

Abstract
Identification of proteins and their modifications via liquid chromatography-tandem mass spectrometry is an important task for the field of proteomics. However, because of the complexity of tandem mass spectra, the majority of the spectra cannot be identified. The presence of unanticipated protein modifications is among the major reasons for the low spectral identification rate. The conventional database search approach to protein identification has inherent difficulties in comprehensive detection of protein modifications. In recent years, increasing efforts have been devoted to developing unrestrictive approaches to modification identification, but they often suffer from their lack of speed. This paper presents a statistical algorithm named DeltAMT (Delta Accurate Mass and Time) for fast detection of abundant protein modifications from tandem mass spectra with high-accuracy precursor masses. The algorithm is based on the fact that the modified and unmodified versions of a peptide are usually present simultaneously in a sample and their spectra are correlated with each other in precursor masses and retention times. By representing each pair of spectra as a delta mass and time vector, bivariate Gaussian mixture models are used to detect modification-related spectral pairs. Unlike previous approaches to unrestrictive modification identification that mainly rely upon the fragment information and the mass dimension in liquid chromatography-tandem mass spectrometry, the proposed algorithm makes the most of precursor information. Thus, it is highly efficient while being accurate and sensitive. On two published data sets, the algorithm effectively detected various modifications and other interesting events, yielding deep insights into the data. Based on these discoveries, the spectral identification rates were significantly increased and many modified peptides were identified.
Liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS)1 is currently the predominant technology used to identify proteins and their modifications (1–3). The most successful approach for interpreting tandem mass spectra involves searching the database of known protein sequences (4–9). Other approaches include the database-independent de novo peptide sequencing (10–13) and the hybrid sequence tag-based approach (14–16). However, in a typical shotgun proteomics experiment, only ∼10–30% of the tandem mass spectra can be successfully identified, and the remaining majority is discarded (17). Many factors contribute to the complexity of protein digests and the low identification rate of tandem mass spectra (18). Understanding the origin and identity of the unidentified spectra is of great importance in expanding our knowledge about biological systems and sample processing protocols. Nesvizhskii et al. (19) have shown that by properly mining the unidentified spectra, insights can be gained that are of interest to biologists, such as identification of post-translational modifications, sequence polymorphisms, and novel peptides. The presence of unanticipated protein modifications being a crucial reason for the low spectral identification rate has been demonstrated by many studies (20–22). A recent study of human proteome samples estimated that there are 8–12 modified versions for each unmodified tryptic peptide (23). Efficient and comprehensive detection of protein modifications has become one of the most important and challenging problems in MS/MS-based proteomics.
Conventional database search engines for protein identification, e.g. SEQUEST (4), were originally designed to identify unmodified peptides. To identify peptides with dynamic modifications, the search mode that allows variable modifications was introduced by Yates et al. (24, 25), in which a list of variable modifications are specified by the user and all possible forms of modified peptides are exhaustively enumerated and scored against the input mass spectrum. This is the so-called restrictive approach to modification identification, which has been very successful in identifying proteins with limited modifications and has greatly accelerated the development of proteomics in the past decade. However, as the research focus of the field shifts from identification of protein sequences to characterization of protein modifications, the restrictive approach begins to show problems. For instance, unless protein enrichment is conducted for certain targeted modifications, it is difficult to guess which types of modifications are actually present in a sample. In most cases, oxidation of methionine is the only additional variable modification specified for database searches. On the other hand, considering a large number of variable modifications simultaneously in a search exponentially increases the number of candidate peptides, and thus dramatically degrades the search speed and raises the level of random matches. As of November 22, 2010, there are 660 entries for known modifications in the Unimod database (26). However, for the reason above, popular search engines such as SEQUEST (4) and Mascot (5) allow no more than ten variable modification types in one search.
To alleviate the exponential explosion problem of restrictive modification identification, an approach involving iterative database searches was proposed (27, 28). In this approach, a basic search is first performed against the whole protein database of interest with as few variable modifications specified as possible. Then, a refinement search considering an extensive list of variable modifications is performed against a much smaller database that consists of proteins identified by the basic search. Using this strategy, the search speed and the number of allowed modifications increase considerably. However, if a protein fails to be identified in the basic search, its potential modifications will be missed. More importantly, this approach still requires the user to specify a list of modifications. Modifications not included in this list, e.g. novel ones, cannot be detected.
To overcome the above shortcomings of restrictive modification identification, various unrestrictive approaches have been devised in recent years. The most straightforward approach is the modification-tolerant database search, in which all peptides in a database, rather than only the ones matching the observed precursor mass, are compared with the input spectrum, and putative modifications are introduced to account for the offset of precursor masses. The algorithms or tools belonging to this approach include MS-alignment (20, 29, 30), Protein Prospector (22, 31), P-Mod (32), Interrogator (33), TwinPeaks (34), SeMoP (35), and PTMap (36). Obviously, because removal of the restriction on precursor masses greatly enlarges the search space, this approach is generally applicable to very small databases.
Another unrestrictive approach is based on sequence tags. With this approach, a partial sequence of a possibly modified peptide is first recovered by de novo sequencing. Then, the partial sequence is used as a tag to locate the full peptide sequence. Finally, one or more unanticipated modifications may be inferred according to the remaining mass difference between the full peptide sequence and the observed species. Examples of this approach include OpenSea (37), SPIDER (38), MODi (39), UStag (40), and the Point process model (41). Although this approach is very promising, its applicability is limited because it completely relies on the accuracy of sequence tags, which in turn rely on the quality of spectra. Indeed, de novo sequencing still has limited capabilities and is not as practical as the conventional database search approach.
