

Abstract
Advances in intensive care and antibiotics have prevented the spread of some infections, though sepsis mortality rates remain high. With failure of over thirty clinical trials, sepsis remains a scientific and clinical challenge in modern medicine. Sepsis is defined by the clinical signs of a systemic inflammatory response to infection. “Severe sepsis” is when these symptoms are associated with multiple organ dysfunction. These definitions of sepsis may be too broad and common to heterogeneous groups of patients who do not necessarily have the same disorder. This consideration has become especially evident in the clinical trials that have failed to obtain consistent results in similar studies of patients diagnosed with severe sepsis. In these trials, patients with infections caused by different microorganisms, and affecting different organs, have been combined under the general diagnosis of severe sepsis. The situation is analogous to attempting a clinical trial based on the general definition of cancer, combining all patients with tumor independent of the type of malignancy. In this consideration, it would not be very surprising that activated protein C, the only treatment in sepsis approved by the Food and Drug Administration, is projected for use in only a small subset of patients with severe sepsis. This article reviews novel inflammatory molecular aspects and the experimental anti-inflammatory strategies for sepsis, as they may represent particular pathological processes in specific subsets of patients.
INTRODUCTION
Sepsis is the leading cause of mortality in intensive care units and accounts for roughly 9.3% of overall deaths, killing approximately 250,000 patients annually in the United States alone [1–9]. Despite recent improvements in intensive care and antibiotics, sepsis remains associated with a high mortality rate possibly because the etiology of sepsis can be diverse. Sepsis is defined according to the clinical signs of a systemic response to infection [10]. However, the clinical symptoms of sepsis including hypotension, tachycardia, tachypnea, hypoperfusion, lactic acidosis, and altered body temperature (>38.3°C or <36°C), are not exclusive to infection and can also be triggered by shock, trauma, or severe injury [2, 11]. The term “severe sepsis” refers to the sepsis-associated failure of multiple organ systems [10]. Sepsis is characterized by an overzealous production of inflammatory cytokines such as tumor necrosis factor (TNF), interleukins, macrophage migration inhibitory factor (MIF), and high mobility group box-1 (HMGB1) [2]. During a normal inflammatory response the production of these cytokines is beneficial, and is required for the repair of tissue damage resulting from infection or injury. However, an excessive inflammatory response can cause lethal multiple organ failure and become more dangerous than the initial insult [2]. Current strategies for severe sepsis are largely supportive and include the maintenance of systemic perfusion and the eradication of infection. Still, sepsis remains a major scientific and clinical challenge, characterized by a high mortality rate possibly because these strategies have shown limited therapeutic value to prevent systemic inflammation [8, 11]. An apparent confounding factor is that the definition of sepsis might be too broad, encompassing heterogeneous groups of patients who might not have the same clinical immunological disorder [2, 10].
The heterogeneity of the etiology of sepsis has become an important consideration to explain why strategies targeting inflammatory mediators like TNF or IL-1 have produced modest effects in clinical trials in spite of their effectiveness in experimental models [9]. Since cytokine-blockade-based strategies have not been beneficial in clinical trials, none have gained unconditional approval from the Food and Drug Administration in the United States for the treatment of sepsis [12–14]. Another important unresolved consideration is whether severe sepsis results from hyperactive immune responses or from immunosuppression. Due in part to this controversy, a focus of current investigations is the identification of therapeutic targets that involve endogenous immunomodulatory mechanisms. In contrast to previous strategies that depended primarily on the blockade of secreted cytokines, a potential advantage to targeting endogenous immunomodulatory mechanisms is the ability to temper systemic inflammatory responses but prevent immunosuppresion. A classical example is the hypothalamic-pituitary-adrenal axis that controls the release of glucocorticoids from the adrenal cortex [15, 16]. Activation of the hypothalamic-pituitary-adrenal axis in response to immune stimulation causes the release of adrenal corticotropic hormone from the pituitary gland, which increases secretion of cortisol from the adrenal cortex. Cortisol exerts anti-inflammatory effects on a variety of immune cells including macrophages, monocytes, and neutrophils [15, 16]. Recent studies have identified the anti-inflammatory potential of the vagus nerve to restrain systemic inflammation in sepsis. Acetylcholine, the principal neurotransmitter of the vagus nerve, controls cytokine production from macrophages via nicotinic receptors. This “cholinergic anti-inflammatory mechanism” is mediated by the alpha7-nicotinic acetylcholine receptor (alpha7nAChR) and modulates cytokine production through NF-kB [2, 17–19]. The characterization of this nicotinic anti-inflammatory pathway is of great interest because selective nicotinic agonists for the alpha7nAChR represent a promising pharmacological strategy for infectious and inflammatory disorders. Efficacious treatments for sepsis are a major priority of public health. This article reviews experimental strategies and new targets currently under investigation for the treatment of sepsis and related infectious and inflammatory disorders.
COAGULATION: A LINK WITH INFLAMMATION IN SEPSIS
Recombinant human activated protein C (APC, also known as Drotrecogin alpha-activated) is the only intervention approved by the Food and Drug Administration for the treatment of severe sepsis [20]. Protein C is a serum protease produced by the liver as an inactive precursor that is activated via cleavage by thrombin, an end-product of the coagulation protease cascade (Fig. 1) [21]. Activated protein C proteolytically inactivates factors Va and VIIIa, proteases upstream of thrombin in the coagulation cascade. Thus, activated protein C is part of a classical negative feedback mechanism that blunts the coagulation cascade by preventing further thrombin generation [21]. By preventing thrombin generation, activated protein C blocks thrombin-induced platelet activation and quenches inflammatory responses from immune cells and endothelial cells (Fig. 1) [22]. Activated protein C inhibits the production of inflammatory cytokines in human umbilical vein endothelial cells (HUVEC) by blocking NF-κB activation [23]. Although activated protein C is the first intervention that improves the clinical outcome of severe sepsis, the benefits are limited with an absolute mortality reduction of 6.1% [20]. A deleterious side effect is that activated protein C blunts the blood clotting and significantly increases the risk of severe hemorrhage in patients [20, 24]. Activated protein C treatment is approved only for the small subset of patients with a low risk for hemorrhage. This limitation is particularly relevant to sepsis because most of the patients have an increased risk of internal hemorrhage. In fact, more recent non-industry-supported prospective [25], and retrospective [26] studies, concluded that due to continual bleeding problems, for most patients, the risks associated with activated protein C treatment seem to outweigh the benefits [27, 28]. As this controversy persists, two new placebo-controlled trials are planned to assess the efficacy of recombinant human activated protein C (rhAPC) in septic shock which may resolve the issue. One is PROWESS-SHOCK to study persistent vasopressor-dependent septic shock [29]. The other, APROCCHS, will run in parallel to compare efficacy and safety of rhAPC to that of low dose of corticosteroids, investigating the interaction of these drugs in managing septic shock (http://clinicaltrials.gov/ct2/show/NCT00625209. January, 2008). Recently, a recombinant variant of mouse activated protein C (5A-APC) has been produced to retain the anti-inflammatory and anti-apoptotic ability, but had a reduced anticoagulant activity by 90%. 5A-APC was effective in reducing mortality and increasing survival in mouse models of endotoxemia, peritonitis, and polymicrobial peritoneal sepsis [30].
Fig. (1). APC and the feed-back regulation of Thrombin.

Thrombin activates protein C, which prevents further thrombin generation. Bacterial endotoxin stimulates the production of tissue factor on endothelium. This tissue factor triggers the proteolytic cascade resulting in thrombin generation and subsequent blood clotting/coagulation. Endotoxin also induces the release of a zymogen form of protein C from the liver into the systemic blood. Thrombin cleavage converts protein C into activated protein C, a highly active serine protease. Activated protein C cleaves several substrates including factor Va and factor VIIIa, which are proteases that process pro-thrombin to generate thrombin. Activated protein C therefore functions as a classical feed-back regulator of thrombin generation and coagulation. As such, activated protein C also attenuates inflammation induced by thrombin receptor-mediated platelet activation, and NF-κB activation in blood and endothelial cells.
Disseminated intravascular coagulation (DIC) is a serious condition of simultaneous vascular coagulation and bleeding due to consumption of platelets, which is a typical consequence of severe sepsis. Promising results with high doses of antithrombin were found both in experimental and clinical trials of septic shock. A large, double blind, placebo-controlled multicenter trial (High-Dose Antithrombin III in Severe Sepsis [KyberSept] trial [31]) investigated the effect of antithrombin in sepsis. Although high levels of antithrombin (180% at 24 hrs) were produced, no difference in mortality between the placebo and experimental group was noted. Antithrombin treated patients had significantly higher bleeding complications than the placebo group. Subgroup analysis showed that patients receiving higher doses of antithrombin (>60%) had a beneficial effect on 90-day mortality. Also those patients receiving protective heparin simultaneously had higher mortality (and bleeding) than those not on heparin. These results may be due to heparin interfering with the ability of antithrombin to bind to glycosaminoglycans on endothelial cells, and heparin may also abolish the anti-inflammatory effects of antithrombin. They concluded that antithrombin should not be used with patients on heparin. Further studies of heparin and antithrombin are needed before antithrombin can be recommended for sepsis.
Tissue factor pathway inhibitor (TFPI) or tifacogin is an endogenous anticoagulant secreted by endothelial cells. TFPI abrogated the coagulant response, attenuated the inflammatory response, and improved survival in baboons even when administered several hours after septic challenge [32, 33]. Tifacogin also increased survival in a rabbit model of peritonitis [34]. Tifacogin confirmed the anticoagulant response in humans challenged with endotoxin, but did not inhibit the inflammatory response [35, 36]. Based on the positive results of a small dose-finding trial where anti-inflammatory and coagulant reductions occurred in patients with severe sepsis, a large randomized, double-blind placebo-controlled multi-center trial (the Optimized Phase 3 Tifacogin in Multicenter International Sepsis Trial) was undertaken to test the efficacy of TFPI in sepsis [37]. Two groups of patients were studied. One with international normalized ratio (INR, measure of blood clotting) ≥1.2, and another <1.2. Both groups had significantly reduced coagulatory responses, however no effect of TFPI on mortality was found in the ≥1.2 group. There was also higher risk for bleeding complications in both groups. Mortality rates in both treatment groups were similar regardless of the use of heparin. So unlike the KyberSept trial with antithrombin (described above) where heparin may have interfered with antithrombin binding, heparin may not explain the failure of TFPI in these trails as displacement of TFPI binding to endothelial cells. In discussions regarding the three anticoagulants, activated protein C, antithrombin, and TFPI, since only drotrecogin was effective in reducing mortality in a restricted group of patients, it is proposed that clinical trials should be done with heparin, since it is most available, inexpensive, and most easily used anticoagulant [38].
GLUCOCORTICOIDS: ENDOCRINE-MEDIATED SYSTEMIC IMMUNE SUPPRESSION
Glucocorticoids are anti-inflammatory corticosteroid hormones modulating inflammation during injury and infection [16, 39]. Inflammatory cytokines activate the hypothalamic-pituitary-adrenal axis, a physiological neuro-endocrine mechanism that leads to the release of adrenal corticotropic hormone (ACTH) from the pituitary gland into the circulation [15]. Adrenal corticotropic hormone diffuses to the adrenal cortex and stimulates the release of cortisol, which functions as a potent systemic immunomodulator [40]. Glucocorticoids like cortisol have a dual mechanism of action; they block the production of inflammatory cytokines in many cell types including leukocytes, endothelial, and epithelial cells, while enhancing the production of the anti-inflammatory cytokine IL-10 from T-cells [15, 41, 42].
Glucocorticoids exert their immunosuppressive effects by binding to intracellular receptors, ligand-activated transcription factors of the nuclear hormone receptor superfamily (Fig. 2). Glucocorticoid receptors are expressed in virtually every cell type, and are maintained in the cytosol in an inactive form [43]. Upon activation by hormone, the receptor translocates to the nucleus where it binds to and interferes with the activity of transcriptional activators including AP-1 (c-Fos/c-Jun heterodimer) and NF-κB (p50/p65 heterodimer) [44–47]. This ability to interfere with the activity of other transcriptional activators is specific to glucocorticoid receptors as other classes of steroid receptors such as androgen and estrogen receptors fail to affect inflammatory cytokine production [41]. DNA binding and transcriptional activation are not required for the immunosuppressive effects of activated glucocorticoid receptors [48]. Homozygotic transgenic mice carrying a dimerization domain mutant allele of the glucocorticoid receptor (GRdim A485T) that blocks DNA-binding, and transcription, still show efficient repression of inflammatory responses [48–51]. The basis for the anti-inflammatory activity of the DNA binding-defective glucocorticoid receptor was suggested to be its ability to bind to NF-κB and repress the transcription of inflammatory mediators (Fig. 2) [48]. These studies collectively indicate that transcriptional interference with NF-κB by glucocorticoid receptors is critical for the anti-inflammatory effects of glucocorticoids [15, 41].
Fig. (2). Glucocorticoids: endocrine-mediated systemic immune suppression.

In the absence of glucocorticoid steroids, the glucocorticoid receptor is maintained in the cytosol as part of a large multi-subunit complex associated with the heat-shock protein HSP90. Glucocorticoid causes the dissociation of this complex, releasing the activated glucocorticoid receptor. The anti-inflammatory therapeutic effects of glucocorticoids are predominantly mediated by transrepression of characteristic transcription factors including NF-KB and AP1, while many side effects are mediated by transactivation of target genes. Current efforts are directed to design synthetic selective glucocorticoid receptor agonists (SEGRAs) to enhance this transrepression while preventing transactivation. Transactivation of the glucocorticoids induces the expression of factors like macrophage migration inhibitory factor (MIF), which counteracts the anti-inflammatory potential of the glucocorticoids.
Glucocorticoid therapy improved the outcome in experimental models of lethal sepsis [52–54], however early clinical trials reported that glucocorticoids cause deleterious immunosuppression [55–57]. These trials used short treatment courses with high doses of steroids (up to 600 mg/kg of steroid over the course of 24 hours), which upon meta-analysis were found to be detrimental to the patients receiving treatment [57, 58]. In some cases, the harmful effects of high doses of steroids correlated with an increase in the occurrence of secondary infections [59, 60]. This correlation could reflect glucocorticoid-induced lymphocyte apoptosis, which causes T- and B-cell deficiencies leading to hyper-immunosuppression and an inability of the patient to clear an infection. The loss of T-cell immuno-regulatory function can significantly contribute to the morbidity and mortality of sepsis, resulting in exacerbated inflammation and a worsening of the clinical outcome [16].
Adrenal insufficiency and glucocorticoid hypo-responsiveness are contributing factors in the lethal outcome of sepsis [61–64]. Adrenal insufficiency is prevalent in sepsis and is found in about 50% of the patients with septic shock. Inflammatory cytokines such as TNF suppress cortisol secretion from the adrenal glands, which can result in adrenal insufficiency [65, 66]. Adrenal insufficiency is typically detected by measuring the serum levels of cortisol following the administration of 250 μg syntropin, a synthetic peptide corresponding to the first 24 amino acids of adrenocorticotropic hormone, which contains the biological activity required to evoke cortisol secretion from the adrenal gland [67]. The stress-induced cortisol level should be roughly 25 μg/dl in patients with sepsis [61], and diagnostic criteria for adrenal insufficiency include a maximal response of <20 μg/dl, or an increase <10 μg/dl over the base line [67]. However, 250 μg of syntropin is a dose >100-fold higher than the normal stress-induced adrenocorticotropic hormone level found in patients, and this excessive dose can overcome adrenal hypo-responsiveness and lead to an inaccurate diagnosis. The prevalence of adrenal insufficiency in patients with sepsis suggests that treatment with physiological doses of glucocorticoids could be beneficial [15].
