

Abstract
Alzheimer’s disease (AD) is a slowly progressing form of dementia characterized in its earliest stages as a loss of memory. Individuals with amnestic mild cognitive impairment (aMCI) may be in the earliest stages of the disease and represent an opportunity to identify pathological changes related to the progression of AD. Synaptic loss is one of the hallmarks of AD and associated with cognitive impairment. The inferior temporal gyrus (ITG) plays an important role in verbal fluency, a cognitive function affected early in the onset of AD. Unbiased stereology coupled with electron microscopy was used to quantify total synaptic numbers in lamina 3 of the ITG from short postmortem autopsy tissue harvested from subjects who died at different cognitive stages during the progression of AD. Individuals with aMCI had significantly fewer synapses (36%) compared to individuals with no cognitive impairment (NCI). Individuals with AD showed a loss of synapses very similar to the aMCI cohort. Synaptic numbers correlated highly with Mini Mental State Examination scores and a test of category verbal fluency. These results demonstrate that the inferior temporal gyrus is affected during the prodromal stage of the disease and may underlie some of the early AD-related clinical dysfunctions.
Keywords: synapses, dementia, verbal fluency, language
INTRODUCTION
Alzheimer’s disease (AD) is recognized as the most common type of dementia in the aging population. Recent epidemiologic studies report that approximately 14% of the population over the age of 65 has AD and this number is a staggering 40% for individuals over the age of 85. This particular form of dementia appears clinically as a change in recent memory and some language dysfunction [1] followed by changes in cognition that interfere with functions of daily living [2]. Individuals with AD show difficulty not only in word finding but also disturbances in verbal fluency, which appears to be highly sensitive in the clinical characterization of the disease [3] and appears to be particularly prominent in the early stages of the disease progression [4, 5]. Some individuals have a slowly progressing dementia that appears unrelated to an acute or vascular neurologic insult. They present with a gradual cognitive decline and on neuropsychological testing are determined to have amnestic mild cognitive impairment (aMCI) [6, 7], which is considered an intermediate or prodromal stage of AD [6, 8]. This prodromal stage of dementia occurs in the presence of a high density of cortical lesions: senile plaques (SP) and neurofibrillary tangles (NFT). Over the past two decades numerous studies have indicated that synaptic loss in the limbic regions and neocortex is a consistent pathological defect [9], which correlates best with cognitive decline in AD [10–12]. Although clinical neuropathologic studies have demonstrated AD type lesions in the brain of individuals with aMCI [7, 8, 13, 14], relatively few studies have directly examined synaptic loss in the aMCI neocortex [15–17].
For the most part, studies of changes in synaptic number have focused on the hippocampal formation, which is associated with memory impairment and is affected early in the disease process. However, loss of synapses in the hippocampal formation can only account for a minor part of the AD clinical profile. For example, synaptic dysfunction in the hippocampus cannot explain the ensuing extensive intellectual decline that appears related to neocortical dysfunction as the disease progresses. An area of the neocortex that plays an important role in mediation of verbal fluency, a cognitive function that is affected early in the onset of AD [18], is important for semantic language and has important neural interconnections with medial temporal cortex structures especially the parahippocampal gyrus [19] is the inferior temporal gyrus (ITG; Brodmann area 20). The present study investigated possible changes in ITG synaptic numbers in a cohort of individuals who came to autopsy with a premortem clinical diagnosis of aMCI, mild to moderate AD, or no cognitive impairment (NCI).
MATERIALS AND METHODS
Postmortem human brains
Tissue was examined from 27 right handed individuals (mean age 86.4 ± 6.8 years; range 75 to 99 years; Table 1) who were participants in either the Religious Orders Study (ROS), a longitudinal clinical-pathologic study of aging and Alzheimer’s disease composed of older Catholic nuns, priests, and brothers [20–22], or the University of Kentucky Alzheimer’s Disease Center (UKADC) normal aging cohort [23]. The Human Investigations Committee of Rush University Medical Center and the University of Kentucky College of Medicine approved the studies. Individuals included in these studies agreed to annual clinical evaluation and brain donation at the time of death. For all subjects, cognitive test scores were available within the last year of life; the average interval from last evaluation to time of death was 7.0 ±3.6 months, with no differences among the three diagnostic groups (p < 0.1). Subjects were categorized as NCI (n = 9), aMCI (n = 10), or AD (n = 8) based on cognitive testing prior to death according to well established criteria for each clinical category [6]. The NCI subjects were without a history of dementia or other neurological disorders. Standard criteria for exclusion were the presence of 1) significant cerebral stroke regardless of antemortem date, 2) large cortical infarcts identified in the postmortem neuropathologic evaluation, 3) significant trauma within 12 months before autopsy, 4) individuals on a respirator longer than 12 hours before death, 5) individuals in a coma longer than 12 hours immediately before death, 6) individuals currently undergoing radiation/chemo therapy for tumors, or 7) individuals with Lewy bodies in the cortex.
Table 1.
