

Abstract
Fibroblasts are at the heart of cardiac function and are the principal determinants of cardiac fibrosis. Nevertheless, cardiac fibroblasts remain poorly characterized in molecular terms. Evidence is evolving that the cardiac fibroblast is a highly heterogenic cell population, and that such heterogeneity is caused by the distinct origins of fibroblasts in the heart. Cardiac fibroblasts can derive either from resident fibroblasts, from endothelial cells via an endothelial–mesenchynmal transition or from bone marrow-derived circulating progenitor cells, monocytes and fibrocytes. Here, we review the function and origin of fibroblasts in cardiac fibrosis.NB. The information given is correct.
Most cardiac diseases are associated with fibrosis in the heart. Fibrosis, in general, is a scarring process which is characterized by fibroblast accumulation and excess deposition of extracellular matrix (ECM) proteins, which leads to distorted organ architecture and function (Weber, 2000). The development of cardiac fibrosis is similar to fibrosis in other organs, such as the liver, lungs, and the kidney (Weber, 1997). The contribution of fibrogenesis to impaired cardiac function is increasingly recognized (Espira and Czubryt, 2009). The fibrotic ECM causes increased stiffness and induces pathological signaling within cardiomyocytes resulting in progressive cardiac failure. Also, the excessive ECM impairs mechano-electric coupling of cardiomyocytes and increases the risk of arrhythmias (de Bakker et al., 1996; Spach and Boineau, 1997). Fibroblasts are principally responsible for deposition of the excessive fibrotic ECM and activated fibroblasts may directly cause hypertrophy of cardiomyocytes via paracrine mechanisms further contributing to impaired cardiac function (Gray et al., 1998; Jiang et al., 2007).
Fibrosis manifests in two forms, that is, reactive interstitial fibrosis or replacement fibrosis (Anderson et al., 1979; Weber, 1989). In animal models of left ventricular pressure overloading, reactive interstitial fibrosis is observed which progresses without loss of cardiomyocytes. This initial reactive interstitial fibrosis is an adaptive response aimed to preserve the pressure generating capacity of the heart but will progress into a state of replacement fibrosis, characterized by cardiomyocyte hypertrophy and necrosis (Isoyama and Nitta-Komatsubara, 2002). On the other hand, in animal models of acute myocardial infarction, an initial inflammatory reaction is followed exclusively by myocyte death and replacement fibrosis (Hasenfuss, 1998). Although both animal models represent certain stages and mechanisms of human cardiopathy, they also show distinct and non-overlapping fibroblast reactions (Hasenfuss, 1998). Hence, researchers should be cautious when generalizing results obtained by the use of a single animal model and should validate their findings on human tissue samples. These prerequisites have to be met, if we are to unravel the definite contribution of cardiac fibroblasts (CF) to human cardiopathy, which at present remains elusive.
Fibroblasts, and related myofibroblasts, are the principle producers of ECM and contribute significantly to fibrosis in the heart (Eghbali and Weber, 1990; Carver et al., 1993). However, the source of these myofibroblasts is not fully resolved and remains an area of active research (Hinz et al., 2007; Wynn, 2008). Typically, myofibroblasts are thought to be derived through the activation of resident CF. However, this limited view has been challenged by the demonstration of phenotypic heterogeneity among fibroblasts (Chang et al., 2002), not only between organs, but also within the same organ during health and disease (Fries et al., 1994; Jelaska et al., 1999).
So, what exactly is a fibroblast? Fibroblasts are cells of mesenchymal origin that produce a wide variety of matrix proteins and biochemical mediators, such as growth factors and proteases (Souders et al., 2009). Although synthesis and deposition of ECM are key features of fibroblasts, they are not commonly assessed in the identification of fibroblasts. This implies that the characterization of fibroblasts in general relies on morphological, proliferative, and phenotypical characteristics. Morphologically, fibroblasts are flat spindle shaped cells with multiple processes originating from their cell body. In the cardiac tissue, fibroblasts are the only cell type that are not associated with a basement membrane.
Although much research has been performed examining the fibroblast phenotype in various organs, no marker proteins have been identified that are exclusively expressed by fibroblasts (Table 1). However, some discriminative markers exist for organ-specific fibroblast subsets. For example, in the human and mouse cardiac tissue, the collagen-activated receptor tyrosine kinase discoidin domain receptor 2 (DDR2) and the intermediate-filament associated calcium-binding protein S100A4 (or fibroblast-specific protein 1 (FSP-1)) are expressed primarily by fibroblasts in the heart (Camelliti et al., 2005; Banerjee et al., 2007).
TABLE 1. Commonly used fibroblast markers.
| Protein | Function | Expressed by other cell type | Refs. |
|---|---|---|---|
| α-Smooth muscle actin (αSMA) | Intermediate-filament associated protein | Smooth muscle cells, pericytes, myoepithelial cells | Akpolat et al. (2005); Azuma et al. (2009) |
| Cadherin-9 | Ca-dependent adhesion molecule | Neurons; tumor vasculature | Thedieck et al. (2007); Hirano et al. (2003) |
| CD40 | TNFα receptor family member | Various antigen presenting cells | Smith (2004) |
| CD248 (TEM1) | Collagen receptor | Pericytes, endothelial cells | Bagley et al. (2008); MacFadyen et al. (2005) |
| Col1a1 | Collagen type I biosynthesis | Osteoblasts, chondroblasts | Liska et al. (1994) |
| Discoidin domain receptor 2 (DDR2) | Collagen-binding tyrosine kinase receptor | Smooth muscle cells, hepatic stellate cells, endothelial cells | Vogel et al. (2006); Olaso et al. (2001); Mohan et al. (2001) |
| Fibroblast activation protein-1 (FAP1) | Serine protease (gelatinase) | Activated melanocytes | Rettig et al. (1993); Ramirez-Montagut et al. (2004) |
| Fibroblast-specific protein-1 (FSP1/S100A4) | Intermediate-filament associated Ca-binding protein | Smooth muscle cells, invasive carcinoma cells | Strutz et al. (1995); Sugimoto et al. (2006) |
| Fibroblast surface antigen (FSA) | Fibronectin-binding molecule | Monocytes/macrophages | Wartiovaara et al. (1974) |
| Heat shock protein-47 (HSP47) | Collagen-binding serpin chaperone | Monocytes/macrophages, various collagen-producing cells | Shioshita et al. (2000); Sauk et al. (2005) |
| Platelet-derived growth factor receptor-β (PDGFRb) | Receptor tyrosine kinase | Smooth muscle cells, pericytes | Lindahl et al. (1997); Kaur et al. (2009) |
| Prolyl-4-hydroxylase | Collagen biosynthesis | Endothelial cells, epithelial cells | Mussini et al. (1967); Langness and Udenfriend (1974) |
| Thymus cell antigen-1 (THY1/CD90) | Cell adhesion molecule | Leukocytes, endothelial cells, various progenitor cells | Wetzel et al. (2006); Dezso et al. (2007) |
| Vimentin | Intermediate-filament associated protein | Endothelial cells, smooth muscle cells, pericytes, myoepithelial cells | Franke et al. (1979); Mork et al. (1990) |
Ca, calcium; TEM, tumor endothelial marker; TNF, tumor necrosis factor.
The distinction between fibroblasts and myofibroblasts is commonly based solely on the expression of contractile proteins, which might be a principally unreliable determinant. It is important to note that both the fibroblast and the myofibroblasts are in principle motile cells that contain actins and myosins, albeit at dissimilar amounts (Eyden, 2001). Fibroblasts are pleiomorphic cells and it seems natural that their expression and arrangement of contractile proteins depends more on their microenvironment, for example, cytokine milieu and mechanical parameters, than on their differentiation to another distinctive cell type. Hence in this review, fibroblasts and myofibroblasts will not be regarded as separate entities.