Meta-analysis was performed of clinical studies testing glucocorticoids on survival and shock reversal in sepsis [68]. Results showed that studies done before 1989 used short period administration with high doses harmed patients, and worsened survival. Studies after 1989 used longer times 5 to 7 day courses of low physiologic doses (equivalent to 200 or 300 mg of hydrocortisone daily), with subsequent tapering off period (5 to 7 days), increased survival rate and shock reversal in patients with vasopressor-dependent septic shock. Glucocortecoids were beneficial when started as late as 72 hrs after the initiation of vasopressors. Classical clinical trials using glucocorticoids are based on prolonged treatment courses (5–7 days) with doses that reproduce the relative cortisol level found in healthy individuals undergoing vigorous exercise. These studies showed an improvement in the clinical outcome of severe sepsis, including patients with adrenal insufficiency. Sepsis patients with documented adrenal insufficiency received either hydrocortisone (50 mg intravenous bolus every 6 hours) and fludrocortisone (50 μg tablet once daily) (n = 114) or physiological saline placebo (n = 115) for 7 days. There were 73 deaths (63%) in the placebo group and 60 deaths (53%) in the corticosteroid group (hazard ratio, 0.67; 95% confidence interval, P =.02) [69]. In contrast to the detrimental effects of high dose short course glucocorticoid treatment [57, 58], therapeutic benefits may be achieved with low doses and prolonged therapy in patients with adrenal insufficiency. However, caution must be exercised, because prolonged glucocorticoid therapy can bear significant deleterious side effects depending upon the strength of the steroid, the dosing regimen, and the duration of treatment [70]. Glucocorticoid-induced side effects can include skin atrophy, wound healing disorders, osteoporosis, glaucoma, diabetes mellitus (hyperglycemia), adrenal insufficiency, hypertension, and immunodeficiency caused by excessive lymphocyte apoptosis [70]. These side effects can contribute to morbidity and mortality, which limits the clinical value of glucocorticoids for the treatment of severe sepsis. After many controversial earlier studies the net result being that steroids were out of favor, a low dose hydrocortisone regimen for septic shock patients in a large multicenter, placebo-controlled randomized double-blind study, where lungs were the primary source of infection, shock was reversed, and mortality decreased [69]. Along with two meta-analyses at that time, low dose steroid therapy for septic shock was again recommended. However, a more recent CORTICUS study where gastrointestinal tract was the primary source of infection, there were no differences in mortality, hydrocortisone did not increase the percentage of patients exhibiting shock reversal (although it did so more quickly), besides patients receiving hydrocortisone had more episodes of superinfection [71]. As a result of these two opposing studies, current recommendations are to use low dose hydrocortisone therapy only in adult septic shock patients who are poorly responsive to resuscitation and vasopressor therapy [64, 72]. The anti-inflammatory therapeutic effects of glucocorticoids are predominantly mediated by transrepression of other transcription factors (Fig. 2) [48], while many side effects are mediated by transactivation of target genes [48, 70]. Current efforts are directed to design synthetic selective glucocorticoid receptor agonists (SEGRAs) to enhance this transrepression while preventing transactivation [70, 73]. Further studies and clinical trials will be required to gauge their potential therapeutic benefits over glucocorticoids as a treatment for severe sepsis.
SEPSIS AND APOPTOSIS: CELL DEATH’S CONTRIBUTION TO SEPSIS MORTALITY
Lymphocyte apoptosis is accelerated during sepsis, and can be exacerbated by glucocorticoid treatment. Patients who die from sepsis-associated multiple organ failure show extensive lymphocyte apoptosis in the spleen, lymph nodes, and intestinal lamina propria [74]. Apoptosis is regulated by a cascade of cysteine-aspartyl proteases (caspases) that are critical effector molecules in programmed cell death because their activation results in the disassembly of the cell through the degradation of various structural proteins [75]. Prevention of sepsis-induced lymphocyte apoptosis by treatment with 10 mg/Kg mouse of broad-spectrum caspase inhibitors (z-VAD or M920) improved the survival rate from about 30% in control mice to roughly 75% in treated mice in cecal ligation and puncture [76, 77]. The effect of caspase inhibitors on the outcome of sepsis correlated with improved lymphocyte survival because RAG-1-deficient mice, which lack mature lymphocytes, did not benefit from treatment with caspase inhibitors [77]. In addition, lymphocytes from transgenic mice that over-express the human anti-apoptotic protein Bcl-2 were resistant to sepsis-induced apoptosis. Adoptive transfer of 1×107 T-cells from these Bcl-2 transgenic mice improved the survival rate of non-transgenic animals from about 20% to roughly 75% in experimental sepsis [77]. These studies suggested that lymphocytes, and particularly T-cells, had a critical role in the resolution of sepsis. T-cells produce interferon-γ and TNF, which activate macrophages and neutrophils. Secretion of TNF and interferon-γ from T-cells happened within 6 hours of activation, before sufficient T-cell clonal expansion could occur, suggesting that interactions between the adaptive and innate immune systems are both rapid and essential for survival in the early stages of sepsis [77–80].
Treatment with 10mg/Kg mouse of a specific inhibitor (M-791) of the effector caspase, caspase-3, also prevented lymphocyte apoptosis and improved survival [77]. There was no difference in long term survival between mice treated with M-920, the broad-spectrum caspase inhibitor, and M-791 [77]. The ability of a specific inhibitor to improve survival in experimental sepsis was significant since there are three classes of caspases; initiator caspases that trigger the apoptotic program, effector caspases that execute apoptosis, and inflammatory caspases that process inflammatory cytokines like IL-1 and IL-18 to their mature secreted form [81]. This result suggested that caspase inhibition improved the survival of mice in experimental sepsis by blocking apoptosis. Consistent with the ability of caspase-3 blockade to prevent lymphocyte apoptosis during sepsis, septic caspase-3−/− mice showed a roughly 3.5-fold reduction in the level of thymocyte apoptosis and a roughly 2-fold decrease in splenocyte apoptosis when compared to septic wild-type mice [77]. Collectively, these studies indicated that caspase-3 is a key component of the sepsis-mediated lymphocyte cell death program. It is noteworthy that NF-κB activity protects lymphocytes against apoptosis [82], and that the NF-κB p50 and p65 subunits are both sensitive caspase-3 substrates that are inactivated during apoptosis [83]. Also, caspase-3 cleavage in the regulatory region of the IκBα inhibitor of NF-κB protects IκBα from proteasome-mediated degradation, and creates a natural “super-repressor” that facilitates apoptosis by constitutively blocking NF-κB translocation [82, 84]. Therefore, caspase-3-mediated NF-κB destruction might represent an important step in the apoptotic loss of lymphocytes during sepsis. Taken together, these studies indicate that caspase-3 is a potential therapeutic target in sepsis.
Caspases can also contribute to the pathogenesis of sepsis by directly regulating cytokine production through the proteolytic processing of cytokine precursors [81, 85]. As a consequence, genetic polymorphisms in caspases can alter the production of cytokines. For example, a well documented human polymorphism in the gene encoding caspase-12 increases susceptibility to sepsis [86]. The analysis of over 1100 human genomic DNA sequences encoding caspase-12 revealed a single nucleotide polymorphism (SNP) in the stop codon of exon 4 (Stop (TGA) to Arg (CGA)), causing a read-through that results in the production of a ‘long’ isoform of caspase-12 (caspase-12-L). Although the caspase-12-L product did not produce a catalytically competent caspase, it acted as a dominant-negative regulator of inflammatory responses that prevented the activation of the NF-κB pathway and attenuated the production of cytokines during sepsis [86]. The polymorphism leading to the production of caspase-12-L was found only in subjects of African descent, and patients carrying this polymorphism had increased susceptibility to infection and a three-fold higher mortality rate in sepsis [86]. The results of this study suggested that individuals homozygous for the caspase-12-L isoform were more susceptible to infection because they produced an inadequate cytokine response, facilitating bacterial dissemination into the bloodstream, which contributed to severe sepsis [86]. A general discussion of genetic polymorphisms in human susceptibility to infection is outside of the scope of this review, however it has become increasingly evident that genetic differences in inflammatory responses exist within the human population, and that these differences may play key roles in determining the clinical outcome of infectious and inflammatory diseases [87, 88].
MACROPHAGE MIGRATION INHIBITORY FACTOR (MIF): COUNTERACTS THE ANTI-INFLAMMATORY EFFECTS OF GLUCOCORTICOIDS
Macrophage migration inhibitory factor (MIF) is a potent inflammatory cytokine that limits the anti-inflammatory effects of glucocorticoids [89, 90]. MIF induces the production of TNF and other pro-inflammatory mediators from macrophages, which in turn elicit MIF production, establishing a positively reinforcing autocrine loop that can limit the immunosuppressive effects of glucocorticoid treatment [91, 92]. MIF is the only inflammatory cytokine that is not inhibited by the glucocorticoids. Indeed, glucocorticoid stimulated MIF release in macrophages (7). The release of MIF in response to glucocorticoid treatment was dose-dependent, and followed a bell-shaped dose-response curve [92]. Secretion of MIF was induced at concentrations of dexamethasone or hydrocortisone as low as 10−16 M, with a peak between 10−14 M and 10−12 M, and was suppressed at doses above 10−10 M, which corresponds to stress-induced levels where cortisol functions as a potent immune suppressant [92]. MIF overcame the inhibitory effects of glucocorticoids (10−9 M dexamethasone), and restored the production of TNF, IL-1, IL-6, and IL-8 by monocytes stimulated with bacterial endotoxin (1μg/ml) in vitro [92]. Likewise, treatment with recombinant MIF (0.6 mg/Kg) plus dexamethasone (1.25 mg/Kg) two hours prior to the administration of a lethal dose of endotoxin to mice (22.5 mg/Kg) ablated the protective effects of dexamethasone alone [89]. As such, MIF remains unique among inflammatory cytokines in its ability to counter-regulate the anti-inflammatory effects of glucocorticoids.
MIF contributes to the pathogenesis of sepsis and other infectious and immune disorders [90]. Blockade of MIF action with specific neutralizing monoclonal antibodies protected mice from lethal sepsis induced by cecal ligation and puncture (survival was 81% in antibody treated mice and 31% in control mice). This treatment was successful even when the antibodies were administered 4.5 hours after the cecal ligation and puncture (survival was 61% in antibody treated mice and 5% in control mice) [93]. Recently, a DNA vaccine that generates anti-MIF antibodies increased survival and decreased levels of TNF, IL-1β, IL-6, TLR-4 in endotoxemic (LPS) or cecal ligation and puncture mouse models of sepsis [94]. Crystallographic studies on recombinant human MIF identified a tautomerase enzymatic activity that was reported to be important for the counter-regulation of glucocorticoid action by MIF [95]. A physiological substrate for MIF enzymatic activity has not yet been identified, however the insertion of an alanine residue between proline 1 and methionine 2 of MIF abolishes its tautomerase activity against the substrate L-dopachrome methyl ester by blocking the active site centering around proline 1 [96]. This alanine insertion also blocked the ability of the mutant MIF protein to act as a glucocorticoid counter-regulator, which suggested an important role for the N-terminal catalytic region in the biological activity of MIF [95]. Consistent with a role for enzymatic activity, the potential MIF tautomerase inhibitor (S, R)-3-(4-hydroxyphenyl)-4, 5-dihydro-5-isoxazole acetic acid methyl ester (ISO-1) was able to dose-dependently inhibit its ability to counteract the anti-inflammatory effects of 10−8 M dexamethasone on cultured monocytes that were treated with bacterial endotoxin (0.5 μg/ml) [95]. ISO-1 attenuates but did not inhibit TNF production in primary culture of peritoneal macrophages form wild-type but not from MIF-knockout mice. Such MIF inhibitors could potentially inactivate its inflammatory activity and could enhance the effectiveness of glucocorticoids in the treatment of infectious and inflammatory diseases.
LYSOPHOSPHATIDYLCHOLINE: A LIPID PHARMACOLOGICAL IMMUNOMODULATOR OF NEUTROPHILS
Plasma levels of lysophosphatidylcholine (LPC), a major component of the oxidized low-density lipoprotein, are decreased in septic patients, and these levels are especially lower in patients that succumb to the septic episode [97]. Administration of lysophosphatidylcholine protects animals from experimental sepsis, prevents lethality during bacteremia and endotoxemia, and improves survival in polymicrobial peritonitis, even when the treatment is delayed up to 10 hours after cecal ligation and puncture [98, 99]. Different molecular species of lysophosphatidylcholine differ in their activities, and 18: 0 and 18: 1 lysophosphatidylcholine were the most effective at promoting survival of mice during experimental sepsis [98, 99]. Other lysophospholipids including lysophosphatidylserine, lysophosphatidylethanolamine, lysophosphatidylinositol, lysoplatelet activating factor (L-PAF) and sphingosylphosphorylcholine did not improve survival in sepsis, which demonstrated the specificity of lysophosphatidylcholine action [99]. Lysophosphatidylcholine exerted its action through at least two distinct mechanisms; it enhanced neutrophilmediated bacterial clearance and inhibited the systemic inflammatory effects of bacterial endotoxin [98, 99]. Therefore, the stimulating effect of lysophosphatidylcholine on neutrophil bactericidal activity could be useful in treating patients with microbial infections that have not yet progressed to sepsis. Such an approach could be important, especially considering the continuous appearance of new pathogenic microbes that are resistant to currently available antibiotics.
Defects in innate immunity, particularly the dysfunction of neutrophils and macrophages, are believed to contribute to sepsis-induced mortality [100, 101]. Neutrophils derived from septic rats are impaired for hydrogen peroxide production in response to phorbol 12-myristate 13-acetate (PMA) [102]. These findings are significant because invading microbes are predominantly cleared by neutrophils, and lysophosphatidylcholine inhibits their sepsis-induced inactivation, promoting hydrogen peroxide production and bacterial clearance in vitro [99]. These findings suggest that neutrophils may be the principal target cells that govern the lysophosphatidylcholine-mediated survival in sepsis. However, lysophosphatidylcholine also affects the inflammatory responses of endothelial cells, T-cells, dendritic cells, smooth muscle cells, and monocytes [103–109]. Therefore, it is possible that the beneficial effects of lysophosphatidylcholine are the culmination of many signaling events in multiple cell types.
