

Abstract
In endothelial cells there remain uncertainties in the details of how Ca2+ signals are generated and maintained, especially in intact preparations. In particular the role of the sarco-endoplasmic reticulum Ca2+-ATPase (SERCA), in contributing to the components of agonist-induced signals is unclear.
The aim of this work was to increase understanding of the detailed mechanism of Ca2+ signalling in endothelial cells using real time confocal imaging of Fluo-4 loaded intact rat tail arteries in response to muscarinic stimulation. In particular we have focused on the role of SERCA, and its interplay with capacitative Ca2+ entry (CCE) and ER Ca2+ release and uptake. We have determined its contribution to the Ca2+ signal and how it varies with different physiological stimuli, including single and repeated carbachol applications and brief and prolonged exposures.
In agreement with previous work, carbachol stimulated a rise in intracellular Ca2+ in the endothelial cells, consisting of a rapid initial phase, then a plateau upon which oscillations of Ca2+ were superimposed, followed by a decline to basal Ca2+ levels upon carbachol removal. Our data support the following conclusions: (i) the size (amplitude and duration) of the Ca2+ spike and early oscillations are limited by SERCA activity, thus both are increased if SERCA is inhibited. (ii) SERCA activity is such that brief applications of carbachol do not trigger CCE, presumably because the fall in luminal Ca2+ is not sufficient to trigger it. However, longer applications sufficient to deplete the ER or even partial SERCA inhibition stimulate CCE. (iii) Ca2+ entry occurs via STIM-mediated CCE and SERCA contributes to the cessation of CCE. In conclusion our data show how SERCA function is crucial to shaping endothelial cell Ca signals and its dynamic interplay with both CCE and ER Ca releases.
Keywords: Endothelial cells, Ca2+ signalling, SERCA pump, Capacitative Ca2+ entry, STIM
1. Introduction
Endothelial cells play a variety of important physiological roles, including influencing vascular tone, permeability, inflammation and platelet aggregation [1–5]. There is structural and functional heterogeneity in the endothelium depending on the size and location of the blood vessel it lines. However elevation of intracellular Ca2+ in endothelial cells is key to their physiological functions, including regulation of vascular tone, inter-endothelial cell gap formation, angiogenesis, immune defense, gene expression and cell growth [6,7]. In endothelial cells Ca2+ can be released from the intracellular store in the ER. Depletion of the ER Ca2+ store induces Ca2+ entry across the plasma membrane, in a process known as capacitative or store operated Ca2+ entry (CCE or SOCE) [8–11]. Coupling of agonists to receptors can open receptor operated channels, through which Ca2+ can also enter the cell [12–15]. Details of the interplay between these elements and the characteristics of the resulting Ca2+ signals are still being elucidated. For example cytosolic calcium signalling plays an important role in the generation of endothelium derived relaxing factors (NO, EDHF), which contributes to vascular smooth muscle relaxation, and in turn, reduces blood pressure. However, the mechanisms controlling the spatial and temporal characteristics of the Ca2+ signalling involved in the activation of eNOS and other processes, are still poorly understood. Recently, the proteins STIM-1 and Orai 1 have emerged as candidate components mediating CCE [16–20]. Stim-1 responds to the depletion of Ca2+ stores activating CCE via interaction with ORAI. However, it has also been reported that unlike macrovessels, CCE is not important in microvascular endothelial cell [21,22]. We have recently shown that in ureteric precapillary arterioles (microvessels), Ca2+ signalling in endothelial cells does not require Ca2+ entry and consists mainly of asynchronous, localized, high frequency Ca2+ oscillations [23].
From the above it can be appreciated that the SR/ER Ca-ATPase (SERCA) will be intimately involved in governing the characteristics and kinetics of agonist-induced Ca signals, through binding Ca, influencing Ca releases through IP3-dependent Ca2+ channels and instigating and terminating CCE via effects on luminal Ca levels [24]. However the information concerning SERCA's role in the endothelium is limited. Fierro and Parekh found that SERCA pumps were remarkably effective in RBL-1 cells and could prevent ICRAC from activating, despite significant Ca2+ leakage from the stores [25]. Patch clamp studies in other types of cells [26,27] suggest however that SERCA should be able to terminate CCE as it refills the store.
Thus there is a need to systematically examine all these components in one cell type and endothelium from a single type of blood vessel. Furthermore there is also a lack of clarity around the roles of SERCA and CCE in the Ca2+ signals occurring in intact endothelium. While there may be advantages to studying cultured endothelial cells, recent research has highlighted the importance of studying the intact endothelium e.g. [28,29]. In the current study therefore we have determined the temporal and spatial organization of Ca2+ signals induced by muscarinic stimulation in situ in Fluo-4 loaded endothelial cells of rat tail artery using confocal microscopy, to better define the mechanisms controlling the characteristics of the Ca signals in response to muscarinic stimulation and the exact role of SERCA during different parameters of stimulations.
We demonstrate that the initial Ca2+ wave and the early Ca2+ oscillations originated from the ER by the interplay of repetitive releases and re-uptake of Ca2+ by IP3-dependent Ca2+ channels and SERCA pump activity, respectively. In contrast, interplay between Ca2+ release, Ca2+ influx through CCE channels and SERCA was required to sustain the late Ca2+ oscillations and plateau.
2. Materials and methods
2.1. Ca2+ measurements
Tail was removed from rat humanely killed by cervical dislocation under CO2 anesthesia in accordance with Home Office legislation Artery was dissected from the ventral grove, cleaned of fat and kept in physiological saline before use. They were loaded with Fluo-4 acetoxymethyl ester (Invitrogen, UK, 15 μmol/L; dissolved in DMSO with pluronic acid) for 2–3 h at 20 °C and then transferred to indicator-free solution for 30 min. Small segments of Fluo-4 loaded tail artery (3–4 mm in length) were cut open and fixed to the bottom of the chamber by aluminum foil clips, to minimize movement, with endothelium facing down. All experiments were performed at 30 °C. We used a Nipkow disc based, confocal microscope (Perkin Elmer), connected to a sensitive iXon cooled charge-coupled device camera (Andor). Images were collected at 30 frames per second using a 60× water objective (NA 1.20) for best spatial resolution or dry (20×, 0.70 NA) for a larger field of view.
2.2. Solutions
Physiological saline of the following composition was used (mmol/L): NaCl 120.4, KCl 5.9, MgSO4 1.2, CaCl2 2.0, glucose 8, and HEPES 11. In some experiments, Ca2+-free solution (2 mmol/L EGTA) was used. 2-Aminoethoxydiphenyl borate (2-APB), ryanodine, and U-73122 (to inhibit phospholipase C), were from Calbiochem (Nottingham, UK), all other chemicals were from Sigma (UK). Phenylephrine, endothelin-1 (ET-1), ryanodine, and carbachol were dissolved in water; 2-APB, cyclopiazonic acid (CPA, to inhibit SERCA), U-73122 in DMSO.
