin) does not [35].
Regarding binding, our flow cytometry data indicated that the bispecific drug was superior to its monospecific counterparts because it bound better. These data correlated with our in vivo studies which showed that the monospecific forms were not as effective at dosages comparable to the bispecific drug. ATFKDEL did not induce complete remissions in 4 of 5 or 80% of the mice, and EGFKDEL killed all of the mice. We have observed that monospecific EGFKDEL is at least a log more toxic than bispecific EGFATFKDEL and we are currently determining whether this is related to the smaller size of EGFKDEL and consequent kidney filtration.
In summary, we have shown that EGFATFKDEL 7mut is effective in inducing complete remissions against GBM tumors. In vitro, the drug is effective in the picomolar range against tumor cells and against HUVEC cells which proves that it binds vascular cells. Attempts at deimmunization of the drug have been successful with a reduction of 80–90% anti-toxin antibodies. Based on its reduced immunogenicity and in vivo efficacy, the drug warrants further consideration for clinical development.
Acknowledgments
This work was supported in part by the US Public Health Service Grants RO1-CA36725 and RO1-CA082154 awarded by the NCI and the NIAID, DHHS and the Martha L. Kramer Fund. We thank Travis M. Spangler and Zintis Inde for assistance. This manuscript partially fulfilled requirements for the Master of Science degree for A. Tsai, CLS program, University of Minnesota.
Contributor Information
Alexander K. Tsai, Department of Therapeutic Radiology-Radiation Oncology, Section on Molecular Cancer Therapeutics, University of Minnesota Masonic Cancer Center, MMC: 367, Minneapolis, MN 55455, USA
Seunguk Oh, Department of Therapeutic Radiology-Radiation Oncology, Section on Molecular Cancer Therapeutics, University of Minnesota Masonic Cancer Center, MMC: 367, Minneapolis, MN 55455, USA.
Hua Chen, Department of Therapeutic Radiology-Radiation Oncology, Section on Molecular Cancer Therapeutics, University of Minnesota Masonic Cancer Center, MMC: 367, Minneapolis, MN 55455, USA.
Yanqun Shu, Department of Therapeutic Radiology-Radiation Oncology, Section on Molecular Cancer Therapeutics, University of Minnesota Masonic Cancer Center, MMC: 367, Minneapolis, MN 55455, USA.
John R. Ohlfest, Department of Pediatrics, University of Minnesota Masonic Cancer Center, Minneapolis, MN, USA
Daniel A. Vallera, Email: valle001@umn.edu, Department of Therapeutic Radiology-Radiation Oncology, Section on Molecular Cancer Therapeutics, University of Minnesota Masonic Cancer Center, MMC: 367, Minneapolis, MN 55455, USA
References
- 1.Lowe S, Schmidt U, Unterberg A, Halatsch ME. The epidermal growth factor receptor as a therapeutic target in glioblastoma multiforme and other malignant neoplasms. Anticancer Agents Med Chem. 2009;9:703–715. doi: 10.2174/187152009788680019. [DOI] [PubMed] [Google Scholar]
- 2.Kioi M, Husain SR, Croteau D, Kunwar S, Puri RK. Convection-enhanced delivery of interleukin-14 receptor-directed cytotoxin for malignant glioma therapy. Technol Cancer Res Treat. 2006;5:239–250. doi: 10.1177/153303460600500307. [DOI] [PubMed] [Google Scholar]
- 3.Laske DW, Youle RJ, Oldfield EH. Tumor regression with regional distribution of the targeted toxin TF-CRM 107 in patients with malignant brain tumors. Nat Med. 1997;3:1362–1368. doi: 10.1038/nm1297-1362. [DOI] [PubMed] [Google Scholar]
- 4.Hall WA, Vallera DA. Efficacy of antiangiogenic targeted toxins against glioblastoma multiforme. Neurosurg Focus. 2006;20:E23. doi: 10.3171/foc.2006.20.4.15. [DOI] [PubMed] [Google Scholar]
- 5.Fuchs H, Bachran C. Targeted tumor therapies at a glance. Curr Drug Targets. 2009;10:89–93. doi: 10.2174/138945009787354557. [DOI] [PubMed] [Google Scholar]
- 6.Stish BJ, Oh S, Chen H, Dudek AZ, Kratzke RA, Vallera DA. Design and modification of EGF4KDEL 7mut, a novel bispecific ligand-directed toxin, with decreased immunogenicity and potent anti-mesothelioma activity. Br J Cancer. 2009;101:1114–1123. doi: 10.1038/sj.bjc.6605297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vallera DA, Chen H, Sicheneder AR, Panoskaltsis-Mortari A, Taras EP. Genetic alteration of a bispecific ligand-directed toxin targeting human CD19 and CD22 receptors resulting in improved efficacy against systemic B cell malignancy. Leuk Res. 2009;33:1233–1242. doi: 10.1016/j.leukres.2009.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stish BJ, Chen H, Shu Y, Panoskaltsis-Mortarti A, Vallera DA. A bispecific recombinant cytotoxin (DTEGF13) targeting human IL-13 and EGF receptors in a mouse xenograft model of prostate cancer. Clin Cancer Res. 2007;13:6486–6493. doi: 10.1158/1078-0432.CCR-07-0938. [DOI] [PubMed] [Google Scholar]