G2A, OGR1, and GPR4, members of the 7 transmembrane domain-containing G protein-coupled receptor superfamily, were identified as lysophosphatidylcholine receptors [110]. The G2A receptor is a key immuno-regulator [111] that has an important role in the protective effects of lysophosphatidylcholine during experimental polymicrobial sepsis- and bacterial endotoxin-induced lethality [99]. Genetic ablation of G2A in mice resulted in the development of a progressive autoimmune wasting syndrome, characterized by lymphocyte infiltration into various tissues, glomerular immune complex deposition, and anti-nuclear autoantibodies [112]. G2A-deficient T-cells were self-reactive and hyper-responsive to T-cell receptor stimulation, exhibiting enhanced proliferation and a lower threshold for activation [112]. Activation of G2A by lysophosphatidylcholine increased intracellular calcium concentration, activated the ERK mitogen-activated protein kinase pathway, and modified migratory responses of Jurkat T-cells [113], and inhibited NF-κB activation in a variety of cell types [114]. Specific antibodies that block activation of the G2A receptor inhibited the stimulating effect of lysophosphatidylcholine on hydrogen peroxide production from neutrophils in vitro, and blocked its protective effect on mice during experimental sepsis [99]. Although the G2A receptor plays an important role in mediating the anti-inflammatory effects of lysophosphatidylcholine, it is likely that other receptors like GPR4 and OGR1 also function in an overlapping manner to modulate cellular immune responses [111]. Future work should resolve the relative contributions of these receptors to the action of lysophosphatidylcholine, and their suitability as potential drug targets for the treatment of severe sepsis.
PHARMACOLOGICAL INHIBITION OF HIGH MOBILITY GROUP BOX 1 (HMGB1)
Neutralization of TNF or IL-1 can prevent the development of septic shock in animals [115], but these strategies produced modest clinical effects in critically ill patients [12–14]. Several studies suggest that other “late” downstream pro-inflammatory cytokines may contribute to the progression of sepsis. In agreement with this hypothesis, high doses of endotoxin are lethal to TNF-deficient mice, and death from endotoxemia and sepsis often occurs days after the “early” TNF production, long after serum TNF had returned to basal levels [2, 116]. These “late” downstream mediators hypothetically present a clinical advantage for the treatment of sepsis, because they expand the therapeutic time window [2, 116]. Unlike “early” cytokines, High Mobility Group Box1 (HMGB1) acts as a “late” mediator of lethal systemic inflammation in sepsis [116]. Originally described as an intracellular DNA-binding protein, HMGB1 is released into the extracellular milieu by activated macrophages. Extracellular HMGB1 acts as a pro-inflammatory cytokine; it induces tissue-type plasminogen activator, stimulates the production of pro-inflammatory cytokines (TNF, IL-1β and IL-8), and promotes epithelial cell permeability and neutrophil accumulation [117–120]. HMGB1 is a sufficient and necessary mediator for sepsis: (A) Systemic HMGB1 is found in animal models of sepsis and in the serum of patients with bacteremia and sepsis-induced organ dysfunction [116]; (B) Administration of recombinant HMGB1 to mice recapitulates the most characteristic clinical signs of sepsis including fever, malaise, derangement of the intestinal barrier function, acute lung injury and lethal multiple organ failure [119–121]; (C) Anti-HMGB1 antibodies prevent lethal endotoxemia and reverse the pathological course of established sepsis [122]. HMGB1 was defined as a “late” mediator of sepsis because endotoxin induces HMGB1 release from macrophages approximately 24 hours after the production of TNF. This delayed release is also observed in vivo, where HMGB1 is released into the serum approximately 24 hours after the induction of endotoxemia, bacteremia or peritonitis [116, 117, 122]. As a consequence, anti-HMGB1 therapeutics are successful even if administered after the appearance of clinical signs of sepsis [117, 122]. The late role for HMGB1 in mediating severe sepsis is akin to the early role for TNF as a prototype mediator of “septic shock”. In contrast to severe sepsis, “septic shock” is a highly lethal acute syndrome that kills within 24–48 hours, and is invariably accompanied by ischemic necrosis and cardiovascular collapse [2]. TNF administration can trigger the entire pathological spectrum of septic shock including hypotension, intravascular coagulopathy and hemorrhagic necrosis, and neutralization of TNF prevents endotoxic- or bacteremic-induced shock, even when endotoxins or bacteria persist in the circulation [123]. Therefore, it is significant that ethyl pyruvate was effective for rescuing mice from lethal sepsis even when its administration was delayed one day after cecal ligation and puncture [117, 124]. On the other hand, HMGB1 appears to be a prototype mediator for “severe sepsis”. Severe sepsis is a protracted inflammatory syndrome that kills over the course of several days or weeks [2]. Elevated systemic HMGB1 levels are found in patients and in experimental models of sepsis-induced organ dysfunction [125]. The administration of recombinant HMGB1 to mice recapitulates the characteristic organ dysfunction of severe sepsis [118, 122, 126–128], and independent strategies that inhibit either HMGB1 activity [122, 125], or its secretion prevent multiple organ failure and rescue mice in experimental models of severe sepsis [17, 98, 99, 117, 124].
Since many strategies based on antibodies neutralizing particular cytokines have failed in sepsis, a significant effort was made to design pharmacological inhibitors for HMGB1 secretion in sepsis. Ethyl pyruvate was one of the first pharmacological inhibitors described for HMGB1 secretion [117, 124]. These studies were of significant interest because ethyl pyruvate provides wider pharmacological time window for experimental sepsis [1, 2, 5, 6, 129]. Ethyl pyruvate was initially designed as a pyruvate-like product to mimic the therapeutic potential of pyruvate while avoiding its collateral toxicity. Pyruvate is a physiological product of glucose metabolism that has been shown to be beneficial in experimental models of stroke, hemorrhagic shock, and cardiac, hepatic or mesenteric ischemia and reperfusion [130–132]. The beneficial effects of pyruvate are based on its positive myocardial inotropic effects by acting as an energy source, and a potential antioxidant and anti-inflammatory capability [131–133]. Despite its potential benefits, pyruvate is not stable in solution and rapidly undergoes an aldol-like condensation reaction to form parapyruvate, which can spontaneously convert to 2, 4-dihydroxy-2-methylglutarate, a mitochondrial poison [134, 135]. Ethyl pyruvate is a more stable simple aliphatic ester derivative of pyruvic acid, classified as a GRAS (Generally Recognized As Safe) according to the guidelines of the FDA [136]. Ethyl pyruvate provides therapeutic protection in experimental models of hemorrhagic shock [137, 138], ischemia/reperfusion injury [130, 139, 140], acute pancreatitis [141, 142], endotoxemia and polymicrobial sepsis [117, 143, 144]. However, a recent study indicates that a single dose of ethyl pyruvate administered at the same time as LPS may worsen survival in endotoxemia [145] depending on the dose and time of administration [146]. These studies are of particular interest since a recent phase II trial using ethyl pyruvate showed no therapeutic benefits in cardiovascular bypass surgery [147]. Despite these clinical results, the anti-inflammatory mechanism of ethyl pyruvate shed light on the pharmacological inhibition of HMGB1 in sepsis.
The durability of ethyl pyruvate protection argues that its principal anti-inflammatory effect involves the persistent modification of a intracellular component in order to inhibit HMGB1 secretion [148]. Since pyruvate is known to form reversible covalent bonds with the sulfur moiety of cysteine residues [149], one possible target for ethyl pyruvate is the cysteine-containing tri-peptide redox sensor glutathione [148], whose oxidation state influences NF-κB activity [149]. The ability of glutathione to influence NF-κB activity involves a key cysteine residue (Cys 62) in the p50 (c-Rel) subunit that is essential for DNA binding [150]. This cysteine residue is in a sequence context that makes it susceptible to oxidation, resulting in S-glutathionylation of the p50 subunit [151]. Likewise, treatment of 293 cells with ethyl pyruvate ablated the binding of p65 (Rel-A) homodimers to DNA [152]. This effect of ethyl pyruvate on p65 DNA binding required a cysteine residue at position 38 of p65 because treatment of 293 cells transfected with an expression vector carrying a cys38-ser substitution with ethyl pryuvate showed no decrease in p65 DNA binding [152]. This result indicated that ethyl pyruvate might covalently modify cys38 of the p65 subunit and effectively block its ability to bind DNA. Consistent with this hypothesis, the crystal structure of NF-κB p50/p65 hetero-dimers complexed with DNA showed that cys38 in the p65 subunit participates directly in DNA binding [153]. Collectively, these studies suggest that ethyl pyruvate does not exert its anti-inflammatory effects through a single mechanism, but rather that multiple modes of action converge on the inhibition of NF-κB activity [154]. Regardless of the underlying mechanism, ethyl pyruvate is an effective pharmacological inhibitor of NF-κB activity that conveys durable protection in experimental models of infectious and inflammatory diseases.
One of the most controversial and significant scientific challenges is the mechanism of HMGB1 production and its biological activity. Initial studies indicated that Extracellular HMGB1 binds to the cellular receptor for advanced glycation end-products (RAGE) in a concentration-dependent manner [155]. RAGE is a transmembrane protein that belongs to the immunoglobulin super-family, and acts as a receptor for diverse ligands including advanced glycation end-products, amyloid peptide, members of the S100 family of inflammatory-mediating peptides, and is currently the best characterized cellular receptor for HMGB1 [156, 157]. Recent studies suggest that in addition to RAGE, TLR-2 and TLR-4 also mediate the cytokine activity of HMGB1 [6, 17]. The specific contributions of RAGE, TLR-2 and TLR-4 to the cytokine activity of HMGB1 remain unclear in part because these receptors share common downstream effector molecules (especially NF-KB, JNK, and p38 MAPK) that stimulate similar intracellular signaling events [6, 17]. This sharing of signaling components makes genetic studies using dominant-negative constructs and heterologous expression systems difficult to interpret, and has lead to contradictory conclusions in the literature regarding the roles of RAGE, TLR-2 and TLR-4 in executing the cytokine activity of HMGB1 [17, 157, 158]. In addition to the receptor, recent studies debate the biological activity of highly pure HMGB1, suggesting that HMGB1 may rather serve as a carrier for other more potent proinflammatory products. Along this line, recent studies suggest that redox reactions cause misfolding and inactivation of DAMPs. The recent reported oxidation-dependent inactivation of HMGB1 may modulate its inflammatory potential and it may also contribute to HMGB1 inactivation during extensive purification processing. One significant consideration is whether these controversial results may be explained in part by the mechanisms of HMGB1 production. There appears to be two potential mechanisms for cells to liberate HMGB1 into the extracellular milieu (Fig. 3). The first mechanism is a “passive release” of HMGB1 from damaged or necrotic cells. In this context, the extracellular HMGB1 released during necrosis acts as an immune-stimulatory signal that indicates the extent of tissue injury [159]. The second mechanism is by “active secretion” of HMGB1 from immune cells. During an immunological challenge, extracellular HMGB1 secreted by immune cells acts as a conventional pro-inflammatory cytokine [119, 160]. Both mechanisms result in significant levels of extracellular HMGB1 that can trigger a systemic inflammatory response to ischemia, trauma, burn, infection, or sepsis. Future studies are needed to further determine the mechanisms of HMGB1 production and its biological activity [161].
Fig. (3). HMGB1: from the septic DAMP to the immune secretion.

There are two potential mechanisms for cells to liberate HMGB1 into the extracellular milieu. Somatic cells contain large amounts of HMGB1 that is “passively released” into the extracellular milieu following cell membrane perturbation during cellular damage, unresolved apoptosis or necrosis. In this scenario, HMGB1 represents an intracellular protein adopted by the innate immune system to recognize tissue damage and initiate reparative response. Recent studies suggest that the immune system has copied this mechanism to activate innate responses and initiate tissue repair. The second mechanism can be an “active secretion” of HMGB1 from immune cells to mimic the necrotic process and activate innate immune response during an immunological challenge. From an immunological perspective, HMGB1 represents a characteristic “necrotic marker” or damage-associated molecular pattern (DAMP) molecule selected to activate the immune system.
CHOLINERGIC ANTI-INFLAMMATORY PATHWAY: A NERVOUS GRIP ON THE ANTI-INFLAMMATORY REIGNS
The central nervous system can modulate innate immune responses and prevent systemic inflammation via the vagus nerve. Unlike the systemic endocrine mechanism of glucocorticoids [15], the vagus nerve represents a direct physical link between an actual neuronal network and the immune system [1, 2, 162]. This characteristic may provide two major advantages: 1) it is more precise than systemic endocrine mechanisms and is able to produce a “real time” immunomodulatory effect, 2) the vagus nerve can generate an integrated host response that incorporates diverse inputs and signals. Precise regulation of the inflammatory cytokine response to infection or injury is critical because deficient responses can cause a failure in clearing an infection, while excessive responses can be more injurious than the initial insult [1, 2]. The anti-inflammatory effects of vagus nerve stimulation are most likely mediated by acetylcholine, the principal neurotransmitter of the vagus nerve. Acetylcholine blocks the release of inflammatory cytokines from cultured primary human macrophages and murine macrophages through an alpha7nAChR-dependent mechanism (Fig. 4) [1, 17, 19, 162]. Similar to acetylcholine, treatment of cultured macrophages with nicotine, a more selective alpha7nAChR-agonist, dose-dependently inhibited inflammatory cytokine secretion to a maximum of about 90% compared to vehicle controls [163]. The inhibitory concentration of nicotine that blocked 50% of the TNF response (IC50) was lower than that required for acetylcholine (IC50 8.3 +/− 7.1 nM and 20.2 +/− 8.7 nM, respectively), indicating that nicotine was a more potent inflammatory inhibitor [17, 19, 163]. Treatment of cultured macrophages with nicotine specifically prevented endotoxin-induced NF-κB activation, as the p38, JNK, and ERK signaling pathways were not significantly affected (Fig. 4) [17]. NF-κB activity is critical for the production of inflammatory cytokines by macrophages [47], and nicotine treatment dose-dependently blocked expression of an NF-κB-dependent luciferase reporter construct in bacterial endotoxin challenged macrophages [17]. The suppression of NF-κB activity by nicotine required the alpha7nAChR because specific antisense oligos against the alpha7 subunit blocked its expression and restored luciferase production even in the presence of nicotine or acetylcholine [17]. These results indicate that nicotine blocks inflammatory cytokine production by inhibiting the NF-κB pathway through an alpha7nAChR-dependent mechanism. This physiological mechanism, termed the “nicotinic anti-inflammatory pathway” because it is activated by nicotine and is dependent on the alpha7nAChR can be exploited therapeutically as a treatment for infectious and inflammatory disorders [1, 17, 164]. Nicotine also inhibits the nuclear translocation of NF-κB in microvascular endothelial cells. This inhibition of NF-κB resulted in diminished TNF-induced expression of leukocyte adhesion molecules (ICAM-1, VCAM-1 and E-selectin) and pro-inflammatory chemokines (IL-8, MCP-1, and RANTES) [165]. Similar anti-inflammatory effects of nicotine have been reported from studies using an experimental model system of spinal cord injury [166]. Therefore, nicotine-induced inhibition of NF-κB activity may represent a general mechanism for its anti-inflammatory action. Indeed, similar regulation of the NF-kB pathway by nicotine has been confirmed in other cell lines including human monocytes (U937) and microvascular endothelial (HuMVEC) cells. However, it is unknown whether NF-κB inhibition is a direct target of the “nicotinic anti-inflammatory pathway”, as the molecular components of this signaling pathway remain largely uncharacterized. These results suggest that pharmacological manipulation of the “nicotinic anti-inflammatory pathway” can be exploited therapeutically for the treatment of infectious and inflammatory disorders [1, 17, 164].
Fig. (4). Nicotinic anti-inflammatory potential in sepsis.