2.3. Immunohistochemistry
Specimens of dissected tail artery in the absence of stimulation (control) or following two consecutive applications of carbachol in the presence of CPA to deplete Ca2+ stores and maximally activate CCE (see Fig. 11 for details), were fixed with 4% paraformaldehyde in 0.1 M phosphate-buffered saline (PBS), pH 7.4 for 1.5 h at 23 °C, and then in 25% sucrose in 0.1 M PBS overnight at 4 °C, cut on a cryostat at 10 μm and mounted onto poly-lysine coated microscope slides. Cryostat sections were rinsed in 0.1 M PBS, permeabilized in alcohol and processed for single labelling immunofluorescence. In brief, tissue sections were pre-blocked in 10% normal donkey serum in 0.1 M PBS and incubated with rabbit polyclonal STIM1 (C-terminal) antibody (ProSci Incorporated, Nottingham, UK; 1:300) overnight at 4 °C. The secondary antibody was Texas red-conjugated (AffiniPure) donkey anti-rabbit IgG (Jackson ImmunoResearch Laboratories; West Grove, PA, USA) and sections of tail artery were mounted under Vectashield containing 4′6-diamidino-2-phenylindole (DAPI) (Vector Laboratories) to preserve fluorescence and stain cell nuclei. Specificity of immunostaining was determined by omitting the primary antibody. Sections were examined using an Axioplan Universal microscope, and images were processed using the Axio Vision 3.0 Imaging system with deconvolution options (Carl Zeiss Vision, Jena, Germany).
Fig. 11.

Effect of combined action of CPA and CCh on distribution of STIM 1 in ECs of intact rat tail artery. (A) Representative images of EC labelled by anti-STIM1 antibody (Texas red) and DAPI (blue) before and after stimulation with CCh in the presence of CPA (10 μM) to maximally deplete the ER. (B) Representative graph showing activation of Ca2+ entry induced by combined action of CPA and CCh in the presence of external Ca2+. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of the article.)
2.4. Statistics
All analysis was performed using Microcal Origin 8.0 (Massachusetts, USA). Quantification of calcium records from confocal imaging were made by measuring the peak amplitude, frequency and duration of calcium transients. The results are given as percentage of control value unless otherwise stated. Values are means ± SEM, and n is number of vessels or cells. Between 3 and 5 animals were used for each series of experiments. Differences were taken as significant at P < 0.05 in Student's t-test.
3. Results
3.1. Live morphology of the ECs in situ
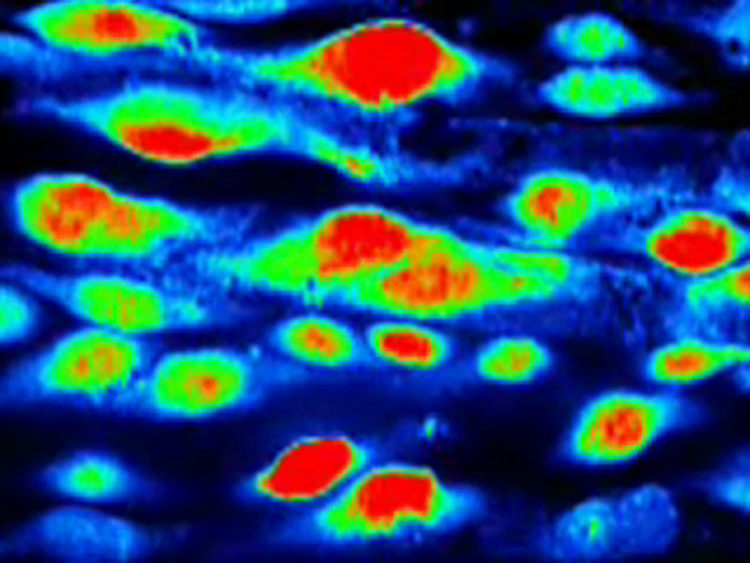
By taking a series of optical sections in the Z direction it was possible to recreate a three-dimensional image of the endothelium of intact rat tail artery loaded with Fluo 4 (Fig. 1, Video 1). The endothelial cells (EC) appeared as monolayer of cells lining the luminal surface of the elastic lamina with variable length and width (Fig. 1, Video 1).
Fig. 1.

Live morphology of endothelial cells (ECs) of intact rat tail artery in situ. 3-Dimensional confocal image of intact ECs seen in x–y projection observed using 60× objective (Video 1).
3.2. Temporal and spatial characteristics of the Ca2+ transients induced by carbachol
Initially we used three concentrations of CCh (0.1, 1 and 10 μM) to assess the sensitivity of the intact endothelium. We observed variation between endothelial cells, ranging from transient localized Ca2+ events (puffs) seen mainly with low concentration of [CCh] to complex global Ca2+ transients, with an initial spike, then a plateau component, which usually had Ca2+ oscillations superimposed on it (Fig. 2, Video 2). The percentage of cells responding with global Ca2+ transient to CCh was concentration dependent. Thus about 40–60% of cells responded to 0.1 μM, 90–92% to 1 μM and 99–100% to 10 μM CCh. The initial spike component was a regenerative propagating (14–44 μm/s) Ca2+ wave, initiated at one or both ends of the cells (Fig. 2A and C, Video 2). As shown in Fig. 2C, where traces from the images in Fig. 2A have been superimposed, to show how the initial wave from either region 1 or 3, propagates to region 2 in the centre of the cell. The frequency of Ca2+ oscillations was also variable and concentration dependent (Fig. 2B and D). Ca2+ oscillations at 0.1 μM CCh ranged between 0.02 and 0.08 Hz (n = 51 cells, 5 vessels), at 1 μM they were 0.08–0.33 Hz (n = 63 cells, 7 vessels), and at10 μM CCh they were 0.18–0.36 Hz (n = 77 cells, 11 vessels). Since 1 μM CCh induced reproducible Ca2+ transients with distinct Ca2+ oscillations seen in the majority of cells, this concentration was used for the remainder of the study.
Fig. 2.

Temporal and spatial characteristics of the Ca2+ transient induced by CCh in EC of intact rat tail artery. (A) Stack of pseudo-colour images showing propagating Ca2+ wave initiated at two ends of the EC seen as an initial spike component. (B) Graph showing time dependent changes of Fluo-4 fluorescence induced by CCh in three parts of EC where region 1 and 3 are initiation sites. (C) Superimposed traces of initial Ca spike seen as propagating Ca wave initiated in region 1 (top panel) and region 3 (bottom panel) propagating to region 2 (centre of the cell). Arrows in A and C show direction of the wave propagation. (D) Patterns of the typical Ca2+ transients induced by 1 μM CCh in intact EC recorded by placing a region of interest in central part of the cell (Video 2). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of the article.)
3.3. Contribution of ER
To identify the ER's role in the complex Ca2+ responses induced by CCh, the effects of removal of extracellular Ca2+ for variable periods of times on the parameters of the Ca2+ transient induced by an application of CCh were studied. We found that the removal of external Ca2+ for 1–3 min had little or no effect on the amplitude of the initial spike and the onset of Ca2+ oscillations (Fig. 3Ai). However, when the endothelial cells were exposed to Ca2+-free solution for 30 min or more, this resulted in suppression of Ca2+ oscillations and reduction of all components of the Ca2+ transient (Fig. 3Aii). The amplitude of the initial spike component after 30 min exposure to Ca2+-free solution was reduced to 78.0 ± 2.5% (n = 27, P < 0.05) and after 60 min to 62 ± 4.2% (n = 35, P < 0.05) of control, respectively (Fig. 3B).