Characteristics of NCI, aMCI, AD subjects
| Subjects | Age (y) | Gender | Brain wt (g) | Education (y) | MMSE | CERAD | Braak | PMI (h) | Last DX (y) |
|---|---|---|---|---|---|---|---|---|---|
| NCI | 90 | M | 1310 | 18 | 30 | None | 0 | 4.0 | 14 |
| NCI | 90 | F | 1110 | 16 | 29 | None | 2 | 4.0 | 6 |
| NCI | 75 | M | 1240 | 16 | 28 | Probable | 5 | 4.0 | 8 |
| NCI | 80 | F | 1150 | 16 | 27 | Possible | 1 | 4.8 | 5 |
| NCI | 85 | F | 1200 | 20 | 30 | Possible | 3 | 6.3 | 5 |
| NCI | 76 | F | 1360 | 20 | 28 | None | 3 | 8.0 | 10 |
| NCI | 86 | M | 1160 | 21 | 27 | None | 5 | 7.0 | 9 |
| NCI | 75 | M | 1150 | 16 | 30 | None | 1 | 4.0 | 7 |
| NCI | 87 | M | 1170 | 18 | 27 | None | 1 | 6.2 | 10 |
| Mean ± SD | 82.7 ± 6.2 | 1205 ± 83 | 17.9 ± 2.0 | 28.4 ± 1.3 | 5.4 ± 1.5 | ||||
| aMCI | 90 | F | 1150 | 18 | 30 | Definite | 5 | 4.6 | 2 |
| aMCI | 79 | M | 1140 | 18 | 23 | Possible | 3 | 4.9 | 4 |
| aMCI | 97 | M | 1308 | 16 | 22 | None | 1 | 4.0 | 3 |
| aMCI | 95 | F | 1130 | 18 | 26 | Possible | 0 | 4.0 | 1 |
| aMCI | 99 | F | 930 | 18 | 21 | Possible | 5 | 2.0 | 4 |
| aMCI | 82 | M | 1495 | 18 | 26 | None | 3 | 7.3 | 1 |
| aMCI | 88 | F | 1080 | 16 | 28 | Definite | 5 | 3.0 | 3 |
| aMCI | 93 | F | 1050 | 18 | 27 | Probable | 3 | 4.0 | 3 |
| aMCI | 83 | M | 1060 | 15 | 22 | None | 3 | 7.6 | 2 |
| aMCI | 87 | M | 1200 | 16 | 24 | Definite | 4 | 3.0 | 3 |
| Mean ± SD | 89.3 ± 6.7 | 1154 ± 156 | 17.1 ± 1.2 | 24.9 ± 3.0 | 4.4 ± 1.8 | ||||
| AD | 89 | F | 1190 | 20 | 3 | Probable | 5 | 7.3 | 5 |
| AD | 97 | M | 1080 | 12 | 9 | Probable | 4 | 4.8 | 4 |
| AD | 91 | F | 1050 | 12 | 9 | Definite | 5 | 5.0 | 4 |
| AD | 88 | F | 1190 | 15 | 21 | Possible | 4 | 4.0 | 3 |
| AD | 90 | M | 1230 | 14 | 21 | Definite | 4 | 5.0 | 5 |
| AD | 86 | F | 1070 | 16 | 14 | Definite | 6 | 5.0 | 3 |
| AD | 78 | F | 1080 | 16 | 11 | Definite | 6 | 4.0 | 3 |
| AD | 78 | F | 1190 | 13 | 23 | Definite | 5 | 4.0 | 3 |
| Mean ± SD | 87.1 ± 6.5 | 1135 ± 71 | 14.8 ± 2.7 | 13.9 ± 7.2 | 2.4 ± 0.9 |
NCI (no cognitive impairment); aMCI (amnestic mild cognitive impairment); AD (Alzheimer’s disease); M (Male); F (Female); MMSE (Mini Mental State Examination); CERAD (Consortium to Establish a Registry for Alzheimer’s Disease); PMI (Post mortem interval); DX (Final clinical diagnosis); SD (standard deviation)
Clinical Evaluations
Details of the ROS and UKADC methods have been published elsewhere [20, 24]. All subjects have detailed annual mental status testing as well as neurologic and physical examinations. Subjects were followed for 3 to 14 years (median 9 years). Once a subject transitioned to having aMCI or AD, they received the mental status test battery and neurologic evaluation every 6–9 months. The 10 subjects with aMCI and 8 with AD were initially without cognitive impairment on enrollment into the longitudinal study and later developed aMCI and AD during follow-up. Duration in years of final clinical diagnosis prior to death is shown in Table 1. All MCI subjects were amnestic without multi domain involvement. Questionable cases were not included in the study.
Pathologic Evaluation and Electron Microscopy
At autopsy, brains were processed as previously described [13, 22]. The postmortem interval (PMI) did not differ across groups (p = 0.488; table 1). A neuropathologist conducted a gross examination of brain neuropathology and cases were excluded if they exhibited non-AD type pathologic conditions (e.g. brain tumors, encepthalitis, large strokes, etc.). A pathological diagnosis was made as previously described [25, 26] with the neuropathologist blinded to the clinical diagnosis. Neuropathological designations were based on the NIA Reagan criteria [27, 28]. In addition, each case received a Braak score [29]. APOE genotyping was performed by PCR analysis [30].
The procedure used for ultrastructural assessment of synapses was identical to that described previously [15, 16]. In brief, at the time of autopsy, the entire left ITG was removed in toto, and within the first 0.5 cm a random starting point was chosen according to unbiased stereologic sampling methods [31]. The remaining entire gyrus was subsequently sectioned into 0.5 cm coronal slabs and every other slab (five to eight slabs per subject) was immediately immersion fixed for 24 hours in 4% paraformaldehyde with 1% glutaraldehyde. Slabs were exhaustively sectioned at 100μm with a vibratome (Vibratome Co., St. Louis, MO), and a random number table used to identify sections for ultrastructural investigation. Designated sections were postfixed in 1% osmium tetroxide (OsO4), stained en bloc with 0.5% uranyl acetate, dehydrated in a graded series of ethanol, infiltrated with epoxy embedding resin, and flat embedded in circular molds (Ted Pella, Redding, CA). Sections immediately adjacent to those used for ultrastructure were designated for determination of the reference volume and treated as previously described [16]. The analysis was confined to lamina 3 because previous investigations have shown this lamina to exhibit significant synaptic loss in various regions of the neocortex [32].
For all subsequent tissue processing and evaluation, the experimenters were blinded to the clinical diagnosis. On each section that contained the ITG, the length of lamina 3 was determined with the Bioquant image analysis system and this length portioned into a minimum of 6 equal segments. A random number table was used to determine which portions of the lamina 3 length to analyze. Blocks containing the randomly selected portion of the ITG were trimmed to the appropriate region. Ribbons of six to eight ultrathin sections (silver-gold interface range) were taken and collected on formvar-coated, carbon-stabilized slot grids. Sections were stained with uranyl acetate (3%) and Reynolds lead citrate. A total of 15 to 24 different regions of the ITG were assessed depending on brain size. Larger brains have a larger ITG and generated more regions.
The physical disector method [33] was used to approximate the total number of synapses per unit volume (Nv). Electron micrographs were taken with a Zeiss EM-902 (Oberkochen, West Germany) at X4,400 and photographically enlarged to approximately X20,000. Every synaptic profile on each micrograph was identified by the presence of the postsynaptic density in association with the postsynaptic element and synaptic vesicles in a presynaptic terminal and labeled (Figure 1). An unbiased counting frame was randomly superimposed over the micrographs. Only those synaptic profiles observed on the reference micrograph within the counting frame that did not violate the counting frame rules [31] and were not on the look-up micrograph were counted [34]. To increase efficiency, the look-up and reference sections were reversed, and the counting frame was again applied in a random fashion. There were between 30 and 48 disectors (38 ± 6) for each ITG. The thickness of the ultrathin sections was estimated with the Small method of minimal folds [35].
Figure 1.