The vagus nerve can modulate the innate immune response and restrain inflammation through a physiological mechanism that can be translated into a pharmacological strategy. Since acetylcholine, the principal neuro-transmitter of the vagus nerve, signals through either muscarinic or nicotinic receptor, selective agonists (atropine, conotoxin, or mecamylamine) were used to identify the receptors involved in the control of macrophages. The vagus nerve and acetylcholine can restrain the production of pro-inflammatory cytokines from macrophages through a nicotinic acetylcholine receptor. Nicotine, a more selective cholinergic agonist, is more efficient than acetylcholine, and inhibits the production of pro-inflammatory cytokines from macrophages through a mechanism dependent on the alpha7nAChR.
Vagus nerve stimulation attenuates inflammatory responses and protects mice in experimental models of endotoxemia [18, 162]. The protective effect of vagus nerve stimulation was recapitulated by treatment of endotoxemic mice with 400 μg nicotine/Kg. This treatment improved their survival to 81% as compared to 44% for vehicle treated mice (P<0.05) [17]. Nicotine was also effective for rescuing mice with established sepsis when treatment began one day after the induction of lethal peritonitis by cecal ligation and puncture. Nicotine therapy improved the survival rate from roughly 50% in control mice to 85% in nicotine-treated animals [17]. Rescue of these mice correlated with the attenuation of systemic HMGB1 levels [17]. This effect is of special interest because HMGB1 appears to be a necessary and sufficient mediator of lethal sepsis [3, 116], and treatments that block HMGB1 cytokine activity rescue animals from experimental sepsis [5, 6]. These studies indicated that alpha-7nAChR–agonists can be effective therapeutic agents for preventing HMGB1 secretion, and improving survival in sepsis (Fig. 4).
Recent studies indicate that alpha7nAChR signaling can protect neurons against β-amyloid protein in a neuronal cell culture model system of Alzheimer’s disease [167]. Treatment of PC12 cells with nicotine leads to the activation of the tyrosine kinase Jak2 and its downstream signaling cascades, resulting in neuroprotection. The neuroprotective effects of nicotine were lost upon pre-treatment of the cells with AG490, a specific Jak2 kinase inhibitor [167]. A study using a mouse model of intestinal post-operative ileus indicated that a similar pathway can function in macrophages downstream of the alpha7nAChR to govern the anti-inflammatory effects of nicotine [168]. Jak2 was constitutively associated with the alpha7nAChR in mouse peritoneal macrophages, and it was activated upon treatment with nicotine. Activated Jak2 triggered the activity of signal transducer and activator of transcription (STAT)-3 in these cells [168]. The anti-inflammatory effect of vagus nerve stimulation on macrophages required STAT-3 in vivo because vagus nerve stimulation in mice defective in STAT-3 expression specifically in macrophages and granulocytes (LysM-Stat3fl/− mice) did not attenuate intestinal inflammation or peritoneal IL-6 levels compared to control mice (LysM-Stat3fl/+ mice) that underwent the same intestinal manipulation [168]. These studies suggested that the “nicotinic anti-inflammatory path- way” triggered by alpha7nAChR stimulation includes the Jak2/STAT-3 pathway in macrophages. Current studies are oriented towards determining whether Jak2/STAT-3 pathway action in response to nicotine causes suppression of the NF-κB pathway in macrophages, and whether other alpha7nAChR signaling mechanisms, such as those controlled by intracellular calcium, contribute to the protective effects of the “nicotinic anti-inflammatory pathway”.
The discovery of the “nicotinic anti-inflammatory pathway” has opened new avenues for the development of pharmacological therapies for the treatment of sepsis and other inflammatory disorders. Indeed, nicotine has been successfully used in clinical trials for the treatment of ulcerative colitis, an inflammatory bowel disorder. However, nicotine has clinical limitations because it is not a specific alpha- 7nAChR-agonist, and it has potentially toxic effects when administered at clinically relevant doses [1, 169]. Therefore, more selective alpha7nAChR agonists including GTS21 ((3-[(2, 4-dimethoxy)benzylidene]-anabaseine dihydrochloride) [170] and TC-1698 (2-(3-Pyridyl)-1-azabicyclo[3.2.2]nonane) [171] could overcome clinical limitations of nicotine and be more effective therapeutic agents for the treatment of sepsis. For example, in contrast to nicotine, GTS21 had no effect on locomotor activity in mice or dopamine turnover in rats [172], and was well tolerated at doses up to 450 mg/day in clinical trials for the treatment of Alzheimer’s disease-induced dementia [173]. These results provided evidence that selective nicotinic agonists could be less toxic than nicotine, and could be administered at clinically relevant doses. The efficacy of GTS-21 and TC-1698 for the treatment of sepsis remains to be determined, however their reduced toxicity and ability to more selectively target the alpha7nAChR indicates that they might be useful novel therapeutic agents for the treatment of infectious and inflammatory diseases.
DID WE MISS THE INTERPRETATION OF THE EXPERIMENTAL RESULTS OR THE DESIGN OF THE CLINICAL TRIALS?
Notwithstanding more than several decades of research progress, sepsis remains a major scientific and clinical challenge in modern medicine [8, 174]. Therapeutic attempts have focused on inflammatory mediators that seem to work well in experimental models but fail to translate efficacy in clinical trials [9, 175, 176]. An obvious debate includes the characteristic of the experimental models of sepsis including endotoxemia, bacteremia, pneumonia, polymicrobial sepsis and cecal ligation and puncture (CLP) [176, 177]. Among them, bacterial endotoxemia and cecal ligation and puncture are the most popular. Bacterial lipopolysaccharide (LPS) or peptidoglycan (PGN) infusions employ large amounts of toxic bacterial cell wall components and clinical symptoms develop within a few hours as opposed to the protracted progression of cecal ligation and puncture (CLP) [176]. Endo-toxemia is normally associated with an acute response with very high levels of inflammatory cytokines (e.g., IL-1β, TNF-α, IL-6), while cecal ligation and puncture has more moderate levels of cytokines and significant production of C5a (anaphylatoxin) [178]. In the LPS model there are protective effects with anti-TNF, but not in the CLP model [176], as well as very low or lack of LPS levels in CLP, and anti-C5a increased survival in CLP rats [100]. Humans have a different profile of cytokine release than found in LPS models, and cytokines peaked much later, and were found at lower levels than in experimental models. Overall, CLP has been the “gold standard” and represents human sepsis more closely than LPS, which at higher doses is a better model for endotoxic shock [179, 180]. However, consistency can be a problem in CLP. The length of the ligated cecum was a major determinant of mortality [181]. Serum levels of IL-6 and TNF increased with increasing ligated cecum length. The needle size and number of punctures, fluid resuscitation, and antibiotic treatment in CLP led to variability of results [178]. Consistency and reproducibility (requiring large number of animals for statistical significance) need to be considered in their evaluation. LPS or mass infusion of live organisms may not be relevant to human sepsis. Even CLP may not be a relevant surrogate of human sepsis, since there is increasing evidence that human sepsis may also be caused by Gram positive bacteria, fungi and parasites [182–184]. By either method, excessive proinflammatory release can result in consumptive depletion of the clotting system. Excessive release of anti-inflammatory cytokines can lead to immunosuppression or anergy. A more reproducible variation involving intraperitoneal implantataion of fibrin clots embedded with live E. coli in a model using conscious dogs resembled human sepsis [185] and septic shock [186]. This model improves precision but is a less accurate simulation of peritonitis than CLP. Although Baboons have been used [187, 188], whether non-human primates may provide different information than that already gained from studies with septic rodents is debatable and involves ethical considerations. The expense, animal welfare concerns, and need to have ICU facilities make this difficult to achieve. A more recent and successful attempt to make these models less expensive and more practical using conscious rats [178], still has not brought us any closer to solving the translational problems leading to successful clinical trials.
A fundamental difference between classical experimental models and clinical trials is the incidence of age in sepsis. Mice used in experiments are usually young animals <3 months old, average life-span of 24 months, some inbred, same weight, healthy; whereas humans with sepsis are typically > 60 years old [8], have different ethnicities or predisposing genetic and variable physical makeup, with comorbidities (hypertension, pre-existing immunosuppressions, diabetes, or other diseases common to old age). There are also significant differences at the molecular level. Humans have C-reactive protein (CRP), an acute-phase marker for inflammation, which activates the complement system. Rat CRP does not influence the complement system [189]. Also, there is a difference in toll-like receptors (TLRs) in mice and humans [190]. For instance, TLR10 appears to be produced in human but not mice, and TRL11 is represented in humans only by a pseudogene. Animals typically receive anesthesia (morphine), inotropic drugs during sepsis-inducing procedures which can lead to immunosuppression, and therefore invalidate conclusions.
Pneumonia is the leading cause of infectious deaths in the developing world and the lungs are the most common site of infection in the septic patients. During sepsis and severe infection, the lungs are the most frequently affected organs. Community-acquired pneumonia is commonly caused by Streptococcus pneumoniae, whereas ventilator-associated pneumonia is usually due to Pseudomonas aeruginosa. Pneumonia frequently leads to sepsis and organ failure due to the lung being the first structural respiratory barrier breached. However, cytokine-mediated breach of the gut also causes lung injury and lung “systemic pneumonia” by factors produced in the lymph and contributing to acute respiratory distress syndrome (ARDS) [191]. ARDS and pneumonia are associated with local activation of coagulation within the airways mediated by the tissue factor pathway. In experimental models of ARDS, intratracheal administration of recombinant HMGB1 led to neutrophil accumulation, lung edema and increased levels of proinflammatory cytokines MIP-2, TNF and IL-1β [192]. HMGB1 antibodies decreased neutrophil accumulation, edema and lung permeability. In another model, instillation of endotoxin in mice caused inflammatory cell migration to the lungs, destruction of pulmonary parenchyma cells, pulmonary hemorrhage, and acute lung injury. High levels of HMGB1 were found in bronchiolar fluid lavage implicating macrophage involvement. At first, macrophages are activated and attempt to localize or ward off foreign pathogens under normal circumstances. In ARDS or pneumonia, macrophages become overwhelmed. Uncontrolled activation of macrophages (and breached intestinal barrier function) have been implicated in the development of ARDS and multiple organ failure. Toxic effects of zymosan and organ damage in macrophage- depleted mice were due to excessive activation of macrophages rather than systemic spread [193]. Lung defense against pathogens involves innate and acquired immune responses. Pathogen-associated molecular pattern recognition molecules (PAMPS) such as Toll-like receptors (TLR) play a pivotal role of the dendritic cell in linking the innate and adaptive immune response. Experimental models include TLR-knockout mice for (a) TLR4, which recognize LPS, and Gm- bacteria [194], (b) TLR2, which recognize peptidoglycan, and Gm+ bacteria [195], and (c) TLR2+TRL6 together, which recognize diacetylated lipoproteins (mycoplasma), and zymosan (fungal cell protein) [196, 197].
Another critical consideration is the scientific design of the clinical trial and the selection of the patients. Inflammatory cytokines represent molecular messages that codify for a precise immune response against infection, trauma or injury. Similar to a molecular fingerprint, a “pathological” profile of cytokine production results in a characteristic constellation of clinical symptoms. The characterization of this putative “cytokine code” will allow the translation of a specific group of proinflammatory cytokines into a specific constellation of clinical symptoms, and thus, immune disorders can be associated with the causative molecular derangements (Fig. 5). The translation of this code represents a major advantage for the future design of clinical trials against sepsis and other inflammatory disorders. In agreement with this concept, recent studies in sepsis suggest that TNF is a mediator of “septic shock”, and HMGB1 is a prototype mediator of “severe sepsis”. This fundamental approach provides possible reasons for the failure of previous clinical trials against sepsis. For example, the majority of patients with “severe sepsis” in large-scale trials of anti-TNF did not have shock and they were enrolled many hours or days into their clinical course [21, 22]. At this time, the majority of the patients had almost non-detectable TNF levels, and in consequence, it would not be very surprising that these patients with “severe sepsis” have not received significant benefit from a therapy that specifically targeted shock-induced TNF levels. As similarly required in the experimental models, a major advantage in the design of the clinical trials will be the shift from clinical to molecular determinants of patient selection.
Fig. (5). The Cytokine Code.
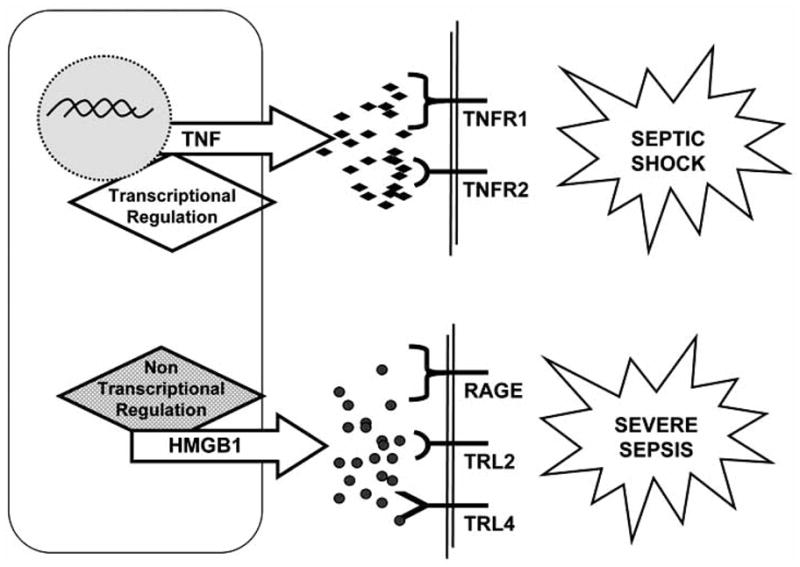
The definition of sepsis is too broad and may not necessary represent a particular clinical entity. The situation is analogous to attempting a clinical trial based on the general definition of cancer, combining all patients with tumor independent of the type of malignancy. Inflammatory cytokines represent molecular messages that codify for a precise immune response against infection, trauma or injury. Similar to a molecular fingerprint, a “pathological” profile of cytokines production results in a characteristic constellation of clinical symptoms. Under this consideration, TNF represents a characteristic mediator of “septic shock”, whereas HMGB1 has been proposed as a prototype for severe sepsis. The characterization of this “cytokine code” will allow to associate particular molecular patterns for the characterization of discreet groups of patients or particular clinical entities generally lumped under the general definition of sepsis.
Although we have learned a great deal of information, promising experimental strategies did not efficiency translate to clinical trials [176, 198]. Since new causes of sepsis are emerging, one might use live Candida [182] or fungal extracts, or Gram-positive bacteria [184, 199], live methicillin-resistant Staph. aureus MRSA [200] or staphylococcal toxins [176], with or without Gram negative bacteria [201, 202] or LPS, for currently relevant models of endotoxemia. Although Baboons, and human volunteers [203, 204], have been used in preliminary immunological assessments of cytokine levels in endotoxemia, this poses ethical or practical concerns for routine or large scale realistic experimental sepsis models. Possibly using older mice or rats, especially with appropriate known human disease models (e.g., diabetetic mice) might be helpful. It would be extremely difficult to perform the cecal ligation and puncture procedure without using anesthesia. Also, disconnects betweens experimental models and humans (e.g., C-reactive protein function, different TLRs, or genetics, or different signaling pathways, or known physiological differences) represent insurmountable obstacles. Still, in spite of these hurdles, animal models of sepsis have provided a great deal of potential translational knowledge, and are probably the only tools we have for eventually achieving clinical success in the study of inflammation and sepsis.
