

Abstract
Microvessels, the main components of the blood-brain barrier (BBB) are vulnerable to oxidative damage during alcohol-induced stress. Alcohol produces oxidative damage within the vessels and in the brain. Using our animal model of catheter implant into the common carotid artery (CCA), we trace the footprints of alcohol-induced oxidative damage and inflammatory process at the BBB and into the brain. The uniqueness of the finding is that ethanol causes oxidative damage in all neurovascular components by activating NADPH oxidase and inducible nitric oxide synthase in the brain. It is not the oxidants but the ethanol that traverses through the BBB because we found that the highly reactive peroxynitrite does not cross the BBB. Thus, oxidative damage is caused at the site of oxidant production in the microvessels and in the brain. Our data indicate that acetaldehyde (the primary metabolite of ethanol) is the inducer/activator of these enzymes that generate oxidants in brain neurovascular cells. Evidence for alcohol-induced BBB damage is indicated by the alterations of the tight junction protein occludin in intact microvessels. Importantly, we demonstrate that the site of BBB oxidative damage is also the site of immune cells aggregation in the microvessels, which paves the path for inflammatory footprints. These findings reveal the underlying mechanisms that ethanol-elicited BBB oxidative damage initiate the brain vascular inflammatory process, which ultimately leads to neuroinflammation.
Keywords: Blood-brain barrier, microvessels, oxidative damage, tight junction, neuroinflammation
INTRODUCTION
Immediate inflammation is a defense mechanism of immune cells for elimination of foreign bodies from the site of tissue injury. In the brain microvessels (the main components of the blood-brain barrier, BBB), vessel tissue injury is caused by reactive oxidative damage during stress. Brain cells are extremely susceptible to oxidative injury because exogenous and endogenous antioxidant levels are not abundant in the brain (Lakhan et al., 2009; Won et al., 2002).
Immune cells aggregate at the luminal site of vessel injury where they become highly activated and begin to infiltrate into the brain tissue. Chronic infiltration of immune cells into the brain as a result of pro-inflammatory mediators (cytokines and chemokines) secretion together with oxidative injury leads to the pathology of neuroinflammation. Leakiness of BBB and infiltration of immune cells was reported in acute autoimmune encephalomylitis (Floris et al., 2004). Neurovascular inflammation is integrated by a complex activation of brain endothelial cells, microglia, astrocytes and neuronal degeneration. In this chaotic inflammatory process, neuronal cells are vulnerable to secreted toxic agents such as cytokines and oxidative products. Oxidative-mediated neuroinflammation is a common signature for many neurological diseases such as Alzheimer, Parkinson, multiple sclerosis and stroke (Lin and Beal, 2006; Maracchioni et al., 2007), which may be due to a consequence of BBB dysfunction (Daneman and Rescigno, 2009). In fact, development of inflammatory multiple sclerosis characterized by multi-focal perivascular infiltration of mononuclear cells in BBB breakdown has been reported recently (Sospedra and Martin, 2005).
Initiation of alcohol-induced neuroinflammation and neurodegeneration follow the similar pattern of tissue injury and immune adaptive response. There is growing evidence that alcohol mediates the production of cytokines in all major organs such as the livers, lungs, intestine and brain causing respective tissue damage (Crews et al., 2006; McClain et al., 2004). Interestingly, the intestinal microflora-derived lipopolysaccharide translocation seems to act as the axis of gut-liver-brain interactions in tissue damage and disease development in alcohol abuse (Wang et al., 2010). In deed, Fulton Crews lab demonstrated this interconnected proinflammatory path, in which secretion of proinflammatory mediators (TNF α, LPS and MCP-1) at systemic site and in glial cells leads to neuroinflammation and neuronal degeneration in chronic alcohol ingestion (Qin et al., 2008; Qin et al., 2007). Interestingly, neuroadaptations and neuroprotective response in human chronic alcoholics appear to correlate with dysregulation of NF-kB system in neuroinflammation and neurodegeneration (Okvist et al., 2007; Yakovleva et al., 2010). Potula et al. (2006) also showed that alcohol abuse enhances neuroinflammation and impairs immune responses in an animal model of human immunodeficiency virus-1 encephalitis (Potula et al., 2006).
Recently, we reported a novel finding that alcohol-induced oxidative production triggers the interactive phosphorylation of non-receptor protein tyrosine Src kinase and toll-like receptor-4 (TLR4) protein in primary human astrocytes (Floreani et al., 2010). It revealed that induction of phospholipase A2 and cyclooxygenase-2 by ethanol leads to secretion of inflammatory mediator through an interactive phosphorylation of Src kinase-TLR4 dependent manner. Similar to this finding, Alfonso-Loeches et al. (2010) showed the pivotal role of TLR4 in alcohol-induced neuroinflammation and neuronal degeneration in TLR4-deficient mice (Alfonso-Loeches et al., 2010). They alluded that chronic ethanol intake fails to activate astroglial cells for induction of inflammatory mediators in TLR4-deficient mice, suggesting that the presence of TLR4 response is essential in ethanol-induced neuroinflammation. In a review article, Gill et al. (2010) described the putative mechanism that stimulation of TLR connects the link between oxidative stress and inflammatory pathways (Gill et al., 2010).