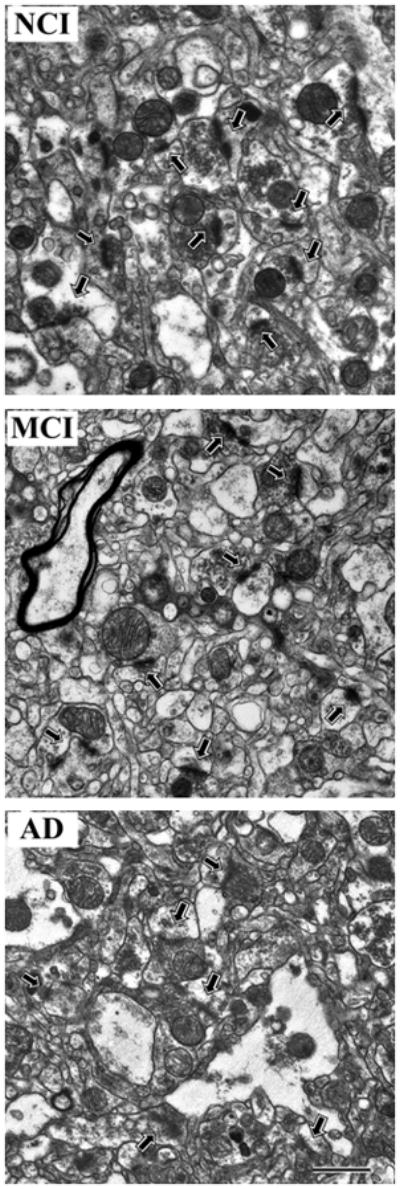
Representative electron micrographs of lamina 3 of the inferior temporal gyrus showing synaptic complexes (arrows) in tissue from the three different clinical groups A) no cognitive impairment (NCI), B) amnestic mild cognitive impairment (MCI), C) Alzheimer disease (AD). In all tissue, the synaptic complexes appeared normal with synaptic vesicles observed in the presynaptic component and a synaptic density observed in the postsynaptic component. Calibration bar = 1.0 μm.
The numerical density of synapses per unit volume, Nv, was calculated using the following formula: Nv = Q−/Vdis, where Q− is the mean number of synapses counted in each disector and Vdis is the mean disector volume. The total number of synapses, Nsyn, was calculated for each case using the following formula Nsyn = Nv • Vref, where Vref is the reference volume.
Statistical Analysis
The relationship between dependent variables and clinical group was examined with an analysis of variance (ANOVA) using Statview 5.0 (SAS Institute, Cary, NC). If a significant ANOVA was found, post-hoc tests (Student Newman-Keuls) controlling for multiple comparisons were used to identify pairs of diagnostic groups that differed significantly. Because the diagnostic groups were heterogeneous in their clinical features, the relationship between total synaptic counts and performance on neuropsychological tests at last clinical evaluation was examined using Spearman correlation with the InStat program (GraphPad, San Diego, CA). Level of significance was set at p < 0.05.
RESULTS
Demographics
Table 1 shows characteristics of the sample population by diagnostic group. The NCI, aMCI, and AD groups were found to be similar in age, PMI, and brain weight (p > 0.05). An ANOVA revealed an expected difference in Mini Mental State Examination (MMSE) between groups [F(2,24) = 25.701; p < 0.0001]. Post hoc analysis showed no difference between NCI and aMCI groups (p > 0.05) but did reveal a difference between AD and the other two diagnostic groups (p < 0.05). An ANOVA [F(2,24) = 5.631; p < 0.01] revealed a significant difference in level of education between the groups. The AD group had significantly less (p < 0.05) years of education (14 ± 2.6) compared to NCI (17.9 ± 2.0) and aMCI (17.1 ± 1.2), which did not differ from each other.
ITG Total Synaptic Number and Volume in Lamina 3
The total synaptic counts for lamina 3 of the ITG for each subject in each of the three diagnostic groups are shown in Figure 2A. An ANOVA revealed a significant difference between group means [F(2,24) = 13.265; p < 0.0001]. Post hoc analyses showed that the synaptic counts for the AD (−42%) and aMCI (−36%) groups were significantly lower than the NCI cases (p < 0.005). Post hoc analysis showed no difference in total synaptic numbers between aMCI and AD groups (p > 0.05). An ANOVA revealed a significant difference in group means for the volume of lamina 3 of the ITG [F(2,24) = 23.033; p < 0.0001]. Post hoc analysis showed that both the aMCI (−15%) and AD (−44%) groups had a significantly (p < 0.05, p < 0.005) smaller volume (Figure 2B) compared to the NCI group. The analyses also revealed that the aMCI and AD were significantly different (p < 0.005).
Figure 2.

Estimate of the total number of synapses (A) in lamina 3 of the inferior temporal gyrus. Subjects were categorized clinically as no cognitive impairment (NCI), amnestic mild cognitive impairment (aMCI), or mild to moderate Alzheimer’s disease (AD). Estimates were obtained using unbiased stereology coupled with electron microscopic imaging of synapses. The total volume (B) of lamina 3 of the inferior temporal gyrus was estimated with the Cavalieri method directly from tissue sections immediately adjacent to regions used for synaptic counts. Single points represent individual subjects. Horizontal lines indicates group median. *p < 0.05; **p < 0.005 compared to NCI; #p < 0.005 compared to aMCI.
Neuropsychological Test Scores and Lamina 3 Synaptic Counts
We evaluated the association between the total number of synapses in lamina 3 of the ITG and scores achieved on neuropsychological tests obtained during the subject’s last clinical evaluation. As total synaptic counts in lamina 3 declined, we observed a significant decline in MMSE scores (r = .554; p < 0.005) (Figure 3A). There was also a significant association between total synaptic numbers and both word recognition (r = .444; p < 0.05) and animal naming (r = .552; p < 0.005) (Figure 3B). Other standard neuropsychological scores failed to demonstrate a significant relationship: immediate word list recall (r = .371; p > 0.05), delayed word list recall (r = .282; p > 0.1); Boston Naming Test (BNT) (r = .027; p > 0.1). The subjects’ years of education (r = .307; p > 0.1) was not significantly associated with total synaptic numbers or with the neuropsychological test scores (r = .237; p > 0.1). Correlational analysis revealed a significant association between age at time of death and total synaptic number (r = .615; p < 0.005) (Figure 4).
Figure 3.

Relationship between estimates of total number of synapses in lamina 3 of the inferior temporal gyrus and the subjects score on the Mini Mental Status Examination (MMSE) (A) and a test of verbal fluency using an animal naming test (B). Subjects were categorized clinically as no cognitive impairment (NCI), amnestic mild cognitive impairment (aMCI), or mild to moderate Alzheimer’s disease (AD). As the total number of synapses increased, so did the subjects’ performance on these two cognitive tests. Lines are used to represent the direction of the association and do not indicate a line of regression. Single points represent individual subjects’ scores for each group. **p < 0.005
Figure 4.