It becomes clear that fibroblasts are not a static cell population but rather display a large heterogeneity (Fries et al., 1994; Jelaska et al., 1999; Sugimoto et al., 2006). This heterogeneity may derive from different origins of fibroblast subtypes and may contribute to the cardiac structure and function during health and disease. Better understanding of these fibroblast subtypes, as well as of the factors that regulate their function, provides new insight into the development of cardiac fibrosis and potentially identifies novel therapeutic targets for its treatment. In this review we will address the heterogeneity of CF origin and function during health and disease. Furthermore, we will highlight emerging hypotheses for further research.
The Origin of Cardiac Fibroblasts During Embryonic Development
Although it is generally assumed that non-myocytes represent the largest cell population in the mammalian heart, the actual number of fibroblasts in the cardiac tissue remains unknown. It has been stated that the adult mouse heart contains approximately 55% myocytes and only 45% non-myocyte cells (∼27% fibroblasts). In contrast, the adult rat heart consists of 30% myocyte cells and 70% non-myocyte cells (∼67% fibroblasts) (Vliegen et al., 1991; Banerjee et al., 2007). Although there is no comparable human study, these data show that the cardiac cellular makeup varies greatly between species and one could speculate that these differences in cellular buildup underlie differences in cardiac pressure generating capacity, cardiac collagen content, heart rate, or cardiac conductance (Banerjee et al., 2007).
Because of the lack of a robust CF specific marker (Table 1), the late embryonic development of CF after the gross cardiac morphogenesis, and the variability among current lineage tracking tools, it is still conceivable that there are multiple spatiotemporal sources of CF. Nonetheless, fibroblasts in the cardiac interstitium and the annulus fibrosis are thought to derive principally from mesenchymal cells in the embryonic proepicardium (Norris et al., 2008) (Fig. 1). These cells migrate over the surface of the embryonic heart and form the epicardium, which in turn gives rise to the epicardium-derived cells (EPDC) (Lie-Venema et al., 2007). EPDC in the cardiac wall undergo epithelial–mesenchymal transition (EMT) (Munoz-Chapuli et al., 2001) and progressively differentiate into a fibroblast phenotype (Gittenberger-de Groot et al., 1998; Zhou et al., 2010) under the influence of growth factors, including platelet-derived growth factor (PDGF), fibroblast growth factor (FGF), and transforming growth factor (TGF) (Olivey et al., 2006).
Fig. 1.
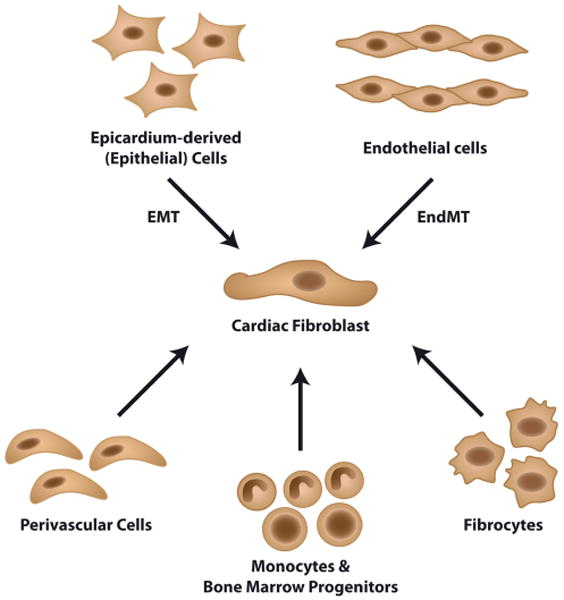
Sources of cardiac fibroblasts. In the developing embryo, cardiac fibroblast originate from the epithelial cells of the proepicardium through a process termed epithelial–mesenchymal transition (EMT). Valvular fibroblast arise through endothelial–mesenchymal transition (EndMT) of the endocardium. The development of fibroblast during cardiopathy involves considerably more cell plasticity, with the pro-fibrotic cells being derived from the endothelium and epithelium through mesenchymal transition (EMT and EndMT). Also fibroblast may be derived from perivascular cells, circulating monocytes and bone marrow-derived progenitor cells and circulating fibrocytes.
In addition to the development of cardiac interstitial fibroblasts, valvular fibroblasts originate from the cardiac endothelium (de Lange et al., 2004). Endothelial cells in the region of the forming cardiac cushion delaminate and undergo endothelial–mesenchymal transformation (EndMT) under the influence of various cytokines such as TGF-β, PDGF, and Wnt. Thereafter, transformed cells invade the cardiac jelly and mature into a fibroblastic phenotype (Armstrong and Bischoff, 2004; de Lange et al., 2004).
In summary, CF are interspersed in the collagen network and the differential expression of motile and contractile proteins observed between fibroblast and related myofibroblasts represents a pleiomorphic continuum. The origin of CF, however, remains to be elucidated more comprehensively.
Pleiotropic Functions of Cardiac Fibroblasts
Although CF are mostly known for their role in the synthesis and degradation of the cardiac ECM, it must be noted that CF are more than matrix producing cells. Fibroblast sense changes in their microenvironment and react to these changes to preserve organ function. As such, CF contribute to the structural, mechanical, biochemical, and electrical properties of the heart.
Homeostasis of the ECM
CF maintain the ECM, which includes the interstitial collagens, proteoglycans, glycoproteins, cytokines, growth factors, and proteases (Corda et al., 2000; Bowers et al., 2010). The ECM serves multiple purposes; (1) it forms an organizational network that surrounds and interconnects cells and provides scaffold for cardiac cell types, (2) it distributes mechanical forces throughout the cardiac tissue and conveys mechanical signals to individual cells, and (3) electrically separates the atria and the ventricles to facilitate proper cardiac contraction.
CF are at the heart of ECM homeostasis because of their ability to secrete and breakdown the proteins that form the ECM (Fig. 2). In response to several growth factors (e.g., TGFβ, PDGF), cytokines (e.g., TNFα, IL1β, IL6) or mechanical stimulation (e.g., stretch), CF produce the fibrillar type collagens I and type III that together comprise approximately 90% of all collagen in the heart, as well as the less abundant ECM molecules collagen types IV, V, VI, elastin, and laminin (Bosman and Stamenkovic, 2003). Alternatively, CF modulate the degradation of ECM breakdown by modulating the expression of the matrix metalloproteinases (MMP) and their natural inhibitors (tissue inhibitor of MMP; TIMP) (Tsuruda et al., 2004).
Fig. 2.
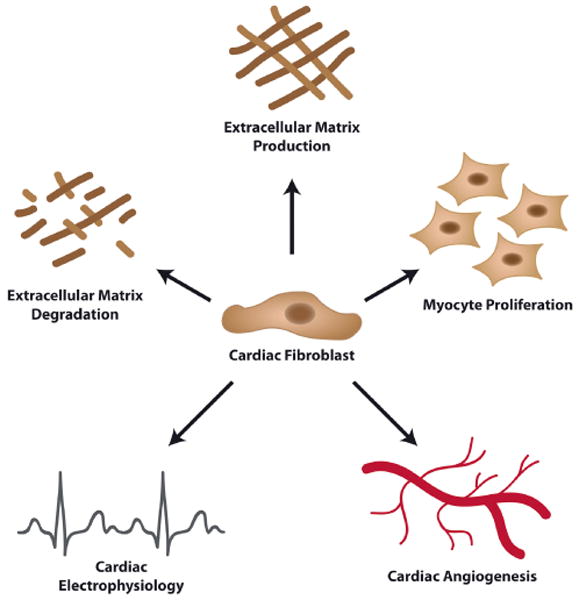
Pleiotropic functions of cardiac fibroblasts. Cardiac fibroblasts are at the heart of cardiac development and function. Cardiac fibroblasts are involved in the production and degradation of the cardiac extracellular matrix through the production of amongst others collagens, proteoglycans, matrix metalloproteinases and TIMPs. Additionally, cardiac fibroblasts secrete various bioactive mediators (e.g., VEGFa, FGFs, TGFβ, PDGF) which influence cardiac angiogenesis and myocyte proliferation. Furthermore, cardiac fibroblasts influence cardiac electrophysiology by insulating myocyte bundles, transmitting electrical signals, and converting mechanical stimuli into electronic signals.
