

Abstract
Understanding genome and chromosome evolution is important for understanding genetic inheritance and evolution. Universal events comprising DNA replication, transcription, repair, mobile genetic element transposition, chromosome rearrangements, mitosis, and meiosis underlie inheritance and variation of living organisms. Although the genome of a species as a whole is important, chromosomes are the basic units subjected to genetic events that coin evolution to a large extent. Now many complete genome sequences are available, we can address evolution and variation of individual chromosomes across species. For example, “How are the repeat and nonrepeat proportions of genetic codes distributed among different chromosomes in a multichromosome species?” “Is there a general rule behind the intuitive observation that chromosome lengths tend to be similar in a species, and if so, can we generalize any findings in chromosome content and size across different taxonomic groups?” Here, we show that chromosomes within a species do not show dramatic fluctuation in their content of mobile genetic elements as the proliferation of these elements increases from unicellular eukaryotes to vertebrates. Furthermore, we demonstrate that, notwithstanding the remarkable plasticity, there is an upper limit to chromosome-size variation in diploid eukaryotes with linear chromosomes. Strikingly, variation in chromosome size for 886 chromosomes in 68 eukaryotic genomes (including 22 human autosomes) can be viably captured by a single model, which predicts that the vast majority of the chromosomes in a species are expected to have a base pair length between 0.4035 and 1.8626 times the average chromosome length. This conserved boundary of chromosome-size variation, which prevails across a wide taxonomic range with few exceptions, indicates that cellular, molecular, and evolutionary mechanisms, possibly together, confine the chromosome lengths around a species-specific average chromosome length.
Keywords: chromosome size, genome evolution, evolutionary modeling
Introduction
Genome sequencing has revealed detailed information on the genetic content of genomes and chromosomes for more than a 100 species across different phyla. It is now not only possible to answer questions concerning metagenomics of environmental samples and the molecular and evolutionary basis of speciation but also to ask many more questions in biology and evolution (Tringe and Rubin 2005; Misteli 2007; Metzker 2010; Presgraves 2010). Although the genome size of eukaryotes varies over five orders of magnitude, the distribution is skewed toward small values (Oliver et al. 2007). Overall, genome size and complexity clearly have increased during evolution from archaea and bacteria to eukaryota (Lynch and Conery 2003), but the network of mechanisms of the many competing processes that either expand or shrink the genome remain to be discovered in detail (Lynch and Conery 2003; Whitney et al. 2010). Previous research, based on estimated genome size across 20 eukaryotic clades, found that variation of genome size within a clade increases with the average genome size of the clade (Oliver et al. 2007). Based on genome size values measured by flow cytometry, a recent study demonstrated that there is a significant correlation between genome size and meiotic recombination rate (Whitney et al. 2010). Given the relative abundance of completed genome sequences, we can address the evolutionary dynamics of genome size and variation of chromosome size across species with base pair numbers. In particular, detailed sequence information allows us to characterize features and variations of chromosomes across multiple species, which was not possible with previous overall genome size estimation. In this study, we specifically address the following major questions, “How are the repeat and nonrepeat proportions of genetic codes distributed among different chromosomes in a multichromosome species?” “Is there a general rule behind the intuitive observation that chromosome lengths tend to be similar in a species, and if so, can we generalize any findings in chromosome content and size across different taxonomic groups?”
In eukaryota, DNA repeats increase chromosome size, as do intron size and gene duplication (Lynch and Conery 2003). Changes in chromosome number reflect the balance between forces that increase chromosome number (such as chromosome fission, chromosome missegregation, as well as allopolyploidization or autopolyplodization) and those that decrease it (such as chromosome fusion or missegregation). Some of these events also lead to changes in chromosome size. A systematic examination of repeat proportion at the genome level and chromosome level across taxonomic groups should provide further insight into genome and chromosome evolutions.
The transition from circular to linear chromosomes is one prerequisite for increases in individual chromosome size and chromosome number (Schubert 2007). In a seminal paper using field bean, it was demonstrated experimentally that there is an upper boundary of chromosome size for normal development of an organism (Schubert and Oud 1997). Sterility was mediated by chromosomes with arms exceedingly long via disturbance of meiotic division. This phenomenon was confirmed for barley, a monocot with a large genome (Hudakova et al. 2002). On the other hand, chromosomes of a much smaller size than average frequently do not segregate correctly during meiosis (Schubert 2001; Murata et al. 2006). Taken together, experimental research in individual species suggested a limit of chromosome-size variation, and a generalization of this finding to a wide range of species should provide an insight regarding genome and chromosome-size evolution, mechanisms involved in mitosis and meiosis, and genetic stability of natural or artificial minichromosomes.
Many evolutionary alterations affect chromosome number and/or chromosome size including reciprocal translocations, deletions and insertions, unequal crossover, dispersion of repetitive sequences, genome duplication, and chromosome fusion and fission and missegregation (Schubert 2007). Among these factors, reciprocal translocations have been considered one of the major forces to shape chromosome-size variation (Bickmore and Teague 2002; Schubert 2007) and were incorporated in previous evolutionary modeling studies (Sankoff and Ferretti 1996; De et al. 2001). These studies primarily considered individual species with specific numbers of chromosomes, and the comparisons were made to chromosome size estimated from karyotpes.