Fig. 3.

Effects of removal of extracellular Ca2+ for different periods of time on the Ca2+ transient induced by CCh in the ECs of intact rat tail artery. (A(i–ii)) Original records of CCh induced Ca2+ transients in control conditions and at different times of exposure (i) 1–3 min, (ii) 30 min to Ca2+-free solution containing 2 mM EGTA. B(a–d) Percentage of inhibition of the initial phasic component of the Ca2+ transient induced by CCh after exposure of the ECs to 30 s (b), 5 min (c) and 30 min (d). Peak Ca2+ transient in control condition was taken for 100% (a). (c and d) P < 0.05, significantly different from control.
We next investigated the ability of the ER to sustain Ca2+ signalling responses to CCh in the absence of extracellular Ca2+. To do this cells in Ca2+-free solution were stimulated by a series of applications of CCh of different durations; 10, 60 or 120 s. The endothelial cells were exposed to Ca2+-free solution for 3 min and then stimulated by a series of CCh applications (in 0-Ca2+ with 2 mM EGTA solution) and Ca2+ transients were recorded from a large number of cells using 20× objective lens.
When EC were stimulated by brief (10 s) applications of CCh, Ca2+ transients and oscillations could be observed for 15.0 ± 2.3 min (Fig. 4A). When the CCh application was increased to 60 s, EC quickly lost their ability to generate normal Ca2+ signalling after 3–4 CCh applications (Fig. 4B). It can also be seen that the first application of CCh induced a normal Ca2+ spike and initial Ca2+ oscillations but the sustained component gradually relaxed and late Ca2+ oscillations were abolished (Fig. 4B). Calcium transients induced by the 2nd and 3rd applications of CCh showed a marked reduction in the amplitude of the initial spike and plateau, and Ca2+ oscillations were not observed (Fig. 4B), In Ca2+-free solution and longer application of CCh (2 min), only one response was produced and the Ca2+ transients spontaneously relaxed to baseline (Fig. 4C).
Fig. 4.

Effects of removal of extracellular Ca2+ on Ca2+ transients induced by series of CCh applications for different lengths of time. (A, B and C) Representative traces of the Ca2+ transients induced by a series of 10, 60 and 120 s applications of CCh, respectively.
The ability of the EC to respond with regular Ca2+ transients for 15–20 min to short applications of CCh in Ca2+-free solution (Figs. 4A and 5A), suggests that under these conditions there is a minimal loss of ER Ca2+ and, that most of the Ca2+ released from the ER is quickly re-sequestered into the store by SERCA activity. To investigate this, the effects of the SERCA inhibitor CPA on the Ca2+ transients induced by brief (10 s) applications of CCh in Ca2+-free solution, were investigated. Exposure of the EC to CPA (20 μM to maximally inhibit SERCA), produced a large increased in Ca2+ which spontaneously relaxed to the baseline. Subsequent applications of CCh were ineffective, suggesting depletion of the Ca2+ store (Fig. 5B).
Fig. 5.

Effects of SERCA pump inhibition by cyclopiazonic acid (CPA) on the Ca2+ transients induced by brief (10 s) applications of CCh in Ca2+-free solution. (A) Ca2+ transients induced by a series of 10 s applications of CCh in Ca2+ free solution (control experiments); (B) inhibition of CCh-induced Ca2+ transients induced by CCh in Ca2+ free solution by CPA (20 μM). In these protocol EC were first pre-exposed to Ca2+-free solution with 2 mM EGTA for 3 min to fully remove external Ca2+.
3.4. Role of phospholipase C and ER release channels
The involvement of phospholipase C (PLC) in the transduction pathway leading to CCh evoked Ca2+ transients in EC in Ca2+-free solution was studied by using U-73122, an inhibitor of PLC activity. Pretreatment of the EC for 30 min with U-73122 (50 μM) abolished Ca2+ transients induced by CCh in Ca2+-free solution (n = 7 vessels, Fig. 6A). The possible roles of IP3 receptor (IP3R) and ryanodine receptor (RyR) channels in the ER CCh-induced Ca2+ release in Ca2+-free solution was studied by using the (non-selective) inhibitor of IP3R, 2-APB and the (selective) inhibitor of RyR channels, ryanodine. In these experiments EC were pretreated with 2-APB (50 μM) or ryanodine (50 μM) for 30 min in Ca2+-free solution and then stimulated with CCh. Since 2-APB is known to also block Ca2+ influx from the extracellular space [30,31], its effect on Ca2+ release was studied in Ca2+-free solution i.e. a protocol similar to that used in Fig. 3. Pretreatment of the ECs in Ca2+-free solution with 2-APB for 30 min abolished CCh-induced Ca2+ transients (Fig. 6B). Application of CCh following 30 min ryanodine pretreatment had little or no effect on CCh-evoked Ca2+ transient (n = 11 cells, 3 vessels, Fig. 6C). This suggests that RyR channels are not involved in CCh-induced Ca2+ signalling in these EC (Fig. 6C). To confirm this the efficacy of inhibiting RyR channels was tested using caffeine (5 mM) and focusing confocally on the smooth muscle cell layer of the artery. Here caffeine induced a large Ca2+ transient associated with vasoconstriction (not shown), which was abolished by the 30 min pre-treatments with ryanodine, suggesting that RyR channels under these conditions are indeed blocked (Fig. 6D).
Fig. 6.

Effects of the PLC inhibitor U-73122, 2-APB and ryanodine on the Ca2+ transient induced by CCh in the EC. (A and B) Full inhibition of Ca2+ transients induced by CCh in Ca2+-free solution by U-73122 (50 μM) or 2-APB (50 μM), respectively; (C and D) effects of ryanodine (50 μM) on Ca2+ transients induced by CCh in EC and smooth muscle cells (SMC) of the same intact rat tail artery, respectively. In C and D, CCh and caffeine were applied in the presence of external Ca2+, respectively.
3.5. Contribution of extracellular calcium
To study the role of Ca2+ influx in the control of Ca2+ signalling in the intact EC, we used two (non-selective) blockers of CCE, La3+ and Gd3+. Lanthanum (10 μM) in the presence of external Ca2+ had little or no effect on Ca2+ transients induced by a series of short (10 s) applications of CCh, suggesting that Ca2+ influx was not required to maintain Ca2+ signals, within this period (Fig. 7A,i). In contrast, Ca2+ transients induced by 1 min applications of CCh showed a time dependent inhibition by La3+ (Fig. 7A,ii); late Ca2+ oscillations were inhibited and the plateau was markedly reduced, even in the first CCh application. Subsequent applications of CCh showed inhibition of all components of the Ca2+ transient (n = 49 cells, 7 vessels, Fig. 7A,ii). A 2 min application of CCh in the presence of La3+ evoked Ca2+ transients which spontaneously relaxed to the baseline (Fig. 7A,iii). Again, the initial Ca2+ spike and early Ca2+ oscillations were not affected but late Ca2+ oscillations were suppressed (Fig. 7A,iii) (n = 21 cell, 5 vessels). Similar effects were obtained with Gd3+ (data not shown).