Production of bioactive molecules
A second major function of CF is to produce and secrete growth factors, cytokines, and other signaling molecules. These bioactive molecules can subsequently exert autocrine and paracrine effects on the cardiac cell types, thereby directing cell proliferation, contractions, and apoptosis, amongst others.
Homeostasis of cardiac vessels
Significantly, CF contribute to cardiac vessel homeostasis. Angiogenesis (i.e., the formation of capillaries from pre-existing blood vessels) depends on environmental signals that modulate endothelial cell behavior (Risau, 1997). The interaction between fibroblasts and endothelial cells during vessel formation was already reported several years back (Villaschi and Nicosia, 1994), but remains to be fully resolved. FGFs and vascular endothelial growth factor (VEGF) are potent inducers of angiogenesis produced and secreted by CF (Zhao and Eghbali-Webb, 2001; Chintalgattu et al., 2003). Contrary, the expression and secretion of connective tissue growth factor (CTGF) and PDGF by CF poses an antiangiogenic effect (Inoki et al., 2002; Zhao and Eghbali-Webb, 2001). These data indicate that CF have the ability to either induce, or inhibit the formation of new blood vessels (Fig. 2). However, the exact functions of CF during cardiac vessel formation need further investigation.
Also, CF produce mitogens that play a major role in myocyte development and maintenance of the adult phenotype. As such, CF may orchestrate the proliferation of cardiomyocytes directly. Although cardiomyocytes were thought to be terminally differentiated and incapable of proliferation, recent data indicate that adult cardiomyocytes in fact proliferate slowly, which may be an essential part of cardiac homeostasis and turnover (Bergmann et al., 2009). CF produce factors such as FGF and periostin which were shown to induce adult myocyte proliferation in vitro as well as in vivo (Engel et al., 2005; Kuhn et al., 2007). Hence, it is tempting to speculate that CF modulate the turnover of myocytes in a paracrine manner (Fig. 2).
Cardiac electrophysiology
CF are non-excitable cells, however CF do contribute to cardiac electrophysiology (Fig. 2). Passively, CF can obstruct the orderly spread of electrical stimuli by producing an insulating layer of ECM which physically separates groups of myocytes. This can clearly be observed in the annulus fibrosis, which electrically separates the atria and ventricles and thus allows for the sequential contraction of these structures which is needed for proper cardiac function (Zhou et al., 2010).
The possibility that CF may also contribute actively to cardiac electrophysiology has emerged only recently. CF have a high cell membrane resistance, which makes them good conductors for electrical signals (Kohl, 2003). Also, CF are coupled to myocytes through connexins-43 and connexin-45 (Kohl, 2003; Chilton et al., 2007) and recent in vitro evidence shows that this coupling of myocytes and fibroblasts allows for electrical signal transduction (Gaudesius et al., 2003; Miragoli et al., 2006). These data suggest that CF can form bridges that link regions of myocytes that would normally be separated by an insulating layer of ECM. Also, interconnectivity between myocytes and fibroblasts may provide synchronization of spontaneous activity in distant cardiac myocytes (Rohr, 2004). However, the in vivo relevance of active CF contribution to electrical signaling needs further elucidation.
Another active contribution of CF to the cardiac electrophysiology may be found in their mechano-sensitivity. CF express multiple stretch-activated ion channels that are permeable to Na+, K+, and Ca+ (Hu and Sachs, 1997; Li et al., 2009). In response to mechanical stimuli, imposed by the contractile activity of the surrounding myocardium, these ion channels open and lower fibroblast membrane potential making the fibroblast an efficient mechano-electrical transducer (Isenberg et al., 2003; Kamkin et al., 2003). The physiological relevance of this mechano-electrical transduction, however, needs further investigation.
In summary, the CF is a multi-functional cell that is at the heart of cardiac development and function (Fig. 2). The CF not only provides the cardiac scaffold for all the cardiac cell types, it also orchestrates myocyte growth and cardiac vessel formation. Furthermore, CF are actively involved in cardiac electrophysiology, allowing for proper functioning of the heart.
The Cardiac Fibroblast in Cardiopathy
In general, net deposition of collagen in the healthy heart is limited. However during cardiopathy, collagen deposition is dramatically increased in response to injury, which results in distorted organ architecture and function. Although collagen-producing fibroblasts undergo apoptosis and leave a mature scar composed of cross-linked collagen and other matrix components during wound healing (Gurtner et al., 2008), following cardiopathy CF overcome this regulatory mechanism and cardiac fibrosis often becomes a persistent process which progresses into cardiac failure over time. To date, it is unclear why CF seem insensitive to this regulatory mechanism and persist in the heart.
Fibrosis of the cardiac tissue has significant consequences on cardiac function. Increased EMC synthesis and decreased degradation result in increased mechanical stiffness and diastolic dysfunction (Chaturvedi et al., 2010). Moreover, increased ECM deposition between layers of cardiomyocytes may disrupt their electrical coupling, leading to impaired cardiac contraction (Spach and Boineau, 1997). Furthermore, inflammation and fibrosis in the perivascular areas may decrease the flow of oxygen and nutrients and increase the pathological remodeling response (Kai et al., 2006).
As previously mentioned, the phenotypes of the fibrous tissue can be divided into two distinct types: (1) replacement fibrosis, which occurs throughout the myocardium and is associated with loss of cardiomyocyte mass and (2) reactive interstitial fibrosis, which originates from areas surrounding the microvasculature and spreads throughout the myocardium (Anderson et al., 1979; Weber, 1989). Since both types of fibrosis originate at distinct sites in the cardiac tissue, it is tempting to speculate that fibroblasts of distinct origins are involved in the development of these cardiopathies. The origin of proliferating collagen-producing fibroblasts in the heart during cardiopathy is currently an area of active research.
Heterogeneous Origins of (Myo)Fibroblasts During Cardiopathy
The resident cardiac fibroblast and its progenitor cells
The traditional view is that activated fibroblasts in fibrotic hearts derive from resident fibroblasts through proliferation and activation. Such belief is based on the observation that cardiac fibroblasts are sensitive to circulating signaling molecules that may affect their proliferative response towards a pathologic stimulus (Fredj et al., 2005; Lucas et al., 2010). This response could, theoretically, take place at any injury site in the cardiac tissue, making resident fibroblasts a highly feasible source of matrix-producing cells during replacement fibrosis. However, investigations that tracked proliferating cell populations during cardiac hypertrophy (i.e., during reactive interstitial fibrosis) showed only proliferating fibroblast-like cells in the vicinity of the blood vessels (Ljungqvist and Unge, 1973; Mandache et al., 1973). These data suggest that proliferating pro-fibrotic cells in this context may not be derived from cardiac resident fibroblasts, but are actively recruited from other cellular sources during reactive interstitial fibrosis.
Endothelial–mesenchymal transition
In experimental models of cardiac fibrosis, about 30% of activated fibroblasts are generated from endothelial cells via a cellular transition which is referred to as endothelial–mesenchymal transition (EndMT) (Zeisberg et al., 2007). Endothelial cells, under pressure of pro-fibrotic stimuli (e.g., TGFβ and hypoxia) respond by acquiring a fibroblast-like phenotype while losing characteristics of endothelial cells (Krenning et al., 2008; Moonen et al., 2010). Upon phenotypic conversion these cells leave the microvascular bed and enter the interstitium where they appear as fibroblasts. Hence, EndMT contributes to cardiac fibrosis both by contributing to fibroblast accumulation and also to microvascular rarefication (Zeisberg et al., 2007).