The third unrestrictive approach is spectral matching, i.e. spectral clustering or library search. This approach takes advantage of the important fact that the modified and unmodified versions of the same peptide often exist simultaneously in a protein sample (21). Although the mass spectra produced by the modified and unmodified versions are different, they are often similar to each other, especially when the peak shifts caused by the modification are taken into account. The Spectral-Networks algorithm of Bandeira et al. (42, 43) uses spectral alignment (29) to detect spectral pairs produced from unmodified and modified versions of peptides. These spectral pairs are then constructed into networks of related spectra, from which more informative virtual spectra can be generated to facilitate de novo peptide sequencing, and also unanticipated modifications can be identified by propagation of database search results. Similarly, the Bonanza algorithm of Falkner et al. (44) uses an improved version of the common spectral-dot-product similarity with shifted mass matching window to cluster related spectral pairs. Ahrne et al. (45) proposed to construct an online library of spectra confidently identified by sequence database search and subsequently search the remaining unidentified spectra against the spectral library with a very large precursor mass tolerance. However, the library search tool that they used, SpectraST (46), was not designed for this open search mode, and thus its ability to identify unanticipated modifications was limited. Recently, Ye et al. (47) reported a deliberate open library search tool, pMatch, which took into account several factors to tolerate unanticipated modifications and showed much better performance than SpectraST.
Although the above approaches to unrestrictive modification identification are attractive and useful, they have two disadvantages. First, they involve open search or matching of the fragmentation spectra and are thus computationally expensive. Moreover, their performances greatly depend on the quality of fragmentation spectra. Some modifications, however, can significantly change the fragmentation patterns of peptide ions, resulting in spectra that are either of low quality or very different from those of unmodified peptides. For example, phosphorylated peptides often undergo insufficient backbone fragmentation under collision-induced disassociation, resulting in spectra dominated by neutral-loss peaks of precursors. Second, the above approaches completely ignore the retention time information provided by LC-MS/MS. The retention time of a peptide is mainly determined by the hydrophobicity of the peptide and thus has discriminative power in peptide identification. Smith et al. have shown that it is feasible to identify peptides directly using their accurate mass and time (AMT) tags (48, 49). For most modifications, the modified peptides tend to consistently elute earlier or later from LC columns than do their unmodified counterparts. Therefore, the retention time shift caused by a modification can serve as orthogonal evidence for the occurrence of this modification (21).
In this paper, we present a statistical algorithm named DeltAMT (Delta Accurate Mass and Time) for efficient detection of abundant modifications by taking full advantage of the precursor information from LC-MS/MS with high precursor mass accuracy. The algorithm aims at finding pairs of modification-related spectra and thus might be classified into the spectral matching approach. However, the fragmentation spectra are never matched with each other. Therefore, modifications can be detected with an extremely fast speed. The underlying principle of DeltAMT is based on the simple fact that an abundantly present modification in a sample usually leads to observation of many spectral pairs with nearly identical precursor mass differences and similar retention time distances. The main steps of DeltAMT are as follows. First, each pair of spectra from an LC-MS/MS run is represented by a vector, called a delta mass and time vector, with two dimensions being the differences in precursor mass and retention time, respectively. Then, bivariate Gaussian mixture models are fitted to the complete set of observed delta vectors. Fitted distribution components are scored to discriminate modification-induced distributions from random ones. Finally, putative modifications are reported with the estimated mass and retention time shifts as well as modification-related spectral pairs. To identify the modified peptides, the modifications detected by DeltAMT can be included into sequence database search as variable modification parameters. Alternatively, peptide identifications obtained in some way can be propagated among the modification-related spectral pairs.
Previous algorithms that are most closely related to ours are the ModifiComb and the Mass Distance Fingerprint (MDF) algorithms. The ModifiComb algorithm of Savitski et al. (21) pioneered the use of retention time as complementary information to detect modifications. However, ModifiComb builds histograms of differences in precursor masses and retention times between detected similar spectral pairs only, rather than simply between all possible pairs of spectra as is done in DeltAMT. It is featured in the combined use of two complementary fragmentation modes and post-processing of data to produce semi-interpreted fragmentation spectra (peaks with inferred fragment ion types). In addition, ModifiComb determines spectral pairs empirically instead of using a statistical framework. The MDF algorithm proposed by Potthast et al. (50) is a close analog of DeltAMT but it makes use of the precursor mass information only, hence limiting the accuracy and sensitivity of modification detection. Moreover, MDF does not address the pseudo-modification problem associated with unrestrictive modification identification algorithms (20, 43). Consequently, many reported mass shifts could not be properly interpreted. For example, the mysterious 25.0252 Da that was repeatedly detected by MDF was simply a pseudo-modification corresponding to the combination of oxidation (15.9949 Da) and ICAT label (9.0302 Da). Most other unknown modifications reported by MDF (50) can be interpreted in the same fashion.