Here, we examined genome complexity by coupling information about evolutionary mechanisms and genome sequence information, thus revealing a general increase in genome size, chromosome size, and variability of chromosome characteristics from prokaryotes to unicellular eukaryotes, invertebrates, vascular plants, and vertebrates. Systematic analyses and computer simulations using genome sequence information from various species revealed that chromosome-size expansion in the course of evolution follows a stochastic process constrained by an upper limit to chromosome-size variation in many diploid eukaryotic genomes. Despite the dramatic differences in cellular and organismal complexity, the common pattern of chromosome-size variation in different eukaryotic genomes suggests a conserved constraint to chromosome evolution.
Materials and Methods
Genomes and Chromosomes
Genome and chromosome data of 128 genomes (68 eukaryotes and 60 prokaryotes) with multiple chromosomes were obtained from different databases including GenBank, Ensembl, JGI, and Phytozome as well as individual species’ genome databases (supplementary tables 1 and 2, Supplementary Material online). Sequences unanchored to chromosomes were not included in tabulating the base pair length. For species with more than one strain sequenced, we randomly selected one strain to represent the species. Chromosome sizes within each species were listed in ascending order in base pair units. Common name groups were assigned using the literature and database information. Accession number or version of genome assembly was provided. The sex chromosomes of 14 species were excluded from the analysis because of their unique evolutionary processes (Charlesworth D and Charlesworth B 2005; Charlesworth et al. 2005). For species without masked-ready genome sequence information, we identified the repetitive sequences with RepeatMasker 3.2.8 by using the library identified by RepeatScout 1.0.5 to mask the repetitive regions (Smit et al. 2010; verified on May 11, 2010). Because our focus was to obtain the general pattern of repeat proportion of the genomes and chromosomes rather than exact values for a certain species, we chose this more extensively used library-based program (Lerat 2010). Repeat and nonrepeat regions of chromosomes were obtained after the masking process.
The common theme of the current study was to examine genome size and chromosome size across different species. Variations of genome size increased as the average genome size increased across different common name groups (i.e., prokaryotes, unicellular eukaryotes, invertebrates, vascular plants, and vertebrates). For chromosome size in diploid eukaryotes, we further demonstrated that the standard deviation (SD) of chromosome size increased as the average chromosome size increased and that a common coefficient of variation (CV) existed. Further model fitting and computer simulations revealed that common distribution of chromosome-size variation can be modeled with a Gamma distribution.
Data Analysis and Statistical Modeling
Data of genome size and chromosome size were analyzed with SAS and R following standard procedures of correlation, regression, and plotting (fig. 1; supplementary figs. 1 and 2, Supplementary Material online). Because circular chromosomes in prokaryotes have different mechanisms for replication and separation in cell cycles (Schubert 2007), we focused only on eukaryotes with linear chromosomes. We used two approaches to conduct statistical modeling of chromosome-size variation. In the first approach, we fit an intuitive cubic function to capture the relationship between chromosome size and chromosome index. Chromosome size was calculated as the ratio of base pair length of a chromosome to average base pair length of chromosome of the species, 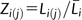 where Li(j) is the base pair chromosome length for the jth chromosome of a species i;
where Li(j) is the base pair chromosome length for the jth chromosome of a species i; 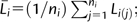 ni is the total chromosome number; and i = 1, 2, …, n species. Chromosome index was calculated as (j − 0.5)/ni. The fitted function was
ni is the total chromosome number; and i = 1, 2, …, n species. Chromosome index was calculated as (j − 0.5)/ni. The fitted function was
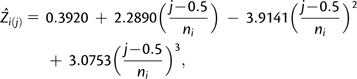 |
where 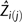 is the predicted chromosome size for the jth chromosome of a species i, and ni is the total chromosome number. Subtracting 0.5 in chromosome index was justified, because we used a continuous distribution to model the discrete chromosome number; this is a standard practice.
is the predicted chromosome size for the jth chromosome of a species i, and ni is the total chromosome number. Subtracting 0.5 in chromosome index was justified, because we used a continuous distribution to model the discrete chromosome number; this is a standard practice.
FIG. 1.

(A) Genome size in Mb of sequenced prokaryotes, unicellular eukaryotes, invertebrates, vascular plants, and vertebrates. (B) Boxplot of genome size in Log10 scale. The F test for genome size in Log10 scale among groups is highly significant (P = 2.3 × 10−57), and all pairwise group comparisons are significant. (C) The SD of genome size within each group positively correlates with genome size (r = 0.92; P = 0.025). Values are in Log10 scale for plotting. (D) After the dependency of SD on genome size is removed with Log10 transformation, the SD of genome size within the groups shows no correlation (r = −0.05; P = 0.93) with genome size. (E) Boxplot of the repeat proportions of genomes. The overall F test for repeat proportions among groups is highly significant (P = 3.0 × 10−26), and all pairwise group comparisons are significant except prokaryotes–unicellular eukaryotes and vascular plants–vertebrates.
The second approach was more systematic and aimed to model chromosome-size variation from statistical distributions. We used iteratively reweighted least square method to derive the parameter estimate. Four distributions commonly used in biology were considered: Gamma distribution, Normal distribution, Truncated Normal distribution (truncation at zero), and Lognormal distribution. Gamma distribution was chosen for four reasons. First, Zi(j) were all nonnegative. Second, the histogram of Zi(j) was skewed right and can be modeled by a Gamma distribution. Third, unlike Lognormal distribution, Gamma distribution is a member of the exponential family and permits a generalized linear model (Schabenberger and Pierce 2002). Fourth, model fitting showed that Gamma distribution had the best model fit. Model fitting statistics were calculated for mean square error (MSE), R2, and Akaike’s information criterion (AIC). 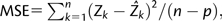 where Zk is the kth observed data point;
where Zk is the kth observed data point; 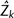 is the predicted value; k = 1, … ,n; n = 886 chromosomes, and p is the number of parameters in the model. The original definition of R2 was used, that is, R2 = 1 − (SSE/SST), where
is the predicted value; k = 1, … ,n; n = 886 chromosomes, and p is the number of parameters in the model. The original definition of R2 was used, that is, R2 = 1 − (SSE/SST), where  and AIC = nln(SSE) − nln(n) + 2p.
and AIC = nln(SSE) − nln(n) + 2p.