It is now widely accepted that BBB (brain microvessels) is vulnerable to alcohol-induced oxidative damage and neuroinflammation via alcohol metabolism (Haorah et al., 2005a; Haorah et al., 2005b; Haorah et al., 2007b). Ethanol metabolizing enzymes such as alcohol dehydrogenase, cytochrome P450 2E1 and catalase have been shown to localize in brain microvessel endothelium (Haorah et al., 2005b; Martinez et al., 2001; Zimatkin et al., 2006). Initially, metabolism of alcohol in brain endothelium generates oxidative and nitrosative products that cause oxidative damage to BBB and neuronal cells (Haorah et al., 2005a; Haorah et al., 2005b; Haorah et al., 2008a). Oxidative disruption of BBB enhances permeability of toxic agents and migration of immune cells across the BBB and into the brain (Haorah et al., 2007a; Haorah et al., 2007b; Haorah et al., 2008b). The uniqueness of alcohol-induced oxidative damage is that it occurs in all neurovascular cell types because each of cell type is capable of generating free radicals via the metabolism of ethanol (Floreani et al., 2010; Haorah et al., 2008a; Haorah et al., 2007b). Interestingly, alcohol intake produces differential oxidative products and oxidative damage in these cell types, in which nitric oxide is detected in inducible nitric oxide synthase expressing neurons while ROS is detected predominantly in NADPH oxidase expressing astroglial cells (Rump et al., 2010).
The present studies demonstrated the underlying mechanisms of alcohol-induced oxidative damage in the microvessel (BBB) as the initial event for imprinting the inflammatory footprints. Using our animal model of catheter implantation into the common carotid artery (CCA), we are able to trace the oxidative damage in the microvessel caused by the infusion of 3-morpholinosydnonimine (SIN-1, spontaneous peroxynitrite donor), acetaldehyde into the CCA or in chronic alcohol intake animal. We also show the evidence that adhesion and migration of immune cells occur at these sites of oxidative damage in BBB vessels.
MATERIALS AND METHODS
Chemicals and antibodies
We purchased the antibodies to 3-nitrotyrosine (3-NT), NADPH oxidase 1 (NOX1) and inducible nitric oxide synthase (iNOS) from Abcam (Cambridge, MA) and antibody to 4HNE (4-hydroxynononal) from Alpha Diagnostic, San Antonia, TX. The antibody to α-actin was from Millipore (Billerica, MA). The primary antibody occludin and all secondary Alexa fluor antibodies and Fluo-3 were from Invitrogen (Carlsbad, CA, USA). Acetaldehyde (Ach) and 3-morpholinosydnonimine (SIN-1) were from Sigma-Aldrich (St. Louis, MO) and Lieber DeCarli liquid-diets were from Dyets, Inc (Bethlehem, PA).
Animals and treatments
Six wks old male Sprague Dawley rats purchased from Jackson Laboratory (Bar Harbor, ME) were housed in UNMC animal facility following NIH Guide for Care and Use of Laboratory Animals. Rats weighing about 255±12.5 gram were acclimated to Lieber DeCarli control and 29 % calorie (5% vol/vol) ethanol (EtOH) liquid-diets from Dyets Inc for 1 wk prior to weight-match pair feeding regimens for 8–9 weeks. Pair feeding of control animals (12 rats, 4 rats for control, 4 rats for SIN-1 infusion and 4 rats for Ach infusion) was based on the amount of ethanol-liquid diets consumed by ethanol animals (6 rats). The macronutrient composition of control-liquid diets as percent of total calories is 47% carbohydrate, 35% fat, and 18% protein; and that of ethanol-liquid diets is 35% fat, 18% protein, 19% carbohydrate and 29% ethanol caloric intake. Daily food intake and weekly body weights were recorded. After week 8, animals were anesthetized with ketamine (100 mg/kg body weight) and xylazine (10 mg/kg body weight) ip injection as approved by AVMA Panel on Euthanasia. At the time of sacrifice, the average body weights were 357.6 ± 7.5 gram for pair-fed control and 441.5±10.5 gram for ethanol-liquid diets respectively. SIN1 (200 μM) or acetaldehyde (200 μM) in 100 μL volume was infused into the right common carotid artery was performed in control liquid-diets intake rats. Two hours after infusion of SIN1 or acetaldehyde (Ach), rats were euthanized and then microvessels were isolated and brain tissues were dissected.
Immunohistochemistry
Brain tissue sections (8 μm thickness) derived from chronic EtOH intake and pair-fed control rats or acute infusion of SIN1 or Ach in control rats were washed with PBS and fixed in acetone-methanol (1:1 v/v) fixative. Tissues antigen blocked with 3% bovine serum albumin at room temperature for 1 hr in the presence of 0.4% Triton X-100 were incubated with respective primary antibodies such as mouse anti-3NT (Cat No: ab61392; 1:250 dilution), rabbit anti-NOX1 (Cat No: ab55831; 1:150 dilution), rabbit anti-4HNE (Cat No: HNE-11S; 1:500 dilution), rabbit anti-iNOS (Cat No: ab3523; 1:50 dilution) and rabbit anti-occludin (Cat No: 71–1500; 1:60 dilution, without Triton X-100) for overnight at 4°C. After washing with PBS, the tissues were incubated for 1 hr with secondary antibody: anti-mouse-IgG Alexa fluor 594 for 3NT and anti-rabbit-IgG Alexa fluor 594 for 4HNE, NOX1 and iNOS. Then tissue slides were mounted with immunomount containing DAPI (Invitrogen). Fluorescence microphotographs were captured by fluorescent microscopy (Eclipse TE2000-U, Nikon microscope, Melville, NY) using NIS elements (Nikon, Melville, NY) software.