Unlike previous methods for unrestrictive modification identification, which are based on the fragment information and the mass dimension in LC-MS/MS, the DeltAMT algorithm presented in this paper is the first attempt to make full use of the precursor information in a rigorous statistical framework to rapidly and accurately detect abundant modifications present in a protein sample. Although the algorithm is not designed to detect low-abundance modifications, it should be noted that even for methods using fragmentation data, it is usually the abundant modifications that are accepted as confident results (20–22). Compared with previous approaches for modification detection, DeltAMT possesses several advantages. First, it is very fast, thus able to overcome the speed bottleneck encountered by previous methods. To analyze the mass spectra from one LC-MS/MS run, it only takes DeltAMT several minutes to complete the entire process from reading raw data to reporting putative modifications. Unwanted chemical modifications or contaminants introduced by sample processing can be rapidly reported, providing real-time measurements of the quality of experiments. Second, a unique feature of DeltAMT is that it is not sensitive to the quality of fragmentation spectra. It is known that modified peptides often have complex fragmentation patterns, but DeltAMT is naturally immune to this problem. Third, the use of retention time provides an independent source of evidence for the detected modifications. Two dimensions are more discriminative than one in detecting modification-related spectral pairs. Fourth, because the mass differences are computed between experimental precursor masses, the estimated modification masses are not sensitive to systematic mass errors of mass spectrometers. Finally, the problem of pseudo-modifications is, for the first time, carefully addressed.
On a standard protein data set and a proteome quantification data set, our method successfully detected many chemical modifications, metal adducts, isotope labels, as well as nonspecific digestion and even nonpeptide contaminants, yielding deep insights into the data. By incorporating these discoveries into database search or performing peptide propagation among detected spectral pairs, the spectral identification rates were significantly increased, and many modified peptides were identified.
MATERIALS AND METHODS
The flowchart of the DeltAMT algorithm for detection and identification of protein modifications is illustrated in Fig. 1 Details of each step are described below.
Fig. 1.
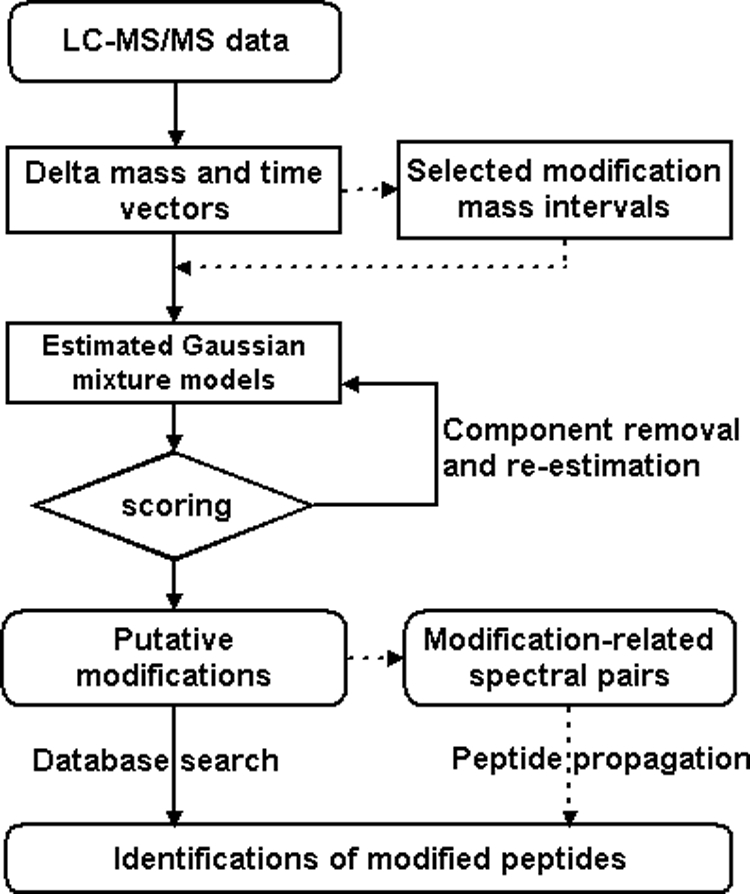
Flowchart of the DeltAMT algorithm (dotted lines indicate optional steps).
Delta Mass and Time Vectors (Δ)
For most modifications, the modified and unmodified versions of a peptide often exist simultaneously. A modification abundantly present in a sample will lead to many mass spectral pairs, with their precursor masses differing by the modification mass. In addition to peptide masses, peptide retention times provide another dimension of information available in LC-MS/MS experiments. The modified and unmodified versions of a peptide share the same amino acid sequence and differ by a modification group. Thus, they often exhibit slight difference in physicochemical properties and LC behavior. A modification usually has a relatively consistent effect on the retention times of peptides carrying it. For example, it is known that peptides containing oxidized methionine(s) usually elute earlier (51, 52), whereas deamidation of asparagine tends to slightly increase the retention time of peptides (21, 53) on reversed-phase high performance LC. Therefore, the retention time is an orthogonal source of evidence for the existence of a modification. To detect potential modifications in a data set of tandem mass spectra with the DeltAMT algorithm, every pair of spectra is represented by a two-dimensional vector, called a delta mass and time vector or delta vector for short (denoted by Δ):
where Δm and Δt represent the differences in precursor mass and retention time between the two spectra, respectively. A high-frequency Δm value accompanied by a concentrated Δt indicates a potential modification. The aim of DeltAMT is to find all such Δm values by statistical analysis of the empirical distribution of Δ.
In tandem mass spectrometry, abundant peptides can produce many copies of spectra, resulting in data redundancy. There are two main disadvantages of spectral redundancy in our situation. First, spectral redundancy brings a high computational burden, because the number of calculated Δ instances increases quadratically with the number of spectra. Second and no less important, spectral redundancy may cause an unexpected effect on the distribution of Δ. To mitigate the problems arising from spectral redundancy, we utilize a simple strategy for efficiency consideration. Among the spectra with precursor masses in close proximity, e.g. less than 5 ppm for Fourier transform mass spectrometers, the one with the median retention time is reserved as the representative. Although such a simple processing step may remove some nonredundant spectra, we find that a representative subset of spectra is sufficient to reveal the dominant modifications. Moreover, when we reach the spectral pair detection step, all spectra will be considered.