- 9.Stish BJ, Chen H, Shu Y, Panoskaltsis-Mortari A, Vallera DA. Increasing anticarcinoma activity of an anti-erbB2 recombinant immunotoxin by the addition of an anti-EpCAM sFv. Clin Cancer Res. 2007;15:3058–3067. doi: 10.1158/1078-0432.CCR-06-2454. [DOI] [PubMed] [Google Scholar]
- 10.Pastan I, Chaudhary V, FitzGerald DJ. Recombinant toxins as novel therapeutic agents. Annu Rev Biochem. 1992;61:331–354. doi: 10.1146/annurev.bi.61.070192.001555. [DOI] [PubMed] [Google Scholar]
- 11.Kreitman RJ, Pastan I. Accumulation of a recombinant immunotoxin in a tumor in vivo: fewer than 1000 molecules per cell are sufficient for complete responses. Cancer Res. 1998;58:968–975. [PubMed] [Google Scholar]
- 12.Kreitman RJ, Pastan I. Importance of the glutamate residue of KDEL in increasing the cytotoxicity of Pseudomonas exotoxin derivatives and for increased binding to the KDEL receptor. Biochem J. 1995;307:29–37. doi: 10.1042/bj3070029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol. 2001;2:127–137. doi: 10.1038/35052073. [DOI] [PubMed] [Google Scholar]
- 14.Pytel P, Lukas RV. Update on diagnostic practice: tumors of the nervous system. Arch Pathol Lab Med. 2009;133:1062–1077. doi: 10.5858/133.7.1062. [DOI] [PubMed] [Google Scholar]
- 15.Wang MY, Lu KV, Zhu S, et al. Mammalian target of rapamycin inhibition promotes response to epidermal growth factor receptor kinase inhibitors in PTEN-deficient and PTEN-intact glioblastoma cells. Cancer Res. 2006;66:7864–7869. doi: 10.1158/0008-5472.CAN-04-4392. [DOI] [PubMed] [Google Scholar]
- 16.Haas-Kogan DA, Prados MD, Lamborn KR, Tihan T, Berger MS, Stokoe D. Biomarkers to predict response to epidermal growth factor receptor inhibitors. Cell Cycle. 2005;4:1369–1372. doi: 10.4161/cc.4.10.2105. [DOI] [PubMed] [Google Scholar]
- 17.Rich JN, Reardon DA, Peery T, et al. Phase II trial of gefitinib in recurrent glioblastoma. J Clin Oncol. 2004;22:133–142. doi: 10.1200/JCO.2004.08.110. [DOI] [PubMed] [Google Scholar]
- 18.Mori T, Abe T, Wakabayashi Y, et al. Up-regulation of urokinase-type plasminogen activator and its receptor correlates with enhanced invasion activity of human glioma cells mediated by transforming growth factor-a or basic fibroblast growth factor. J Neurooncol. 2000;46:115–123. doi: 10.1023/a:1006339717748. [DOI] [PubMed] [Google Scholar]
- 19.Grondahl-Hansen J, Peters HA, van Putten WL, et al. Prognostic significance of the receptor for urokinase plasminogen activator in breast cancer. Clin Cancer Res. 1995;1:1079–1087. [PubMed] [Google Scholar]
- 20.Verspaget HW, Sier CF, Ganesh S, Griffoen G, Lamers CB. Prognostic value of plasminogen activators and their inhibitors in colorectal cancer. Eur J Cancer. 1995;31:1105–1109. doi: 10.1016/0959-8049(95)00170-n. [DOI] [PubMed] [Google Scholar]
- 21.Prager GW, Breuss JM, Steurer S, Mihaly J, Binder BR. Vascular endothelial growth factor (VEGF) induces rapid prourokinase (pro-uPA) activation on the surface of endothelial cells. Blood. 2004;103:955–962. doi: 10.1182/blood-2003-07-2214. [DOI] [PubMed] [Google Scholar]
- 22.Choong PF, Nadesapillai AP. Urokinase plasminogen activator system: a multifunctional role in tumor progression and metastasis. Clin Orthop Relat Res. 2003;415(Supple):S46–S58. doi: 10.1097/01.blo.0000093845.72468.bd. [DOI] [PubMed] [Google Scholar]
- 23.Rustamzadeh E, Hall WA, Todhunter DA, et al. Intracranial therapy of glioblastoma with the fusion protein DTAT in immunodeficient mice. Int J Cancer. 2006;120:411–419. doi: 10.1002/ijc.22278. [DOI] [PubMed] [Google Scholar]
- 24.Hassan R, Bullock S, Premkumar A, et al. Phase I study of SS1P, a recombinant anti-mesothelin immunotoxin given as a bolus I.V. infusion to patients with mesothelin-expressing mesothelioma, ovarian, and pancreatic cancers. Clin Cancer Res. 2007;13:5144–5149. doi: 10.1158/1078-0432.CCR-07-0869. [DOI] [PubMed] [Google Scholar]