Western blotting
Brain tissue homogenates from respective experimental conditions were lysed with CellLytic-M buffer (Sigma) for 30 min at 4°C, centrifuged at 14000 × g. Total lysates protein extracts were estimated by BCA for protein concentrations (Thermo Scientific, Rockford, IL). We loaded 20 μg protein/lane and resolved the various molecular weight proteins by SDS-PAGE on gradient gels (Thermo Scientific) and then transferred the protein onto nitrocellulose membranes. After blocking, membranes were incubated with primary antibodies against mouse 3NT (Cat No. ab52309; 1:1000), rabbit 4HNE (Cat No: HNE-11S; 1:1000), rabbit NOX1 (Cat No: ab55831; 1:1000), and rabbit iNOS (Cat No: ab3523; 1:200) proteins for overnight at 4°C, followed by 1 hr incubation with horse-radish peroxidase conjugated secondary antibodies. Immunoreactive bands were detected by West Pico chemiluminescence substrate (Thermo Scientific). Data were quantified as arbitrary densitometry intensity units using the Gelpro32 software package (Version 3.1, Media Cybernetics, Marlow, UK).
Isolation and differentiation of macrophages
Femoral bones from euthanized rat were dissected out under sterile condition and washed in PBS. Bone marrow was flushed out repeatedly with 1X Hanks’ Balanced Salt Solution (HBSS) through the cut ends of the bones using 1 ml syringe. The bone marrow suspension was filtered through a 40 μm cell strainer. Filtrate was collected and centrifuged at 1, 800 rpm for 5 minutes at 4°C. The pellet was suspended in 1 ml of DMEM/F-12 media containing 10% fetal bovine serum (FBS), penicillin and streptomycin (100 μg/ml each, Invitrogen) and 0.001% MCSF (500 μl in 500 mL media). Then the cells were dissociated by trituration (10–15 times) and counted with Trypan Blue using Hemocytometer. Bone marrow-derived cells were differentiated to macrophages in culture containing the macrophage-colony stimulating factor (MCSF) in the culture medium (plating 2× 106 cells/T-75 flask). Cells were fed every third day.
Cells labeling and infusion
Bone marrow cells differentiated to macrophages for 6 days were detached by cell scraper, suspended in 1 ml HBSS and centrifuged at 1800 rpm for 5 min at 4°C. Cell pellets were dissociated by repeated trituration, counted and were labeled with 5 μM Fluo-3 (Molecular probes, Invitrogen) in 1 ml of HBSS at 37°C for 15 minutes. Excess unbound Fluo-3 was washed out by centrifugation and cell pellets re-suspended in 100 μl HBSS (2×106 cells) was infused through the common carotid artery using 27.5 G needle. Animal was euthanized 1 hr after the cell infusion and brain microvessels were surgically removed for observation of Fluo-3 labeled macrophages under fluorescent microscope.
Statistical analysis
Values are expressed as the mean ± SD and N = number of experiments. In each individual experiment, the mean data was determined from 4 replicates. We used the GraphPad Prism V5 (Sorrento Valley, CA) for statistical analysis of the data. Comparisons between the experimental conditions were performed by one/two-way ANOVA with Dunnett’s post-hoc test. Differences were considered significant at P values ≤ 0.05.
RESULTS
Tracking the oxidative footprints in brain microvessel
In order to trace the oxidative footprints in the brain, we utilized our animal model of catheter implantation into the right common carotid artery (CCA), the detail surgical procedure is shown in Fig 1. To study the involvement of acetaldehyde (Ach, ethanol metabolite) for the activation of free radical generating enzymes, 200 μM of Ach (inducer of NOX and iNOS) was infused directly into the CCA of pair-fed control rats via the surgically implanted catheter. Activation of NOX and iNOS is expected to generate ROS and NO, and reaction of superoxide and NO produces the highly reactive peroxynitrite. As a proof-of-concept for peroxynitrite formation, 200 μM of SIN-1 (spontaneous donor of peroxynitrite) was infused directly into the CCA of pair-fed control rats via the surgically implanted catheter. Results obtain from SIN-1 infusion can trace whether SIN-1 by itself or the peroxynitrite can cross the BBB, and compare with those of ethanol intake and Ach infusion for inference. Rats were euthanized at 2 hrs after infusion of SIN-1 or Ach. Intact microvessels and brain tissues were collected for analyses of NOX1, iNOS, 4HNE and 3NT respectively.
Figure 1. Animal model of catheter implantation into the carotid artery.