Fig. 7.

Effects of La3+ on Ca2+ transients induced by CCh and readmission of external Ca2+ to Ca2+-free solution following ER Ca2+ depletion. (A) Effects of La3+ (10 μM) on Ca2+ transients induced by CCh applied for 10 (i), 60 (ii) or 120 (iii) s, respectively. (B) Effects of La3+ (10 μM) on Ca2+ transient induced by readmission of external Ca2+ to Ca2+-free solution following ER Ca2+ depletion by either 20 μM CPA (i) or 1 μM CCh (ii) applied in Ca2+ free solution.
Using either a CPA 20 μM (17 cells, 5 vessels) or long exposure to CCh in Ca2+-free solution to deplete the store, produced a substantial sustained rise of intracellular Ca2+ upon readmission of external Ca2+. This Ca2+ influx was inhibited by 10 μM La3+ (n = 16 cells, 5 vessels, Fig. 7Bi,ii) or 1 μM Gd3+ (data not shown).
3.6. Effects of low [CPA] on Ca2+ transients induced by brief applications of carbachol
As brief applications of CCh in Ca2+-free solution or in La3+ produced regular Ca2+ transients, it suggests that the endothelial cells under these conditions maintain normal Ca2+ signalling, as long as SERCA is functional (see Figs. 5A and 7A,i). Thus with brief applications of CCh in Ca2+-free solution, either the amount of Ca2+ released from the ER is small and/or SERCA activity is high enough so that most of the Ca2+ released is taken back into the ER, keeping luminal level of Ca2+ above the threshold of activation of CCE. To test this hypothesis in the next series of experiments the effects of short application of CCh on Ca2+ transients in the presence of low concentrations of CPA (5 μM) were studied, in the presence and the absence of external Ca2+ and in the presence of CCE blockers. Fig. 8 shows Ca2+ transients produced by these conditions, with (Fig. 8A) and without (Fig. 8B) external Ca2+. As was shown previously (e.g. Fig. 4) and in Fig. 8A, applications of CCh produced Ca2+ transients the duration of which correlated well with the duration of the CCh pulse. The EC were then exposed to 5 μM CPA for 30 s and then in the continuous presence of CPA, stimulated further by brief applications of CCh. We found that even after this brief exposure to CPA the first application of CCh evoked larger Ca2+ transients (Fig. 8A). With CPA the duration of the Ca2+ transient was no longer limited by the time of application of CCh. After removal of the agonist Ca2+ remained elevated for 30–40 s before relaxing to a new steady state baseline (Fig. 8A). In the presence of CPA the duration of the plateau, measured at 50% of the peak amplitude, was increased 2.1 ± 0.4 times and its amplitude to 2.2 ± 0.44 times (n = 31, 5 vessels, P < 0.05) relative to control (Fig. 8A). The amplitude of the spike component evoked by the first application of CCh in the presence of CPA was also increased 1.2 ± 0.1 times, P < 0.05. These data show that a large part of the sustained component of the CCh-induced Ca2+ transient was observed after removal of CCh from the media, which in the absence of CPA produced a quick relaxation of the Ca2+ transient (Fig. 8A). This suggests that this part of the Ca2+ transient could be due to activation of CCE, which remains open for longer period of times due to the slowed operation of the SERCA. To test this we performed the same experiments but in Ca2+-free solution or in the presence of CCE blockers. Fig. 8B shows typical traces of the Ca2+ transients induced by brief applications of CCh in Ca2+-free solution (n = 27 cells, 5 vessel) or in the presence of 10 μM La3+ (n = 18 cells, 4 vessels, Fig. 8C). It illustrates that in the absence of external Ca2+ or in the presence of La3+, CCh stimulation of the EC in the presence of CPA, produced brief spike-like Ca2+ transients with no plateau component (Fig. 8B, C). Also, EC now were unable to respond with Ca2+ transients when stimulated by a series of brief CCh applications. It was also noticeable that the amplitude of the spike component evoked by the first application of CCh in the presence of CPA, was still increased, 1.23 ± 0.12 times in Ca2+ free solution (n = 21 cells, 3 vessels, P < 0.05) and 1.37 ± 0.21 times in the presence of La3+ (n = 33 cells, 7 vessels, P < 0.05).
Fig. 8.

Effects of low concentration of CPA (5 μM) on temporal characteristics of Ca2+ transients induced by brief applications of CCh under different experimental conditions. (A and B) Effects of CPA (5 μM) on Ca2+ transients induced by applications of CCh in the presence and the absence of external Ca2+, respectively; (C) effects of La3+ (10 μM) on Ca2+ transients induced by CCh in the presence of CPA in normal physiological solution.
3.7. Role of SERCA in control of Ca2+ spike and Ca2+ oscillations
The data obtained in the presence of low concentration of [CPA] showed that the amplitude of the spike component induced by the first application of CCh, in the presence or the absence of external Ca2+, was always increased and Ca2+ oscillations abolished (Fig. 8). These data suggest that SERCA acts as a negative feedback mechanism controlling the characteristics of Ca2+ spikes and oscillations, by quickly buffering cytoplasmic Ca2+ released from the ER. To address this important point we studied the effects of SERCA inhibition on Ca2+ oscillations induced by longer (30 s) applications of CCh in the presence and the absence of external Ca2+. Data are presented in Fig. 9.
Fig. 9.

Effects of CPA on Ca2+ transients induced by 30 s applications of CCh in the presence and the absence of external Ca2+. (A) Typical records of Ca2+ transients induced by 30 s application of CCh in the presence of external Ca2+ in two EC with high (Cell 1) and low (Cell 2) frequency of Ca2+ oscillations recorded before (left panel) and 30 s after (right panel) addition of CPA (5 μM); (B) typical records of Ca2+ transients induced by 30 s application of CCh in EC exposed to Ca2+ free solution for 2 min in the absence (Cell 1) and the presence of 5 μM CPA (Cell 2) added 30 s before application of an agonist. Note that CPA fully blocked Ca2+ oscillations in the presence and the absence of external Ca2+.
As already described (Figs. 2 and 4) CCh produced Ca2+ transients with oscillations superimposing on the plateau component, in the presence and absence of external Ca2+. Fig. 9A shows that in the presence of external Ca2+ brief (20–30 s) pre-treatment of the EC with 5 μM CPA produced an increase in the amplitude and duration of the initial Ca2+ spike and plateau and inhibition of Ca2+ oscillations. The amplitude of the initial component was increased 1.2 ± 0.1times control and plateau 2.9 ± 0.4 times (n = 37 cells, 7 vessels, P < 0.05). Also, as was the case with brief applications of CCh (Fig. 8A), its removal did not terminate the Ca2+ transient; these lasted for an additional 30–50 s before gradually relaxing to new baseline (Fig. 9A). As already discussed, EC pre-exposed to Ca2+-free solution for 1–3 min retained their ability to generate early Ca2+ oscillations (Fig. 9B, top trace). However, if EC were pretreated for 30 s with CPA before CCh stimulation, Ca2+ oscillations in Ca2+-free solution were also abolished (n = 45 cells, 5 vessels, Fig. 9B, bottom trace). Again the amplitude of the spike component was increased 1.19 ± 0.12 times (n = 17 cells, 3 vessels, P < 0.05).