The concept of EndMT emerged from fate mapping studies which demonstrated that up to 30% of fibroblasts originate from endothelial cells. Interestingly, none of the genetic fate mapping studies which utilized different endothelial-specific reporter genes (i.e., Tie1, Flk1, VE-Cadherin) showed significant endothelial contribution to the fibroblast population in the normal heart (Kisanuki et al., 2001; Alva et al., 2006; Lugus et al., 2009), but only in the damaged myocardium (Zeisberg et al., 2007), indicating distinct origins of fibroblasts under physiological and pathological conditions.
Recent studies suggested that epicardial epithelium may similarly contributes to accumulation of fibroblasts by undergoing an EMT (van Tuyn et al., 2007; Zhou et al., 2010). Both EndMT and EMT are considered to follow similar pathways and are considered closely related cellular events. During development, EndMT contributes to formation of the AV-canal (Arciniegas et al., 2005; Niessen et al., 2008) and EMT generates mesenchyme of the annulus fibrosis (Zhou et al., 2010), hence both events are considered remnants of persisting embryonic pathways which can be activated in adult cardiopathy. Both EndMT and EMT are considered economical means of the body to recruit substantial numbers of fibroblasts to perivascular and subendocardial areas during injury as opposed to recruitment and proliferation of distant fibroblasts.
Perivascular cells
Another fibroblast cell source may lie in the perivascular space of the cardiac vessels. Pericytes have been shown to differentiate into collagen-producing cells in models of dermal scarring (Sundberg et al., 1996). Furthermore, retinal pericytes were shown to display a large phenotypical and functional overlap with fibroblast in vitro (Covas et al., 2008) and recent lineage tracing studies in the kidney revealed that CD73+ pericytes were a source of fibroblast in the injured kidney (Humphreys et al., 2010). However, in the absence of definite pericyte and fibroblast markers (Table 1) and of specific fate mapping techniques, the discussion on pericyte contribution to cardiac fibrosis remains active.
Circulating bone marrow-derived progenitor cells, monocytes and fibrocytes
Bone marrow-derived progenitor cells are considered another substantial source of fibroblasts in the fibrotic heart (Fig. 1). Such thinking is based on studies in which green fluorescent protein (GFP)-expressing cells were found in the fibrotic cardiac tissue of mice that received a transplant with GFP-expressing bone marrow cells prior to myocardial infarction (van Amerongen et al., 2007a; Kania et al., 2009) or aortic banding (Zeisberg et al., 2007). Although it is reported that these bone marrow-derived fibroblasts can represent 25–60% of all fibroblasts at the site of cardiac injury, it is unlikely that these fibroblasts contribute significantly to the formation of a persistent fibrotic reaction, since their number is highly reduced 14 days post-myocardial infarction (van Amerongen et al., 2007a). Furthermore, there is a debate if the bone marrow-derived cells are fibroblasts or present a specific phenotype of inflammatory cells.
Monocytes (Fig. 1) have also been suggested as potential source of pathology-associated fibroblasts. Invading fibroblast-like cells in the infarcted cardiac tissue were found to co-express monocytic (CD45; CD11b) and myofibroblast markers (S100A4, αSMA) (Haudek et al., 2006). Moreover, inhibition of monocyte recruitment diminished the CF population and myocardial remodeling following myocardial infarction (van Amerongen et al., 2007b).
Fibrocytes represent a unique fibroblast progenitor population in the circulation that co-express markers of the mesenchyme and hematopoietic system (CD45, CD34, procollagen 1, vimentin) (Abe et al., 2001). Circulating fibrocytes originate from the hematopoietic stem cells in the bone marrow and display phenotypic similarities to other leukocytes such as CD14 expressed by monocytes (Ogawa et al., 2006). However, if this phenotypic overlap represent common descent (Niedermeier et al., 2009) or represents functional overlap remains unclear.
Taken together, the origin of fibroblasts during cardiopathy is currently not established beyond doubt. Following cardiopathy, activated fibroblasts are found in the cardiac tissue which originate in the endothelium, bone marrow, circulation or perivascular spaces. The relative contribution of each of these compartments to the collagen-producing proliferating fibroblasts remains elusive, as is the cellular function of these fibroblast from different origins.
Conclusions
Recent investigations have yielded remarkable insights into the development of the cardiac tissue. Although CF have long been regarded as a uniform and static cell population, recent evidence has revealed it as a complex and diverse cell population with multiple origins and functions. Understanding how the complex process of CF differentiation and function is regulated during health and cardiopathy are hampered by the lack of suitable fibroblast markers and appropriate lineage mapping tools. Hence, processes toward understanding the molecular, cellular, and morphological events required to make the cardiac interstitium function continue to be a challenge for investigators (see also Box 1).
Open issues on cardiac fibroblast biology.
What is the number of cardiac fibroblasts in the developing and adult mammalian heart?
Do differences in pressure generating capacity underlie differences in cellular buildup of the cardiac tissue?
Are there multiple subtypes of cardiac fibroblast progenitors involved in the generation of the adult mammalian heart?
Do different subtypes of fibroblast have distinct physiological functions in the mammalian heart?
Why do (myo)fibroblasts in the mammalian heart persist in the normal heart and not in other normal tissues?
Are distinct fibroblast subtypes involved in the different fibrosis processes (i.e., reactive interstitial and reparative fibrosis)?
What is the contribution of the cardiac epithelium to the pool of pro-fibrotic fibroblast during cardiopathy?
A challenge facing cardiac scientists alike is the characterization and isolation of the key progenitor cells of the CF lineage. The identification of unique cell surface markers of CF and their progenitors will be invaluable in these investigations and a prerequisite for understanding CF heterogeneity. Current results show that the CF is a pleiotropic cell that is involved in the vast majority of cardiac functions. Moreover, evidence is emerging that fibroblasts of different origin show distinct and overlapping functions during cardiac physiology and pathology. Better understanding of the CF biology in general would also allow for the study of CF function during cardiopathy and may shed insights essential for the development of novel therapies.
Acknowledgments
GK is supported by a research stipend from the Niels Stensen Foundation, The Netherlands (NSF). EMZ is supported by a Scientist Development Grant (SDG0735602T) from the American Heart Association and the Mentored Clinical Scientist Development Award (1K08 CA129204) from the National Institute of Health. RK is supported by grants DK 55001 and CA 125550 from the National Institute of Health, and the research funds from the Division of Matrix Biology at the Beth Israel Deaconess Medical Center.
Contract grant sponsor: National Institute of Health;
Contract grant numbers: DK 55001, CA 125550.
Contract grant sponsor: Division of Matrix Biology at the Beth Israel Deaconess Medical Center.
Contract grant sponsor: National Institute of Health;
Contract grant number: 1K08 CA129204.
Contract grant sponsor: American Heart Association and the Mentored Clinical Scientist Development Award;
Contract grant number: SDG0735602T.
Contract grant sponsor: Niels Stensen Foundation (NSF), the Netherlands.