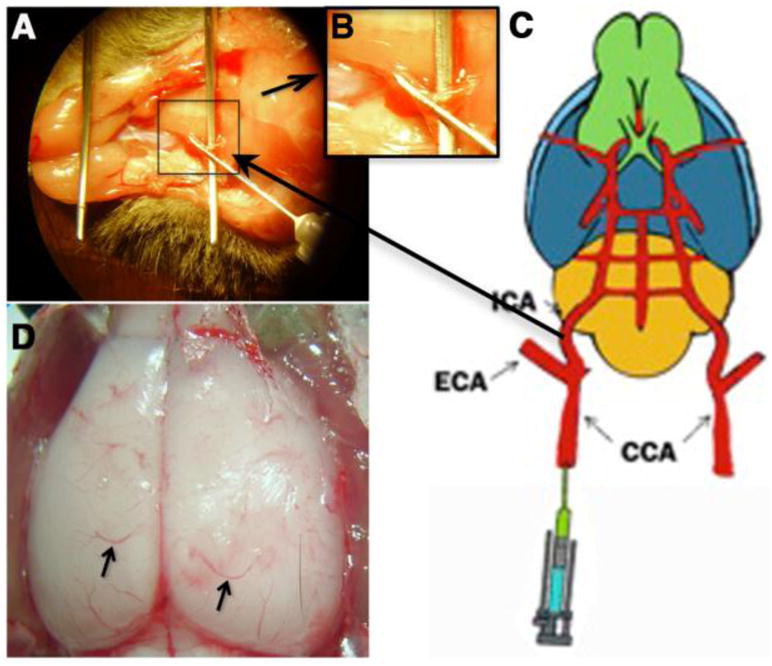
(A) Infusion of SIN1/Ach into the common carotid artery though implanted catheter. (B) Inlet; enlarged view of marked portion of catheter implanted part. (C) Sketch for the model of a rat for infusion of SIN1/Ach through catheter implanted common carotid artery. (D) Exposed dorsal view of rat brain where arrow shows the microvessels used for the study.
As expected, infusion of SIN-1 up-regulated the expression of 3NT protein around the luminal walls of microvessels, but not inside the basolateral side of the microvessels as detected by immunohistochemistry (Fig 2A–D). The expression of 3NT within the luminal microvessel after SIN-1 infusion was further confirmed by staining of 3NT in microvessels of midbrain cortical tissue section (Fig 2E). These observations were then validated by Western blot analyses of the 3NT protein levels in protein extracts from isolated brain microvessels and cortical tissues (without microvessels), wherein, 3NT was not detected in brain tissue protein extract (see Fig 2F). Antibody to 3NT detected three immunoreactive proteins bands with sizes of 25, 55 and 160 kDa. Since the immunoreactive bands for 25 kDa and 160 kDa were relatively fade (not quantifiable), the data presented here was obtained from the 55 kDa immunoreactive protein band only. These findings indicated that SIN-1 as well as peroxynitrite does not cross the BBB or biological membrane.
Figure 2. Tracking the SIN-1 oxidative damage in the brain microvessel.

(A–B) Immunohistochemistry of 3-NT (green) in control (A) and SIN1 treated (B) rat brain intact microvessels. (C–D) Immunohistochemistry of 3-NT (green) in cross section of the brain microvessels of control (C) and SIN1 treated (D) rat. (E) Expression of 3-NT (green) in brain cortical tissue section of SIN1 treated rat. (F) Western blotting analysis of 3-NT protein in rat brain microvessels and cortical tissues. The bar graph shows the results, which are expressed as the ratio of 3-NT to α-actin bands and presented as mean values (± SD; n = 4). **Statistically significant (p<0.01) compared with control. Scale bar indicates 40 μm in panels A–B; 10 μm in C–D and 20 μm in E.
Interestingly, infusion of Ach markedly increased the formation of 3NT mostly around the microvessels of midbrain cortical tissue section and spread inside the brain tissue very similar to that of 3NT staining in midbrain tissue section of chronic ethanol intake (see Fig 3A–F). Detection of 3NT adduct around the microvessel (not in brain tissue) of pair-fed control brain tissue sections was attributed to endogenous circulation of oxidants in the vascular system. Western blot analyses also confirmed a significant increase (p<0.01) in 3NT protein levels by EtOH and Ach (see Fig 3G). Taken together, our data indicate that SIN-1 or the peroxynitrite does not cross the BBB, while EtOH or Ach crosses the BBB. Thus, we suggest that oxidative damage observed in alcohol intake and Ach infusion occurred at the site of oxidative production within the neurovascular components, and not that oxidants traversed from one membrane to the other. These findings suggest that ethanol metabolite initiates oxidative production in microvessel and inside the brain during alcohol ingestion.
Figure 3. Tracking the EtOH/Ach-induce oxidative footprints in the brain microvessel.

(A–F) Immunohistochemistry of 3-NT (red) merged with DAPI (blue) in control (A–B), EtOH (C–D) and Ach (E–F) treated brain cortical tissue sections. The results show the expression of 3-NT tissue portion of the brain cortical tissue sections of EtOH and Ach treated animals. The rectangle indicates the microvessels portion in the tissue section in C and D panels. (G) Western blot analysis of 3-NT protein in rat brain cortical tissues. The bar graph shows the results, which are expressed as the ratio of 3-NT to α-actin bands and presented as mean values (± SD; n = 3). **Statistically significant (p<0.01) compared with control. Scale bar indicates 10 μm in panels of A–F.
We also examined the marker for ROS mediated lipid peroxidation product 4-hydroxynenonal (4HNE) because in ethanol or Ach condition, 3NT can be formed only if ethanol metabolism can lead to the production of NO and ROS. We observed a remarkable increase for 4HNE staining in microvessel area and in midbrain cortical tissue of chronic alcohol consumption and in acute infusion of Ach compared with controls (see Fig 4A–F). Once again, Western blot also confirmed the significant increase in the intensity of 156 kDa immunoreactive bands of 4HNE levels in brain tissue protein extracts from alcohol intake and Ach infusion compared with controls (see Fig 4G).
Figure 4. EtOH/Ach-induced oxidative marker 4-HNE in the brain.