Although it is not possible to prove statistically that chromosome size must follow a Gamma distribution, our analysis proved that Gamma distribution was the best candidate of the distributions examined. We present the modeling steps for the Gamma distribution in supplementary materials (Supplementary Material online); similar steps were derived for three other distributions.
For crossvalidation, the observed data were randomly split into two parts: model fitting and validation. We then conducted computer simulations to further prove that Gamma distribution viably describes chromosome size and that numbers drawn from the Gamma distribution with the identified parameter Gamma (7.0438, 1/7.0438) can reproduce the pattern from observed data. Details for these two sections are provided in supplementary materials (Supplementary Material online).
Reciprocal Translocation
Among many evolutionary events, reciprocal translocation is a good starting point for understanding the dynamics of chromosome-size variation through modeling (Sankoff and Ferretti 1996; De et al. 2001; Imai et al. 2001; Mazowita et al. 2006). Simulations tested whether reciprocal translocation is partly responsible for observed chromosome-size variation. Numbers obtained through simulation (see supplementary materials [Supplementary Material online] for details) were then plotted against the chromosome index to show whether the resulting line approximates the predicted line from the inverse of the Gamma cumulative distribution function.
Four simulation schemes were carried out: 1) no constraints on chromosome size, 2) a lower threshold, 3) an upper threshold, and 4) both lower and upper thresholds (Sankoff and Ferretti 1996; De et al. 2001; Imai et al. 2001; Mazowita et al. 2006). We incorporated constraints on the smallest and largest chromosomes in the modeling process because 1) chromosome size below a certain threshold will prevent any translocation events; 2) at the cytogenetic level, viable and functional chromosomes must contain at least a centromere and two telomeres to maintain purely structural basis; and 3) each chromosome must have a length sufficient for at least one crossover among the four aligned sister chromatids in meiosis. Moreover, as shown experimentally, if one arm of the chromosome is >21.7% of the total length of all chromosomes, most offspring are sterile (Schubert 2007). The lower threshold was set for the smallest observed chromosome size (Sankoff and Ferretti 1996), and the upper threshold was set using a fitness function (De et al. 2001). In addition, we implemented a constraint in all simulations that resulting chromosomes from reciprocal translocation must have a centromere (De et al. 2001).
Details for reciprocal translocation simulation, confirming outlier species with known reasons, and estimating genome sizes for a much large sample of vascular plants and vertebrates are given in supplementary materials (Supplementary Material online).
Results
Is Average Genome Size of a Taxonomic Group Related to Variation within That Group?
We collected information on genome size, chromosome number, individual chromosome size, repeat-masked chromosome size (without repeat proportion), and common name groupings for 128 species with sequenced genomes, including prokaryotes, unicellular eukaryotes, invertebrates, vascular plants, and vertebrates (supplementary tables 1 and 2, Supplementary Material online). Across all sequenced prokaryotic and diploid eukaryotic species, genome size correlated with chromosome number and average chromosome size. Genome size varied considerably among species with similar levels of cellular and organismal complexity, but there was a general increase in genome size from prokaryotes to unicellular eukaryotes to multicellular eukaryotes (fig. 1). In addition, continuities in the scale of genome size across different groups of organisms indicate that organismal differences in cell/tissue anatomical structure or metabolism are unlikely to be the primary forces driving the evolution of genomic architecture (Lynch and Conery 2003).
Using these base pair data for genome size, we tested whether variation in genome size within each group was proportional to average genome size of the group. Given the sample size of available genomes, we focused our analysis on five phylogenetic branches (i.e., prokaryotes, unicellular eukaryotes, invertebrates, vascular plants, and vertebrates) rather than other finer taxonomic levels. Clearly, variation in genome size (measured as SD) significantly correlated with the average genome size (fig. 1). After we removed the dependency with Log10 transformation (a method to break the association between average of a group of numbers and the variation of these numbers; Oliver et al. 2007), the variation within each group showed no correlation with the average genome size. Groups with a larger average genome size obviously also had a larger variation in genome size. Variation of genome size of each group is the numerator in the calculation of rate of genome size evolution and could provide an approximation if the denominator, evolutionary distance or time, does not differ across groups on the same order of magnitude as the numerator. Interestingly, our findings regarding genome size showed a similar pattern with the previous research in which the rate of genome size evolution was found to be proportional to the average genome size of a clade when the estimated genome size based on C-value was examined across 20 eukaryotic clades and evolutionary distance was obtained from phylogenetic analysis of 18S rDNA (Oliver et al. 2007).
How Are the Repeat and Nonrepeat Proportions of Genetic Codes Distributed among Different Chromosomes in a Multichromosome Species?