Literature cited
- Abe R, Donnelly SC, Peng T, Bucala R, Metz CN. Peripheral blood fibrocytes: Differentiation pathway and migration to wound sites. J Immunol. 2001;166:7556–7562. doi: 10.4049/jimmunol.166.12.7556. [DOI] [PubMed] [Google Scholar]
- Akpolat N, Yahsi S, Godekmerdan A, Yalniz M, Demirbag K. The value of alpha-SMA in the evaluation of hepatic fibrosis severity in hepatitis B infection and cirrhosis development: A histopathological and immunohistochemical study. Histopathology. 2005;47:276–280. doi: 10.1111/j.1365-2559.2005.02226.x. [DOI] [PubMed] [Google Scholar]
- Alva JA, Zovein AC, Monvoisin A, Murphy T, Salazar A, Harvey NL, Carmeliet P, Iruela-Arispe ML. VE-Cadherin-Cre-recombinase transgenic mouse: A tool for lineage analysis and gene deletion in endothelial cells. Dev Dyn. 2006;235:759–767. doi: 10.1002/dvdy.20643. [DOI] [PubMed] [Google Scholar]
- Anderson KR, Sutton MG, Lie JT. Histopathological types of cardiac fibrosis in myocardial disease. J Pathol. 1979;128:79–85. doi: 10.1002/path.1711280205. [DOI] [PubMed] [Google Scholar]
- Arciniegas E, Neves CY, Carrillo LM, Zambrano EA, Rameirez R. endothelial–mesenchymal transition occurs during embryonic pulmonary artery development. Endothelium. 2005;12:193–200. doi: 10.1080/10623320500227283. [DOI] [PubMed] [Google Scholar]
- Armstrong EJ, Bischoff J. Heart valve development: Endothelial cell signaling and differentiation. Circ Res. 2004;95:459–470. doi: 10.1161/01.RES.0000141146.95728.da. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azuma K, Ichimura K, Mita T, Nakayama S, Jin WL, Hirose T, Fujitani Y, Sumiyoshi K, Shimada K, Daida H, Sakai T, Mitsumata M, Kawamori R, Watada H. Presence of alpha-smooth muscle actin-positive endothelial cells in the luminal surface of adult aorta. Biochem Biophys Res Commun. 2009;380:620–626. doi: 10.1016/j.bbrc.2009.01.135. [DOI] [PubMed] [Google Scholar]
- Bagley RG, Honma N, Weber W, Boutin P, Rouleau C, Shankara S, Kataoka S, Ishida I, Roberts BL, Teicher BA. Endosialin/TEM 1/CD248 is a pericyte marker of embryonic and tumor neovascularization. Microvasc Res. 2008;76:180–188. doi: 10.1016/j.mvr.2008.07.008. [DOI] [PubMed] [Google Scholar]
- Banerjee I, Fuseler JW, Price RL, Borg TK, Baudino TA. Determination of cell types and numbers during cardiac development in the neonatal and adult rat and mouse. Am J Physiol Heart Circ Physiol. 2007;293:H1883–H1891. doi: 10.1152/ajpheart.00514.2007. [DOI] [PubMed] [Google Scholar]
- Bergmann O, Bhardwaj RD, Bernard S, Zdunek S, Barnabe-Heider F, Walsh S, Zupicich J, Alkass K, Buchholz BA, Druid H, Jovinge S, Frisen J. Evidence for cardiomyocyte renewal in humans. Science. 2009;324:98–102. doi: 10.1126/science.1164680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosman FT, Stamenkovic I. Functional structure and composition of the extracellular matrix. J Pathol. 2003;200:423–428. doi: 10.1002/path.1437. [DOI] [PubMed] [Google Scholar]
- Bowers SL, Banerjee I, Baudino TA. The extracellular matrix: At the center of it all. J Mol Cell Cardiol. 2010;48:474–482. doi: 10.1016/j.yjmcc.2009.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camelliti P, Borg TK, Kohl P. Structural and functional characterisation of cardiac fibroblasts. Cardiovasc Res. 2005;65:40–51. doi: 10.1016/j.cardiores.2004.08.020. [DOI] [PubMed] [Google Scholar]
- Carver W, Terracio L, Borg TK. Expression and accumulation of interstitial collagen in the neonatal rat heart. Anat Rec. 1993;236:511–520. doi: 10.1002/ar.1092360311. [DOI] [PubMed] [Google Scholar]
- Chang HY, Chi JT, Dudoit S, Bondre C, van de RM, Botstein D, Brown PO. Diversity, topographic differentiation, and positional memory in human fibroblasts. Proc Natl Acad Sci USA. 2002;99:12877–12882. doi: 10.1073/pnas.162488599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaturvedi RR, Herron T, Simmons R, Shore D, Kumar P, Sethia B, Chua F, Vassiliadis E, Kentish JC. Passive stiffness of myocardium from congenital heart disease and implications for diastole. Circulation. 2010;121:979–988. doi: 10.1161/CIRCULATIONAHA.109.850677. [DOI] [PubMed] [Google Scholar]
- Chilton L, Giles WR, Smith GL. Evidence of intercellular coupling between co-cultured adult rabbit ventricular myocytes and myofibroblasts. J Physiol. 2007;583:225–236. doi: 10.1113/jphysiol.2007.135038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chintalgattu V, Nair DM, Katwa LC. Cardiac myofibroblasts: A novel source of vascular endothelial growth factor (VEGF) and its receptors Flt-1 and KDR. J Mol Cell Cardiol. 2003;35:277–286. doi: 10.1016/s0022-2828(03)00006-3. [DOI] [PubMed] [Google Scholar]
- Corda S, Samuel JL, Rappaport L. Extracellular matrix and growth factors during heart growth. Heart Fail Rev. 2000;5:119–130. doi: 10.1023/A:1009806403194. [DOI] [PubMed] [Google Scholar]
- Covas DT, Panepucci RA, Fontes AM, Silva WA, Jr, Orellana MD, Freitas MC, Neder L, Santos AR, Peres LC, Jamur MC, Zago MA. Multipotent mesenchymal stromal cells obtained from diverse human tissues share functional properties and gene-expression profile with CD146+ perivascular cells and fibroblasts. Exp Hematol. 2008;36:642–654. doi: 10.1016/j.exphem.2007.12.015. [DOI] [PubMed] [Google Scholar]
- de Bakker JM, van Capelle FJ, Janse MJ, Tasseron S, Vermeulen JT, de Jonge N, Lahpor JR. Fractionated electrograms in dilated cardiomyopathy: Origin and relation to abnormal conduction. J Am Coll Cardiol. 1996;27:1071–1078. doi: 10.1016/0735-1097(95)00612-5. [DOI] [PubMed] [Google Scholar]
- de Lange FJ, Moorman AF, Anderson RH, Manner J, Soufan AT, de Gier-de VC, Schneider MD, Webb S, van den Hoff MJ, Christoffels VM. Lineage and morphogenetic analysis of the cardiac valves. Circ Res. 2004;95:645–654. doi: 10.1161/01.RES.0000141429.13560.cb. [DOI] [PubMed] [Google Scholar]
- Dezso K, Jelnes P, Laszlo V, Baghy K, Bodor C, Paku S, Tygstrup N, Bisgaard HC, Nagy P. Thy-1 is expressed in hepatic myofibroblasts and not oval cells in stem cell-mediated liver regeneration. Am J Pathol. 2007;171:1529–1537. doi: 10.2353/ajpath.2007.070273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eghbali M, Weber KT. Collagen and the myocardium: Fibrillar structure, biosynthesis and degradation in relation to hypertrophy and its regression. Mol Cell Biochem. 1990;96:1–14. doi: 10.1007/BF00228448. [DOI] [PubMed] [Google Scholar]
- Engel FB, Schebesta M, Duong MT, Lu G, Ren S, Madwed JB, Jiang H, Wang Y, Keating MT. p38 MAP kinase inhibition enables proliferation of adult mammalian cardiomyocytes. Genes Dev. 2005;19:1175–1187. doi: 10.1101/gad.1306705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espira L, Czubryt MP. Emerging concepts in cardiac matrix biology. Can J Physiol Pharmacol. 2009;87:996–1008. doi: 10.1139/Y09-105. [DOI] [PubMed] [Google Scholar]