In order to study the effects of SERCA on the parameters of the Ca2+ spike, EC were stimulated in Ca2+-free solution with brief (3–5 s) applications of CCh. Although the number of cells responding to CCh was decreased, we could still identify cells producing Ca2+ transients consisting of initial Ca2+ spikes (Fig. 10A). The advantage of these experiments was that in the absence of external Ca2+, EC could respond with regular Ca2+ spikes for 20–25 min with minimal decrease in their amplitude (Fig. 10A). Addition of 5 μM CPA to Ca2+-free solution 30 s before the next brief application of CCh, increased the amplitude and duration of the Ca2+ spike (Fig. 10A and B). The duration of the Ca2+ spike measured at 50% of the peak amplitude was increased by CPA 3.4 ± 0.3 times (n = 9 cells, 2 vessels, P < 0.05). The duration of the Ca2+ spike evoked by the second application of CCh, in the presence of CPA, was increased further (Fig. 10A). Fig. 10B shows superimposed records of the last and the first Ca2+ spikes recorded before and after CPA, on an expanded time scale. It can be clearly seen that the amplitude and duration of the Ca2+ spike induced by the brief application of CCh in Ca2+-free solution, in the presence of CPA, were increased.
Fig. 10.

Effect of CPA on the temporal characteristics of the initial Ca2+ spike induced by brief application of CCh in Ca2+ free solution. (A) A series of Ca2+ spikes induced by a series of brief (3 s) application of CCh in Ca2+ free solution before and after addition of 5 μM CPA; (B) superimposed records of Ca2+ spike extracted from A recorded 30 s before (i) and 30 s after (ii) addition of CPA to Ca2+ free solution. Note that CPA increased the amplitude and duration of initial Ca2+ spike measured at 50% of peak amplitude to 3.4 ± 0.3 times (n = 9, 2 vessels, P < 0.05, significantly different from control).
3.8. Evidence for the presence of STIM 1
Our data suggest involvement of a Ca2+ release/entry coupling mechanism in the control of Ca2+ signalling in EC of tail artery. Although we did not undertake a systematic study on the physiological conditions involving STIM/Orai interaction, we used antibodies against STIM 1 for immunohistochemistry. We found (Fig. 11A) that STIM 1 is present in the EC of these vessels. It was homogenously distributed in the EC before stimulation (Fig. 11A, left hand side), but appears as puncta following depletion of the ER by a combined action of CPA and CCh (Fig. 11A, right hand side). The combined action of CCh and CPA (bottom panel) was used in these experiments to maximally deplete the store and activate Ca entry via SOC (Fig. 11B).
4. Discussion
The intact endothelium of tail artery was amenable to study with confocal microscopy and produced reproducible Ca2+ signal for 2–3 h. Clear Ca2+ signals were obtained without contamination from myocytes. Given the caveats that have arisen about using isolated or cultured endothelial cells for studying Ca2+ signalling [32–35], we suggest this is a good model for studying intact endothelium of macrovessels.
Our data reveal that, in the intact rat tail artery preparation, many aspects of the complex Ca2+ signal in EC produced in response to agonist stimulation are generated without the need for Ca2+ entry, as continuous cycles of IP3-induced ER Ca2+ release and SERCA uptake occur. We discuss our findings in the context of previous data.
4.1. Importance of SERCA
Our major finding concerns the large role of SERCA in governing the responses of the endothelium to CCh and switching CCE on and off. SERCA binds and hence buffers cytoplasmic Ca2+ and acts as a powerful negative feedback mechanism controlling the amplitude and duration of the initial Ca2+ spike and early Ca2+ oscillations. Even brief exposure of EC to low concentration of the SERCA pump inhibitor CPA, markedly increases the amplitude of the Ca2+ spike and blocks Ca2+ oscillations. This buffering by SERCA prevents activation of CCE during brief applications of CCh, allowing EC to sustain Ca2+ oscillations in Ca2+-free solution for 15–20 min. A similar conclusion was reached by Kwan et al., 1990 in lacrimal acinar cells [49]. However, longer application of CCh (>20 s) causes partial depletion of the ER, resulting in activation of CCE. Inactivation of CCE is fast as SERCA takes up Ca2+, as also shown in patch clamp studies in other types of cells [26,27]. SERCA effectively buffers this Ca2+ entry by binding (and then transporting it into the ER lumen) [36] so that when the agonist is removed, CCE is terminated and there is rapid relaxation of the Ca2+ signalling. When SERCA activity is decreased by CPA, stimulation of EC, even with brief applications of CCh, now results in activation of CCE, and the channels remain open after removal of CCh.
4.2. SERCA and CCE
The finding that low [CPA] potentiated the initial spike and blocked Ca2+ oscillations suggests that Ca2+ is continuously cycling across the ER. The Ca2+ efflux through IP3R is most likely countered by reuptake via SERCA and thus short applications of CCh are not enough to reach the threshold for CCE activation. Our results indicate that it is SERCA activity that prevents CCE activation. It might seem surprising that SERCA can maintain a sufficient Ca2+ store content to prevent CCE activation in the face of continuous Ca2+ release via open IP3R. Our data are again in good agreement with those of Parekh and his group who found that SERCA pumps were remarkably effective in RBL-1 cells and could prevent ICRAC from activating, despite significant Ca2+ leakage from the stores and a cytosolic Ca2+ binding ratio of between 2000 and 5000 (more than two orders of magnitude greater than within the stores) [25].
Small changes in ambient luminal and cytosolic Ca2+ could also have significant effects on SERCA pump activity as was shown in pancreatic acinar cells [37]. Small (two fold) rises of cytosolic Ca2+ stimulated SERCA activity almost 10 fold [38,39]. Thus, changes in SERCA-mediated uptake will have quite dramatic effects on the activation of CCE. Our data are in good agreement with finding that SERCA is the most important component in the control of the kinetics of Ca2+ wave propagation in Xenopus oocytes, where it was shown that the frequency of IP3-induced Ca2+ waves is controlled by SERCA [40].
4.3. Calcium oscillations
The Ca2+ oscillations in the intact EC in tail artery seen in Ca2+-free solution are best explained as follows: the IP3 receptor channels need to be sensitized to release Ca2+ periodically by cyclical fluctuations of Ca2+ within the lumen of the ER. Each pulse of released Ca2+ leads to a decrease in luminal Ca2+ which has to be replenished before the IP3R are re-sensitized to deliver the next pulse of Ca2+. It is this loading of the ER that explains why the early and the late Ca2+ oscillations were quickly abolished even by low [CPA]. Entry of external Ca2+, which occurs mainly through CCE channels, combined with SERCA activity, determines the rate of store re-loading responsible for adjusting the sensitivity of the IP3 receptors to maintain late Ca2+ oscillations. The expression and distribution of different isoforms of IP3R and SERCA, as well as possible sub-divisions within the ER, are probably important for the fine tuning and kinetics of these events [41–44], but were not part of the present study.