Another important issue associated with high-resolution mass spectrometers is the determination of accurate precursor masses. In tandem mass spectrometry, the measured precursor masses are prone to errors, such as misidentified mono-isotopic peaks. Identifying the mono-isotopic peak in a weak ion cluster of a large peptide or within overlapping ion clusters has remained a basic and not very well-resolved problem of mass spectrometry. However, as a statistical algorithm, DeltAMT computes from a large number of data points and can tolerate a certain degree of random data errors. The precursor masses used in this paper were directly exported by the instrument control software. Though DeltAMT will benefit from a better third-party peak-picking algorithm, e.g. MaxQuant (54), we did not employ such an algorithm here for the purpose of simplicity, independence and efficiency.
Random and Modification-induced Distributions of Δ
All instances of Δ can be categorized into two groups: random and modification-induced. A random instance of Δ is derived from two spectra that are produced from two independent peptides. A modification-induced instance of Δ, as its name implies, results from two modification-related spectra that are produced by two peptides with the same sequence but different modification states.
In the vicinity (about ±0.5 Da) of a modification mass, the distribution of Δ is assumed to be a mixture of multiple components, one of which is produced by random spectral pairs and the others by modification-related spectral pairs. The probability density function (pdf) of Δ is
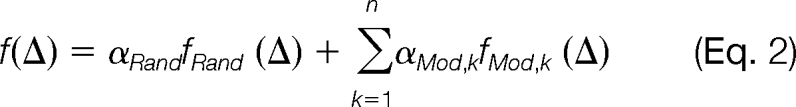 |
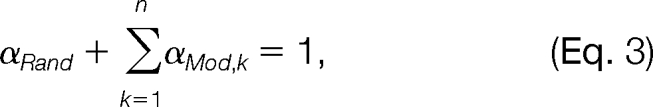 |
where fRand (Δ) represents the pdf of the random distribution in this mass interval, fMod,k (Δ) represents the pdf of the k-th modification-induced distribution, n is the total number of modifications in this mass interval, and αRand and αMod,k are mixing coefficients. We further assume that both the random distribution and the modification-induced distributions are Gaussian. That is, both fRand (Δ) and fMod,k (Δ) are of the form
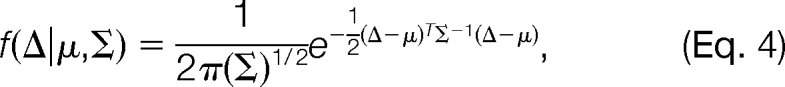 |
where parameters μ and Σ denote the mean and covariance of the Gaussian distribution, respectively. The reasons for the choice of the Gaussian Mixture Model are twofold: Gaussian Mixture Model can be efficiently estimated using the Expectation-Maximization algorithm, and the real data illustrate good fitness to the model. Fig. 2 presents a real example of the distribution of Δ obtained on the ISB standard protein mix data set (see the Results section for details). We can see that there are two modification-induced distributions (n = 2) of Δ in the mass interval from 15.5 to 16.5 Da, one of which is induced by the oxidation modification and the other by a subtractive pseudo-modification. There are occasionally such cases, as described here, in which more than one modification exists within a small mass interval. Another example is the combination of deamidation (0.98402 Da) and precursor isotope (1.0072 Da). These mass-alike modifications can be better discriminated from each other from the retention time dimension, as illustrated by Fig. 2.
Fig. 2.

An example of the distribution of Δ obtained from the ISB standard protein mix data set. In this example, three distribution components were automatically detected by the DeltAMT algorithm. One of them is the random distribution marked by the large dashed square, and the other two are modification-induced distributions marked by the small solid squares. The two modifications are oxidation (right) and the (calcium - sodium) subtractive pseudo-modification (left). These two modifications are well discriminated from each other by the mass and time dimensions.
After the random and modification-induced distributions of Δ are estimated, modification-related spectral pairs can be identified at a required confidence level. Given the observed Δ from a pair of spectra, the posterior probability of this spectral pair being related by the k-th modification is
 |
Then, the posterior error probability (PEP) is (1 - Pr(Modk|Δ)). Given a cut-off for PEP, a list of modified/unmodified spectral pairs can be obtained for each modification detected.
Scoring Modification-induced Distributions of Δ
To evaluate the reliability of a distribution component as being modification-induced, a scoring function, named density score (d-score), is defined as
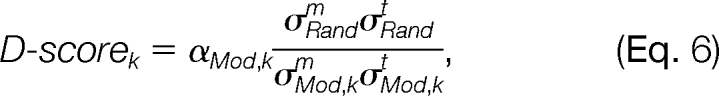 |
where αMod,k is the mixing coefficient of the k-th modification-induced component in the mixture distribution, σMod,km and σMod,kt are the standard deviations of the Δm and Δt elements of the k-th modification-induced component, respectively, and σRandm and σRandt are the standard deviations of the Δm and Δt elements of the random distribution component, respectively. d-score indicates that the more samples a distribution component contains and the more compact the distribution is, the more likely this distribution component is induced by a modification.