Relationship between estimates of the total number of synapses in lamina 3 of the inferior temporal gyrus and an individual’s age at the time of death. Subjects were categorized clinically as no cognitive impairment (NCI), amnestic mild cognitive impairment (aMCI), or mild to moderate Alzheimer’s disease (AD). There was a significant association with decreased synaptic numbers with increased subject’s age. Lines are used to represent the direction of the association and do not indicate a line of regression. Single points represent individual subjects’ scores for each group. **p < 0.005
APOE Genotype and Lamina 3 Synaptic Counts
The apolipoprotien E (APOE) genotype (ε2/2, ε2/3, ε3/3, ε3/4, ε4/4) was determined for each of the subjects and the number of synapses in each group analyzed (Table 2). An ANOVA showed no statistically significant difference between groups [F(3,23) = 1.112, p > 0.3]. Total synaptic counts were further grouped by whether or not the individual had any APOEε4 allele. An unpaired t-test failed to detect a significant difference between these two groups [t(25) = 1.754, p > 0.05]. These results indicate that in the present study APOE genotype did not influence total synaptic numbers in lamina 3 of the ITG.
Table 2.
Distribution of APOE categories by diagnosis
| APOE | NCI | aMCI | AD |
|---|---|---|---|
| ε2/2 | 0 | 0 | 0 |
| ε2/3 | 3 (33%) | 2 (20%) | 0 |
| ε3/3 | 6 (67%) | 5 (50%) | 4 (50%) |
| ε3/4 | 0 | 3 (30%) | 3 (38%) |
| ε4/4 | 0 | 0 | 1 (12%) |
NCI (no cognitive impairment); aMCI (amnestic mild cognitive impairment); AD (Alzheimer’s disease)
NIA-Reagan Criteria, Plaques, Braak Score and Lamina 3 Synaptic Counts
The present study examined the possible relationship between neuropathological diagnosis by the NIA-Reagan criteria [36] (no AD, low, intermediate, or high likelihood of AD) in the different clinical diagnostic groups and the total number of synapses in lamina 3 of the ITG (Table 3). Most cases in the NCI group were either low likelihood or “not AD” (89%) with only a single case being rated as high likelihood of AD. In contrast, the AD group was rated as either high or intermediate likelihood (88%). An ANOVA failed to reveal a difference in synaptic numbers as a function of the NIA-Reagan classification [F(3,23) = 1.654, p > 0.1]. The packing density (#/mm2) of plaques (diffuse and neuritic) was determined for lamina 3 of the ITG using Bielschowsky stained sections. The analysis revealed a very weak and non-significant association between the packing density of plaques and synapses in the ITG (r = .122, p > 0.1). Overall Braak staging was not significantly (r = .166, p > 0.1) associated with total number of synapses. We also evaluated a possible relationship by grouping the data into three different levels of Braak stages (0-II; III-IV; V-VI). An ANOVA showed no statistically significant difference between these different Braak groupings and total synaptic numbers [F(2,23) = .236, p > 0.5].
Table 3.
Distribution of clinical diagnoses by NIA-Reagan criteria
| Clinical Diagnosis | NCI | aMCI | AD | |
|---|---|---|---|---|
| 1. | High likelihood | 1 (11%) | 1 (11%) | 5 (63%) |
| 2. | Intermediate likelihood | 0 | 2 (22%) | 2 (25%) |
| 3. | Low likelihood | 6 (67%) | 5 (56%) | 1 (12%) |
| 4. | Not AD | 2 (22%) | 1 (11%) | 0 |
NCI (no cognitive impairment); aMCI (amnestic mild cognitive impairment); AD (Alzheimer’s disease)
DISCUSSION
This is the first study to estimate the total number of synapses in the human ITG lamina 3 using unbiased stereology coupled with quantitative electron microscopy. All individuals used in these studies were followed longitudinally and initially enrolled as cognitively normal. Compared to age- and PMI-matched individuals with no cognitive impairment, subjects who transitioned to aMCI demonstrated a statistically significant 36% loss of synaptic contacts in the ITG. This decline in cortical connectivity was very similar to the 42% loss observed in the mAD cohort. These results support the idea that synaptic loss in the ITG is an important pathological defect in the early stages of the disease process. The total number of synapses strongly associated not only with the subject’s MMSE but also animal naming, a direct test of semantics access suggesting an underlying mechanism for some verbal fluency dysfunction in early AD. The present findings have both clinical and theoretical implications.
Previous studies assessing synaptic loss [9] have focused primarily on the hippocampus and the more prominent areas of the frontal, temporal, and parietal lobes. Very few investigations have assessed synaptic change in the early progression of the disease such as aMCI [15–17]. Most descriptions of neuropathological alternations have concentrated on the loss of neurons [37–39] and the presence of NFT [13, 14, 24, 40]. Masliah and colleagues reported the loss of synaptophysin staining in the frontal cortex of individuals with a CDR of 0.5 [41]. Counts et al [17] reported a decline in the postsynaptic marker drebrin in the temporal lobe in aMCI. Two previous AD studies evaluated changes in synaptophysin as a marker of synaptic connectivity in the ITG. The earliest study [42] reported a significant AD-related loss of staining, while the second study [43] failed to find a difference between individuals with AD and a cohort of controls. The excessively long PMI of the tissue used in that study may account for the observed differences. The decline in synaptic connectivity in the aMCI cohort observed in the present study is very striking considering previous investigations that evaluated possible synaptic loss early in the disease process. Ultrastructral data derived from the hippocampus shows an area in transition with many of the aMCI subjects within the lower range of the NCI cohort [15, 16]. By contrast, only a single aMCI subjects in the present ITG study was within the NCI range with most of the aMCI cohort overlapping with the mAD values, supporting the idea that this cortical structure is involved early in the disease. Several investigators have suggested that the ITG should be included in the routine assessment of cortical involvement in the CERAD determination of AD [44, 45] along with other heavily involved structures [18] because of the accumulation of AD-related lesions in this region of the temporal lobe.
Studies in primates have demonstrated interconnections between the inferior and superior temporal gyrus and also between the ITG and perirhinal and parahippocampal cortex [46]. While it is clear that the ITG projects to entorhinal cortex, whether or not there are reciprocal connections is unclear [47]. The ITG can be considered as a tertiary visual association cortex as part of the pathway that originates in the primary visual cortex (BA17) and progresses through secondary visual areas BA18, 19 and BA37 and then B20, with some information relayed to the prefrontal cortex [48]. Ablation studies in primates have identified B20 as being involved in learning tasks that require visual recognition of objects [49, 50]. Several studies have linked regions of the ITG to the amygdala and nucleus accumbens suggesting the addition of an emotional tone to learning of cognitive tasks [51, 52]. The temporal lobe is strongly associated with language [53] with its anterior regions implicated in semantic memory [54]. Language deficits are important manifestations of cognitive disability associated with AD with verbal fluency (semantic/category) performance a key indicator of the progression of dementia [55].