Acknowledgments
This review focuses on some of the most significant concepts of inflammation in sepsis. However, the field is very broad and we apologize to those authors whose work couldn’t be cited due to space limitations. LU is supported by the faculty program of the Department of Surgery of the New Jersey Medical School, and grants from the US Army Medical Research Command (USAMRMC#05308004), the American Heart Association (AHA06352230N), and the NIH (RO1-GM084125).
References
- 1.Ulloa L. The vagus nerve and the nicotinic anti-inflammatory pathway. Nat Rev Drug Discov. 2005;4:673–84. doi: 10.1038/nrd1797. [DOI] [PubMed] [Google Scholar]
- 2.Ulloa L, Tracey KJ. The “cytokine profile”: a code for sepsis. Trends Mol Med. 2005;11:56–63. doi: 10.1016/j.molmed.2004.12.007. [DOI] [PubMed] [Google Scholar]
- 3.Ulloa L, Messmer D. High-mobility group box 1 (HMGB1) protein: friend and foe. Cytokine Growth Factor Rev. 2006;17:189–201. doi: 10.1016/j.cytogfr.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 4.Deitch EA, Dayal SD. Intensive care unit management of the trauma patient. Crit Care Med. 2006;34:2294–301. doi: 10.1097/01.CCM.0000233857.94604.73. [DOI] [PubMed] [Google Scholar]
- 5.Mantell LL, Parrish WR, Ulloa L. Hmgb-1 as a therapeutic target for infectious and inflammatory disorders. Shock. 2006;25:4–11. doi: 10.1097/01.shk.0000188710.04777.9e. [DOI] [PubMed] [Google Scholar]
- 6.Lotze MT, Tracey KJ. High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat Rev Immunol. 2005;5:331–42. doi: 10.1038/nri1594. [DOI] [PubMed] [Google Scholar]
- 7.Angus DC, Linde-Zwirble WT, Lidicker J, Clermont G, Carcillo J, Pinsky MR. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit Care Med. 2001;29:1303–10. doi: 10.1097/00003246-200107000-00002. [DOI] [PubMed] [Google Scholar]
- 8.Martin GS, Mannino DM, Eaton S, Moss M. The epidemiology of sepsis in the United States from 1979 through 2000. N Engl J Med. 2003;348:1546–54. doi: 10.1056/NEJMoa022139. [DOI] [PubMed] [Google Scholar]
- 9.Riedemann NC, Guo RF, Ward PA. Novel strategies for the treatment of sepsis. Nat Med. 2003;9:517–24. doi: 10.1038/nm0503-517. [DOI] [PubMed] [Google Scholar]
- 10.Abraham E, Matthay MA, Dinarello CA, Vincent JL, Cohen J, Opal SM, et al. Consensus conference definitions for sepsis, septic shock, acute lung injury, and acute respiratory distress syndrome: time for a reevaluation. Crit Care Med. 2000;28:232–5. doi: 10.1097/00003246-200001000-00039. [DOI] [PubMed] [Google Scholar]
- 11.Riedemann NC, Guo R-F, Ward PA. The enigma of sepsis. J Clin Invest. 2003;112:460–7. doi: 10.1172/JCI19523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fisher CJJ, Dhainaut JF, Opal SM, Pribble JP, Balk RA, Slotman GJ, et al. Recombinant human interleukin 1 receptor antagonist in the treatment of patients with sepsis syndrome. Results from a randomized, double-blind, placebo-controlled trial. Phase III rhIL-1ra Sepsis Syndrome Study Group. JAMA. 1994;271:1836–43. [PubMed] [Google Scholar]
- 13.Abraham E, Anzueto A, Gutierrez G, Tessler S, San Pedro G, Wunderink R, et al. Double-blind randomised controlled trial of monoclonal antibody to human tumour necrosis factor in treatment of septic shock. NORASEPT II Study Group. Lancet. 1998;351:929–33. [PubMed] [Google Scholar]
- 14.Abraham E, Laterre PF, Garbino J, Pingleton S, Butler T, Dugernier T, et al. Lenercept (p55 tumor necrosis factor receptor fusion protein) in severe sepsis and early septic shock: a randomized, double-blind, placebo-controlled, multicenter phase III trial with 1,342 patients. Crit Care Med. 2001;29:503–10. doi: 10.1097/00003246-200103000-00006. [DOI] [PubMed] [Google Scholar]
- 15.Smoak KA, Cidlowski JA. Mechanisms of glucocorticoid receptor signaling during inflammation. Mech Ageing Dev. 2004;125:697–706. doi: 10.1016/j.mad.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 16.Yeager MP, Guyre PM, Munck AU. Glucocorticoid regulation of the inflammatory response to injury. Acta Anaesthesiol Scand. 2004;48:799–813. doi: 10.1111/j.1399-6576.2004.00434.x. [DOI] [PubMed] [Google Scholar]
- 17.Wang H, Liao H, Ochani M, Justiniani M, Lin X, Yang L, et al. Cholinergic agonists inhibit HMGB1 release and improve survival in experimental sepsis. Nat Med. 2004;10:1216–21. doi: 10.1038/nm1124. [DOI] [PubMed] [Google Scholar]
- 18.Czura CJ, Tracey KJ. Autonomic neural regulation of immunity. J Intern Med. 2005;257:156–66. doi: 10.1111/j.1365-2796.2004.01442.x. [DOI] [PubMed] [Google Scholar]
- 19.Wang H, Yu M, Ochani M, Amella CA, Tanovic M, Susarla S, et al. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature. 2003;421:384–8. doi: 10.1038/nature01339. [DOI] [PubMed] [Google Scholar]
- 20.Bernard GR, Vincent JL, Laterre PF, LaRosa SP, Dhainaut JF, Lopez-Rodriguez A, et al. Efficacy and safety of recombinant human activated protein C for severe sepsis. N Engl J Med. 2001;344:699–709. doi: 10.1056/NEJM200103083441001. [DOI] [PubMed] [Google Scholar]
- 21.Esmon CT. The protein C pathway. Chest. 2003;124:26S–32S. doi: 10.1378/chest.124.3_suppl.26s. [DOI] [PubMed] [Google Scholar]
- 22.Coughlin SR. Thrombin signalling and protease-activated receptors. Nature. 2000;407:258–64. doi: 10.1038/35025229. [DOI] [PubMed] [Google Scholar]
- 23.Joyce DE, Gelbert L, Ciaccia A, DeHoff B, Grinnell BW. Gene expression profile of antithrombotic protein C defines new mechanisms modulating inflammation and apoptosis. J Biol Chem. 2001;276:11199–203. doi: 10.1074/jbc.C100017200. [DOI] [PubMed] [Google Scholar]
- 24.Bernard GR. Drotrecogin alfa (activated) (recombinant human activated protein C) for the treatment of severe sepsis. Crit Care Med. 2003;31:S85–93. doi: 10.1097/00003246-200301001-00012. [DOI] [PubMed] [Google Scholar]
- 25.Bertolini G, Rossi C, Anghileri A, Livigni S, Addis A, Poole D. Use of Drotrecogin alfa (activated) in Italian intensive care units: the results of a nationwide survey. Intens Care Med. 2007;33:426–34. doi: 10.1007/s00134-007-0554-x. [DOI] [PubMed] [Google Scholar]
- 26.Kanji S, Perreault MM, Chant C, Williamson D, Burry L. Evaluating the use of Drotrecogin alfa (activated) in adult severe sepsis: a Canadian multicenter observational study. Intensive Care Med. 2007;33:517–23. doi: 10.1007/s00134-007-0555-9. [DOI] [PubMed] [Google Scholar]
- 27.Williams G. Recently published papers: therapies failed, disputed, and beneficent. Crit Care. 2007;11:143. doi: 10.1186/cc5931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eichacker PQ, Natanson C. Increasing evidence that the risks of rhAPC may outweigh its benefits. Intensive Care Med. 2007;33:396–9. doi: 10.1007/s00134-007-0556-8. [DOI] [PubMed] [Google Scholar]
- 29.Finfer S, Ranieri VM, Thompson BT, Barie PS, Dhainaut JF, Douglas IS, et al. Design, conduct, analysis and reporting of a multi-national placebo-controlled trial of activated protein C for persistent septic shock. Intensive Care Med. 2008;34:2319. doi: 10.1007/s00134-008-1266-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kerschen EJ, Fernandez JA, Cooley BC, Yang XV, Sood R, Mosnier LO, et al. Endotoxemia and sepsis mortality reduction by non-anticoagulant activated protein C. J Exp Med. 2007;204:2439–48. doi: 10.1084/jem.20070404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Warren BL, Eid A, Singer P, Pillay SS, Carl P, Novak I, et al. Caring for the critically ill patient. High-dose antithrombin III in severe sepsis: a randomized controlled trial. JAMA. 2001;286:1869–78. doi: 10.1001/jama.286.15.1869. [DOI] [PubMed] [Google Scholar]
- 32.Creasey AA, Chang AC, Feigen L, Wun TC, Taylor FB, Jr, Hinshaw LB. Tissue factor pathway inhibitor reduces mortality from Escherichia coli septic shock. J Clin Invest. 1993;91:2850–60. doi: 10.1172/JCI116529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Carr C, Bild GS, Chang AC, Peer GT, Palmier MO, Frazier RB, et al. Recombinant E. coli-derived tissue factor pathway inhibitor reduces coagulopathic and lethal effects in the baboon gram-negative model of septic shock. Circ Shock. 1994;44:126–37. [PubMed] [Google Scholar]
- 34.Camerota AJ, Creasey AA, Patla V, Larkin VA, Fink MP. Delayed treatment with recombinant human tissue factor pathway inhibitor improves survival in rabbits with gram-negative peritonitis. J Infect Dis. 1998;177:668–76. doi: 10.1086/514246. [DOI] [PubMed] [Google Scholar]
- 35.de Jonge E, Dekkers PE, Creasey AA, Hack CE, Paulson SK, Karim A, et al. Tissue factor pathway inhibitor dose-dependently inhibits coagulation activation without influencing the fibrinolytic and cytokine response during human endotoxemia. Blood. 2000;95:1124–9. [PubMed] [Google Scholar]
- 36.de Jonge E, Dekkers PE, Creasey AA, Hack CE, Paulson SK, Karim A, et al. Tissue factor pathway inhibitor does not influence inflammatory pathways during human endotoxemia. J Infect Dis. 2001;183:1815–8. doi: 10.1086/320723. [DOI] [PubMed] [Google Scholar]
- 37.Abraham E, Reinhart K, Opal S, Demeyer I, Doig C, Rodriguez AL, et al. Efficacy and safety of tifacogin (recombinant tissue factor pathway inhibitor) in severe sepsis: a randomized controlled trial. JAMA. 2003;290:238–47. doi: 10.1001/jama.290.2.238. [DOI] [PubMed] [Google Scholar]
- 38.Angus DC, Crowther MA. Unraveling severe sepsis: why did OPTIMIST fail and what’s next? JAMA. 2003;290:256–8. doi: 10.1001/jama.290.2.256. [DOI] [PubMed] [Google Scholar]
- 39.Munck A, Naray-Fejes-Toth A. Glucocorticoids and stress: permissive and suppressive actions. Ann NY Acad Sci. 1994;746:115–30. doi: 10.1111/j.1749-6632.1994.tb39221.x. [DOI] [PubMed] [Google Scholar]
- 40.Perlstein R, Whitnall M, Abrams J, Mougey E, Neta R. Synergistic roles of interleukin-6, interleukin-1, and tumor necrosis factor in the adrenocorticotropin response to bacterial lipopolysaccharide in vivo. Endocrinology. 1993;132:946–52. doi: 10.1210/endo.132.3.8382602. [DOI] [PubMed] [Google Scholar]
- 41.Almawi WY, Melemedjian OK. Molecular mechanisms of glucocorticoid antiproliferative effects: antagonism of transcription factor activity by glucocorticoid receptor. J Leukoc Biol. 2002;71:9–15. [PubMed] [Google Scholar]
- 42.Marik PE, Zaloga GP. Adrenal insufficiency in the critically ill: a new look at an old problem. Chest. 2002;122:1784–96. doi: 10.1378/chest.122.5.1784. [DOI] [PubMed] [Google Scholar]
- 43.Yang J, DeFranco DB. Assessment of glucocorticoid receptor-heat shock protein 90 interactions in vivo during nucleocytoplasmic trafficking. Mol Endocrinol. 1996;10:3–13. doi: 10.1210/mend.10.1.8838140. [DOI] [PubMed] [Google Scholar]
- 44.Schmidt C, Hocherl K, Kurt B, Bucher M. Role of nuclear factor-kappaB-dependent induction of cytokines in the regulation of vasopressin V1A-receptors during cecal ligation and puncture-induced circulatory failure. Crit Care Med. 2008;36:2363–72. doi: 10.1097/CCM.0b013e318180b51d. [DOI] [PubMed] [Google Scholar]
- 45.Gottlicher M, Heck S, Herrlich P. Transcriptional cross-talk, the second mode of steroid hormone receptor action. J Mol Med. 1998;76:480–9. doi: 10.1007/s001090050242. [DOI] [PubMed] [Google Scholar]
- 46.Hawiger J. Innate Immunity and Inflammation: A Transcriptional Paradigm. Immunol Res. 2001;23:99–110. doi: 10.1385/IR:23:2-3:099. [DOI] [PubMed] [Google Scholar]
- 47.Li Q, Verma IM. NF-kappaB regulation in the immune system. Nat Rev Immunol. 2002;2:725–34. doi: 10.1038/nri910. [DOI] [PubMed] [Google Scholar]
- 48.Reichardt HM, Tuckermann JP, Gottlicher M, Vujic M, Weih F, Angel P, et al. Repression of inflammatory responses in the absence of DNA binding by the glucocorticoid receptor. EMBO J. 2001;20:7168–73. doi: 10.1093/emboj/20.24.7168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Heck S, Kullmann M, Gast A, Ponta H, Rahmsdorf HJ, Herrlich P, et al. A distinct modulating domain in glucocorticoid receptor monomers in the repression of activity of the transcription factor AP-1. EMBO J. 1994;13:4087–95. doi: 10.1002/j.1460-2075.1994.tb06726.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Heck S, Bender K, Kullmann M, Gottlicher M, Herrlich P, Cato AC. I kappaB alpha-independent downregulation of NF-kappaB activity by glucocorticoid receptor. EMBO J. 1997;16:4698–707. doi: 10.1093/emboj/16.15.4698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Reichardt HM, Kaestner KH, Wessely O, Gass P, Schmid W, Schutz G. Analysis of glucocorticoid signalling by gene targeting. J Steroid Biochem Mol Biol. 1998;65:111–5. doi: 10.1016/s0960-0760(97)00181-7. [DOI] [PubMed] [Google Scholar]
- 52.Hinshaw LB, Archer LT, Beller-Todd BK, Coalson JJ, Flournoy DJ, Passey R, et al. Survival of primates in LD100 septic shock following steroid/antibiotic therapy. J Surg Res. 1980;28:151–70. doi: 10.1016/0022-4804(80)90158-4. [DOI] [PubMed] [Google Scholar]