The estimation of the mixture model is performed in an iterative manner. When the d-score of a modification-induced distribution component is below a cut-off value, this component is removed from the mixture model and the model is re-estimated. This is repeated until the d-scores of all remaining modification-induced components are no less than the cut-off value or no modification-induced components are left. The cut-off value for d-score controls the trade-off between the accuracy and the sensitivity of modification detection. A low cut-off value increases the sensitivity but also increases the level of false positives. According to our experience, 10 is an appropriate cut-off value that can lead to interpretable modifications and reliable spectral pairs. If interested in lower-abundance modifications, one can use lower cut-off values, but this may necessitate the use of additional procedures to verify the findings.
Selection of Potential Modification Mass Intervals
In principle, the above procedure of distribution estimation should be performed on every potential modification mass interval, which is defined as a 1-Da window around each nominal mass value. However, for general protein samples, only a few mass intervals are expected to contain modification masses. To avoid unnecessary computation, it is desirable to select a few potential modification mass intervals that are likely to contain modifications. We use a simple method here to accomplish this purpose. The random Δm within each potential modification mass interval is assumed to come from a Gaussian distribution, which is estimated based on the observed Δm data points. If any values of Δm are observed with frequencies significantly exceeding their estimated random probabilities, then the mass intervals containing these values are considered to contain potential modifications and are subjected to the two-dimensional mixture distribution estimation of Δ. This is illustrated by Fig. 3. A parameter RΔm = o(Δm)/e(Δm) is calculated for Δm to quantify the extent to which the observed frequency of Δm exceeds its expected random probability, where o(Δm) and e(Δm) are the occurrence number of Δm and the number of Δm expected by chance, respectively. The mass bin size used is 0.01 Da by default. According to our experience, a value of 1.3 for RΔm can serve as a good cut-off for removing unproductive mass intervals without losing sensitivity.
Fig. 3.

An example of the distribution of Δm. Within each potential modification mass interval, random Δm is assumed to come from a Gaussian distribution. Those mass intervals that contain Δm values of unexpectedly high frequencies (peaks marked by stars) are selected and subjected to two-dimensional (mass and time) distribution fitting for modification detection. In this example, the highest peak corresponds to a real modification (acetaldehyde), and the other three are random signals.
Recognition of Additive and Subtractive Pseudo-modifications
Some pseudo-modifications may be reported by DeltAMT as computational products of real modifications (mono-modifications). There are two possible types of such pseudo-modifications. The first one is produced by the combination of two modifications and is, therefore, called additive pseudo-modification. For example, many spectra carrying two modifications B and C may lead to reporting a modification A with a mass shift corresponding to the sum of the mass shifts of modifications B and C. The second type of pseudo-modification is produced by the difference between two modifications and is, therefore, called subtractive pseudo-modification. For example, two groups of spectra carrying modifications A and B, respectively, may lead to reporting a modification C with a mass shift corresponding to the mass difference between modifications A and B. Although additive and subtractive pseudo-modifications are not real independent modifications, they are useful for recognizing modification-related spectral pairs and identifying peptides with multiple modifications.
To recognize these two types of pseudo-modifications, three criteria are used:
(1) The masses must be consistent; i.e.
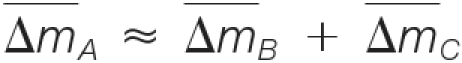 , where
, where  denotes the estimated mass shift caused by the putative modification A, B, or C.
denotes the estimated mass shift caused by the putative modification A, B, or C.(2) The retention times must be consistent; i.e.
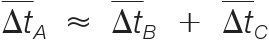 , where
, where 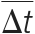 denotes the estimated retention time shift caused by the putative modification A, B, or C.
denotes the estimated retention time shift caused by the putative modification A, B, or C.- (3) The detected spectral pairs must be consistent. Specifically, the following criteria are used:
where |P| denotes the number of elements in set P, T1 and T2 are two thresholds, and
and so forth. The (si, sj) in the above formulae denotes spectral pairs and PEPT denotes the threshold of PEP. Simply put, PB is the set of spectral pairs related by the putative modification B, and SB1 is the set of spectra with modification B in PB. The other set symbols are defined by analogy. PBAC is a subset of PB consisting of the spectral pairs, in which the spectra with modification B also belong to PA, and spectra without modification B also belong to PC. PCAB is defined in a similar way. If PBAC is relatively larger enough than PCAB, B is recognized as the subtractive pseudo-modification of A and C, and vice versa. Otherwise, if both are large enough, A is recognized as the additive pseudo-modification of B and C.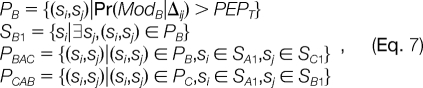
Identification of Modified Peptides
A straightforward way to make use of the modification detection results is to incorporate the detected modification types into ordinary sequence database search. Alternatively, peptides identified by database search or other approaches (e.g. de novo sequencing) can be propagated from the modification-free spectra to the modification-containing spectra based on the detected spectral pairs (43). If the spectra are of good qualities and the amino acid specificities of the detected modifications are known, incorporation of these modifications into sequence database search will be appropriate. Otherwise, peptide propagation may be advantageous. It is known that some modifications, e.g. phosphorylation, suppress peptide fragmentation, resulting in mass spectra with low signal-to-noise ratios. Moreover, for novel modifications, peptide propagation helps determine their amino acid specificities. To locate the modification site of a propagated peptide, the modification is assigned to each amino acid in the peptide, and theoretical spectra are predicted for all assignments and are scored by comparison with the experimental spectrum. The highest-scoring site is considered the modification site. The scoring function used here is the one employed by the pFind search engine (7). Obviously, the accuracy of peptide propagation relies on both the accuracy of input peptide identifications and the reliability of detected spectral pairs. Moreover, modification site assignment is especially difficult for spectra of low qualities. Therefore, the propagation results should be carefully validated. In propagation, two kinds of conflicts may occur. The first is called edge conflict, in which the two spectra S1 and S2 in a spectral pair (S1,S2) are identified as different peptide sequences. The second is called node conflict, in which the two spectra S1 and S2 in two spectral pairs (S1,S3) and (S2,S3) are identified as different peptide sequences. In any case, such spectral pairs are removed.