Intracellular Ca2+ oscillations commonly results from lower (physiological) concentrations of agonists in many kinds of cells [45–48]. These oscillations depend upon complex mechanisms of regenerative intracellular signalling and generally do not persist in the absence of extracellular Ca2+, leading to the conclusion that entry of Ca2+ across the plasma membrane plays a significant role in either their initiation or maintenance. In this study, we have demonstrated that early Ca2+ oscillations in ECs of intact rat tail artery are not driven by Ca2+ entry, but rather depend upon Ca2+ release/Ca2+ uptake coupling while late Ca2+ oscillations require Ca2+ entry for their maintenance. The present study demonstrates that CCE is the underlying mechanism of calcium entry that is required to sustain late CCh-induced [Ca2+]i oscillations in ECs of intact rat tail artery. The observation that CCh-induced Ca2+ spike and early Ca2+ oscillations can be observed in Ca2+-free solution suggests that the mechanisms triggering initial spike and early calcium oscillations are intrinsic to the intracellular milieu and that the role of calcium entry is primarily to replenish any ions lost from the cytoplasm. Late Ca2+ oscillations are more complex and involve interplay between Ca2+ release and Ca2+ entry, working in series with SERCA. However, with long applications of CCh EC lose their ability to sustain late Ca2+ oscillations which suggests that under these conditions Ca2+ influx is required to maintain normal Ca2+ signalling. In agreement with other studies we find CCE is the only physiological mechanism of calcium entry supporting late Ca2+ oscillations in intact ECs of large arteries [49,50].
4.4. Capacitative calcium entry
These data indicate that in the presence of low [CPA], a Ca2+ influx pathway is activated by brief applications of CCh and remained functional for a considerable time after agonist removal, and thus suggests it is not directly regulated by a receptor operated channel. Selective inhibition of the CCE pathway by La3+ or Gd3+ relative specific blockers of CCE in EC [51], suggest that the sustained component of the Ca2+ transient, seen after removal of CCh from the media was due to Ca2+ influx via CCE. We found that with low [CPA], EC in Ca2+-free solution could generate only 3–4 Ca2+ transients. This suggests that some SERCA activity remained, but was not high enough to counter Ca2+ release and Ca2+ leak from the ER, and thus eventually the ER Ca2+ store becomes depleted. The ability of EC to regularly respond to short applications of CCh in the presence of low [CPA] and external Ca2+, suggest that the ER or some of its portions are refilled and could be released by subsequent applications of an agonist. Because there is continued partial inhibition a new steady state base line is set which reflects the new equilibrium between Ca2+ leak and Ca2+ uptake. It is interesting to also note that TRCP4, one of the main candidates ROC channel in EC [52] is potentiated by La3+ [53]. This discrepancy with the inhibition of Ca signals which we saw, may be due to differences in preparations, as we used intact endothelium, but may also suggest that in our preparation, with muscarinic activation, most of the Ca entry is via CCE.
4.5. STIM
Recent data indicate that activation of CCE (CRAC) channels by store depletion involves the redistribution of the ER Ca2+ sensor, stromal interaction molecule 1 (STIM1), to peripheral sites where it co-clusters with the channel subunit, Orai1 [18–20]. After ER Ca2+ store depletion, STIM clusters are found in the ER, close enough to the cell surface to allow for direct interaction with plasma membrane proteins. The CRAC channel activation involves physical migration of ER-resident STIM to ER–plasma membrane junctions and subsequent aggregation of the Ca2+ influx channel. So STIM serves as a Ca2+ sensor and messenger at the same time as it also organizes Orai subunits into adjacent plasma membrane clusters [54]. The clusters of messenger STIM and corresponding Orai channel proteins have been termed the ‘elementary unit’ of CCE and CRAC channel activation, as Ca2+ influx through CRAC channels occurs precisely at STIM–Orai clusters [1]. In ECs CCE has been reported for several different vascular beds [51,55,56], although as with many other cell types, recording of their inward current (ICRAC) are scarce, due presumably to its small size. Although there is still some debate over the molecular identity of CCE channels, we were able to demonstrate expression and redistribution of STIM1 in native EC. This is consistent with the recent findings of [57] in proliferating EC who established the requirement of STIM1/Orai1 for CCE. Levels of Stim1 were reported by this group to quite low, but they were able to also measure ICRAC.
5. Summary
In conclusion we have been able to study Ca2+ signalling in response to agonist in intact endothelium from tail artery. We demonstrate alterations in the components of the Ca2+ signals depending upon concentration and duration of agonists and describe the mechanisms involved in generating the different components, spike, oscillations and plateau, of the Ca2+ signal. Importantly we have found that SERCA activity plays an important role in shaping the Ca2+ signals but also in turning CCE on and off. Thus agonist modulation of SERCA activity may be a mechanism of affecting endothelium function in vivo.
Conflict of interest
The authors do not have any conflict of interest.
Acknowledgements
This work was supported by Higher Education Commission of Pakistan and British Heart Foundation.
Footnotes
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.ceca.2010.11.010.
Appendix A. Supplementary data
Live 3-dimensional image showing EC of intact rat tail artery loaded with Fluo-4 in situ. EC are seen as monolayer of cells lining up elastic lamina.
{kind=link}
Real time confocal imaging of Ca2+ signalling in Fluo-4 loaded EC of intact artery stimulated with 1 μM CCh. Images are displayed at a speed of 30 fps.