- 53.Fabian TC, Patterson R. Steroid therapy in septic shock. Survival studies in a laboratory model. Am Surg. 1982;48:614–7. [PubMed] [Google Scholar]
- 54.Beller BK, Archer LT, Passey RB, Flournoy DJ, Hinshaw LB. Effectiveness of modified steroid-antibiotic therapies for lethal sepsis in the dog. Arch Surg. 1983;118:1293–9. doi: 10.1001/archsurg.1983.01390110045011. [DOI] [PubMed] [Google Scholar]
- 55.Sprung CL, Caralis PV, Marcial EH, Pierce M, Gelbard MA, Long WM, et al. The effects of high-dose corticosteroids in patients with septic shock. A prospective, controlled study. N Engl J Med. 1984;311:1137–43. doi: 10.1056/NEJM198411013111801. [DOI] [PubMed] [Google Scholar]
- 56.Lefering R, Neugebauer EA. Steroid controversy in sepsis and septic shock: a meta-analysis. Crit Care Med. 1995;23:1294–303. doi: 10.1097/00003246-199507000-00021. [DOI] [PubMed] [Google Scholar]
- 57.Cronin L, Cook DJ, Carlet J, Heyland DK, King D, Lansang MA, et al. Corticosteroid treatment for sepsis: a critical appraisal and meta-analysis of the literature. Crit Care Med. 1995;23:1430–9. doi: 10.1097/00003246-199508000-00019. [DOI] [PubMed] [Google Scholar]
- 58.Zeni F, Freeman B, Natanson C. Anti-inflammatory therapies to treat sepsis and septic shock: a reassessment. Crit Care Med. 1997;25:1095–100. doi: 10.1097/00003246-199707000-00001. [DOI] [PubMed] [Google Scholar]
- 59.Bone RC, Fisher CJ, Jr, Clemmer TP, Slotman GJ, Metz CA, Balk RA. A controlled clinical trial of high-dose methylprednisolone in the treatment of severe sepsis and septic shock. N Engl J Med. 1987;317:653–8. doi: 10.1056/NEJM198709103171101. [DOI] [PubMed] [Google Scholar]
- 60.Klastersky J, Cappel R, Debusscher L. Effectiveness of betamethasone in management of severe infections. A double-blind study. N Engl J Med. 1971;284:1248–50. doi: 10.1056/NEJM197106032842206. [DOI] [PubMed] [Google Scholar]
- 61.Marik PE, Zaloga GP. Adrenal insufficiency during septic shock. Crit Care Med. 2003;31:141–5. doi: 10.1097/00003246-200301000-00022. [DOI] [PubMed] [Google Scholar]
- 62.Zaloga GP. Sepsis-induced adrenal deficiency syndrome. Crit Care Med. 2001;29:688–90. doi: 10.1097/00003246-200103000-00051. [DOI] [PubMed] [Google Scholar]
- 63.Soni A, Pepper GM, Wyrwinski PM, Ramirez NE, Simon R, Pina T, et al. Adrenal insufficiency occurring during septic shock: incidence, outcome, and relationship to peripheral cytokine levels. Am J Med. 1995;98:266–71. doi: 10.1016/S0002-9343(99)80373-8. [DOI] [PubMed] [Google Scholar]
- 64.Marik PE, Pastores SM, Annane D, Meduri GU, Sprung CL, Arlt W, et al. Recommendations for the diagnosis and management of corticosteroid insufficiency in critically ill adult patients: consensus statements from an international task force by the American College of Critical Care Medicine. Crit Care Med. 2008;36:1937–49. doi: 10.1097/CCM.0b013e31817603ba. [DOI] [PubMed] [Google Scholar]
- 65.Jaattela M, Carpen O, Stenman UH, Saksela E. Regulation of ACTH-induced steroidogenesis in human fetal adrenals by rTNF-alpha. Mol Cell Endocrinol. 1990;68:R31–6. doi: 10.1016/0303-7207(90)90196-f. [DOI] [PubMed] [Google Scholar]
- 66.Jaattela M, Ilvesmaki V, Voutilainen R, Stenman UH, Saksela E. Tumor necrosis factor as a potent inhibitor of adrenocorticotropin-induced cortisol production and steroidogenic P450 enzyme gene expression in cultured human fetal adrenal cells. Endocrinology. 1991;128:623–9. doi: 10.1210/endo-128-1-623. [DOI] [PubMed] [Google Scholar]
- 67.Shenker Y, Skatrud JB. Adrenal insufficiency in critically ill patients. Am J Respir Crit Care Med. 2001;163:1520–3. doi: 10.1164/ajrccm.163.7.2012022. [DOI] [PubMed] [Google Scholar]
- 68.Minneci PC, Deans KJ, Banks SM, Eichacker PQ, Natanson C. Meta-analysis: the effect of steroids on survival and shock during sepsis depends on the dose. Ann Intern Med. 2004;141:47–56. doi: 10.7326/0003-4819-141-1-200407060-00014. [DOI] [PubMed] [Google Scholar]
- 69.Annane D, Sebille V, Charpentier C, Bollaert PE, Francois B, Korach JM, et al. Effect of treatment with low doses of hydrocortisone and fludrocortisone on mortality in patients with septic shock. JAMA. 2002;288:862–71. doi: 10.1001/jama.288.7.862. [DOI] [PubMed] [Google Scholar]
- 70.Schacke H, Docke WD, Asadullah K. Mechanisms involved in the side effects of glucocorticoids. Pharmacol Ther. 2002;96:23–43. doi: 10.1016/s0163-7258(02)00297-8. [DOI] [PubMed] [Google Scholar]
- 71.Sprung CL, Annane D, Keh D, Moreno R, Singer M, Freivogel K, et al. Hydrocortisone therapy for patients with septic shock. N Engl J Med. 2008;358:111–24. doi: 10.1056/NEJMoa071366. [DOI] [PubMed] [Google Scholar]
- 72.Dellinger RP, Levy MM, Carlet JM, Bion J, Parker MM, Jaeschke R, et al. Surviving Sepsis Campaign: international guidelines for management of severe sepsis and septic shock: 2008. Crit Care Med. 2008;36:296–327. doi: 10.1097/01.CCM.0000298158.12101.41. [DOI] [PubMed] [Google Scholar]
- 73.Song IH, Gold R, Straub RH, Burmester GR, Buttgereit F. New glucocorticoids on the horizon: repress, don’t activate. J Rheumatol. 2005;32:1199–207. [PubMed] [Google Scholar]
- 74.Hotchkiss RS, Swanson PE, Freeman BD, Tinsley KW, Cobb JP, Matuschak GM, et al. Apoptotic cell death in patients with sepsis, shock, and multiple organ dysfunction. Crit Care Med. 1999;27:1230–51. doi: 10.1097/00003246-199907000-00002. [DOI] [PubMed] [Google Scholar]
- 75.Thornberry NA, Lazebnik Y. Caspases: Enemies within. Science. 1998;281:1312–6. doi: 10.1126/science.281.5381.1312. [DOI] [PubMed] [Google Scholar]
- 76.Hotchkiss RS, Tinsley KW, Swanson PE, Chang KC, Cobb JP, Buchman TG, et al. Prevention of lymphocyte cell death in sepsis improves survival in mice. PNAS. 1999;96:14541–6. doi: 10.1073/pnas.96.25.14541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hotchkiss RS, Chang KC, Swanson PE, Tinsley KW, Hui JJ, Klender P, et al. Caspase inhibitors improve survival in sepsis: a critical role of the lymphocyte. Nat Immunol. 2000;1:496–501. doi: 10.1038/82741. [DOI] [PubMed] [Google Scholar]
- 78.Badami CD, Senthil M, Caputo FJ, Rupani BJ, Doucet D, Pisarenko V, et al. Mesenteric lymph duct ligation improves survival in a lethal shock model. Shock. 2008;30:680–5. doi: 10.1097/SHK.0b013e318173edd1. [DOI] [PubMed] [Google Scholar]
- 79.Barlos D, Deitch EA, Watkins AC, Caputo FJ, Lu Q, Abungu B, et al. Trauma hemorrhagic shock-induced pulmonary epithelial and endothelial cell injury utilizes different programmed cell death signaling pathways. Am J Physiol Lung Cell Mol Physiol. 2009;296(3):L404–17. doi: 10.1152/ajplung.00491.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Xu DZ, Zaets SB, Chen R, Lu Q, Rajan H, Yang X, et al. Elimination of C5ar Prevents Intestinal Mucosal Damage and attenuates neutrophil infiltration in local and remote organs. Shock. 2008 doi: 10.1097/SHK.0b013e318188b3cc. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Martinon F, Tschopp J. Inflammatory caspases: linking an intracellular innate immune system to autoinflammatory diseases. Cell. 2004;117:561–74. doi: 10.1016/j.cell.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 82.Barkett M, Gilmore TD. Control of apoptosis by Rel/NF-kappaB transcription factors. Oncogene. 1999;18:6910–24. doi: 10.1038/sj.onc.1203238. [DOI] [PubMed] [Google Scholar]
- 83.Ravi R, Bedi A, Fuchs EJ, Bedi A. CD95 (Fas)-induced caspase-mediated proteolysis of NF-kappaB. Cancer Res. 1998;58:882–6. [PubMed] [Google Scholar]
- 84.Reuther JY, Baldwin AS., Jr Apoptosis promotes a caspase-induced amino-terminal truncation of Ikappa Balpha that functions as a stable inhibitor of NF-kappa B. J Biol Chem. 1999;274:20664–70. doi: 10.1074/jbc.274.29.20664. [DOI] [PubMed] [Google Scholar]
- 85.Nicholson DW. Caspase structure, proteolytic substrates, and function during apoptotic cell death. Cell Death Differ. 1999;6:1028–42. doi: 10.1038/sj.cdd.4400598. [DOI] [PubMed] [Google Scholar]
- 86.Saleh M, Vaillancourt JP, Graham RK, Huyck M, Srinivasula SM, Alnemri ES, et al. Differential modulation of endotoxin responsiveness by human caspase-12 polymorphisms. Nature. 2004;429:35–7. doi: 10.1038/nature02451. [DOI] [PubMed] [Google Scholar]
- 87.Texereau J, Pene F, Chiche JD, Rousseau C, Mira JP. Importance of hemostatic gene polymorphisms for susceptibility to and outcome of severe sepsis. Crit Care Med. 2004;32:S313–9. doi: 10.1097/01.ccm.0000126363.46191.dc. [DOI] [PubMed] [Google Scholar]
- 88.Villar J, Maca-Meyer N, Perez-Mendez L, Flores C. Bench-to-bedside review: understanding genetic predisposition to sepsis. Crit Care. 2004;8:180–9. doi: 10.1186/cc2863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bucala R. MIF rediscovered: cytokine, pituitary hormone, and glucocorticoid- induced regulator of the immune response. FASEB J. 1996;10:1607–13. doi: 10.1096/fasebj.10.14.9002552. [DOI] [PubMed] [Google Scholar]
- 90.Calandra T, Roger T. Macrophage migration inhibitory factor: a regulator of innate immunity. Nat Rev Immunol. 2003;3:791–800. doi: 10.1038/nri1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Calandra T, Bernhagen J, Mitchell RA, Bucala R. The macrophage is an important and previously unrecognized source of macrophage migration inhibitory factor. J Exp Med. 1994;179:1895–902. doi: 10.1084/jem.179.6.1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Calandra T, Bernhagen J, Metz CN, Spiegel LA, Bachner M, Donnelly T, et al. MIF as a glucocorticoid-induced modulator of cytokine production. Nature. 1995;377:68–71. doi: 10.1038/377068a0. [DOI] [PubMed] [Google Scholar]
- 93.Calandra T, Echtenacher B, Roy DL, Pugin J, Metz CN, Hultner L, et al. Protection from septic shock by neutralization of macrophage migration inhibitory factor. Nat Med. 2000;6:164–70. doi: 10.1038/72262. [DOI] [PubMed] [Google Scholar]
- 94.Tohyama S, Onodera S, Tohyama H, Yasuda K, Nishihira J, Mizue Y, et al. A novel DNA vaccine-targeting macrophage migration inhibitory factor improves the survival of mice with sepsis. Gene Ther. 2008 doi: 10.1038/gt.2008.112. [DOI] [PubMed] [Google Scholar]
- 95.Lubetsky JB, Dios A, Han J, Aljabari B, Ruzsicska B, Mitchell R, et al. The Tautomerase Active Site of Macrophage Migration Inhibitory Factor Is a Potential Target for Discovery of Novel Anti-inflammatory Agents. J Biol Chem. 2002;277:24976–82. doi: 10.1074/jbc.M203220200. [DOI] [PubMed] [Google Scholar]
- 96.Lubetsky JB, Swope M, Dealwis C, Blake P, Lolis E. Pro-1 of macrophage migration inhibitory factor functions as a catalytic base in the phenylpyruvate tautomerase activity. Biochemistry. 1999;38:7346–54. doi: 10.1021/bi990306m. [DOI] [PubMed] [Google Scholar]
- 97.Drobnik W, Liebisch G, Audebert F-X, Frohlich D, Gluck T, Vogel P, et al. Plasma ceramide and lysophosphatidylcholine inversely correlate with mortality in sepsis patients. J Lipid Res. 2003;44:754–61. doi: 10.1194/jlr.M200401-JLR200. [DOI] [PubMed] [Google Scholar]
- 98.Chen G, Li J, Qiang X, Czura CJ, Ochani M, Ochani K, et al. Suppression of HMGB1 release by stearoyl lysophosphatidylcholine: an additional mechanism for its therapeutic effects in experimental sepsis. J Lipid Res. 2005;46:623–7. doi: 10.1194/jlr.C400018-JLR200. [DOI] [PubMed] [Google Scholar]
- 99.Yan JJ, Jung JS, Lee JE, Lee J, Huh SO, Kim HS, et al. Therapeutic effects of lysophosphatidylcholine in experimental sepsis. Nat Med. 2004;10:161–7. doi: 10.1038/nm989. [DOI] [PubMed] [Google Scholar]
- 100.Czermak BJ, Sarma V, Pierson CL, Warner RL, Huber-Lang M, Bless NM, et al. Protective effects of C5a blockade in sepsis. Nat Med. 1999;5:788–92. doi: 10.1038/10512. [DOI] [PubMed] [Google Scholar]
- 101.Muller Kobold AC, Tulleken JE, Zijlstra JG, Sluiter W, Hermans J, Kallenberg CG, et al. Leukocyte activation in sepsis; correlations with disease state and mortality. Intens Care Med. 2000;26:883–92. doi: 10.1007/s001340051277. [DOI] [PubMed] [Google Scholar]
- 102.Huber-Lang MS, Younkin EM, Sarma JV, McGuire SR, Lu KT, Guo RF, et al. Complement-induced impairment of innate immunity during sepsis. J Immunol. 2002;169:3223–31. doi: 10.4049/jimmunol.169.6.3223. [DOI] [PubMed] [Google Scholar]
- 103.Coutant F, Perrin-Cocon L, Agaugue S, Delair T, Andre P, Lotteau V. Mature dendritic cell generation promoted by lysophosphatidylcholine. J Immunol. 2002;169:1688–95. doi: 10.4049/jimmunol.169.4.1688. [DOI] [PubMed] [Google Scholar]
- 104.Coutant F, Agaugue S, Perrin-Cocon L, Andre P, Lotteau V. Sensing environmental lipids by dendritic cell modulates its function. J Immunol. 2004;172:54–60. doi: 10.4049/jimmunol.172.1.54. [DOI] [PubMed] [Google Scholar]