(A–F) Immunohistochemistry of 4-HNE (red) merged with DAPI (blue) in control (A–B), EtOH (C–D) and Ach (E–F) treated brain cortical tissue sections. The results show the expression of 4-HNE in tissue portion of the brain cortical tissue sections of EtOH and Ach treated animals. (G) Western blot analysis of 4-HNE adduct in rat brain cortical tissues. The bar graph shows the results, which are expressed as the ratio of 4-HNE to α-actin bands and presented as mean values (± SD; n = 3). **Statistically significant (p<0.01) compared with control. Scale bar indicates 10 μm in panels of A–F.
Alcohol-induced NO and ROS producing enzymes
We observed that ethanol intake increased the levels of 3NT and 4HNE in the brain microvessels. These observations led us to examine the activation of NO and ROS generating enzymes by alcohol in the microvessels. In all these experiments, Ach is used as positive control to prove the point that metabolism of ethanol exerts the activation of these enzymes. Immunohistochemical detection revealed a high intensity expression of inducible nitric oxide synthase (iNOS) in the brain microvessels and surrounding brain cortical tissue sections from alcohol intake or Ach infused rats compared with controls (Fig 5A–F). Western blotting also confirmed that ethanol or Ach increased the levels of iNOS protein in protein extracts from brain tissues (data not shown).
Figure 5. Alcohol activates NO producing enzyme.

(A–C) Immunohistochemistry of iNOS (red) in control (A), EtOH (B) and Ach (C) treated brain cortical tissue sections. The results show the expression of iNOS in tissue portion of the brain cortical tissue sections of EtOH and Ach treated animals. Scale bar indicates 5 μm in panels A–B and 10 μm in panels C.
Similarly, to assess the association between NADPH oxidase (NOX1) activation and oxidative damage in the brain, we evaluated the expression of NOX1 by immunohistochemistry in the midbrain cortical tissue sections containing microvessels. Immunohistochemical detection and Western blot analyses showed a substantial increase in the levels of NOX1 expression in microvessels and surrounding brain area following chronic alcohol intake and acute Ach infusion compared with control (Fig 6A–G). Antibody to NOX1 detected multiple immunoreactive bands with a prominent band size of 65 kDa. Ethanol and Ach administration caused a significant increase in 65 kDa NOX1 immunoreactive bands and data presented in Figure 6 G was derived from these immunoreactive bands.
Figure 6. Alcohol activates ROS producing enzyme.

(A–F) Immunohistochemistry of NOX1 (red) merged with DAPI (blue) in control (AB), EtOH (C–D) and Ach (E–F) treated brain cortical tissue sections. The results show the expression of NOX1 in tissue portion of the brain cortical tissue sections of EtOH and Ach treated animals. (G) Western blot analysis of NOX1 protein in rat brain cortical tissues. The bar graph shows the results, which are expressed as the ratio of NOX to α-actin bands and presented as mean values (± SD; n = 4). **Statistically significant (p<0.01) compared with control. Scale bar indicates 10 μm in panels of A–F.
Alcohol-induced BBB damage enhances immune cells adhesion
We hypothesized that BBB breakage initiates the neuroinflammatory process. We have shown the alcohol-induced BBB injury by the oxidative damage of the brain microvessels. In chronic alcohol intake, the injury of the brain microvessels was indicated by a hemorrhagic-like blood spread along the external microvessels and thrombus-like in between the cerebral cortex and cerebellum junction (Fig 7A–B). The alteration of BBB integrity was observed by immunohistochemical staining of the tight junction protein occludin in an intact brain microvessels of chronic alcohol ingestion compared with pair-fed control (Fig 7C–D). This is the first demonstration that occludin can be stained very distinctly in intact brain microvessels. As a proof-of-concept for immune cells adhesion and infiltration at this site of BBB oxidative damage, fluo-3 labeled macrophages were infused into common carotid artery through the implanted catheter at week 9 of alcohol ingestion. Fluorescent imaging of the surgically removed microvessels revealed the aggregation of macrophages at the damage sites of the microvessels in chronic alcohol intake compared with pair-fed controls (Fig 7E–F). These results suggest that BBB oxidative injury in deed initiates inflammatory footprints for a more complex neuroinflammation in chronic alcohol abuse.
Figure 7. Alcohol-induced BBB damage enhances immune cells adhesion.
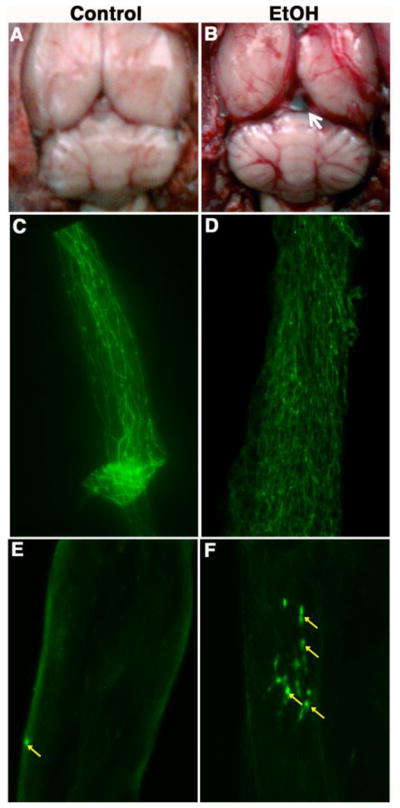
(A) Whole brain structure of control animal. (B) Whole brain structure of ethanol intake animal indicating the breakage of brain microvessels and hemorrhage. The arrow shows the formation of thrombus. (C–D) Immunohistochemical staining of the tight junction protein occludin in an intact brain microvessels of chronic alcohol ingestion compared with pair-fed control. (E–F) Infusion of fluo-3 labeled macrophage into the CCA via implanted catheter. The arrows indicate the aggregations of macrophages at the BBB damage site.