Inference of Modification Types
For mass spectra produced from current popular hybrid instruments (e.g. LTQ-FT or LTQ-Orbitrap), the accuracy of precursor masses is at the ppm level. From such data, even higher accuracy for modification masses can be estimated by DeltAMT. This is because of several reasons. First, the mass shift caused by a modification is estimated from many pairs of spectra rather than one pair of spectra. Second, the estimation is less biased by the systematic error of a mass spectrometer, because it is the mass differences instead of the original mass values that are used. Third, the retention time shift as the second dimension of the distribution of Δ can exclude a substantial number of random spectral pairs. With accurate modification masses, the identities of most modifications can be easily determined by searching a modification database such as Unimod. The estimated retention time shift also helps infer modification types. Different modifications have different effects on the retention time of a peptide. Additionally, amino acid specificity of a modification as revealed by modification site location is also useful. However, fully automated inference of modification types is hardly realistic, so manual examination is necessary. Expertise and a priori knowledge about the source of the protein sample and the sample processing procedure are always important, particularly for novel modifications.
RESULTS
To validate the DeltAMT algorithm, two published data sets of tandem mass spectra were analyzed, with detailed discussions focused on the first data set. For each data set, putative modifications (mass and time shifts) and related spectral pairs were detected by the DeltAMT algorithm. The detected modifications were interpreted according to their mass and retention time shifts as well as their locations on peptides. To identify the peptides carrying these modifications, both database search and peptide propagation were performed. The MS-Alignment algorithm was also run on the first data set and compared with DeltAMT in terms of speed and sensitivity. All data analyses were performed on a personal computer with a 1.8 GHz Intel Core 2 Duo CPU, 2 GB of main memory and the Windows XP operating system.
Software Implementation
The DeltAMT algorithm was implemented in MATLAB and C++ independently (software accessible at http://pfind.ict.ac.cn/pcluster/). The analysis results presented in this paper were obtained with the MATLAB version. The software can directly read the tandem mass spectra in the RAW format from mass spectrometers of Thermo Scientific Corporation (e.g. LTQ-FT/Orbitrap). It also supports data in the DTA format, in which case the scan numbers in file names are used as the substitute for retention times. After all parameters needed by DeltAMT are specified (default values are provided which can be kept unchanged in general use), the software can be executed in a fully automated manner.
Data
One data set used is the ISB standard protein mix data set, a standard protein data set from the Institute for Systems Biology (55), and the other is the MaxQaunt HeLa Proteome data set, a stable isotope labeling with amino acids in cell culture (SILAC)-treated HeLa proteome data set from the Max-Planck Institute for Biochemistry (56). Both data sets are of good quality and are available in the public domain.
ISB Standard Protein Mix Data Set
This data set is currently the largest and most diverse mass spectra data set deliberately designed for the purpose of testing peptide and protein identification software tools (55). Eighteen purified proteins were mixed and digested by trypsin into peptide mixtures, which were then analyzed by LC-MS/MS on diverse mass spectrometers and under various conditions. The analysis of Mixture 3 on a Thermo Scientific LTQ-FT mass spectrometer consisted of 10 LC-MS/MS runs. One of these runs (B06–11073), consisting of a total of 4085 MS/MS scans, was selected at random for detailed analysis in this paper. This data set was downloaded at http://regis-web.systemsbiology.net/PublicData sets/.
MaxQuant HeLa Proteome Data Set
This data set was originally published in (56) and was used for testing the MaxQuant program (54). Proteins from SILAC-treated stimulated HeLa cells were digested with trypsin. The resulting peptides were separated, purified, and analyzed by LC-MS/MS in triplicate on a Thermo Scientific LTQ-Orbitrap mass spectrometer. A total of 72 LC-MS/MS runs were performed. One of these runs (20070522_NH_Orbi2_HelaEpo_10), consisting of a total of 13,572 MS/MS scans, was selected at random for the use in this paper. This data set was downloaded from Tranche at http://tranche.proteomecommons.org/.
Both data sets were in the Thermo Scientific RAW data format, and the precursor information was directly extracted from the RAW files. Note that although we selected only one LC-MS/MS run for each data set, similar results were obtained for other runs (data not shown). For the ISB standard protein mix data set, the other nine runs were used to estimate the variability of the DeltAMT algorithm.