To further examine the role of repeats on genome size and chromosome size, repeat masking of the genome was obtained from either original publications of the sequenced genomes or repeat masking analysis (Lerat 2010; Smit et al. 2010 verified on May 11, 2010). In general, the repeat proportion of the genome increased from prokaryotes (mean: 0.04) to unicellular eukaryotes (0.08), invertebrates (0.14), vascular plants (0.35), and vertebrates (0.38), following the same trend as genome size (fig. 1). For vascular plants with complete genome sequence, the repeat proportion of maize (82.5%) and sorghum (60.9%) skewed distribution to the right side. Overall, repeat proportion of chromosomes increases during evolution from prokaryotes to vertebrates, and this trend may become more evident as large genomes of vascular plants and vertebrates are sequenced.
Following the similar logic in genome size analysis, we also tested whether the SD of chromosome size (in base pair) within each species was proportional to the mean of chromosome size. Because of the difference in response to repeat accumulation between circular and linear chromosomes, we considered only eukaryotes with linear chromosomes in this analysis. There was a significant positive correlation between SD of chromosome size and the average chromosome size of a species (fig. 2). After we removed the magnitude effects with Log10 transformation, however, the SD of chromosome size for all eukaryotic species was bounded in a much smaller region than that for the prokaryotic species. Because 68 diploid eukaryotic species were used and the signal of the relationship between SD and average chromosome size was strong (P = 1.3 × 10−38), we then derived the regression slope (0.3700) of SD on average chromosome size across species. This regression slope provided an ad hoc estimate of a common CV (= SD/mean) for the underlying distributions of chromosome sizes in different species. Although large differences existed for average chromosome size and SD of chromosome size across species, the proportional relationship between them approached a constant. This was further verified by plotting CV, and any deviation was not unexpected because individual CV calculated for each species represented a sample (supplementary fig. 1, Supplementary Material online). On the other hand, there was no significant correlation between variation of chromosome size and total chromosome number of a species (supplementary fig. 1, Supplementary Material online).
FIG. 2.

(A) Chromosome-size variation as measured by SD of chromosome size within species correlates positively with average chromosome size (r = 0.96, P = 1.3 × 10−38). Values are in Log10 scale for plotting. Estimate of a common CV in original scale is 0.3700. (B) Absolute nonrepeat size variation (r = 0.97, P = 5.8 × 10−40). (C) Absolute repeat size variation (r = 0.94, P = 4.8 × 10−31). (D) After the dependency of absolute chromosome-size variation on preceding chromosome size is removed with Log10 transformation, chromosome-size variation within species shows no correlation (r = −0.10, P = 0.43) with average chromosome size. (E) Prior Log10 transformed nonrepeat size variation (r = −0.11, P = 0.37). (F) Prior Log10 transformed repeat size variation (r = −0.02; P = 0.89). Prokaryotic chromosomes are not included in the correlation calculation. Each color-coded dot represents the value for individual species.
Similar to the findings for chromosome size, the SD of nonrepeat size was proportional to the average nonrepeat size and the SD of repeat size proportional to the average repeat size. Although the mechanisms by which nonrepeat and repeat sequences were expanded in eukaryotic genomes are complicated (Lerat 2010), our results suggest that the rate of expansion among chromosomes is proportional to the preceding chromosome size, which indicates a stochastic process (fig. 2). Previous estimations of repeat proportions of the genomes have been species specific or based on extrapolation from a smaller number of species (Lynch and Conery 2003; Lerat 2010) than estimations included in the current study. Our general approach to studying repeat evolution across species with genome sequence data lays the groundwork for detailed studies on evolution of different classes of repeats and their composition among chromosomes, genomes, and taxonomic groups.
Is There a General Rule Behind the Intuitive Observation That Chromosome Lengths Tend to Be Similar in a Species?
We next examined chromosome-size variation in eukaryotes in detail because data available on chromosome length across the sequenced genomes permitted systematic modeling of chromosome size (supplementary fig. 2, Supplementary Material online). In addition to the common CV of chromosome size in eukaryotes, we noted that base pair sizes of the chromosomes within individual species usually have the same order of magnitude; this inspired further investigation of chromosome-size variation. Two transformations made the modeling process statistically possible and biologically sound: relative chromosome size and chromosome index. Relative chromosome size is obtained by dividing chromosome size in base pair by the average chromosome size of the individual species. Using average chromosome size as the unit of measure standardized the original chromosome size (in base pair) in different orders of magnitude for different species into comparable numbers. Chromosome index is obtained by dividing the ascending ranked chromosome number (subtracting a continuity correction factor 0.5) by the total chromosome number of that particular species. For example, for a species with 2 chromosomes, instead of 1 and 2, the chromosome index becomes 0.25 and 0.75. For a species with 5 chromosomes, instead of 1–5, the chromosome index becomes 0.1, 0.3, 0.5, 0.7, and 0.9. Chromosome index is bounded between 0 and 1, which permits modeling of chromosome size across species with different chromosome numbers. Amazingly, the plot of chromosome size against chromosome index revealed a clear pattern and strongly suggested a common curve similar to a cubic function: the incremental change in chromosome size larger at both ends of the curve but smaller in the middle (fig. 3).
FIG. 3.