- Eyden B. The myofibroblast: An assessment of controversial issues and a definition useful in diagnosis and research. Ultrastruct Pathol. 2001;25:39–50. doi: 10.1080/019131201300004672. [DOI] [PubMed] [Google Scholar]
- Franke WW, Schmid E, Osborn M, Weber K. Intermediate-sized filaments of human endothelial cells. J Cell Biol. 1979;81:570–580. doi: 10.1083/jcb.81.3.570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredj S, Bescond J, Louault C, Potreau D. Interactions between cardiac cells enhance cardiomyocyte hypertrophy and increase fibroblast proliferation. J Cell Physiol. 2005;202:891–899. doi: 10.1002/jcp.20197. [DOI] [PubMed] [Google Scholar]
- Fries KM, Blieden T, Looney RJ, Sempowski GD, Silvera MR, Willis RA, Phipps RP. Evidence of fibroblast heterogeneity and the role of fibroblast subpopulations in fibrosis. Clin Immunol Immunopathol. 1994;72:283–292. doi: 10.1006/clin.1994.1144. [DOI] [PubMed] [Google Scholar]
- Gaudesius G, Miragoli M, Thomas SP, Rohr S. Coupling of cardiac electrical activity over extended distances by fibroblasts of cardiac origin. Circ Res. 2003;93:421–428. doi: 10.1161/01.RES.0000089258.40661.0C. [DOI] [PubMed] [Google Scholar]
- Gittenberger-de Groot AC, Vrancken Peeters MP, Mentink MM, Gourdie RG, Poelmann RE. Epicardium-derived cells contribute a novel population to the myocardial wall and the atrioventricular cushions. Circ Res. 1998;82:1043–1052. doi: 10.1161/01.res.82.10.1043. [DOI] [PubMed] [Google Scholar]
- Gray MO, Long CS, Kalinyak JE, Li HT, Karliner JS. Angiotensin II stimulates cardiac myocyte hypertrophy via paracrine release of TGF-β1 and endothelin-1 from fibroblasts. Cardiovasc Res. 1998;40:352–363. doi: 10.1016/s0008-6363(98)00121-7. [DOI] [PubMed] [Google Scholar]
- Gurtner GC, Werner S, Barrandon Y, Longaker MT. Wound repair and regeneration. Nature. 2008;453:314–321. doi: 10.1038/nature07039. [DOI] [PubMed] [Google Scholar]
- Hasenfuss G. Animal models of human cardiovascular disease, heart failure and hypertrophy. Cardiovasc Res. 1998;39:60–76. doi: 10.1016/s0008-6363(98)00110-2. [DOI] [PubMed] [Google Scholar]
- Haudek SB, Xia Y, Huebener P, Lee JM, Carlson S, Crawford JR, Pilling D, Gomer RH, Trial J, Frangogiannis NG, Entman ML. Bone marrow-derived fibroblast precursors mediate ischemic cardiomyopathy in mice. Proc Natl Acad Sci USA. 2006;103:18284–18289. doi: 10.1073/pnas.0608799103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinz B, Phan SH, Thannickal VJ, Galli A, Bochaton-Piallat ML, Gabbiani G. The myofibroblast: One function, multiple origins. Am J Pathol. 2007;170:1807–1816. doi: 10.2353/ajpath.2007.070112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano S, Suzuki ST, Redies C. The cadherin superfamily in neural development: Diversity, function and interaction with other molecules. Front Biosci. 2003;8:d306–d355. doi: 10.2741/972. [DOI] [PubMed] [Google Scholar]
- Hu H, Sachs F. Stretch-activated ion channels in the heart. J Mol Cell Cardiol. 1997;29:1511–1523. doi: 10.1006/jmcc.1997.0392. [DOI] [PubMed] [Google Scholar]
- Humphreys BD, Lin SL, Kobayashi A, Hudson TE, Nowlin BT, Bonventre JV, Valerius MT, McMahon AP, Duffield JS. Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am J Pathol. 2010;176:85–97. doi: 10.2353/ajpath.2010.090517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoki I, Shiomi T, Hashimoto G, Enomoto H, Nakamura H, Makino Ki, Ikeda E, Takata S, Kobayashi Ki, Okada Y. Connective tissue growth factor binds vascular endothelial growth factor (VEGF) and inhibits VEGF-induced angiogenesis. FASEB J. 2002;16:219–221. doi: 10.1096/fj.01-0332fje. [DOI] [PubMed] [Google Scholar]
- Isenberg G, Kazanski V, Kondratev D, Gallitelli MF, Kiseleva I, Kamkin A. Differential effects of stretch and compression on membrane currents and [Na+]c in ventricular myocytes. Prog Biophys Mol Biol. 2003;82:43–56. doi: 10.1016/s0079-6107(03)00004-x. [DOI] [PubMed] [Google Scholar]
- Isoyama S, Nitta-Komatsubara Y. Acute and chronic adaptation to hemodynamic overload and ischemia in the aged heart. Heart Fail Rev. 2002;7:63–69. doi: 10.1023/a:1013701923065. [DOI] [PubMed] [Google Scholar]
- Jelaska A, Strehlow D, Korn JH. Fibroblast heterogeneity in physiological conditions and fibrotic disease. Springer Semin Immunopathol. 1999;21:385–395. [PubMed] [Google Scholar]
- Jiang ZS, Jeyaraman M, Wen GB, Fandrich RR, Dixon IMC, Cattini PA, Kardami E. High- but not low-molecular weight FGF-2 causes cardiac hypertrophy in vivo; possible involvement of cardiotrophin-1. J Mol Cell Cardiol. 2007;42:222–233. doi: 10.1016/j.yjmcc.2006.09.002. [DOI] [PubMed] [Google Scholar]
- Kai H, Mori T, Tokuda K, Takayama N, Tahara N, Takemiya K, Kudo H, Sugi Y, Fukui D, Yasukawa H, Kuwahara F, Imaizumi T. Pressure overload-induced transient oxidative stress mediates perivascular inflammation and cardiac fibrosis through angiotensin II. Hypertens Res. 2006;29:711–718. doi: 10.1291/hypres.29.711. [DOI] [PubMed] [Google Scholar]
- Kamkin A, Kiseleva I, Isenberg G, Wagner KD, Gunther J, Theres H, Scholz H. Cardiac fibroblasts and the mechano-electric feedback mechanism in healthy and diseased hearts. Prog Biophys Mol Biol. 2003;82:111–120. doi: 10.1016/s0079-6107(03)00009-9. [DOI] [PubMed] [Google Scholar]
- Kania G, Blyszczuk P, Stein S, Valaperti A, Germano D, Dirnhofer S, Hunziker L, Matter CM, Eriksson U. Heart-infiltrating prominin-1+/CD133+ progenitor cells represent the cellular source of transforming growth factor beta-mediated cardiac fibrosis in experimental autoimmune myocarditis. Circ Res. 2009;105:462–470. doi: 10.1161/CIRCRESAHA.109.196287. [DOI] [PubMed] [Google Scholar]
- Kaur H, Chaurasia SS, de Medeiros FW, Agrawal V, Salomao MQ, Singh N, Ambati BK, Wilson SE. Corneal stroma PDGF blockade and myofibroblast development. Exp Eye Res. 2009;88:960–965. doi: 10.1016/j.exer.2008.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kisanuki YY, Hammer RE, Miyazaki J, Williams SC, Richardson JA, Yanagisawa M. Tie2-Cre transgenic mice: A new model for endothelial cell-lineage analysis in vivo. Dev Biol. 2001;230:230–242. doi: 10.1006/dbio.2000.0106. [DOI] [PubMed] [Google Scholar]
- Kohl P. Heterogeneous cell coupling in the heart: An electrophysiological role for fibroblasts. Circ Res. 2003;93:381–383. doi: 10.1161/01.RES.0000091364.90121.0C. [DOI] [PubMed] [Google Scholar]