Tests of language dysfunction, in particular those of verbal fluency, are one of the most commonly used diagnostic tools for the assessment of neurologic damage. Both semantic and phonemic fluency are often tested, with semantic fluency more impaired following damage to the temporal lobes [55]. The semantic fluency test used in the present study required individuals to name as many different animals as they could within 60 seconds. The total number of novel animal names generated consisted of that individual’s score. This particular test appears to be resistant to the influence of practice in MCI and early AD [56]. Impairment in animal fluency is strongly associated with early stages of AD [1, 57]. AD patients that develop semantic memory dysfunction also develop significant neuropathology in neocortical association areas linked with semantic language function. Individuals with semantic dementia (SD) differ from AD subjects in that they have preserved episodic memory and perform well on tests such as the BNT. Among the neocortical regions most severely affected in SD is the anterior inferior temporal lobe [58]. In the present study, there was a greater association between animal naming than the BNT, supporting previous findings that deficits in confrontation naming may be linked to a more advanced stage of AD [55]. The connectivity of the ITG appears to be strongly associated not only with semantic fluency but also MMSE and word recognition scores.
The current literature suggests that AD-associated decline in cognition may be an inevitable consequence of the age-associated changes not only in neuron number but synaptic loss. While careful unbiased evaluation of cortical neuronal numbers have failed to support this idea [59, 60], there does appear to be a significant age-related decline in white matter, indicative of a loss of brain connectivity and synapses [61–64]. In the present study, we found a significant age-related decline in total synaptic contacts in lamina 3 of the ITG when all subjects were included. This correlation may not necessarily reflect normal age-related associations since both cognitively normal and demented subjects were included in the analysis. How this impacts on the present loss of synapses in the aMCI and AD groups is unclear since there was no significant difference in the mean age of the groups (see Table 1). Immunohistochemical studies have previously reported an age-related loss of synaptic markers in the ITG and other neocortical regions [42, 65, 66]. This is in contrast to previous ultrastructural studies that demonstrated a preservation of synapses in the superior frontal [67] and the postcentral gyrus [66] across a wide age spectrum in cognitively normal individuals. A previous study that included both NCI and AD subjects investigating the superior and middle temporal cortical regions also failed to find an age-related change in synaptic numbers [68]. The disparity among these studies supports a heterogeneous view of the neocortex and cautions against global statements concerning age-related morphological cortical changes.
Currently, neuroimaging appears to be the easiest way to evaluate possible atrophy in a region of interest and has been limited to estimating gross changes. In the present study, we were able to directly evaluate possible changes in the volume of lamina 3 of the ITG from histological sections coupled with unbiased stereology. We found a significant decline not only in mAD but also in aMCI, although the decline was not as robust as the loss in synapses. This finding also differs from previous hippocampal aMCI studies that evaluated subregion volumetric changes and failed to show a change in either the outer molecular layer of the dentate gyrus [16] or stratum radiatum of the hippocampal region superior [15]. The overall significance of this difference may represent subtle differences in cytoarchitecture between the ITG and hippocampus [69]. As AD progresses, various regions of the neocortex are functionally disconnected from each other due to a loss of corticocortical projections [70]. Numerous studies suggest that some of the neocortical association areas are the most vulnerable in the progression of AD and it may be the case that the dramatic loss of synapses observed in the present study signals the onset of dementia as suggested by others [18, 71–73].
Finally, it is important to note that the present set of experiments consisted of a cross-sectional picture of a relatively small sample size that may reduce the significance when applied to a larger and more diverse population. The present cohort of subjects was well educated and consisted exclusively of white Caucasians and caution should be maintained when extrapolating the results to a more diverse population. Moreover, the MCI subjects we examined were amnestic and may not reflect the clinical and pathology of other types of MCI individuals.
Acknowledgments
This work was supported by the National Institutes of Health grants: AG27219, AG19241, P01 AG14449, AG028383 and P30 AG10161. We are indebted to the altruism of the participants in the Religious Orders Study (http://www.rush.edu/rumc/page-R12394.html) and SBCOA (http://www.mc.uky.edu/coa/clinicalcore/research_opportunities.html). The pathological diagnosis was carried performed by J.A. Schneider (Rush Unvierstiy Medical Center) and M.R. Markesbery and P.T. Nelson (SBCOA). The authors declare that they have no competing financial interests.