- 105.Engelmann B, Zieseniss S, Brand K, Page S, Lentschat A, Ulmer AJ, et al. Tissue factor expression of human monocytes is suppressed by lysophosphatidylcholine. Arterioscler Thromb Vasc Biol. 1999;19:47–53. doi: 10.1161/01.atv.19.1.47. [DOI] [PubMed] [Google Scholar]
- 106.Kume H, Ito S, Ito Y, Yamaki K. Role of lysophosphatidylcholine in the desensitization of beta -adrenergic receptors by Ca2+ sensitization in tracheal smooth muscle. Am J Respir Cell Mol Biol. 2001;25:291–8. doi: 10.1165/ajrcmb.25.3.4364. [DOI] [PubMed] [Google Scholar]
- 107.Legradi A, Chitu V, Szukacsov V, Fajka-Boja R, Szekely Szucs K, Monostori E. Lysophosphatidylcholine is a regulator of tyrosine kinase activity and intracellular Ca(2+) level in Jurkat T cell line. Immunol Lett. 2004;91:17–21. doi: 10.1016/j.imlet.2003.10.009. [DOI] [PubMed] [Google Scholar]
- 108.Murugesan G, Sandhya Rani MR, Gerber CE, Mukhopadhyay C, Ransohoff RM, Chisolm GM, et al. Lysophosphatidylcholine regulates human microvascular endothelial cell expression of chemokines. J Mol Cell Cardiol. 2003;35:1375–84. doi: 10.1016/j.yjmcc.2003.08.004. [DOI] [PubMed] [Google Scholar]
- 109.Takabe W, Kanai Y, Chairoungdua A, Shibata N, Toi S, Kobayashi M, et al. Lysophosphatidylcholine enhances cytokine production of endothelial cells via induction of L-type amino acid transporter 1 and cell surface antigen 4F2. Arterioscler Thromb Vasc Biol. 2004;24:1640–5. doi: 10.1161/01.ATV.0000134377.17680.26. [DOI] [PubMed] [Google Scholar]
- 110.Xu Y. Sphingosylphosphorylcholine and lysophosphatidylcholine: G protein-coupled receptors and receptor-mediated signal transduction. Biochim Biophys Acta. 2002;1582:81–8. doi: 10.1016/s1388-1981(02)00140-3. [DOI] [PubMed] [Google Scholar]
- 111.Kabarowski JH, Xu Y, Witte ON. Lysophosphatidylcholine as a ligand for immunoregulation. Biochem Pharmacol. 2002;64:161–7. doi: 10.1016/s0006-2952(02)01179-6. [DOI] [PubMed] [Google Scholar]
- 112.Le LQ, Kabarowski JH, Weng Z, Satterthwaite AB, Harvill ET, Jensen ER, et al. Mice lacking the orphan G protein-coupled receptor G2A develop a late-onset autoimmune syndrome. Immunity. 2001;14:561–71. doi: 10.1016/s1074-7613(01)00145-5. [DOI] [PubMed] [Google Scholar]
- 113.Radu CG, Yang LV, Riedinger M, Au M, Witte ON. T cell chemotaxis to lysophosphatidylcholine through the G2A receptor. PNAS. 2004;101:245–50. doi: 10.1073/pnas.2536801100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hourton D, Stengel D, Chapman MJ, Ninio E. Oxidized low density lipoproteins downregulate LPS-induced platelet-activating factor receptor expression in human monocyte-derived macrophages: Implications for LPS-induced nuclear factor-{kappa}B binding activity. Eur J Biochem. 2001;268:4489–96. doi: 10.1046/j.1432-1327.2001.02372.x. [DOI] [PubMed] [Google Scholar]
- 115.Tracey KJ, Fong Y, Hesse DG, Manogue KR, Lee AT, Kuo GC, et al. Anti-cachectin/TNF monoclonal antibodies prevent septic shock during lethal bacteraemia. Nature. 1987;330:662–4. doi: 10.1038/330662a0. [DOI] [PubMed] [Google Scholar]
- 116.Wang H, Bloom O, Zhang M, Vishnubhakat JM, Ombrellino M, Che J, et al. HMG-1 as a late mediator of endotoxin lethality in mice. Science. 1999;285:248–51. doi: 10.1126/science.285.5425.248. [DOI] [PubMed] [Google Scholar]
- 117.Ulloa L, Ochani M, Yang H, Tanovic M, Halperin D, Yang R, et al. Ethyl pyruvate prevents lethality in mice with established lethal sepsis and systemic inflammation. Proc Natl Acad Sci U S A. 2002;99:12351–6. doi: 10.1073/pnas.192222999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Andersson U, Wang H, Palmblad K, Aveberger AC, Bloom O, Erlandsson-Harris H, et al. High mobility group 1 protein (HMG-1) stimulates proinflammatory cytokine synthesis in human monocytes. J Exp Med. 2000;192:565–70. doi: 10.1084/jem.192.4.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Bonaldi T, Talamo F, Scaffidi P, Ferrera D, Porto A, Bachi A, et al. Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretion. EMBO J. 2003;22:5551–60. doi: 10.1093/emboj/cdg516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Li J, Kokkola R, Tabibzadeh S, Yang R, Ochani M, Qiang X, et al. Structural basis for the proinflammatory cytokine activity of high mobility group box 1. Mol Med. 2003;9:37–45. [PMC free article] [PubMed] [Google Scholar]
- 121.Bustin M. At the crossroads of necrosis and apoptosis: signaling to multiple cellular targets by HMGB1. Sci STKE 2002. 2002:PE39. doi: 10.1126/stke.2002.151.pe39. [DOI] [PubMed] [Google Scholar]
- 122.Yang H, Ochani M, Li J, Qiang X, Tanovic M, Harris HE, et al. Reversing established sepsis with antagonists of endogenous high-mobility group box 1. Proc Natl Acad Sci USA. 2004;101:296–301. doi: 10.1073/pnas.2434651100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Tracey KJ, Cerami A. Tumor necrosis factor: A pleiotropic cytokine and therapuetic target. Ann Rev Med. 1994;45:491–503. doi: 10.1146/annurev.med.45.1.491. [DOI] [PubMed] [Google Scholar]
- 124.Ulloa L, Fink MP, Tracey KJ. Ethyl pyruvate protects against lethal systemic inflammation by preventing HMGB1 release. Ann NY Acad Sci. 2003;987:319–21. [Google Scholar]
- 125.Wang H, Bloom O, Zhang M, Vishnubhakat JM, Ombrellino M, Che J, et al. HMG-1 as a late mediator of endotoxin lethality in mice. Science. 1999;285:248–51. doi: 10.1126/science.285.5425.248. [DOI] [PubMed] [Google Scholar]
- 126.Degryse B, Bonaldi T, Scaffidi P, Muller S, Resnati M, Sanvito F, et al. The High Mobility Group (HMG) boxes of the nuclear protein HMG1 induce chemotaxis and cytoskeleton reorganization in rat smooth muscle cells. J Cell Biol. 2001;152:1197–206. doi: 10.1083/jcb.152.6.1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Sappington PL, Yang R, Yang H, Tracey KJ, Delude RL, Fink MP. HMGB1 B box increases the permeability of Caco-2 enterocytic monolayers and impairs intestinal barrier function in mice. Gastroenterology. 2002;123:790–802. doi: 10.1053/gast.2002.35391. [DOI] [PubMed] [Google Scholar]
- 128.Li J, Wang H, Mason JM, Levine J, Yu M, Ulloa L, et al. Recombinant HMGB1 with cytokine-stimulating activity. J Immunol Methods. 2004;289:211–23. doi: 10.1016/j.jim.2004.04.019. [DOI] [PubMed] [Google Scholar]
- 129.Parrish W, Ulloa L. High-mobility group box-1 isoforms as potential therapeutic targets in sepsis. Methods Mol Biol. 2007;361:145–62. doi: 10.1385/1-59745-208-4:145. [DOI] [PubMed] [Google Scholar]
- 130.Tsung A, Kaizu T, Nakao A, Shao L, Bucher B, Fink MP, et al. Ethyl pyruvate ameliorates liver ischemia-reperfusion injury by decreasing hepatic necrosis and apoptosis. Transplantation. 2005;79:196–204. doi: 10.1097/01.tp.0000151681.07474.2e. [DOI] [PubMed] [Google Scholar]
- 131.Mallet RT, Sun J, Knott M, Sharma AB, Olivencia-Yurvati AH. Metabolic cardioprotection by pyruvate: Recent progress. Exp Biol Med. 2005;230:435–43. doi: 10.1177/153537020523000701. [DOI] [PubMed] [Google Scholar]
- 132.Fink MP. Ethyl pyruvate: a novel anti-inflammatory agent. Crit Care Med. 2003;31:S51–6. doi: 10.1097/00003246-200301001-00008. [DOI] [PubMed] [Google Scholar]
- 133.Toyosawa T, Suzuki M, Kodama K, Araki S. Highly purified vitamin B2 presents a promising therapeutic strategy for sepsis and septic shock. Infect Immun. 2004;72:1820–3. doi: 10.1128/IAI.72.3.1820-1823.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Montgomery CM, Fairhurst AS, Webb JL. Metabolic studies on heart mitochondria. III. THE action of parapyruvate on agrketoglutaric oxidase. J Biol Chem. 1956;221:369–76. [PubMed] [Google Scholar]
- 135.Montgomery CM, Webb JL. Metabolic studies on heart mitochondria. II. The inhibitory action of parapyruvate on the tricarboxylic acid cycle. J Biol Chem. 1956;221:359–68. [PubMed] [Google Scholar]
- 136.Fink MP. Ethyl pyruvate: a novel treatment for sepsis and shock. Minerva Anestesiol. 2004;70:365–71. [PubMed] [Google Scholar]
- 137.Tawadrous ZS, Delude RL, Fink MP. Resuscitation from hemorrhagic shock with Ringer’s ethyl pyruvate solution improves survival and ameliorates intestinal mucosal hyperpermeability in rats. Shock. 2002;17:473–7. doi: 10.1097/00024382-200206000-00006. [DOI] [PubMed] [Google Scholar]
- 138.Yang R, Gallo DJ, Baust JJ, Uchiyama T, Watkins SK, Delude RL, et al. Ethyl pyruvate modulates inflammatory gene expression in mice subjected to hemorrhagic shock. Am J Physiol Gastrointest Liver Physiol. 2002;283:G212–21. doi: 10.1152/ajpgi.00022.2002. [DOI] [PubMed] [Google Scholar]
- 139.Sims CA, Wattanasirichaigoon S, Menconi MJ, Ajami AM, Fink MP. Ringer’s ethyl pyruvate solution ameliorates ischemia/reperfusion-induced intestinal mucosal injury in rats. Crit Care Med. 2001;29:1513–8. doi: 10.1097/00003246-200108000-00003. [DOI] [PubMed] [Google Scholar]
- 140.Uchiyama T, Delude RL, Fink MP. Dose-dependent effects of ethyl pyruvate in mice subjected to mesenteric ischemia and reperfusion. Intensive Care Med. 2003;29:2050–8. doi: 10.1007/s00134-003-1966-x. [DOI] [PubMed] [Google Scholar]
- 141.Yang R, Uchiyama T, Alber SM, Han X, Watkins SK, Delude RL, et al. Ethyl pyruvate ameliorates distant organ injury in a murine model of acute necrotizing pancreatitis. Crit Care Med. 2004;32:1453–9. doi: 10.1097/01.ccm.0000130835.65462.06. [DOI] [PubMed] [Google Scholar]
- 142.Cheng BQ, Liu CT, Li WJ, Fan W, Zhong N, Zhang Y, et al. Ethyl pyruvate improves survival and ameliorates distant organ injury in rats with severe acute pancreatitis. Pancreas. 2007;35:256–61. doi: 10.1097/MPA.0b013e318064678a. [DOI] [PubMed] [Google Scholar]
- 143.Venkataraman R, Kellum JA, Song M, Fink MP. Resuscitation with Ringer’s ethyl pyruvate solution prolongs survival and modulates plasma cytokine and nitrite/nitrate concentrations in a rat model of lipopolysaccharide-induced shock. Shock. 2002;18:507–12. doi: 10.1097/00024382-200212000-00004. [DOI] [PubMed] [Google Scholar]
- 144.Miyaji T, Hu X, Yuen PS, Muramatsu Y, Iyer S, Hewitt SM, et al. Ethyl pyruvate decreases sepsis-induced acute renal failure and multiple organ damage in aged mice. Kidney Int. 2003;64:1620–31. doi: 10.1046/j.1523-1755.2003.00268.x. [DOI] [PubMed] [Google Scholar]
- 145.Su J, Li X, Cui X, Li Y, Fitz Y, Hsu L, et al. Ethyl pyruvate decreased early nuclear factor-kappaB levels but worsened survival in lipopolysaccharide-challenged mice. Crit Care Med. 2008;36:1059–67. doi: 10.1097/CCM.0B013E318164403B. [DOI] [PubMed] [Google Scholar]
- 146.Tenhunen JJ. Bull’s eye missed by the magic bullet: preclinical investigations, publication bias, and promising new interventions. Crit Care Med. 2008;36:1361–3. doi: 10.1097/CCM.0b013e31816a1414. [DOI] [PubMed] [Google Scholar]
- 147.Fink MP. Ethyl pyruvate. Curr Opin Anaesthesiol. 2008;21:160–7. doi: 10.1097/ACO.0b013e3282f63c2e. [DOI] [PubMed] [Google Scholar]
- 148.Sappington PL, Fink ME, Yang R, Delude RL, Fink MP. Ethyl pyruvate provides durable protection against inflammation-induced intestinal epithelial barrier dysfunction. Shock. 2003;20:521–8. doi: 10.1097/01.shk.0000092697.10326.8b. [DOI] [PubMed] [Google Scholar]
- 149.Droge W, Mihm S, Bockstette M, Roth S. Effect of reactive oxygen intermediates and antioxidants on proliferation and function of T lymphocytes. Methods Enzymol. 1994;234:135–51. doi: 10.1016/0076-6879(94)34084-6. [DOI] [PubMed] [Google Scholar]
- 150.Droge W. Free radicals in the physiological control of cell function. Physiol Rev. 2002;82:47–95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- 151.Pineda-Molina E, Klatt P, Vazquez J, Marina A, Garcia de Lacoba M, Perez-Sala D, et al. Glutathionylation of the p50 subunit of NF-kappaB: a mechanism for redox-induced inhibition of DNA binding. Biochemistry. 2001;40:14134–42. doi: 10.1021/bi011459o. [DOI] [PubMed] [Google Scholar]
- 152.Han Y, Englert JA, Yang R, Delude RL, Fink MP. Ethyl Pyruvate Inhibits Nuclear Factor-{kappa}B-Dependent Signaling by Directly Targeting p65. J Pharmacol Exp Ther. 2005;312:1097–105. doi: 10.1124/jpet.104.079707. [DOI] [PubMed] [Google Scholar]
- 153.Berkowitz B, Huang DB, Chen-Park FE, Sigler PB, Ghosh G. The x-ray crystal structure of the NF-kappa B p50. p65 heterodimer bound to the interferon beta -kappa B site. J Biol Chem. 2002;277:24694–700. doi: 10.1074/jbc.M200006200. [DOI] [PubMed] [Google Scholar]
- 154.Fong CH, Bebien M, Didierlaurent A, Nebauer R, Hussell T, Broide D, et al. An antiinflammatory role for IKKbeta through the inhibition of “classical” macrophage activation. J Exp Med. 2008;205:1269–76. doi: 10.1084/jem.20080124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Hori O, Brett J, Slattery T, Cao R, Zhang J, Chen JX, et al. The receptor for advanced glycation end products (RAGE) is a cellular binding site for amphoterin. Mediation of neurite outgrowth and co-expression of rage and amphoterin in the developing nervous system. J Biol Chem. 1995;270:25752–61. doi: 10.1074/jbc.270.43.25752. [DOI] [PubMed] [Google Scholar]