In summary, metabolism of ethanol in brain microvessels produces the reactive acetaldehyde, which activates the free radical generating enzymes iNOS and NOX. Activation iNOS and NOX generates NO and ROS respectively, which further reacts to form the highly reactive peroxynitrite. Nitration and oxidation of microvessel protein by peroxynitrite and ROS lead to disruption of the BBB, which initiates the brain vascular inflammation and a more complicated neuroinflammation. The putative sequence of events for this inflammatory pathway may be depicted in Fig 8 schematic presentation.
Figure 8.

Schematic representation of alcohol-induced inflammatory pathway.
DISCUSSION
Our hypothesis is that (a) oxidative damage occurs at the site of oxidant production and does not traverse through the BBB, (b) oxidative damage of the BBB initiates the neuroinflammatory footprints, and (c) alcohol induces oxidative damage wherever it traverses.
The uniqueness of alcohol-induced oxidative damage in the brain lies in the ability of ethanol to activate free radical generating enzymes such as NADPH oxidase and inducible nitric oxide. Since ethanol crosses the BBB and reaches the brain very quickly (Griffin et al., 2009), it can induce the generation of reactive species at the BBB and in the brain cells as observed in the present findings. We have shown that brain endothelial cells, astrocytes and neurons can produce ROS and RNS via CYP2E1-catalyzed (Cytochrome P450-2E1) metabolism of ethanol (Floreani et al., 2010; Haorah et al., 2007a; Haorah et al., 2008a; Rump et al., 2010). CYP2E1-mediated metabolism of ethanol produces acetaldehyde and ROS as primary metabolites (Ingelman-Sundberg and Johansson, 1984; Lu and Cederbaum, 2008).
Our data indicate that infusion of acetaldehyde into the CCA causes oxidative damage in the microvessels and in the brain by activating NADPH oxidase and inducible nitric oxide synthase. These results validate the point that it is the metabolism of ethanol and not the parent ethanol (un-metabolized ethanol) that triggers the production of oxidative damage in the brain, which further confirms our previous report (Floreani et al., 2010; Haorah et al., 2007a; Haorah et al., 2008a; Rump et al., 2010). The marked increase in 3NT and 4HNE levels around the vessel and inside the brain tissue indicates the formation of peroxynitrite and ROS following acetaldehyde infusion and ethanol intake. Thus, ethanol is a producer of ROS (via CYP2E1-mediated metabolism) and an inducer of ROS/RNS generation (via the activation of NOX and iNOS by acetaldehyde). Activation of NOX and iNOS in the microvessel is likely to impair the bioavailability of NO from eNOS during alcohol intake. Thus, there is considerable evidence that alcohol intake affect the vasodilation of cerebral arterioles by suppressing eNOS and nNOS through hyper-activation of NADPH oxidase (Sun et al., 2006b), (Sun et al., 2006a), and (Landmesser et al., 2003).
It is noted here that oxidative damage observed at the BBB and in the brain occur at the site of oxidant production because reactive species do not cross the BBB. As a proof-of-concept, infusion of SIN-1 into the CCA up-regulates 3NT adduct formation (tyrosine nitrated protein product of peroxynitrite) around the luminal walls of the microvessel only, but not across the BBB. This finding indicates that the parent compound SIN-1 and the byproduct peroxynitrite do not traverse the BBB or cellular membrane. Impermeability of peroxynitrite cross the blood-brain interface may be contributed to its extremely high reactive nature and a very short half-life of 2 seconds. Therefore, free radical damage is expected to occur at the site of production, with the exception of nitric oxide since it is diffusible across the cellular membrane. These results also suggest that intra-peritoneal injection of SIN-1 even at high concentration may not be a viable positive control for oxidative damage in the brain.
We propose that adhesion of immune cells occurs at the site of oxidative injury in the BBB. In deed we observe a reduced expression of occludin with gap formation in intact microvessel from chronic alcohol intake. Adhesion and infiltration of immune cells appear to aggregate at this site of BBB disruption as indicated by the accumulation of fluo-3 labeled macrophages in the microvessel. To avoid host cells rejection from other species, bone marrow cells isolated from the same rat species were differentiated to macrophages, and were used for cell adhesion and infiltration assays. Deposition of these immune cells at the site of BBB oxidative injury in the microvessel walls is expected to exacerbate the brain vascular inflammation and neuroinflammatory complication in alcohol abuse. Thus, neuroinflammation is a complex integration of inflammatory process involving ingression of immune cells into the brain via endothelium and a subsequent activation of astroglial cells at the expense of neuronal loss that we demonstrated recently in mouse model chronic alcohol intake (Floreani et al., 2010; Haorah et al., 2007a; Haorah et al., 2008a; Rump et al., 2010). Chronic neuroinflammation is a pathologic index of neurological diseases such as multiple sclerosis and stroke. The present findings support the notion that oxidative injury of the BBB initiates the inflammatory footprints and subsequent neuroinflammation in chronic alcohol abusers.