References
- 1.Weiner MF, Neubecker KE, Bret ME, Hynan LS. Language in Alzheimer's disease. J Clin Psychiatry. 2008;69:1223–7. doi: 10.4088/jcp.v69n0804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barberger-Gateau P, Fabrigoule C, Helmer C, Rouch I, Dartigues JF. Functional impairment in instrumental activities of daily living: an early clinical sign of dementia? J Am Geriatr Soc. 1999;47:456–62. doi: 10.1111/j.1532-5415.1999.tb07239.x. [DOI] [PubMed] [Google Scholar]
- 3.Cahn DA, Salmon DP, Bondi MW, Butters N, Johnson SA, Wiederholt WC, Barrett-Connor E. A population-based analysis of qualitative features of the neuropsychological test performance of individuals with dementia of the Alzheimer type: implications for individuals with questionable dementia. J Int Neuropsychol Soc. 1997;3:387–93. [PubMed] [Google Scholar]
- 4.Chertkow H, Bub D. Semantic memory loss in dementia of Alzheimer's type. What do various measures measure? Brain. 1990;113 ( Pt 2):397–417. doi: 10.1093/brain/113.2.397. [DOI] [PubMed] [Google Scholar]
- 5.Monsch AU, Bondi MW, Butters N, Salmon DP, Katzman R, Thal LJ. Comparisons of verbal fluency tasks in the detection of dementia of the Alzheimer type. Arch Neurol. 1992;49:1253–8. doi: 10.1001/archneur.1992.00530360051017. [DOI] [PubMed] [Google Scholar]
- 6.Petersen RC. Mild cognitive impairment as a diagnostic entity. J Intern Med. 2004;256:183–94. doi: 10.1111/j.1365-2796.2004.01388.x. [DOI] [PubMed] [Google Scholar]
- 7.Petersen RC, Parisi JE, Dickson DW, Johnson KA, Knopman DS, Boeve BF, Jicha GA, Ivnik RJ, Smith GE, Tangalos EG, Braak H, Kokmen E. Neuropathologic features of amnestic mild cognitive impairment. Arch Neurol. 2006;63:665–72. doi: 10.1001/archneur.63.5.665. [DOI] [PubMed] [Google Scholar]
- 8.Bennett DA, Schneider JA, Bienias JL, Evans DA, Wilson RS. Mild cognitive impairment is related to Alzheimer disease pathology and cerebral infarctions. Neurology. 2005;64:834–41. doi: 10.1212/01.WNL.0000152982.47274.9E. [DOI] [PubMed] [Google Scholar]
- 9.Scheff SW, Price DA. Alzheimer's disease-related alterations in synaptic density: neocortex and hippocampus. J Alzheimers Dis. 2006;9:101–15. doi: 10.3233/jad-2006-9s312. [DOI] [PubMed] [Google Scholar]
- 10.Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R. Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30:572–80. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- 11.DeKosky ST, Scheff SW. Synapse loss in frontal cortex biopsies in Alzheimer's disease: correlation with cognitive severity. Ann Neurol. 1990;27:457–64. doi: 10.1002/ana.410270502. [DOI] [PubMed] [Google Scholar]
- 12.DeKosky ST, Scheff SW, Styren SD. Structural correlates of cognition in dementia: quantification and assessment of synapse change. Neurodegeneration. 1996;5:417–21. doi: 10.1006/neur.1996.0056. [DOI] [PubMed] [Google Scholar]
- 13.Markesbery WR, Schmitt FA, Kryscio RJ, Davis DG, Smith CD, Wekstein DR. Neuropathologic substrate of mild cognitive impairment. Arch Neurol. 2006;63:38–46. doi: 10.1001/archneur.63.1.38. [DOI] [PubMed] [Google Scholar]
- 14.Saito Y, Murayama S. Neuropathology of mild cognitive impairment. Neuropathology. 2007;27:578–84. doi: 10.1111/j.1440-1789.2007.00806.x. [DOI] [PubMed] [Google Scholar]
- 15.Scheff SW, Price DA, Schmitt FA, DeKosky ST, Mufson EJ. Synaptic alterations in CA1 in mild Alzheimer disease and mild cognitive impairment. Neurology. 2007;68:1501–8. doi: 10.1212/01.wnl.0000260698.46517.8f. [DOI] [PubMed] [Google Scholar]
- 16.Scheff SW, Price DA, Schmitt FA, Mufson EJ. Hippocampal synaptic loss in early Alzheimer's disease and mild cognitive impairment. Neurobiol Aging. 2006;27:1372–84. doi: 10.1016/j.neurobiolaging.2005.09.012. [DOI] [PubMed] [Google Scholar]
- 17.Counts SE, Nadeem M, Lad SP, Wuu J, Mufson EJ. Differential expression of synaptic proteins in the frontal and temporal cortex of elderly subjects with mild cognitive impairment. J Neuropathol Exp Neurol. 2006;65:592–601. doi: 10.1097/00005072-200606000-00007. [DOI] [PubMed] [Google Scholar]
- 18.Bouras C, Hof PR, Giannakopoulos P, Michel JP, Morrison JH. Regional distribution of neurofibrillary tangles and senile plaques in the cerebral cortex of elderly patients: a quantitative evaluation of a one-year autopsy population from a geriatric hospital. Cereb Cortex. 1994;4:138–50. doi: 10.1093/cercor/4.2.138. [DOI] [PubMed] [Google Scholar]
- 19.Suzuki WA, Amaral DG. Perirhinal and parahippocampal cortices of the macaque monkey: cortical afferents. J Comp Neurol. 1994;350:497–533. doi: 10.1002/cne.903500402. [DOI] [PubMed] [Google Scholar]
- 20.Bennett DA, Wilson RS, Schneider JA, Evans DA, Beckett LA, Aggarwal NT, Barnes LL, Fox JH, Bach J. Natural history of mild cognitive impairment in older persons. Neurology. 2002;59:198–205. doi: 10.1212/wnl.59.2.198. [DOI] [PubMed] [Google Scholar]
- 21.DeKosky ST, Ikonomovic MD, Styren SD, Beckett L, Wisniewski S, Bennett DA, Cochran EJ, Kordower JH, Mufson EJ. Upregulation of choline acetyltransferase activity in hippocampus and frontal cortex of elderly subjects with mild cognitive impairment. Ann Neurol. 2002;51:145–55. doi: 10.1002/ana.10069. [DOI] [PubMed] [Google Scholar]
- 22.Mufson EJ, Ma SY, Cochran EJ, Bennett DA, Beckett LA, Jaffar S, Saragovi HU, Kordower JH. Loss of nucleus basalis neurons containing trkA immunoreactivity in individuals with mild cognitive impairment and early Alzheimer's disease. J Comp Neurol. 2000;427:19–30. doi: 10.1002/1096-9861(20001106)427:1<19::aid-cne2>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 23.Davis DG, Schmitt FA, Wekstein DR, Markesbery WR. Alzheimer neuropathologic alterations in aged cognitively normal subjects. J Neuropathol Exp Neurol. 1999;58:376–88. doi: 10.1097/00005072-199904000-00008. [DOI] [PubMed] [Google Scholar]
- 24.Schmitt FA, Davis DG, Wekstein DR, Smith CD, Ashford JW, Markesbery WR. "Preclinical" AD revisited: neuropathology of cognitively normal older adults. Neurology. 2000;55:370–6. doi: 10.1212/wnl.55.3.370. [DOI] [PubMed] [Google Scholar]