(A) Model fitting of chromosome size on chromosome index across 886 chromosomes from 68 diploid eukaryotic species. The blue dotted line is the fitted cubic function, and the red line is the fitted inverse of Gamma cumulative distribution function 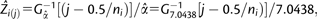 where is the predicted chromosome size for the jth ordered chromosome of a species i with a total of ni chromosomes, and
where is the predicted chromosome size for the jth ordered chromosome of a species i with a total of ni chromosomes, and 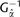 is the inverse of Gamma cumulative distribution function with parameter
is the inverse of Gamma cumulative distribution function with parameter 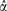 . (B) Histogram of chromosome size distribution with the overlaid probability density functions of Gamma (7.0438, 1/7.0438) and Normal (1.0000, 0.1371). The histogram has a mean of 1.0 and a skewness of 1.0046. Gray bars represent approximately 95% of the chromosome size between 0.3851 and 1.8608, and black bars represent the remaining 5% on both ends. Gamma (7.0438, 1/7.0438) has a mean of 1.0 and a variance of 0.1420. Of the chromosome size from Gamma (7.0438, 1/7.0438), 95% lies between 0.4035 and 1.8626. (C) Predicted chromosome-size proportion versus observed chromosome-size proportion. (D) Predicted chromosome-size proportion for a species with a given number of chromosomes. Predictions are plotted for the low hinge, median, and high hinge of the boxplot of individual common name groups: unicellular eukaryotes, invertebrates, vascular plants, and vertebrates.
. (B) Histogram of chromosome size distribution with the overlaid probability density functions of Gamma (7.0438, 1/7.0438) and Normal (1.0000, 0.1371). The histogram has a mean of 1.0 and a skewness of 1.0046. Gray bars represent approximately 95% of the chromosome size between 0.3851 and 1.8608, and black bars represent the remaining 5% on both ends. Gamma (7.0438, 1/7.0438) has a mean of 1.0 and a variance of 0.1420. Of the chromosome size from Gamma (7.0438, 1/7.0438), 95% lies between 0.4035 and 1.8626. (C) Predicted chromosome-size proportion versus observed chromosome-size proportion. (D) Predicted chromosome-size proportion for a species with a given number of chromosomes. Predictions are plotted for the low hinge, median, and high hinge of the boxplot of individual common name groups: unicellular eukaryotes, invertebrates, vascular plants, and vertebrates.
Further investigation into the potential distribution from which the chromosome sizes (samples) were drawn suggested that a Gamma distribution was a more plausible candidate than other distributions (fig. 3). Gamma distribution is widely used in engineering and science to model continuous variables that are nonnegative but have right-skewed probability densities (Schabenberger and Pierce 2002) and provides a natural framework to model chromosome size that is nonnegative. Indeed, a Gamma distribution approximated a histogram of all chromosome sizes (with a mean of 1 and skewness of 1.0046) better than a Normal distribution. Histograms generated from data of individual species, from the pooled data of species with the same total number of chromosomes, and from the pooled data of each common group corroborated this finding. We then theoretically derived the approximate relationship function between chromosome size and chromosome index as an inverse of a Gamma cumulative distribution function, G(α,1/α) − 1, where α is the parameter. Because no closed form exists for this nonlinear function, we used an iterative procedure (iteratively reweighted least square) that minimizes the influence of variance heterogeneity to obtain the parameter estimate G(7.0438,1/7.0438) − 1with a 95% confidence interval of as (6.6609, 7.4267). Model fitting statistics indicated a better fit with the Gamma distribution than with other distributions or the intuitive cubic function. Notice that the variance (and CV because mean = 1) of G7.0438 − 1 is 0.3768, which is close to the previous ad hoc CV estimate 0.3700 obtained through simple regression analysis. On the basis of G(7.0438,1/7.0438) − 1, 95% of the chromosomes in a species are expected to have a base pair length between 0.4035 and 1.8626 times the average chromosome length; this interval is applicable to chromosomes in diploid eukaryotic species. However, we admit that practically a Normal distribution is almost equally viable in capturing the chromosome-size variation (fig. 3 and supplementary table 3, Supplementary Material online) and is a more general one. The major reason of not choosing Normal distribution is the possible negative values implicated.
Can Prediction Be Made on Chromosome Size?
It follows that, for a given species, chromosome sizes can be predicted by chromosome number. Furthermore, given either genome size or average chromosome base pair length (genome size = average chromosome size × total chromosome number), we can predict the size range of all chromosomes of that species in base pair (fig. 3). Chromosome-size proportion was obtained by dividing chromosome size by genome size; the sum of chromosome-size proportions equaled one. For example, for a species with 15 chromosomes, the shortest and longest chromosomes would be expected to account for 2.87% and 11.99% of the genome, respectively. The predicted ratio of the longest to the shortest chromosome for a given species was 1.68 for a species with two chromosomes and 5.70 for a species with 38 chromosomes. We used this general prediction to confirm the cases in which exceptions occurred for a few outlier species for known reasons: three species known to have macrochromosomes and microchromosomes, one haploid species, and one species with one linear chromosome and one circular chromosome (supplementary tables 1 and 2, supplementary fig. 3, Supplementary Material online).
To show the robustness of the prediction and ensure that we had used an adequate number of genomes (68 diploid eukaryotic genomes), we performed a series of crossvalidation experiments using different proportions of the observed data for function derivation and the rest of the data for validation. Plots of mean square prediction error (MSPE) and parameter estimate indicated that the original sample size was large enough to derive a robust prediction function (supplementary fig. 4, Supplementary Material online). The MSPE decreased as more data points were used to derive the prediction function. Likewise, the parameter estimate (α) approached the value from the whole data set. With about 50% of the data (≈35 species), both MSPE and α started to level off, indicating an adequate sample size in the original data to derive the function and make a prediction. In addition, simulation results reproduced the pattern of the observed data, indicating that Gamma distribution viably describes the chromosome-size variation observed (supplementary fig. 5, Supplementary Material online). Numbers representing chromosome sizes were drawn from Gamma distributions with specific parameters for species having a chromosome number from 2 to 38. Both the dispersion of the scattered points and the fitted curves of the simulated and observed data confirmed that the pattern discovered was reproducible.