- Krenning G, Moonen JRAJ, van Luyn MJA, Harmsen MC. Vascular smooth muscle cells for use in vascular tissue engineering obtained by endothelial-to-mesenchymal transdifferentiation (EnMT) on collagen matrices. Biomaterials. 2008;29:3703–3711. doi: 10.1016/j.biomaterials.2008.05.034. [DOI] [PubMed] [Google Scholar]
- Kuhn B, del Monte F, Hajjar RJ, Chang YS, Lebeche D, Arab S, Keating MT. Periostin induces proliferation of differentiated cardiomyocytes and promotes cardiac repair. Nat Med. 2007;13:962–969. doi: 10.1038/nm1619. [DOI] [PubMed] [Google Scholar]
- Langness U, Udenfriend S. Collagen biosynthesis in nonfibroblastic cell lines. Proc Natl Acad Sci USA. 1974;71:50–51. doi: 10.1073/pnas.71.1.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li GR, Sun HY, Chen JB, Zhou Y, Tse HF, Lau CP. Characterization of multiple ion channels in cultured human cardiac fibroblasts. PLoS ONE. 2009;4:e7307. doi: 10.1371/journal.pone.0007307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lie-Venema H, van den Akker NM, Bax NA, Winter EM, Maas S, Kekarainen T, Hoeben RC, DeRuiter MC, Poelmann RE, Gittenberger-de Groot AC. Origin, fate, and function of epicardium-derived cells (EPDCs) in normal and abnormal cardiac development. ScientificWorldJournal. 2007;7:1777–1798. doi: 10.1100/tsw.2007.294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindahl P, Johansson BR, Leveen P, Betsholtz C. Pericyte loss and microaneurysm formation in PDGF-B-deficient mice. Science. 1997;277:242–245. doi: 10.1126/science.277.5323.242. [DOI] [PubMed] [Google Scholar]
- Liska DJ, Reed MJ, Sage EH, Bornstein P. Cell-specific expression of alpha 1 (I) collagen-hGH minigenes in transgenic mice. J Cell Biol. 1994;125:695–704. doi: 10.1083/jcb.125.3.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ljungqvist A, Unge G. The proliferative activity of the myocardial tissue in various forms of experimental cardiac hypertrophy. Acta Pathol Microbiol Scand A. 1973;81:233–240. doi: 10.1111/j.1699-0463.1973.tb03530.x. [DOI] [PubMed] [Google Scholar]
- Lucas JA, Zhang Y, Li P, Gong K, Miller AP, Hassan E, Hage F, Xing D, Wells B, Oparil S, Chen YF. Inhibition of transforming growth factor-beta signaling induces left ventricular dilation and dysfunction in the pressure-overloaded heart. Am J Physiol Heart Circ Physiol. 2010;298:H424–H432. doi: 10.1152/ajpheart.00529.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lugus JJ, Park C, Ma YD, Choi K. Both primitive and definitive blood cells are derived from Flk-1+ mesoderm. Blood. 2009;113:563–566. doi: 10.1182/blood-2008-06-162750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacFadyen JR, Haworth O, Roberston D, Hardie D, Webster MT, Morris HR, Panico M, Sutton-Smith M, Dell A, van der GP, Wienke D, Buckley CD, Isacke CM. Endosialin (TEM1, CD248) is a marker of stromal fibroblasts and is not selectively expressed on tumour endothelium. FEBS Lett. 2005;579:2569–2575. doi: 10.1016/j.febslet.2005.03.071. [DOI] [PubMed] [Google Scholar]
- Mandache E, Unge G, Appelgren LE, Ljungqvist A. The proliferative activity of the heart tissues in various forms of experimental cardiac hypertrophy studied by electron microscope autoradiography. Virchows Arch B Cell Pathol. 1973;12:112–122. doi: 10.1007/BF02893991. [DOI] [PubMed] [Google Scholar]
- Miragoli M, Gaudesius G, Rohr S. Electronic modulation of cardiac impulse conduction by myofibroblasts. Circ Res. 2006;98:801–810. doi: 10.1161/01.RES.0000214537.44195.a3. [DOI] [PubMed] [Google Scholar]
- Mohan RR, Mohan RR, Wilson SE. Discoidin domain receptor (DDR) 1 and 2: Collagen-activated tyrosine kinase receptors in the cornea. Exp Eye Res. 2001;72:87–92. doi: 10.1006/exer.2000.0932. [DOI] [PubMed] [Google Scholar]
- Moonen JRAJ, Krenning G, Brinker MGL, Koerts JA, van Luyn MJA, Harmsen MC. Endothelial progenitor cells give rise to pro-angiogenic smooth muscle-like progeny. Cardiovasc Res. 2010;86:506–515. doi: 10.1093/cvr/cvq012. [DOI] [PubMed] [Google Scholar]
- Mork C, van Deurs B, Petersen OW. Regulation of vimentin expression in cultured human mammary epithelial cells. Differentiation. 1990;43:146–156. doi: 10.1111/j.1432-0436.1990.tb00441.x. [DOI] [PubMed] [Google Scholar]
- Munoz-Chapuli R, Perez-Pomares JM, Macias D, Garcia-Garrido L, Carmona R, Gonzalez-Iriarte M. The epicardium as a source of mesenchyme for the developing heart. Ital J Anat Embryol. 2001;106:187–196. [PubMed] [Google Scholar]
- Mussini E, Hutton JJ, Jr, Udenfriend S. Collagen proline hydroxylase in wound healing, granuloma formation, scurvy, and growth. Science. 1967;157:927–929. doi: 10.1126/science.157.3791.927. [DOI] [PubMed] [Google Scholar]
- Niedermeier M, Reich B, Rodriguez GM, Denzel A, Schmidbauer K, Gobel N, Talke Y, Schweda F, Mack M. CD4+ T cells control the differentiation of Gr1+ monocytes into fibrocytes. Proc Natl Acad Sci USA. 2009;106:17892–17897. doi: 10.1073/pnas.0906070106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niessen K, Fu Y, Chang L, Hoodless PA, McFadden D, Karsan A. Slug is a direct Notch target required for initiation of cardiac cushion cellularization. J Cell Biol. 2008;182:315–325. doi: 10.1083/jcb.200710067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris RA, Borg TK, Butcher JT, Baudino TA, Banerjee I, Markwald RR. Neonatal and adult cardiovascular pathophysiological remodeling and repair: Developmental role of periostin. Ann N Y Acad Sci. 2008;1123:30–40. doi: 10.1196/annals.1420.005. [DOI] [PubMed] [Google Scholar]
- Ogawa M, LaRue AC, Drake CJ. Hematopoietic origin of fibroblasts/myofibroblasts: Its pathophysiologic implications. Blood. 2006;108:2893–2896. doi: 10.1182/blood-2006-04-016600. [DOI] [PubMed] [Google Scholar]
- Olaso E, Ikeda K, Eng FJ, Xu L, Wang LH, Lin HC, Friedman SL. DDR2 receptor promotes MMP-2-mediated proliferation and invasion by hepatic stellate cells. J Clin Invest. 2001;108:1369–1378. doi: 10.1172/JCI12373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivey HE, Mundell NA, Austin AF, Barnett JV. Transforming growth factor-beta stimulates epithelial–mesenchymal transformation in the proepicardium. Dev Dyn. 2006;235:50–59. doi: 10.1002/dvdy.20593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez-Montagut T, Blachere NE, Sviderskaya EV, Bennett DC, Rettig WJ, Garin-Chesa P, Houghton AN. FAPalpha, a surface peptidase expressed during wound healing, is a tumor suppressor. Oncogene. 2004;23:5435–5446. doi: 10.1038/sj.onc.1207730. [DOI] [PubMed] [Google Scholar]