Results for the ISB Standard Protein Mix Data Set
Overview
The spectra in the ISB standard protein mix data set were analyzed with the DeltAMT algorithm. Following the removal of data redundancy, delta mass and time vectors were calculated for 2619 MS/MS spectra, and these vectors were subjected to mass interval selection and mixture distribution estimation. As a result, a total of 32 putative modifications with d-scores above 10 were reported, among which 13 were mono-modifications, six were additive pseudo-modifications and 13 were subtractive pseudo-modifications. Table I gives their estimated mass shifts (Δm) and retention time shifts (Δt), as well as their d-scores, numbers of spectral pairs, interpretations, and deviations from theoretical masses. Here, the mass and retention time shifts correspond to the estimated means of modification-induced distributions of delta vectors. The estimated standard deviations are given in supplementary Table S1. Note that the numbers of spectral pairs given in Table I contain redundancy, i.e. a spectrum may appear in multiple pairs. The total number of spectral pairs, thereby, may exceed the number of spectra. Larger d-scores indicate higher confidence and usually correspond to more spectral pairs. Normally, spectral pairs with PEP ≤ 0.02 were reported. When no pairs were found at this level, lower PEP thresholds, 0.05 and further 0.1, were used. Among the 32 reported modifications, 31 were successfully interpreted. Their estimated mass values were very close to their theoretical ones (mostly within 0.001 Da). For several putative modifications, e.g. carbamidomethyl dithiotreitol (DTT), oxidation, dehydration and acetaldehyde, their spectral pairs could not be detected at the lowest PEP level (0.02). This was because the delta vector distributions induced by these modifications had not stood out clearly enough from the random distributions, either because of their small mixing coefficients or large variances. However, they were still distinguished by the DeltAMT algorithm and their distribution centers (the mass dimension at least) were accurately estimated. To examine the variability of the algorithm with respect to replicate runs of the same sample, we further analyzed the data from the other nine LC-MS/MS runs in this data set and calculated the means and standard deviations of the estimated mass and time shifts for each putative modification. The results, given in supplementary Table S1, demonstrate the stability of DeltAMT for different LC-MS/MS runs. The detected modifications were incorporated into sequence database search. In parallel, peptide propagation was also conducted based on the identifications from a basic database search without considering any of the detected modifications. Both approaches identified a substantial number of spectra of modified peptides (Table II and supplementary Table S2), and the identifications made by the two approaches showed good agreement with each other. Table III gives the running times for each step of modification detection and peptide propagation. From reading the raw mass spectra to reporting the presence of putative modifications (mass and time shifts), the procedure took less than 4 min.
Table I. Detected modifications for the ISB standard protein mix data set.
|
Mono-modifications | |||||
|---|---|---|---|---|---|
| Δm (Da) | Δt (min) | d-score | Pairs (PEP) | Interpretation | Mass deviation (Da) |
| 37.94689 | 0.020 | 1127.2 | 2,023 (0.02) | Calcium adduct | −0.00005 |
| 21.98167 | 0.020 | 472.2 | 1,358 (0.02) | Sodium adduct | −0.00027 |
| 113.08411 | −0.012 | 151.2 | 48 (0.02) | I/L (amino acid) | 0.00005 |
| 0.98433 | 0.800 | 73.6 | 335 (0.02) | Deamidation | 0.00031 |
| 151.99699 | 1.842 | 34.2 | 125 (0.05) | Carbamidomethyl DTT | 0.00042 |
| 156.09987 | −2.389 | 30.8 | 119 (0.05) | R (amino acid) | −0.00124 |
| 128.09461 | −2.915 | 29.9 | 144 (0.05) | K (amino acid) | −0.00035 |
| 170.10512 | −0.014 | 29.3 | 39 (0.05) | GI/L or AV (amino acids) | −0.00040 |
| 104.09558 | −13.108 | 20.2 | 6 (0.02) | False positive | |
| 15.99421 | −4.101 | 17.3 | 62 (0.05) | Oxidation | −0.00071 |
| 99.06819 | 0.671 | 15.0 | 43 (0.10) | V (amino acid) | −0.00022 |
| 18.00828 | −0.255 | 13.6 | 36 (0.10) | Dehydration/N-term pyro-glutamic acid | −0.00229 |
| 26.01532 | 2.581 | 10.5 | 67 (0.10) | Acetaldehyde(+26) | −0.00033 |
|
Additive pseudo-modifications | |||||
|---|---|---|---|---|---|
| Δm (Da) | Δt (min) | d-score | Pairs (PEP) | Interpretation | Mass deviation (Da) |
| 43.96545 | 0.0717 | 207.2 | 194 (0.02) | Double sodium | 0.00156 |
| 75.89452 | 0.0340 | 175.4 | 185 (0.02) | Double calcium | 0.00063 |
| 59.93015 | 0.0206 | 169.1 | 278 (0.02) | Calcium + sodium | 0.00127 |
| 38.93119 | 0.8715 | 26.7 | 53 (0.05) | Calcium + deamidation | 0.00023 |
| 189.94359 | 1.3348 | 20.8 | 20 (0.05) | Calcium + carbamidomethyl DTT | 0.00008 |
| 22.96927 | 0.4303 | 18.2 | 3 (0.05) | Sodium + deamidation | 0.00331 |
|
Subtractive pseudo-modifications | |||||
|---|---|---|---|---|---|
| Δm (Da) | Δt (min) | d-score | Pairs (PEP) | Interpretation | Mass deviation (Da) |
| 15.96596 | 0.0011 | 483.7 | 792 (0.02) | Calcium − sodium | 0.00096 |
| 6.01759 | 0.0217 | 232.8 | 165 (0.02) | Double sodium − calcium | 0.00064 |
| 53.91554 | 0.0444 | 111.1 | 116 (0.02) | Double calcium − sodium | 0.00360 |
| 31.93087 | 0.0031 | 110.0 | 41 (0.02) | Double calcium − double sodium | 0.00088 |
| 36.96257 | −0.7860 | 47.3 | 34 (0.02) | Calcium − deamidation | −0.00035 |
| 20.99921 | −0.8399 | 27.7 | 73 (0.05) | Sodium − deamidation | 0.00187 |
| 118.15417 | −2.4277 | 25.7 | 70 (0.05) | R − calcium | 0.00000 |
| 114.05048 | 2.3156 | 19.8 | 2 (0.05) | Carbamidomethyl DTT − calcium | 0.00085 |
| 90.14823 | −2.8493 | 18.7 | 42 (0.05) | K − calcium | 0.00021 |
| 15.00951 | −5.0909 | 16.7 | 28 (0.10) | Oxidation − deamidation | −0.00107 |
| 140.10718 | 1.4453 | 14.5 | 26 (0.10) | R − oxidation | 0.00098 |
| 16.94414 | 0.9930 | 11.1 | 24 (0.10) | Calcium + deamidation − sodium | −0.00514 |
| 21.95289 | 4.2442 | 10.5 | 98 (0.10) | Calcium − oxidation | 0.00087 |
Table II. Spectra identified by incorporation of the detected modifications into sequence database search and by peptide propagation among spectral pairs with PEP ≤ 0.02 for the ISB standard protein mix data set. (*For each detected modification, the number of identified spectra is the number of spectra identified as peptides with this modification; for semispecific digestion, it is the number of spectra identified as semitryptic peptides.).