Should Other Evolutionary Alterations Besides Reciprocal Translocation Be Considered in Evolutionary Modeling Studies?
To verify whether reciprocal translocations can adequately model the chromosome-size variation as suggested in previous evolutionary modeling studies (Sankoff and Ferretti 1996; De et al. 2001; Imai et al. 2001; Mazowita et al. 2006), we ran a set of computer simulations to compare the pattern generated by simulations and by our empirical data. Four simulation schemes were carried out: 1) no constraints on chromosome size, 2) a lower threshold, 3) an upper threshold, and 4) both lower and upper thresholds (Sankoff and Ferretti 1996; De et al. 2001; Imai et al. 2001; Mazowita et al. 2006). Notice that these thresholds are for individual chromosome size, not their variations. Simulated chromosome sizes based on the reciprocal translocation model without thresholds showed greater variation than we observed in these sequenced genomes, but simulations with both thresholds had a better approximation (fig. 4, supplementary fig. 6, Supplementary Material online). Our results suggest that reciprocal translocation is likely to be one of the major forces and future modeling procedures that consider other evolutionary alterations (e.g., genome duplications, chromosome fusion, secondary rearrangements) besides reciprocal translocation may lead to even better congruency (The Chimpanzee Sequencing and Analysis Consortium 2005; Schubert 2007). Unlike previous studies in which modeling was conducted for individual species and much smaller numbers of species were examined, the current study with empirical data analyses and computer simulations established a benchmark for future evolutionary modeling research in chromosome size.
FIG. 4.

Simulation using the reciprocal translocation model to test whether it partly explains observed (red line) chromosome-size variations. (A) No constraints on chromosome size. (B) A lower threshold. (C) An upper threshold. (D) Both lower and upper thresholds. Chromosome-size values are not expected to form a single line because the reciprocal translocation model predicts chromosome sizes independently for different total number of chromosomes.
Discussion
Genome and chromosome complexity has been addressed from different perspectives including population genetics and evolution (Lynch and Conery 2003; Oliver et al. 2007), molecular biology and cytogenetics (Schubert 2007), and evolutionary modeling (Sankoff and Ferretti 1996; Ma et al. 2008). In this work, we systematically studied the dynamics of genome and chromosome-size variation. Using a combination of bioinformatics and statistics approaches and available genome sequences across the evolutionary spectrum, we examined genome size evolution, repeat size evolution, chromosome-size variation, and evolutionary modeling. Chromosome size tends to center around the average chromosome length within a species for most diploid eukaryotes, and chromosome-size variation across species can be adequately modeled with a Gamma distribution. Although it may seem to be intuitive or a common place, systematic proof across multiple species is lacking prior to our study. Our findings are in agreement with the long-standing karyotypes in which chromosomes are usually visualized in descending order (Sankoff and Ferretti 1996). This connection assumes that the higher-order structures of linear DNA sequence do not lead to a different pattern of chromatin size (as captured in karyotype) from the chromosome size in base pair (Misteli 2007). In other words, a relatively constant folding ratio ensures that higher base pair length generally corresponds to longer chromatin size. In a cell cycle, the synchrony of chromosome separation must be precisely controlled to correctly separate homologous chromosomes or sister chromatids. Although the exact mechanism of such synchrony is not clear, chromosome-size variation as a basic feature of chromosome architecture deserves more attention. Uniform chromosome length may facilitate the cell achieving synchronized DNA replication time with the same number of replication forks, correct chromosome configuration on equatorial plate, and accurate migration of homologous chromosomes or sister chromatids to opposite poles (Sharp et al. 2000; Misteli 2007).
In the current modeling of chromosome-size variation across 68 eukaryotic species, species with different genome sizes were examined, for example, Bigelowiella natans with 0.37 Mb, Zea mays with 2.05 Gb, Homo sapiens with 2.88 Gb for autosomes, and Monodelphis domestica 3.42 Gb for autosomes. In addition, resampling simulations demonstrated that the major finding in chromosome-size variation based on available data is robust to sampling process. We realized that genome sequences of some vascular plants and vertebrates with very large genome sizes are not available (Whitney et al. 2010). However, with genome sizes estimated from C-values of a much larger number of species in vascular plants (2,757) and vertebrates (3,140), the rate of genome size evolution as measured by SD of genome size within each group remains to be positively correlated with the average genome size (supplementary fig. 7, Supplementary Material online). The boundary discovered for chromosome-size variation, on the other hand, is less likely to be biased because the context is individual genomes. For example, karyotypes of wheat genome (∼16 Gb) (Gill et al. 1991; Sankoff and Ferretti 1996) and barley genome (∼5 Gb) (Lee et al. 2000) strongly suggest a boundary in chromosome-size variation for these two large genomes with a high proportion of repeats, same as discovered in the current study. Taking the general strategies of this cross-species analysis, evidence supporting the current discovery is likely to be further uncovered with more genomes being sequenced. On the other hand, it would be interesting to study the mechanisms of genome and chromosome stabilities with a few outlier species with known reasons shown in our study.