- Rettig WJ, Garin-Chesa P, Healey JH, Su SL, Ozer HL, Schwab M, Albino AP, Old LJ. Regulation and heteromeric structure of the fibroblast activation protein in normal and transformed cells of mesenchymal and neuroectodermal origin. Cancer Res. 1993;53:3327–3335. [PubMed] [Google Scholar]
- Risau W. Mechanisms of angiogenesis. Nature. 1997;386:671–674. doi: 10.1038/386671a0. [DOI] [PubMed] [Google Scholar]
- Rohr S. Role of gap junctions in the propagation of the cardiac action potential. Cardiovasc Res. 2004;62:309–322. doi: 10.1016/j.cardiores.2003.11.035. [DOI] [PubMed] [Google Scholar]
- Sauk JJ, Nikitakis N, Siavash H. Hsp47 a novel collagen binding serpin chaperone, autoantigen and therapeutic target. Front Biosci. 2005;10:107–118. doi: 10.2741/1513. [DOI] [PubMed] [Google Scholar]
- Shioshita K, Miyazaki M, Ozono Y, Abe K, Taura K, Harada T, Koji T, Taguchi T, Kohno S. Expression of heat shock proteins 47 and 70 in the peritoneum of patients on continuous ambulatory peritoneal dialysis. Kidney Int. 2000;57:619–631. doi: 10.1046/j.1523-1755.2000.00883.x. [DOI] [PubMed] [Google Scholar]
- Smith TJ. Novel aspects of orbital fibroblast pathology. J Endocrinol Invest. 2004;27:246–253. doi: 10.1007/BF03345273. [DOI] [PubMed] [Google Scholar]
- Souders CA, Bowers SL, Baudino TA. Cardiac fibroblast: The renaissance cell. Circ Res. 2009;105:1164–1176. doi: 10.1161/CIRCRESAHA.109.209809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spach MS, Boineau JP. Microfibrosis produces electrical load variations due to loss of side-to-side cell connections: A major mechanism of structural heart disease arrhythmias. Pacing Clin Electrophysiol. 1997;20:397–413. doi: 10.1111/j.1540-8159.1997.tb06199.x. [DOI] [PubMed] [Google Scholar]
- Strutz F, Okada H, Lo CW, Danoff T, Carone RL, Tomaszewski JE, Neilson EG. Identification and characterization of a fibroblast marker: FSP1. J Cell Biol. 1995;130:393–405. doi: 10.1083/jcb.130.2.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimoto H, Mundel TM, Kieran MW, Kalluri R. Identification of fibroblast heterogeneity in the tumor microenvironment. Cancer Biol Ther. 2006;5:1640–1646. doi: 10.4161/cbt.5.12.3354. [DOI] [PubMed] [Google Scholar]
- Sundberg C, Ivarsson M, Gerdin B, Rubin K. Pericytes as collagen-producing cells in excessive dermal scarring. Lab Invest. 1996;74:452–466. [PubMed] [Google Scholar]
- Thedieck C, Kalbacher H, Kuczyk M, Muller GA, Muller CA, Klein G. Cadherin-9 is a novel cell surface marker for the heterogeneous pool of renal fibroblasts. PLoS ONE. 2007;2:e657. doi: 10.1371/journal.pone.0000657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuruda T, Costello-Boerrigter LC, Burnett JC., Jr Matrix metalloproteinases: Pathways of induction by bioactive molecules. Heart Fail Rev. 2004;9:53–61. doi: 10.1023/B:HREV.0000011394.34355.bb. [DOI] [PubMed] [Google Scholar]
- van Amerongen MJ, Bou-Gharios G, Popa ER, van Ark J, Petersen AH, van Dam GM, van Luyn MJA, Harmsen MC. Bone marrow-derived myofibroblasts contribute functionally to scar formation after myocardial infarction. J Pathol. 2007a;214:377–386. doi: 10.1002/path.2281. [DOI] [PubMed] [Google Scholar]
- van Amerongen MJ, Harmsen MC, van Rooijen N, Petersen AH, van Luyn MJA. Macrophage depletion impairs wound healing and increases left ventricular remodeling after myocardial injury in mice. Am J Pathol. 2007b;170:818–829. doi: 10.2353/ajpath.2007.060547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Tuyn J, Atsma DE, Winter EM, van der Velde-van Dijke I, Pijnappels DA, Bax NA, Knaan-Shanzer S, Gittenberger-de Groot AC, Poelmann RE, van der Laarse L, van der Wall EE, Schalij MJ, de Vries AA. Epicardial cells of human adults can undergo an epithelial-to-mesenchymal transition and obtain characteristics of smooth muscle cells in vitro. Stem Cells. 2007;25:271–278. doi: 10.1634/stemcells.2006-0366. [DOI] [PubMed] [Google Scholar]
- Villaschi S, Nicosia RF. Paracrine interactions between fibroblasts and endothelial cells in a serum-free coculture model. Modulation of angiogenesis and collagen gel contraction. Lab Invest. 1994;71:291–299. [PubMed] [Google Scholar]
- Vliegen HW, van der LA, Cornelisse CJ, Eulderink F. Myocardial changes in pressure overload-induced left ventricular hypertrophy. A study on tissue composition, polyploidization and multinucleation. Eur Heart J. 1991;12:488–494. doi: 10.1093/oxfordjournals.eurheartj.a059928. [DOI] [PubMed] [Google Scholar]
- Vogel WF, Abdulhussein R, Ford CE. Sensing extracellular matrix: An update on discoidin domain receptor function. Cell Signal. 2006;18:1108–1116. doi: 10.1016/j.cellsig.2006.02.012. [DOI] [PubMed] [Google Scholar]
- Wartiovaara J, Linder E, Ruoslahti E, Vaheri A. Distribution of fibroblast surface antigen: Association with fibrillar structures of normal cells and loss upon viral transformation. J Exp Med. 1974;140:1522–1533. doi: 10.1084/jem.140.6.1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber KT. Cardiac interstitium in health and disease: The fibrillar collagen network. J Am Coll Cardiol. 1989;13:1637–1652. doi: 10.1016/0735-1097(89)90360-4. [DOI] [PubMed] [Google Scholar]
- Weber KT. Monitoring tissue repair and fibrosis from a distance. Circulation. 1997;96:2488–2492. [PubMed] [Google Scholar]
- Weber KT. Fibrosis and hypertensive heart disease. Curr Opin Cardiol. 2000;15:264–272. doi: 10.1097/00001573-200007000-00010. [DOI] [PubMed] [Google Scholar]
- Wetzel A, Wetzig T, Haustein UF, Sticherling M, Anderegg U, Simon JC, Saalbach A. Increased neutrophil adherence in psoriasis: Role of the human endothelial cell receptor Thy-1 (CD90) J Invest Dermatol. 2006;126:441–452. doi: 10.1038/sj.jid.5700072. [DOI] [PubMed] [Google Scholar]
- Wynn TA. Cellular and molecular mechanisms of fibrosis. J Pathol. 2008;214:199–210. doi: 10.1002/path.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisberg EM, Tarnavski O, Zeisberg M, Dorfman AL, McMullen JR, Gustafsson E, Chandraker A, Yuan X, Pu WT, Roberts AB, Neilson EG, Sayegh MH, Izumo S, Kalluri R. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat Med. 2007;13:952–961. doi: 10.1038/nm1613. [DOI] [PubMed] [Google Scholar]
- Zhao L, Eghbali-Webb M. Release of pro- and anti-angiogenic factors by human cardiac fibroblasts: Effects on DNA synthesis and protection under hypoxia in human endothelial cells. Biochim Biophys Acta. 2001;1538:273–282. doi: 10.1016/s0167-4889(01)00078-7. [DOI] [PubMed] [Google Scholar]
- Zhou B, von GA, Ma Q, Hu YW, Pu WT. Genetic fate mapping demonstrates contribution of epicardium-derived cells to the annulus fibrosis of the mammalian heart. Dev Biol. 2010;338:251–261. doi: 10.1016/j.ydbio.2009.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]