- 25.Nelson PT, Jicha GA, Schmitt FA, Liu H, Davis DG, Mendiondo MS, Abner EL, Markesbery WR. Clinicopathologic correlations in a large Alzheimer disease center autopsy cohort: neuritic plaques and neurofibrillary tangles "do count" when staging disease severity. J Neuropathol Exp Neurol. 2007;66:1136–46. doi: 10.1097/nen.0b013e31815c5efb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nelson PT, Kryscio RJ, Abner EL, Schmitt FA, Jicha GA, Mendiondo MS, Cooper G, Smith CB, Markesbery WR. Acetylcholinesterase inhibitor treatment is associated with relatively slow cognitive decline in patients with Alzheimer's disease and AD + DLB. J Alzheimers Dis. 2009;16:29–34. doi: 10.3233/JAD-2009-0926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, Vogel FS, Hughes JP, van Belle G, Berg L. The Consortium to Establish a Registry for Alzheimer's Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology. 1991;41:479–86. doi: 10.1212/wnl.41.4.479. [DOI] [PubMed] [Google Scholar]
- 28.Hyman BT, Trojanowski JQ. Consensus recommendations for the postmortem diagnosis of Alzheimer disease from the National Institute on Aging and the Reagan Institute Working Group on diagnostic criteria for the neuropathological assessment of Alzheimer disease. J Neuropathol Exp Neurol. 1997;56:1095–7. doi: 10.1097/00005072-199710000-00002. [DOI] [PubMed] [Google Scholar]
- 29.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–59. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 30.Mufson EJ, Ma SY, Cochran EJ, Bennett DA, Beckett LA, Jaffar S, Saragovi HU, Kordower JH. Loss of nucleus basalis neurons containing trkA immunoreactivity in individuals with mild cognitive impairment and early Alzheimer's disease. J Comp Neurol. 2000;427:19–30. doi: 10.1002/1096-9861(20001106)427:1<19::aid-cne2>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 31.Mouton PR. Principles and practices of unbiased stereology. Baltimore: The Johns Hopkins University Press; 2002. [Google Scholar]
- 32.Scheff SW, Price DA. Synaptic pathology in Alzheimer's disease: a review of ultrastructural studies. Neurobiol Aging. 2003;24:1029–46. doi: 10.1016/j.neurobiolaging.2003.08.002. [DOI] [PubMed] [Google Scholar]
- 33.Gundersen HJG. Stereology of arbitrary particles. J Microsc. 1986;143:3–45. [PubMed] [Google Scholar]
- 34.Scheff SW, Price DA. Alzheimer's disease-related synapse loss in the cingulate cortex. J Alzheimers Dis. 2001;3:495–505. doi: 10.3233/jad-2001-3509. [DOI] [PubMed] [Google Scholar]
- 35.Weibel E. Point counting method. In: Weibel ER, editor. Stereological Method. Vol. 1. Academic Press; London: 1979. pp. 101–61. [Google Scholar]
- 36.Consensus recommendations for the postmortem diagnosis of Alzheimer's disease. The National Institute on Aging, and Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer's Disease. Neurobiol Aging. 1997;18:S1–2. [PubMed] [Google Scholar]
- 37.Gomez-Isla T, Hollister R, West H, Mui S, Growdon JH, Petersen RC, Parisi JE, Hyman BT. Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer's disease. Ann Neurol. 1997;41:17–24. doi: 10.1002/ana.410410106. [DOI] [PubMed] [Google Scholar]
- 38.Gomez-Isla T, Price JL, McKeel DW, Jr, Morris JC, Growdon JH, Hyman BT. Profound loss of layer II entorhinal cortex neurons occurs in very mild Alzheimer's disease. J Neurosci. 1996;16:4491–500. doi: 10.1523/JNEUROSCI.16-14-04491.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Price JL, Ko AI, Wade MJ, Tsou SK, McKeel DW, Morris JC. Neuron number in the entorhinal cortex and CA1 in preclinical Alzheimer disease. Arch Neurol. 2001;58:1395–402. doi: 10.1001/archneur.58.9.1395. [DOI] [PubMed] [Google Scholar]
- 40.Schneider JA, Arvanitakis Z, Leurgans SE, Bennett DA. The neuropathology of probable Alzheimer disease and mild cognitive impairment. Ann Neurol. 2009;66:200–8. doi: 10.1002/ana.21706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Masliah E, Mallory M, Alford M, DeTeresa R, Hansen LA, McKeel DW, Jr, Morris JC. Altered expression of synaptic proteins occurs early during progression of Alzheimer's disease. Neurology. 2001;56:127–9. doi: 10.1212/wnl.56.1.127. [DOI] [PubMed] [Google Scholar]
- 42.Liu X, Erikson C, Brun A. Cortical synaptic changes and gliosis in normal aging, Alzheimer's disease and frontal lobe degeneration. Dementia. 1996;7:128–34. doi: 10.1159/000106867. [DOI] [PubMed] [Google Scholar]
- 43.Tannenberg RK, Scott HL, Tannenberg AE, Dodd PR. Selective loss of synaptic proteins in Alzheimer's disease: evidence for an increased severity with APOE varepsilon4. Neurochem Int. 2006;49:631–9. doi: 10.1016/j.neuint.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 44.Hyman BT, Trojanowski JQ. Consensus recommendations for the postmortem diagnosis of Alzheimer disease from the National Institute on Aging and the Reagan Institute Working Group on diagnostic criteria for the neuropathological assessment of Alzheimer disease. J Neuropathol Exp Neurol. 1997;56:1095–7. doi: 10.1097/00005072-199710000-00002. [DOI] [PubMed] [Google Scholar]
- 45.Hyman BT. The neuropathological diagnosis of Alzheimer's disease: clinical- pathological studies. Neurobiol Aging. 1997;18:S27–32. doi: 10.1016/s0197-4580(97)00066-3. [DOI] [PubMed] [Google Scholar]
- 46.Saleem KS, Suzuki W, Tanaka K, Hashikawa T. Connections between anterior inferotemporal cortex and superior temporal sulcus regions in the macaque monkey. J Neurosci. 2000;20 :5083–101. doi: 10.1523/JNEUROSCI.20-13-05083.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Saleem KS, Tanaka K. Divergent projections from the anterior inferotemporal area TE to the perirhinal and entorhinal cortices in the macaque monkey. J Neurosci. 1996;16:4757–75. doi: 10.1523/JNEUROSCI.16-15-04757.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ungerleider IG, Pasternak T. Ventral and dorsal cortical processing streams. In: Chalupa LM, Werner JS, editors. The visual neurosciences. MIT Press; Cambridge: 2004. pp. 541–62. [Google Scholar]