An upper limit to chromosome-size variation provides better evolutionary fitness because the limit of the cell dimension and spindle extension do not favor having chromosomes with significantly different lengths (Schubert and Oud 1997; Schubert 2001, 2007). Considering the number of cells and the mitosis events in an organism, the overall energy savings may also be a factor because ATP molecules are required for chromosome velocity (Nicklas 1965). Temporal control of kinetochore–microtubule dynamics may be a mechanism for maintaining genome stability (Bakhoum et al. 2009a, 2009b). Depolymerization of kinetochore microtubules may partly power chromosome movement during mitosis (Molodtsov et al. 2005). Under normal conditions, chromosomes of different sizes in a single cell have a similar chromosome velocity in anaphase (Nicklas 1965; Raj and Peskin 2006). Large variations in chromosome length may decrease the evolutionary fitness of an organism; overly lengthy chromosomes will delay the separation of sister chromatids and homologous chromosomes during mitosis and meiosis, resulting in cell cycle prolongation, sterility, or even death (Schubert 2007). Moreover, meiotic recombination was experimentally demonstrated to depend on chromosome size in Saccharomyces cerevisiae (Kaback et al. 1992) and in humans (Lander et al. 2001). Therefore, chromosome-size variation is a vital factor in cell biology and evolution.
Genome sequences of neopolyploid species have not been reported. After resolving the assembly hurdle, further sequencing of polyploid genomes would allow us to extend this hypothesis beyond diploid genomes. Many current diploid species have undergone a process of polyploidization and diploidization. Detailed examination of available genomes may also reveal the evolutionary significance of ancient genome duplications (Van de Peer et al. 2009). In addition, the locations of centromeres have been studied in only a few species (Henikoff et al. 2001). It is interesting that although chromosome segregation machinery is highly conserved across all eukaryotes, research about DNA and protein components at centromeric chromatin has not been able to readily identify centromeres in nonmodel species. Once the positions of centromeres have been identified in a wide range of species, further study of length variation of the chromosome arm may allow us to understand both the fine control and variation in chromosome segregation machinery.
Supplementary Materials
Supplementary Materials, supplementary figures S1–S7, and supplementary tables S1–S3, are available at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org/).
Acknowledgments
We are very grateful to Dr. Ingo Schubert and Dr. Brandon Gaut for their critical comments of the manuscript. This work is supported by the National Science Foundation (DBI-0820610; IIS-0844945), the National Institute of Health (NIH/NCI U01-CA128535-01), the Department of Defense (W81XWH-08-1-0065), the Purdue University Discovery Park Seed Grant, the National Research Initiative of the USDA-CSREES (2006-03578), and the Targeted Excellence Program of Kansas State University.
References
- Bakhoum SF, Genovese G, Compton DA. Deviant kinetochore microtubule dynamics underlie chromosomal instability. Curr Biol. 2009a;19:1937–1942. doi: 10.1016/j.cub.2009.09.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakhoum SF, Thompson SL, Manning AL, Compton DA. Genome stability is ensured by temporal control of kinetochore-microtubule dynamics. Nat Cell Biol. 2009b;11:27–35. doi: 10.1038/ncb1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bickmore WA, Teague P. Influences of chromosome size, gene density and nuclear position on the frequency of constitutional translocations in the human population. Chromosome Res. 2002;10:707–715. doi: 10.1023/a:1021589031769. [DOI] [PubMed] [Google Scholar]
- Charlesworth D, Charlesworth B. Sex chromosomes: evolution of the weird and wonderful. Curr Biol. 2005;15:R129–R131. doi: 10.1016/j.cub.2005.02.011. [DOI] [PubMed] [Google Scholar]
- Charlesworth D, Charlesworth B, Marais G. Steps in the evolution of heteromorphic sex chromosomes. Heredity. 2005;95:118–128. doi: 10.1038/sj.hdy.6800697. [DOI] [PubMed] [Google Scholar]
- The Chimpanzee Sequencing and Analysis Consortium. Initial sequence of the chimpanzee genome and comparison with the human genome. Nature. 2005;437:69–87. doi: 10.1038/nature04072. [DOI] [PubMed] [Google Scholar]
- De A, Ferguson M, Sindi S, Durrett R. The equilibrium distribution for a generalized Sankoff–Ferretti model accurately predicts chromosome size distribution in a wide variety of species. J Appl Prob. 2001;38:324–334. [Google Scholar]
- Gill BS, Friebe B, Endo TR. Standard karyotype and nomenclature system for description of chromosome bands and structural aberrations in wheat (Triticum Aestivum) Genome. 1991;34:830–839. [Google Scholar]
- Henikoff S, Ahmad K, Malik HS. The centromere paradox: stable inheritance with rapidly evolving DNA. Science. 2001;293:1098–1102. doi: 10.1126/science.1062939. [DOI] [PubMed] [Google Scholar]