- 25.Onda M, Nagata S, FitzGerald DJ, et al. Characterization of the B cell epitopes associated with a truncated form of Pseudomonas exotoxin (PE38) used to make immunotoxins for the treatment of cancer patients. J Immunol. 2006;177:8822–8834. doi: 10.4049/jimmunol.177.12.8822. [DOI] [PubMed] [Google Scholar]
- 26.Vogelbaum MA, Sampson JH, Kunwar S, et al. Convection-enhanced delivery of cintredekin besudotox (interleukin-13-PE38QQR) followed by radiation therapy with and without temozolomide in newly diagnosed malignant gliomas: phase 1 study of final safety results. Neurosurgery. 2007;61:1031–1037. doi: 10.1227/01.neu.0000303199.77370.9e. [DOI] [PubMed] [Google Scholar]
- 27.Vallera DA, Todhunter DA, Kuroki DW, Shu Y, Sicheneder A, Chen H. A bispecific recombinant immunotoxin, DT2219, targeting human CD19 and CD22 receptors in a mouse xenograft model of B-cell leukemia/lymphoma. Clin Cancer Res. 2005;11:3879–3888. doi: 10.1158/1078-0432.CCR-04-2290. [DOI] [PubMed] [Google Scholar]
- 28.Vallera DA, Li C, Jin N, Panoskaltsis-Mortari A, Hall WA. Targeting urokinase-type plasminogen activator receptor on human glioblastoma tumors with diphtheria toxin fusion protein DTAT. J Nat Cancer Inst. 2002;94:597–605. doi: 10.1093/jnci/94.8.597. [DOI] [PubMed] [Google Scholar]
- 29.Vallera DA, Todhunter D, Kuroki DW, Shu Y, Sicheneder A, Panoskaltsis-Mortari A, Vallera VD, Chen H. Molecular modification of a recombinant, bivalent anti-human CD3 immunotoxin (Bic3) results in reduced in vivo toxicity in mice. Leuk Res. 2005;29:331–341. doi: 10.1016/j.leukres.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 30.Vallera DA, Shu Y, Chen H, et al. Genetically designing a more potent anti-pancreatic cancer agent by simultaneously cotargeting human IL-13 and EGF receptors in a mouse xenograft model. Gut. 2008;57:634–641. doi: 10.1136/gut.2007.137802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vallera DA, Ash RC, Zanjani ED, Kersey JH, LeBien TW, Beverley PC, Neville DM, Jr, Youle RJ. Anti-T-cell reagents for human bone marrow transplantation: ricin linked to three monoclonal antibodies. Science. 1983;222:512–515. doi: 10.1126/science.6353579. [DOI] [PubMed] [Google Scholar]
- 32.Vallera DA, Taylor PA, Sprent J, Blazar BR. The role of host T cell subsets in bone marrow rejection directed to isolated major histocompatability complex class I versus class II differences of bm1 and bm12 mutant mice. Transplantation. 1994;57:249–256. doi: 10.1097/00007890-199401001-00017. [DOI] [PubMed] [Google Scholar]
- 33.Nagata S, Pastan I. Removal of B cell epitopes as a practical approach for reducing the immunogenicity of foreign protein-based therapeutics. Adv Drug Deliv Rev. 2009;61:977–985. doi: 10.1016/j.addr.2009.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Onda M, Beers R, Xiang L, Nagata S, Wang Q, Pastan I. An immunotoxin with greatly reduced immunogenicity by identification and removal of B cell epitopes. PNAS. 2008;105:11311–11316. doi: 10.1073/pnas.0804851105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Oh S, Stish BJ, Sachdev D, Chen H, Dudek AZ, Vallera DA. A novel reduced immunogenicity bispecific targeted toxin simultaneously recognizing human epidermal growth factor and interleukin-4 receptors in a mouse model of metastatic breast carcinoma. Clin Cancer Res. 2009;15:6137–6147. doi: 10.1158/1078-0432.CCR-09-0696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Carmeliet P, Jain RK. Angiogenesis in cancer and other diseases. Nature. 2000;407:249–257. doi: 10.1038/35025220. [DOI] [PubMed] [Google Scholar]
- 37.Kreitman RJ, Stetler-Stevenson M, Margulies I, et al. Phase II trial of recombinant immunotoxin RFB4(dsFv)-PE38 (BL22) in patients with hairy cell leukemia. J Clin Oncol. 2009;27:2983–2990. doi: 10.1200/JCO.2008.20.2630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Oh S, Ohlfest JR, Todhunter DA, et al. Intracranial elimination of glioblastoma brain tumors in nude rats using the bispecific ligand-directed toxin, DTEGF13 and convection enhanced delivery. J Neurooncol. 2009;95:331–342. doi: 10.1007/s11060-009-9932-2. [DOI] [PubMed] [Google Scholar]
- 39.Ho M, Nagata S, Pastan I. Isolation of anti-CD22 Fv with high affinity by Fv display on human cells. Proc Natl Acad Sci USA. 2006;103:9637–9642. doi: 10.1073/pnas.0603653103. [DOI] [PMC free article] [PubMed] [Google Scholar]