- 156.Huttunen HJ, Rauvala H. Amphoterin as an extracellular regulator of cell motility: from discovery to disease. J Intern Med. 2004;255:351–66. doi: 10.1111/j.1365-2796.2003.01301.x. [DOI] [PubMed] [Google Scholar]
- 157.Kokkola R, Andersson A, Mullins G, Ostberg T, Treutiger CJ, Arnold B, et al. RAGE is the major receptor for the proinflammatory activity of HMGB1 in rodent macrophages. Scand J Immunol. 2005;61:1–9. doi: 10.1111/j.0300-9475.2005.01534.x. [DOI] [PubMed] [Google Scholar]
- 158.Park JS, Svetkauskaite D, He Q, Kim JY, Strassheim D, Ishizaka A, et al. Involvement of toll-like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J Biol Chem. 2004;279:7370–7. doi: 10.1074/jbc.M306793200. [DOI] [PubMed] [Google Scholar]
- 159.Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418:191–5. doi: 10.1038/nature00858. [DOI] [PubMed] [Google Scholar]
- 160.Gardella S, Andrei C, Ferrera D, Lotti LV, Torrisi MR, Bianchi ME, et al. The nuclear protein HMGB1 is secreted by monocytes via a non-classical, vesicle-mediated secretory pathway. EMBO Rep. 2002;3:995–1001. doi: 10.1093/embo-reports/kvf198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Kazama H, Ricci JE, Herndon JM, Hoppe G, Green DR, Ferguson TA. Induction of immunological tolerance by apoptotic cells requires caspase-dependent oxidation of high-mobility group box-1 protein. Immunity. 2008;29:21–32. doi: 10.1016/j.immuni.2008.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Tracey KJ. The inflammatory reflex. Nature. 2002;420:853–9. doi: 10.1038/nature01321. [DOI] [PubMed] [Google Scholar]
- 163.Borovikova LV, Ivanova S, Zhang M, Yang H, Botchkina GI, Watkins LR, et al. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature. 2000;405:4458–62. doi: 10.1038/35013070. [DOI] [PubMed] [Google Scholar]
- 164.Matthay MA, Ware LB. Can nicotine treat sepsis? Nat Med. 2004;10:1161–2. doi: 10.1038/nm1104-1161. [DOI] [PubMed] [Google Scholar]
- 165.Saeed RW, Varma S, Peng-Nemeroff T, Sherry B, Balakhaneh D, Huston J, et al. Cholinergic stimulation blocks endothelial cell activation and leukocyte recruitment during inflammation. J Exp Med. 2005;201:1113–23. doi: 10.1084/jem.20040463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Ravikumar R, Flora G, Geddes JW, Hennig B, Toborek M. Nicotine attenuates oxidative stress, activation of redox-regulated transcription factors and induction of proinflammatory genes in compressive spinal cord trauma. Brain Res Mol Brain Res. 2004;124:188–98. doi: 10.1016/j.molbrainres.2004.02.018. [DOI] [PubMed] [Google Scholar]
- 167.Shaw S, Bencherif M, Marrero MB. Janus Kinase 2, an Early Target of alpha 7 Nicotinic Acetylcholine Receptor-mediated Neuro-protection against Abeta -(1–42) Amyloid. J Biol Chem. 2002;277:44920–4. doi: 10.1074/jbc.M204610200. [DOI] [PubMed] [Google Scholar]
- 168.de Jonge WJ, van der Zanden EP, The FO, Bijlsma MF, van Westerloo DJ, Bennink RJ, et al. Stimulation of the vagus nerve attenuates macrophage activation by activating the Jak2-STAT3 signaling pathway. Nat Immunol. 2005;6:844–51. doi: 10.1038/ni1229. [DOI] [PubMed] [Google Scholar]
- 169.Metz CN, Gregersen PK, Malhotra AK. Metabolism and biochemical effects of nicotine for primary care providers. Med Clin North Am. 2004;88:1399–413. ix. doi: 10.1016/j.mcna.2004.06.004. [DOI] [PubMed] [Google Scholar]
- 170.Meyer EM, Tay ET, Papke RL, Meyers C, Huang GL, de Fiebre CM. 3-[2, 4-Dimethoxybenzylidene]anabaseine (DMXB) selectively activates rat alpha7 receptors and improves memory-related behaviors in a mecamylamine-sensitive manner. Brain Res. 1997;768:49–56. doi: 10.1016/s0006-8993(97)00536-2. [DOI] [PubMed] [Google Scholar]
- 171.Marrero MB, Papke RL, Bhatti BS, Shaw S, Bencherif M. The neuroprotective effect of 2-(3-pyridyl)-1-azabicyclo[3.2.2]nonane (TC-1698), a novel {alpha}7 ligand, is prevented through angiotensin II activation of a tyrosine phosphatase. J Pharmacol Exp Ther. 2004;309:16–27. doi: 10.1124/jpet.103.061655. [DOI] [PubMed] [Google Scholar]
- 172.Nanri M, Kasahara N, Yamamoto J, Miyake H, Watanabe H. A comparative study on the effects of nicotine and GTS-21, a new nicotinic agonist, on the locomotor activity and brain monoamine level. Jpn J Pharmacol. 1998;78:385–9. doi: 10.1254/jjp.78.385. [DOI] [PubMed] [Google Scholar]
- 173.Kitagawa H, Takenouchi T, Azuma R, Wesnes KA, Kramer WG, Clody DE, et al. Safety, pharmacokinetics, and effects on cognitive function of multiple doses of GTS-21 in healthy, male volunteers. Neuropsychopharmacology. 2003;28:542–51. doi: 10.1038/sj.npp.1300028. [DOI] [PubMed] [Google Scholar]
- 174.Dombrovskiy VY, Martin AA, Sunderram J, Paz HL. Rapid increase in hospitalization and mortality rates for severe sepsis in the United States: a trend analysis from 1993 to 2003. Crit Care Med. 2007;35:1244–50. doi: 10.1097/01.CCM.0000261890.41311.E9. [DOI] [PubMed] [Google Scholar]
- 175.Poli-de-Figueiredo LF, Garrido AG, Nakagawa N, Sannomiya P. Experimental models of sepsis and their clinical relevance. Shock. 2008;30(Suppl 1):53–9. doi: 10.1097/SHK.0b013e318181a343. [DOI] [PubMed] [Google Scholar]
- 176.Rittirsch D, Hoesel LM, Ward PA. The disconnect between animal models of sepsis and human sepsis. J Leukoc Biol. 2007;81:137–43. doi: 10.1189/jlb.0806542. [DOI] [PubMed] [Google Scholar]
- 177.Piper RD, Cook DJ, Bone RC, Sibbald WJ. Introducing Critical Appraisal to studies of animal models investigating novel therapies in sepsis. Crit Care Med. 1996;24:2059–70. doi: 10.1097/00003246-199612000-00021. [DOI] [PubMed] [Google Scholar]
- 178.Mathiak G, Szewczyk D, Abdullah F, Ovadia P, Feuerstein G, Rabinovici R. An improved clinically relevant sepsis model in the conscious rat. Crit Care Med. 2000;28:1947–52. doi: 10.1097/00003246-200006000-00043. [DOI] [PubMed] [Google Scholar]
- 179.Brackett DJ, Schaefer CF, Tompkins P, Fagraeus L, Peters LJ, Wilson MF. Evaluation of cardiac output, total peripheral vascular resistance, and plasma concentrations of vasopressin in the conscious, unrestrained rat during endotoxemia. Circ Shock. 1985;17:273–84. [PubMed] [Google Scholar]
- 180.Fink MP, Heard SO. Laboratory models of sepsis and septic shock. J Surg Res. 1990;49:186–96. doi: 10.1016/0022-4804(90)90260-9. [DOI] [PubMed] [Google Scholar]
- 181.Singleton KD, Wischmeyer PE. Distance of cecum ligated influences mortality, tumor necrosis factor-alpha and interleukin-6 expression following cecal ligation and puncture in the rat. Eur Surg Res. 2003;35:486–91. doi: 10.1159/000073387. [DOI] [PubMed] [Google Scholar]
- 182.Leon C, Alvarez-Lerma F, Ruiz-Santana S, Leon MA, Nolla J, Jorda R, et al. Fungal colonization and/or infection in non-neutropenic critically ill patients: results of the EPCAN observational study. Eur J Clin Microbiol Infect Dis. 2009;28(3):233–42. doi: 10.1007/s10096-008-0618-z. [DOI] [PubMed] [Google Scholar]
- 183.Yin QQ, Zhang YT, Fang Q. Study on the morbidity and pathogens of patients with candidemia at the intensive care unit. Zhonghua Liu Xing Bing Xue Za Zhi. 2008;29:464–8. [PubMed] [Google Scholar]
- 184.Opal SM, Cohen J. Clinical gram-positive sepsis: does it fundamentally differ from gram-negative bacterial sepsis? Crit Care Med. 1999;27:1608–16. doi: 10.1097/00003246-199908000-00039. [DOI] [PubMed] [Google Scholar]
- 185.Fink MP, MacVittie TJ, Casey LC. Inhibition of prostaglandin synthesis restores normal hemodynamics in canine hyperdynamic sepsis. Ann Surg. 1984;200:619–26. doi: 10.1097/00000658-198411000-00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Natanson C, Fink MP, Ballantyne HK, MacVittie TJ, Conklin JJ, Parrillo JE. Gram-negative bacteremia produces both severe systolic and diastolic cardiac dysfunction in a canine model that simulates human septic shock. J Clin Invest. 1986;78:259–70. doi: 10.1172/JCI112559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Taylor FB., Jr Staging of the pathophysiologic responses of the primate microvasculature to Escherichia coli and endotoxin: examination of the elements of the compensated response and their links to the corresponding uncompensated lethal variants. Crit Care Med. 2001;29:S78–89. doi: 10.1097/00003246-200107001-00026. [DOI] [PubMed] [Google Scholar]
- 188.Espat NJ, Cendan JC, Beierle EA, Auffenberg TA, Rosenberg J, Russell D, et al. PEG-BP-30 monotherapy attenuates the cytokine-mediated inflammatory cascade in baboon Escherichia coli septic shock. J Surg Res. 1995;59:153–8. doi: 10.1006/jsre.1995.1147. [DOI] [PubMed] [Google Scholar]
- 189.Pepys MB, Hirschfield GM, Tennent GA, Gallimore JR, Kahan MC, Bellotti V, et al. Targeting C-reactive protein for the treatment of cardiovascular disease. Nature. 2006;440:1217–21. doi: 10.1038/nature04672. [DOI] [PubMed] [Google Scholar]
- 190.Beutler B, Rehli M. Evolution of the TIR, tolls and TLRs: functional inferences from computational biology. Curr Top Microbiol Immunol. 2002;270:1–21. doi: 10.1007/978-3-642-59430-4_1. [DOI] [PubMed] [Google Scholar]
- 191.Abraham E, Arcaroli J, Carmody A, Wang H, Tracey KJ. HMG-1 as a mediator of acute lung inflammation. J Immunol. 2000;165:2950–4. doi: 10.4049/jimmunol.165.6.2950. [DOI] [PubMed] [Google Scholar]
- 192.Lin X, Yang H, Sakuragi T, Hu M, Mantell LL, Hayashi S, et al. Alpha-chemokine receptor blockade reduces high mobility group box 1 protein-induced lung inflammation and injury and improves survival in sepsis. Am J Physiol Lung Cell Mol Physiol. 2005;289:L583–90. doi: 10.1152/ajplung.00091.2005. [DOI] [PubMed] [Google Scholar]
- 193.Nieuwenhuijzen GA, Haskel Y, Lu Q, Berg RD, van Rooijen N, Goris RJ, et al. Macrophage elimination increases bacterial translocation and gut-origin septicemia but attenuates symptoms and mortality rate in a model of systemic inflammation. Ann Surg. 1993;218:791–9. doi: 10.1097/00000658-199312000-00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Hoshino K, Takeuchi O, Kawai T, Sanjo H, Ogawa T, Takeda Y, et al. Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. J Immunol. 1999;162:3749–52. [PubMed] [Google Scholar]
- 195.Myhre AE, Aasen AO, Thiemermann C, Wang JE. Peptidoglycan--an endotoxin in its own right? Shock. 2006;25:227–35. doi: 10.1097/01.shk.0000191378.55274.37. [DOI] [PubMed] [Google Scholar]
- 196.Sugawara I, Yamada H, Li C, Mizuno S, Takeuchi O, Akira S. Mycobacterial infection in TLR2 and TLR6 knockout mice. Microbiol Immunol. 2003;47:327–36. doi: 10.1111/j.1348-0421.2003.tb03404.x. [DOI] [PubMed] [Google Scholar]
- 197.Takeuchi O, Kawai T, Sanjo H, Copeland NG, Gilbert DJ, Jenkins NA, et al. TLR6: A novel member of an expanding toll-like receptor family. Gene. 1999;231:59–65. doi: 10.1016/s0378-1119(99)00098-0. [DOI] [PubMed] [Google Scholar]
- 198.Rittirsch D, Flierl MA, Ward PA. Harmful molecular mechanisms in sepsis. Nat Rev Immunol. 2008;8:776–87. doi: 10.1038/nri2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Dehring DJ, Crocker SH, Wismar BL, Steinberg SM, Lowery BD, Cloutier CT. Comparison of live bacteria infusions in a porcine model of acute respiratory failure. J Surg Res. 1983;34:151–8. doi: 10.1016/0022-4804(83)90054-9. [DOI] [PubMed] [Google Scholar]
- 200.Sligl W, Taylor G, Gibney RN, Rennie R, Chui L. Methicillin-resistant Staphylococcus aureus in a Canadian intensive care unit: Delays in initiating effective therapy due to the low prevalence of infection. Can J Infect Dis Med Microbiol. 2007;18:139–43. doi: 10.1155/2007/120987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Stamme C, Bundschuh DS, Hartung T, Gebert U, Wollin L, Nusing R, et al. Temporal sequence of pulmonary and systemic inflammatory responses to graded polymicrobial peritonitis in mice. Infect Immun. 1999;67:5642–50. doi: 10.1128/iai.67.11.5642-5650.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Parker SJ, Watkins PE. Experimental models of gram-negative sepsis. Br J Surg. 2001;88:22–30. doi: 10.1046/j.1365-2168.2001.01632.x. [DOI] [PubMed] [Google Scholar]
- 203.Martich GD, Danner RL, Ceska M, Suffredini AF. Detection of interleukin 8 and tumor necrosis factor in normal humans after intravenous endotoxin: the effect of antiinflammatory agents. J Exp Med. 1991;173:1021–4. doi: 10.1084/jem.173.4.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Michie HR, Manogue KR, Spriggs DR, Revhaug A, O’Dwyer S, Dinarello CA, et al. Detection of circulating tumor necrosis factor after endotoxin administration. N Engl J Med. 1988;318:1481–6. doi: 10.1056/NEJM198806093182301. [DOI] [PubMed] [Google Scholar]