- Hudakova S, Kunzel G, Endo TR, Schubert I. Barley chromosome arms longer than half of the spindle axis interfere with nuclear divisions. Cytogenet Genome Res. 2002;98:101–107. doi: 10.1159/000068530. [DOI] [PubMed] [Google Scholar]
- Imai HT, Satta Y, Takahata N. Integrative study on chromosome evolution of mammals, ants and wasps based on the minimum interaction theory. J Theor Biol. 2001;210:475–497. doi: 10.1006/jtbi.2001.2327. [DOI] [PubMed] [Google Scholar]
- Kaback DB, Guacci V, Barber D, Mahon JW. Chromosome size-dependent control of meiotic recombination. Science. 1992;256:228–232. doi: 10.1126/science.1566070. [DOI] [PubMed] [Google Scholar]
- Lander ESLM, Linton B, BirrenC, et al. (255 co-authors) Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Lee JH, Arumuganathan K, Chung YS, Kim KY, Chung WB, Bae KS, Kim DH, Chung DS, Kwon OC. Flow cytometric analysis and chromosome sorting of barley (Hordeum vulgare L.) Mol Cells. 2000;10:619–625. doi: 10.1007/s10059-000-0619-y. [DOI] [PubMed] [Google Scholar]
- Lerat E. Identifying repeats and transposable elements in sequenced genomes: how to find your way through the dense forest of programs. Heredity. 2010;104:520–533. doi: 10.1038/hdy.2009.165. [DOI] [PubMed] [Google Scholar]
- Lynch M, Conery JS. The origins of genome complexity. Science. 2003;302:1401–1404. doi: 10.1126/science.1089370. [DOI] [PubMed] [Google Scholar]
- Ma J, Ratan A, Raney BJ, Suh BB, Miller W, Haussler D. The infinite sites model of genome evolution. Proc Natl Acad Sci U S A. 2008;105:14254–14261. doi: 10.1073/pnas.0805217105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazowita M, Haque L, Sankoff D. Stability of rearrangement measures in the comparison of genome sequences. J Comput Biol. 2006;13:554–566. doi: 10.1089/cmb.2006.13.554. [DOI] [PubMed] [Google Scholar]
- Metzker ML. Sequencing technologies–the next generation. Nat Rev Genet. 2010;11:31–46. doi: 10.1038/nrg2626. [DOI] [PubMed] [Google Scholar]
- Misteli T. Beyond the sequence: cellular organization of genome function. Cell. 2007;128:787–800. doi: 10.1016/j.cell.2007.01.028. [DOI] [PubMed] [Google Scholar]
- Molodtsov MI, Grishchuk EL, Efremov AK, McIntosh JR, Ataullakhanov FI. Force production by depolymerizing microtubules: a theoretical study. Proc Natl Acad Sci U S A. 2005;102:4353–4358. doi: 10.1073/pnas.0501142102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murata M, Shibata F, Yokota E. The origin, meiotic behavior, and transmission of a novel minichromosome in Arabidopsis thaliana. Chromosoma. 2006;115:311–319. doi: 10.1007/s00412-005-0045-1. [DOI] [PubMed] [Google Scholar]
- Nicklas RB. Chromosome velocity during mitosis as a function of chromosome size and position. J Cell Biol. 1965;25(Suppl):119–135. doi: 10.1083/jcb.25.1.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver MJ, Petrov D, Ackerly D, Falkowski P, Schofield OM. The mode and tempo of genome size evolution in eukaryotes. Genome Res. 2007;17:594–601. doi: 10.1101/gr.6096207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Presgraves DC. The molecular evolutionary basis of species formation. Nat Rev Genet. 2010;11:175–180. doi: 10.1038/nrg2718. [DOI] [PubMed] [Google Scholar]
- Raj A, Peskin CS. The influence of chromosome flexibility on chromosome transport during anaphase A. Proc Natl Acad Sci U S A. 2006;103:5349–5354. doi: 10.1073/pnas.0601215103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankoff D, Ferretti V. Karyotype distributions in a stochastic model of reciprocal translocation. Genome Res. 1996;6:1–9. doi: 10.1101/gr.6.1.1. [DOI] [PubMed] [Google Scholar]
- Schabenberger O, Pierce FJ. Contemporary statistical models for the plant and soil sciences. Boca Raton (FL): CRC Press; 2002. [Google Scholar]
- Schubert I. Alteration of chromosome numbers by generation of minichromosomes—is there a lower limit of chromosome size for stable segregation? Cytogenet Cell Genet. 2001;93:175–181. doi: 10.1159/000056981. [DOI] [PubMed] [Google Scholar]
- Schubert I. Chromosome evolution. Curr Opin Plant Biol. 2007;10:109–115. doi: 10.1016/j.pbi.2007.01.001. [DOI] [PubMed] [Google Scholar]
- Schubert I, Oud JL. There is an upper limit of chromosome size for normal development of an organism. Cell. 1997;88:515–520. doi: 10.1016/s0092-8674(00)81891-7. [DOI] [PubMed] [Google Scholar]
- Sharp DJ, Rogers GC, Scholey JM. Microtubule motors in mitosis. Nature. 2000;407:41–47. doi: 10.1038/35024000. [DOI] [PubMed] [Google Scholar]
- Smit AFA, Hubley R, Green P. Verified on May 11, 2010. RepeatMasker Open-3.0.Available from: http://www.repeatmasker.org. [Google Scholar]
- Tringe SG, Rubin EM. Metagenomics: DNA sequencing of environmental samples. Nat Rev Genet. 2005;6:805–814. doi: 10.1038/nrg1709. [DOI] [PubMed] [Google Scholar]
- Van de Peer Y, Maere S, Meyer A. The evolutionary significance of ancient genome duplications. Nat Rev Genet. 2009;10:725–732. doi: 10.1038/nrg2600. [DOI] [PubMed] [Google Scholar]
- Whitney KD, Baack EJ, Hamrick JL, et al. (11 co-authors) A role for nonadaptive processes in plant genome size evolution? Evolution. 2010;64:2097–2109. doi: 10.1111/j.1558-5646.2010.00967.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.