{kind=link}
References
- 1.Luik R.M., Wu M.M., Buchanan J., Lewis R.S. The elementary unit of store-operated Ca2+ entry: local activation of CRAC channels by STIM1 at ER-plasma membrane junctions. J. Cell Biol. 2006;174:815–825. doi: 10.1083/jcb.200604015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dejana E., Corada M., Lampugnani M.G. Endothelial cell-to-cell junctions. FASEB J. 1995;9:910–918. [PubMed] [Google Scholar]
- 3.Birch K.A., Pober J.S., Zavoico G.B., Means A.R., Ewenstein B.M. Calcium/calmodulin transduces thrombin-stimulated secretion: studies in intact and minimally permeabilized human umbilical vein endothelial cells. J. Cell Biol. 1992;118:1501–1510. doi: 10.1083/jcb.118.6.1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Peiretti F., Alessi M.C., Henry M., Anfosso F., Juhan-Vague I., Nalbone G. Intracellular calcium mobilization suppresses the TNF-alpha-stimulated synthesis of PAI-1 in human endothelial cells. Indications that calcium acts at a translational level. Arterioscler. Thromb. Vasc. Biol. 1997;17:1550–1560. doi: 10.1161/01.atv.17.8.1550. [DOI] [PubMed] [Google Scholar]
- 5.Adams D.J., Barakeh J., Laskey R., Van Breeman C. Ion channels and regulation of intracellular calcium in vascular endothelial cells. FASEB J. 1989;3:2389–2400. doi: 10.1096/fasebj.3.12.2477294. [DOI] [PubMed] [Google Scholar]
- 6.Nilius B., Droogmans G. Ion channels and their functional role in vascular endothelium. Physiol. Rev. 2001;81:1415–1459. doi: 10.1152/physrev.2001.81.4.1415. [DOI] [PubMed] [Google Scholar]
- 7.Tran Q.K., Watanabe H. Calcium signalling in the endothelium. Handb. Exp. Pharmacol. 2006:145–187. doi: 10.1007/3-540-32967-6_5. [DOI] [PubMed] [Google Scholar]
- 8.Putney J.W., Jr. Capacitative calcium entry revisited. Cell Calcium. 1990;11:611–624. doi: 10.1016/0143-4160(90)90016-n. [DOI] [PubMed] [Google Scholar]
- 9.Dolor R.J., Hurwitz L.M., Mirza Z., Strauss H.C., Whorton A.R. Regulation of extracellular calcium entry in endothelial cells: role of intracellular calcium pool. Am. J. Physiol. 1992;262:C171–C181. doi: 10.1152/ajpcell.1992.262.1.C171. [DOI] [PubMed] [Google Scholar]
- 10.Hoth M., Penner R. Depletion of intracellular calcium stores activates a calcium current in mast cells. Nature. 1992;355:353–356. doi: 10.1038/355353a0. [DOI] [PubMed] [Google Scholar]
- 11.Freichel M., Schweig U., Stauffenberger S., Freise D., Schorb W., Flockerzi V. Store-operated cation channels in the heart and cells of the cardiovascular system. Cell. Physiol. Biochem. 1999;9:270–283. doi: 10.1159/000016321. [DOI] [PubMed] [Google Scholar]
- 12.Broad L.M., Cannon T.R., Taylor C.W. A non-capacitative pathway activated by arachidonic acid is the major Ca2+ entry mechanim in rat A7r5 smooth muscle cells stimulated with low concentrations of vasopressin. J. Physiol. Lond. 1999;517:121–134. doi: 10.1111/j.1469-7793.1999.0121z.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shuttleworth T.J., Thompson J.L. Discriminating between capacitative and arachidonate-activated Ca(2+) entry pathways in HEK293 cells. J. Biol. Chem. 1999;274:31174–31178. doi: 10.1074/jbc.274.44.31174. [DOI] [PubMed] [Google Scholar]
- 14.Luo D., Broad L.M., Bird G.S., Putney J.W., Jr. Mutual antagonism of calcium entry by capacitative and arachidonic acid-mediated calcium entry pathways. J. Biol. Chem. 2001;276:20186–20189. doi: 10.1074/jbc.M100327200. [DOI] [PubMed] [Google Scholar]
- 15.Moneer Z., Taylor C.W. Reciprocal regulation of capacitative and non-capacitative Ca2+ entry in A7r5 vascular smooth muscle cells: only the latter operates during receptor activation. Biochem. J. 2002;362:13–21. doi: 10.1042/0264-6021:3620013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Roos J., DiGregorio P.J., Yeromin A.V. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J. Cell Biol. 2005;169:435–445. doi: 10.1083/jcb.200502019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang S.L., Yu Y., Roos J. STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature. 2005;437:902–905. doi: 10.1038/nature04147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liou J., Kim M.L., Heo W.D. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr. Biol. 2005;15:1235–1241. doi: 10.1016/j.cub.2005.05.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Feske S., Gwack Y., Prakriya M. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature. 2006;441:179–185. doi: 10.1038/nature04702. [DOI] [PubMed] [Google Scholar]
- 20.Vig M., Peinelt C., Beck A. CRACM1 is a plasma membrane protein essential for store-operated Ca2+ entry. Science. 2006;312:1220–1223. doi: 10.1126/science.1127883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chetham P.M., Babal P., Bridges J.P., Moore T.M., Stevens T. Segmental regulation of pulmonary vascular permeability by store-operated Ca2+ entry. Am. J. Physiol. 1999;276:L41–L50. doi: 10.1152/ajplung.1999.276.1.L41. [DOI] [PubMed] [Google Scholar]
- 22.Alvarez D.F., Gjerde E.A., Townsley M.I. Role of EETs in regulation of endothelial permeability in rat lung. Am. J. Physiol. Lung Cell Mol. Physiol. 2004;286:L445–L451. doi: 10.1152/ajplung.00150.2003. [DOI] [PubMed] [Google Scholar]
- 23.Borisova L., Wray S., Eisner D.A., Burdyga T. How structure, Ca signals, and cellular communications underlie function in precapillary arterioles. Circ. Res. 2009;105:803–810. doi: 10.1161/CIRCRESAHA.109.202960. [DOI] [PubMed] [Google Scholar]
- 24.Wray S., Burdyga T. Sarcoplasmic reticulum function in smooth muscle. Physiol. Rev. 2010:113–178. doi: 10.1152/physrev.00018.2008. [DOI] [PubMed] [Google Scholar]
- 25.Fierro L., Parekh A.B. On the characterisation of the mechanism underlying passive activation of the Ca2+ release-activated Ca2+ current ICRAC in rat basophilic leukaemia cells. J. Physiol. 1999;520(Pt 2):407–416. doi: 10.1111/j.1469-7793.1999.00407.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hoth M., Penner R. Calcium release-activated calcium current in rat mast cells. J. Physiol. 1993;465:359–386. doi: 10.1113/jphysiol.1993.sp019681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zweifach A., Lewis R.S. Rapid inactivation of depletion-activated calcium current (ICRAC) due to local calcium feedback. J. Gen. Physiol. 1995;105:209–226. doi: 10.1085/jgp.105.2.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McSherry I.N., Spitaler M.M., Takano H., Dora K.A. Endothelial cell Ca2+ increases are independent of membrane potential in pressurized rat mesenteric arteries. Cell Calcium. 2005;38:23–33. doi: 10.1016/j.ceca.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 29.Sandow S.L., Grayson T.H. Limits of isolation and culture: intact vascular endothelium and BKCa. Am. J. Physiol. Heart Circ. Physiol. 2009;297:H1–H7. doi: 10.1152/ajpheart.00042.2009. [DOI] [PubMed] [Google Scholar]