- 49.Dean P. Effects of inferotemporal lesions on the behavior of monkeys. Psychol Bull. 1976;83:41–71. [PubMed] [Google Scholar]
- 50.Dean P, Cowey A. Inferotemporal lesions and memory for pattern discriminations after visual interference. Neuropsychologia. 1977;15:93–8. doi: 10.1016/0028-3932(77)90118-x. [DOI] [PubMed] [Google Scholar]
- 51.Cheng K, Saleem KS, Tanaka K. Organization of corticostriatal and corticoamygdalar projections arising from the anterior inferotemporal area TE of the macaque monkey: a Phaseolus vulgaris leucoagglutinin study. J Neurosci. 1997;17:7902–25. doi: 10.1523/JNEUROSCI.17-20-07902.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kalivas PW, Nakamura M. Neural systems for behavioral activation and reward. Curr Opin Neurobiol. 1999;9:223–7. doi: 10.1016/s0959-4388(99)80031-2. [DOI] [PubMed] [Google Scholar]
- 53.Spitsyna G, Warren JE, Scott SK, Turkheimer FE, Wise RJ. Converging language streams in the human temporal lobe. J Neurosci. 2006;26:7328–36. doi: 10.1523/JNEUROSCI.0559-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Visser M, Embleton KV, Jefferies E, Parker GJ, Ralph MA. The inferior, anterior temporal lobes and semantic memory clarified: novel evidence from distortion-corrected fMRI. Neuropsychologia. 2010;48:1689–96. doi: 10.1016/j.neuropsychologia.2010.02.016. [DOI] [PubMed] [Google Scholar]
- 55.Henry JD, Crawford JR, Phillips LH. Verbal fluency performance in dementia of the Alzheimer's type: a meta-analysis. Neuropsychologia. 2004;42:1212–22. doi: 10.1016/j.neuropsychologia.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 56.Cooper DB, Lacritz LH, Weiner MF, Rosenberg RN, Cullum CM. Category fluency in mild cognitive impairment: reduced effect of practice in test-retest conditions. Alzheimer Dis Assoc Disord. 2004;18:120–2. doi: 10.1097/01.wad.0000127442.15689.92. [DOI] [PubMed] [Google Scholar]
- 57.Canning SJ, Leach L, Stuss D, Ngo L, Black SE. Diagnostic utility of abbreviated fluency measures in Alzheimer disease and vascular dementia. Neurology. 2004;62:556–62. doi: 10.1212/wnl.62.4.556. [DOI] [PubMed] [Google Scholar]
- 58.Mummery CJ, Patterson K, Price CJ, Ashburner J, Frackowiak RS, Hodges JR. A voxel-based morphometry study of semantic dementia: relationship between temporal lobe atrophy and semantic memory. Ann Neurol. 2000;47:36–45. [PubMed] [Google Scholar]
- 59.Pakkenberg B, Pelvig D, Marner L, Bundgaard MJ, Gundersen HJ, Nyengaard JR, Regeur L. Aging and the human neocortex. Exp Gerontol. 2003;38:95–9. doi: 10.1016/s0531-5565(02)00151-1. [DOI] [PubMed] [Google Scholar]
- 60.Kordower JH, Chu Y, Stebbins GT, DeKosky ST, Cochran EJ, Bennett D, Mufson EJ. Loss and atrophy of layer II entorhinal cortex neurons in elderly people with mild cognitive impairment. Ann Neurol. 2001;49:202–13. [PubMed] [Google Scholar]
- 61.Marner L, Nyengaard JR, Tang Y, Pakkenberg B. Marked loss of myelinated nerve fibers in the human brain with age. J Comp Neurol. 2003;462:144–52. doi: 10.1002/cne.10714. [DOI] [PubMed] [Google Scholar]
- 62.Salat DH, Tuch DS, Greve DN, van der Kouwe AJ, Hevelone ND, Zaleta AK, Rosen BR, Fischl B, Corkin S, Rosas HD, Dale AM. Age-related alterations in white matter microstructure measured by diffusion tensor imaging. Neurobiol Aging. 2005;26:1215–27. doi: 10.1016/j.neurobiolaging.2004.09.017. [DOI] [PubMed] [Google Scholar]
- 63.Guttmann CR, Jolesz FA, Kikinis R, Killiany RJ, Moss MB, Sandor T, Albert MS. White matter changes with normal aging. Neurology. 1998;50:972–8. doi: 10.1212/wnl.50.4.972. [DOI] [PubMed] [Google Scholar]
- 64.Jernigan TL, Archibald SL, Fennema-Notestine C, Gamst AC, Stout JC, Bonner J, Hesselink JR. Effects of age on tissues and regions of the cerebrum and cerebellum. Neurobiol Aging. 2001;22:581–94. doi: 10.1016/s0197-4580(01)00217-2. [DOI] [PubMed] [Google Scholar]
- 65.Masliah E, Mallory M, Hansen L, DeTeresa R, Terry RD. Quantitative synaptic alterations in the human neocortex during normal aging. Neurology. 1993;43:192–7. doi: 10.1212/wnl.43.1_part_1.192. [DOI] [PubMed] [Google Scholar]
- 66.Adams I. Comparison of synaptic changes in the precentral and postcentral cerebral cortex of aging humans: a quantitative ultrastructural study. Neurobiol Aging. 1987;8:203–12. doi: 10.1016/0197-4580(87)90003-0. [DOI] [PubMed] [Google Scholar]
- 67.Scheff SW, Price DA, Sparks DL. Quantitative assessment of possible age-related change in synaptic numbers in the human frontal cortex. Neurobiol Aging. 2001;22:355–65. doi: 10.1016/s0197-4580(01)00222-6. [DOI] [PubMed] [Google Scholar]
- 68.Scheff SW, Price DA. Synapse loss in the temporal lobe in Alzheimer's disease. Ann Neurol. 1993;33:190–9. doi: 10.1002/ana.410330209. [DOI] [PubMed] [Google Scholar]
- 69.Paxionos G, Mai JK. The Human Nervous System. Elsevier Academic Press; San Diego: 2004. [Google Scholar]
- 70.Hof PR, Morrison JH. The cellular basis of cortical disconnection in Alzheimer disease and related dementing conditions. In: Terry RD, Katzman R, Bick KL, Sisodia SS, editors. Alzheimer disease. Lippncott Williams & Wilkins; Philadelphia: 1999. pp. 207–32. [Google Scholar]
- 71.Bouras C, Hof PR, Morrison JH. Neurofibrillary tangle densities in the hippocampal formation in a non-demented population define subgroups of patients with differential early pathologic changes. Neurosci Lett. 1993;153:131–5. doi: 10.1016/0304-3940(93)90305-5. [DOI] [PubMed] [Google Scholar]
- 72.Hof PR, Bierer LM, Perl DP, Delacourte A, Buee L, Bouras C, Morrison JH. Evidence for early vulnerability of the medial and inferior aspects of the temporal lobe in an 82-year-old patient with preclinical signs of dementia. Regional and laminar distribution of neurofibrillary tangles and senile plaques. Arch Neurol. 1992;49:946–53. doi: 10.1001/archneur.1992.00530330070019. [DOI] [PubMed] [Google Scholar]
- 73.Giannakopoulos P, Hof PR, Bouras C. Selective vulnerability of neocortical association areas in Alzheimer's disease. Microsc Res Tech. 1998;43:16–23. doi: 10.1002/(SICI)1097-0029(19981001)43:1<16::AID-JEMT3>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]