Acknowledgments
This work was supported in part by NIH/NIAAA grant AA016403-01A2 (to JH) and by UNMC Faculty Retention Fund.
ABBREVIATIONS
- Ach
acetaldehyde
- BBB
blood-brain barrier
- EtOH
ethanol
- 3-NT
3-nitrotyrosine
- NOX1
NADPH oxidase 1
- 4HNE
4-hydroxynononal
- iNOS
inducible nitric oxide synthase
- SIN-1
morpholinosydnonimine hydrochloride
Footnotes
CONFLICT OF INTEREST
The authors declare that they have no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alfonso-Loeches S, Pascual-Lucas M, Blanco AM, Sanchez-Vera I, Guerri C. Pivotal role of TLR4 receptors in alcohol-induced neuroinflammation and brain damage. J Neurosci. 2010;30:8285–8295. doi: 10.1523/JNEUROSCI.0976-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crews FT, Bechara R, Brown LA, Guidot DM, Mandrekar P, Oak S, Qin L, Szabo G, Wheeler M, Zou J. Cytokines and alcohol. Alcohol Clin Exp Res. 2006;30:720–730. doi: 10.1111/j.1530-0277.2006.00084.x. [DOI] [PubMed] [Google Scholar]
- Daneman R, Rescigno M. The gut immune barrier and the blood-brain barrier: are they so different? Immunity. 2009;31:722–735. doi: 10.1016/j.immuni.2009.09.012. [DOI] [PubMed] [Google Scholar]
- Floreani NA, Rump TJ, Muneer PM, Alikunju S, Morsey BM, Brodie MR, Persidsky Y, Haorah J. Alcohol-Induced Interactive Phosphorylation of Src and Toll-like Receptor Regulates the Secretion of Inflammatory Mediators by Human Astrocytes. J Neuroimmune Pharmacol. 2010;5(4):533–545. doi: 10.1007/s11481-010-9213-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floris S, Blezer EL, Schreibelt G, Dopp E, van der Pol SM, Schadee-Eestermans IL, Nicolay K, Dijkstra CD, de Vries HE. Blood-brain barrier permeability and monocyte infiltration in experimental allergic encephalomyelitis: a quantitative MRI study. Brain. 2004;127:616–627. doi: 10.1093/brain/awh068. [DOI] [PubMed] [Google Scholar]
- Gill R, Tsung A, Billiar T. Linking oxidative stress to inflammation: Toll-like receptors. Free Radic Biol Med. 2010;48:1121–1132. doi: 10.1016/j.freeradbiomed.2010.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin WC, 3rd, Lopez MF, Yanke AB, Middaugh LD, Becker HC. Repeated cycles of chronic intermittent ethanol exposure in mice increases voluntary ethanol drinking and ethanol concentrations in the nucleus accumbens. Psychopharmacology (Berl) 2009;201:569–580. doi: 10.1007/s00213-008-1324-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haorah J, Heilman D, Knipe B, Chrastil J, Leibhart J, Ghorpade A, Miller DW, Persidsky Y. Ethanol-induced activation of myosin light chain kinase leads to dysfunction of tight junctions and blood-brain barrier compromise. Alcohol Clin Exp Res. 2005a;29:999–1009. doi: 10.1097/01.alc.0000166944.79914.0a. [DOI] [PubMed] [Google Scholar]
- Haorah J, Knipe B, Gorantla S, Zheng J, Persidsky Y. Alcohol-induced blood-brain barrier dysfunction is mediated via inositol 1,4,5-triphosphate receptor (IP3R)-gated intracellular calcium release. J Neurochem. 2007a;100:324–336. doi: 10.1111/j.1471-4159.2006.04245.x. [DOI] [PubMed] [Google Scholar]
- Haorah J, Knipe B, Leibhart J, Ghorpade A, Persidsky Y. Alcohol-induced oxidative stress in brain endothelial cells causes blood-brain barrier dysfunction. J Leukoc Biol. 2005b;78:1223–1232. doi: 10.1189/jlb.0605340. [DOI] [PubMed] [Google Scholar]
- Haorah J, Ramirez SH, Floreani N, Gorantla S, Morsey B, Persidsky Y. Mechanism of alcohol-induced oxidative stress and neuronal injury. Free Radic Biol Med. 2008a;45:1542–1550. doi: 10.1016/j.freeradbiomed.2008.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haorah J, Ramirez SH, Schall K, Smith D, Pandya R, Persidsky Y. Oxidative stress activates protein tyrosine kinase and matrix metalloproteinases leading to blood-brain barrier dysfunction. J Neurochem. 2007b;101:566–576. doi: 10.1111/j.1471-4159.2006.04393.x. [DOI] [PubMed] [Google Scholar]
- Haorah J, Schall K, Ramirez SH, Persidsky Y. Activation of protein tyrosine kinases and matrix metalloproteinases causes blood-brain barrier injury: Novel mechanism for neurodegeneration associated with alcohol abuse. Glia. 2008b;56:78–88. doi: 10.1002/glia.20596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingelman-Sundberg M, Johansson I. Mechanisms of hydroxyl radical formation and ethanol oxidation by ethanol-inducible and other forms of rabbit liver microsomal cytochromes P-450. J Biol Chem. 1984;259:6447–6458. [PubMed] [Google Scholar]