- 30.Ma H.T., Patterson R.L., van Rossum D.B., Birnbaumer L., Mikoshiba K., Gill D.L. Requirement of the Inositol Trisphosphate receptor for activation of store-operated Ca2+ channels. Science. 2000;287:1647–1651. doi: 10.1126/science.287.5458.1647. [DOI] [PubMed] [Google Scholar]
- 31.Prakriya M., Lewis R.S. Potentiation and inhibition of Ca(2+) release-activated Ca(2+) channels by 2-aminoethyldiphenyl borate (2-APB) occurs independently of IP(3) receptors. J. Physiol. 2001;536:3–19. doi: 10.1111/j.1469-7793.2001.t01-1-00003.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kumar S., West D.C., Ager A. Heterogeneity in endothelial cells from large vessels and microvessels. Differentiation. 1987;36:57–70. doi: 10.1111/j.1432-0436.1987.tb00181.x. [DOI] [PubMed] [Google Scholar]
- 33.Tracey W.R., Peach M.J. Differential muscarinic receptor mRNA expression by freshly isolated and cultured bovine aortic endothelial cells. Circ. Res. 1992;70:234–240. doi: 10.1161/01.res.70.2.234. [DOI] [PubMed] [Google Scholar]
- 34.Thorin E., Shreeve S.M. Heterogeneity of vascular endothelial cells in normal and disease states. Pharmacol. Ther. 1998;78:155–166. doi: 10.1016/s0163-7258(98)00005-9. [DOI] [PubMed] [Google Scholar]
- 35.Huang T.Y., Chu T.F., Chen H.I., Jen C.J. Heterogeneity of [Ca(2+)](i) signaling in intact rat aortic endothelium. FASEB J. 2000;14:797–804. doi: 10.1096/fasebj.14.5.797. [DOI] [PubMed] [Google Scholar]
- 36.Higgins E.R., Cannell M.B., Sneyd J. A buffering SERCA pump in models of calcium dynamics. Biophys. J. 2006;91:151–163. doi: 10.1529/biophysj.105.075747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yano K., Petersen O.H., Tepikin A.V. Dual sensitivity of sarcoplasmic/endoplasmic Ca2+-ATPase to cytosolic and endoplasmic reticulum Ca2+ as a mechanism of modulating cytosolic Ca2+ oscillations. Biochem. J. 2004;383:353–360. doi: 10.1042/BJ20040629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moccia F., Berra-Romani R., Baruffi S. Ca2+ uptake by the endoplasmic reticulum Ca2+-ATPase in rat microvascular endothelial cells. Biochem. J. 2002;364:235–244. doi: 10.1042/bj3640235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Redondo P.C., Rosado J.A., Pariente J.A., Salido G.M. Collaborative effect of SERCA and PMCA in cytosolic calcium homeostasis in human platelets. J. Physiol. Biochem. 2005;61:507–516. doi: 10.1007/BF03168376. [DOI] [PubMed] [Google Scholar]
- 40.Camacho P., Lechleiter J.D. Increased frequency of calcium waves in Xenopus laevis oocytes that express a calcium-ATPase. Science. 1993;260:226–229. doi: 10.1126/science.8385800. [DOI] [PubMed] [Google Scholar]
- 41.Iino M. Molecular basis of spatio-temporal dynamics in inositol 1,4,5-trisphosphate-mediated Ca2+ signalling. Jpn. J. Pharmacol. 2000;82:15–20. doi: 10.1254/jjp.82.15. [DOI] [PubMed] [Google Scholar]
- 42.Matsumoto M., Nakagawa T., Inoue T. Ataxia and epileptic seizures in mice lacking type 1 inositol 1,4,5-trisphosphate receptor. Nature. 1996;379:168–171. doi: 10.1038/379168a0. [DOI] [PubMed] [Google Scholar]
- 43.Missiaen L., Lemaire F.X., Parys J.B., De S.H., Sienaert I., Casteels R. Initiation sites for Ca2+ signals in endothelial cells. Pflugers Arch. 1996;431:318–324. doi: 10.1007/BF02207268. [DOI] [PubMed] [Google Scholar]
- 44.Searls Y.M., Loganathan R., Smirnova I.V., Stehno-Bittel L. Intracellular Ca2+ regulating proteins in vascular smooth muscle cells are altered with type 1 diabetes due to the direct effects of hyperglycemia. Cardiovasc. Diabetol. 2010;9:8. doi: 10.1186/1475-2840-9-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Woods N.M., Cuthbertson K.S., Cobbold P.H. Repetitive transient rises in cytoplasmic free calcium in hormone-stimulated hepatocytes. Nature. 1986;319:600–602. doi: 10.1038/319600a0. [DOI] [PubMed] [Google Scholar]
- 46.Fewtrell C. Ca2+ oscillations in non-excitable cells. Annu. Rev. Physiol. 1993;55:427–454. doi: 10.1146/annurev.ph.55.030193.002235. [DOI] [PubMed] [Google Scholar]
- 47.Berridge M.J. The AM and FM of calcium signalling. Nature. 1997;386:759–760. doi: 10.1038/386759a0. [DOI] [PubMed] [Google Scholar]
- 48.Shuttleworth T.J. What drives calcium entry during [Ca2+]i oscillations?—challenging the capacitative model. Cell Calcium. 1999;25:237–246. doi: 10.1054/ceca.1999.0022. [DOI] [PubMed] [Google Scholar]
- 49.Kwan C.Y., Takemura H., Obie J.F., Thastrup O., Putney J.W., Jr. Effects of MeCh, thapsigargin, and La3+ on plasmalemmal and intracellular Ca2+ transport in lacrimal acinar cells. Am. J. Physiol. 1990;258:C1006–C1015. doi: 10.1152/ajpcell.1990.258.6.C1006. [DOI] [PubMed] [Google Scholar]
- 50.Chakrabarti R., Chakrabarti R. Calcium signaling in non-excitable cells: Ca2+ release and influx are independent events linked to two plasma membrane Ca2+ entry channels. J. Cell Biochem. 2006;99:1503–1516. doi: 10.1002/jcb.21102. [DOI] [PubMed] [Google Scholar]
- 51.Freichel M., Suh S.H., Pfeifer A. Lack of an endothelial store-operated Ca2+ current impairs agonist-dependent vasorelaxation in TRP4−/− mice. Nat. Cell Biol. 2001;3:121–127. doi: 10.1038/35055019. [DOI] [PubMed] [Google Scholar]
- 52.Yao X., Garland C.J. Recent developments in vascular endothelial cell transient receptor potential channels. Circ. Res. 2005;97:853–863. doi: 10.1161/01.RES.0000187473.85419.3e. [DOI] [PubMed] [Google Scholar]
- 53.Plant T.D., Schaefer M. TRPC4 and TRPC5: receptor-operated Ca2+-permeable nonselective cation channels. Cell Calcium. 2003;33:441–450. doi: 10.1016/s0143-4160(03)00055-1. [DOI] [PubMed] [Google Scholar]
- 54.Clapham D.E. A STIMulus Package puts orai calcium channels to work. Cell. 2009;136:814–816. doi: 10.1016/j.cell.2009.02.022. [DOI] [PubMed] [Google Scholar]
- 55.Vaca L., Kunze D.L. Depletion of intracellular Ca2+ stores activates a Ca(2+)-selective channel in vascular endothelium. Am. J. Physiol. 1994;267:C920–C925. doi: 10.1152/ajpcell.1994.267.4.C920. [DOI] [PubMed] [Google Scholar]
- 56.Fasolato C., Nilius B. Store depletion triggers the calcium release-activated calcium current (ICRAC) in macrovascular endothelial cells: a comparison with Jurkat and embryonic kidney cell lines. Pflugers Arch. 1998;436:69–74. doi: 10.1007/s004240050605. [DOI] [PubMed] [Google Scholar]
- 57.Abdullaev I.F., Bisaillon J.M., Potier M., Gonzalez J.C., Motiani R.K., Trebak M. Stim1 and Orai1 mediate CRAC currents and store-operated calcium entry important for endothelial cell proliferation. Circ. Res. 2008;103:1289–1299. doi: 10.1161/01.RES.0000338496.95579.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Live 3-dimensional image showing EC of intact rat tail artery loaded with Fluo-4 in situ. EC are seen as monolayer of cells lining up elastic lamina.
Real time confocal imaging of Ca2+ signalling in Fluo-4 loaded EC of intact artery stimulated with 1 μM CCh. Images are displayed at a speed of 30 fps.