- Lakhan SE, Kirchgessner A, Hofer M. Inflammatory mechanisms in ischemic stroke: therapeutic approaches. J Transl Med. 2009;7:97. doi: 10.1186/1479-5876-7-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landmesser U, Dikalov S, Price SR, McCann L, Fukai T, Holland SM, Mitch WE, Harrison DG. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J Clin Invest. 2003;111:1201–1209. doi: 10.1172/JCI14172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- Lu Y, Cederbaum AI. CYP2E1 and oxidative liver injury by alcohol. Free Radic Biol Med. 2008;44:723–738. doi: 10.1016/j.freeradbiomed.2007.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maracchioni A, Totaro A, Angelini DF, Di Penta A, Bernardi G, Carri MT, Achsel T. Mitochondrial damage modulates alternative splicing in neuronal cells: implications for neurodegeneration. J Neurochem. 2007;100:142–153. doi: 10.1111/j.1471-4159.2006.04204.x. [DOI] [PubMed] [Google Scholar]
- Martinez SE, Vaglenova J, Sabria J, Martinez MC, Farres J, Pares X. Distribution of alcohol dehydrogenase mRNA in the rat central nervous system. Consequences for brain ethanol and retinoid metabolism. Eur J Biochem. 2001;268:5045–5056. [PubMed] [Google Scholar]
- McClain CJ, Song Z, Barve SS, Hill DB, Deaciuc I. Recent advances in alcoholic liver disease. IV. Dysregulated cytokine metabolism in alcoholic liver disease. Am J Physiol Gastrointest Liver Physiol. 2004;287:G497–502. doi: 10.1152/ajpgi.00171.2004. [DOI] [PubMed] [Google Scholar]
- Okvist A, Johansson S, Kuzmin A, Bazov I, Merino-Martinez R, Ponomarev I, Mayfield RD, Harris RA, Sheedy D, Garrick T, Harper C, Hurd YL, Terenius L, Ekstrom TJ, Bakalkin G, Yakovleva T. Neuroadaptations in human chronic alcoholics: dysregulation of the NF-kappaB system. PLoS One. 2007;2:e930. doi: 10.1371/journal.pone.0000930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potula R, Haorah J, Knipe B, Leibhart J, Chrastil J, Heilman D, Dou H, Reddy R, Ghorpade A, Persidsky Y. Alcohol abuse enhances neuroinflammation and impairs immune responses in an animal model of human immunodeficiency virus-1 encephalitis. Am J Pathol. 2006;168:1335–1344. doi: 10.2353/ajpath.2006.051181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin L, He J, Hanes RN, Pluzarev O, Hong JS, Crews FT. Increased systemic and brain cytokine production and neuroinflammation by endotoxin following ethanol treatment. J Neuroinflammation. 2008;5:10. doi: 10.1186/1742-2094-5-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin L, Wu X, Block ML, Liu Y, Breese GR, Hong JS, Knapp DJ, Crews FT. Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia. 2007;55:453–462. doi: 10.1002/glia.20467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rump TJ, Muneer PM, Szlachetka AM, Lamb A, Haorei C, Alikunju S, Xiong H, Keblesh J, Liu J, Zimmerman MC, Jones J, Donohue TM, Jr, Persidsky Y, Haorah J. Acetyl-L-carnitine protects neuronal function from alcohol-induced oxidative damage in the brain. Free Radic Biol Med. 2010;49:1494–1504. doi: 10.1016/j.freeradbiomed.2010.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sospedra M, Martin R. Immunology of multiple sclerosis. Annu Rev Immunol. 2005;23:683–747. doi: 10.1146/annurev.immunol.23.021704.115707. [DOI] [PubMed] [Google Scholar]
- Sun H, Molacek E, Zheng H, Fang Q, Patel KP, Mayhan WG. Alcohol-induced impairment of neuronal nitric oxide synthase (nNOS)-dependent dilation of cerebral arterioles: role of NAD(P)H oxidase. J Mol Cell Cardiol. 2006a;40:321–328. doi: 10.1016/j.yjmcc.2005.11.004. [DOI] [PubMed] [Google Scholar]
- Sun H, Zheng H, Molacek E, Fang Q, Patel KP, Mayhan WG. Role of NAD(P)H oxidase in alcohol-induced impairment of endothelial nitric oxide synthase-dependent dilation of cerebral arterioles. Stroke. 2006b;37:495–500. doi: 10.1161/01.STR.0000199033.06678.c3. [DOI] [PubMed] [Google Scholar]
- Wang HJ, Zakhari S, Jung MK. Alcohol, inflammation, and gut-liver-brain interactions in tissue damage and disease development. World J Gastroenterol. 2010;16:1304–1313. doi: 10.3748/wjg.v16.i11.1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Won SJ, Kim DY, Gwag BJ. Cellular and molecular pathways of ischemic neuronal death. J Biochem Mol Biol. 2002;35:67–86. doi: 10.5483/bmbrep.2002.35.1.067. [DOI] [PubMed] [Google Scholar]
- Yakovleva T, Bazov I, Watanabe H, Hauser KF, Bakalkin G. Transcriptional control of maladaptive and protective responses in alcoholics: A role of the NF-kappaB system. Brain Behav Immun. 2010 doi: 10.1016/j.bbi.2010.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimatkin SM, Pronko SP, Vasiliou V, Gonzalez FJ, Deitrich RA. Enzymatic mechanisms of ethanol oxidation in the brain. Alcohol Clin Exp Res. 2006;30:1500–1505. doi: 10.1111/j.1530-0277.2006.00181.x. [DOI] [PubMed] [Google Scholar]