| Digestion mode | Considered modification | Number of spectra identified as modified/semitryptic peptides* |
|
|---|---|---|---|
| Database search | Propagation (PEP ≤ 0.02) | ||
| Full-specific | Calcium (D, E and peptide C-terminus) | 142 | 488 |
| Sodium (D, E and peptide C-terminus) | 205 | 372 | |
| Deamidation (N and Q) | 165 | 98 | |
| Carbamidomethyl DTT (C) | 82 | 0 | |
| Dehydration (T, S, D, and E at peptide N-termimus) | 14 | 0 | |
| Acetaldehyde (H, K and peptide N-termimus) | 38 | 0 | |
| Semispecific | None | 218 | 14 |
| Total | 864 | 972 | |
Table III. Running times of modification detection and peptide propagation for the ISB standard protein mix data set.
| Modification detection |
Peptide propagation |
Total | |||
|---|---|---|---|---|---|
| Reading raw data | Reporting modifications | Reporting spectral pairs | Propagation | Basic database search | |
| 0.3 min | 3.4 min | 1.4 min | 5 min | 9.7 min | 19.8 min |
Database Search and Peptide Identification
To identify the peptides carrying the detected modifications, all spectra were searched using the pFind search engine (7, 57) against a yeast protein sequence database concatenated with the sequences of the 18 standard proteins and 82 contaminant proteins (15 suggested by the authors of the data, 53 from the cRAP database, and 14 additional trypsin and keratin proteins extracted from the UniProt database). The yeast sequences were added as background to produce meaningful scoring statistics. To estimate the false discovery rate (FDR), the database was searched in a target-decoy strategy (58), in which the whole sequence database was reversed and combined with the original database. The precursor and fragment mass matching tolerances were ±10 ppm and ±0.5 Da, respectively. Trypsin was used for in silico protein digestion, and up to two missed cleavages were allowed. The modification parameters were set as follows. In a basic search, cysteine carbamidomethylation was set as the fixed modification and methionine oxidation was set as the only variable modification. In modification-oriented searches, each detected modification was individually added as an additional variable modification. We did not incorporate all modifications into a single search in order to avoid combinatorial explosion of search space and increasing level of random matches. A maximum of six variable modifications per peptide were allowed. Following each database search, highest-scoring peptides were filtered by three criteria: 1% FDR at spectrum level, −2 to 6 ppm precursor mass deviation, and at least two peptide identifications for each identified protein. For the basic database search, 1302 spectra were successfully identified. For the modification-oriented searches, 646 spectra of peptides with detected modifications were identified. Table II gives the numbers of spectra identified for each modification. A search with semitryptic digestion was also performed, and 218 spectra of semitryptic peptides were identified. In total, by incorporating the discoveries of DeltAMT into database searches, the spectral identification rate was raised from 32% to 53%. It can be expected that some spectra of multiply modified peptides would be identified if all detected modifications as well as nonspecific digestion were taken into account in a single database search.
Based on the identifications by the basic database search and the detected spectral pairs by DeltAMT, peptide propagation was carried out. All of the peptides identified by the basic database search came from either the eighteen standard proteins or contaminant proteins, and none from the reversed or yeast protein sequences, demonstrating the high confidence of these identifications. Identified peptides were propagated among spectral pairs that were obtained at different PEP levels. Supplementary Table S2 gives the detailed propagation results. In this paper, we focus on the results obtained with PEP ≤ 0.02 only. With this level, a total of 1211 spectra that had not been identified by the basic database search were successfully identified by peptide propagation. During propagation, eight edge conflicts and seven node conflicts occurred, and the involved spectral pairs were discarded. The numbers of spectra identified as modified/semi-tryptic peptides are listed in Table II for comparison with database search results. It should be noted that because the peptides identified by peptide propagation and those by modification-oriented database search had different error rates in theory, the performance comparison of the two methods was not absolutely fair. However, we found that the peptide assignments of the spectra identified by both methods were perfectly consistent, demonstrating the accuracy of these identifications. Further, Table II shows that calcium and sodium adducts were better identified by peptide propagation (one reason may be because of the degraded spectrum quality of adducts). A more important role for peptide propagation is that information about the specificity of a modification can be revealed by locating the modification sites, and sometimes this is indispensable for precise determination of the modification identity or for performing database search. For example, we observed that the ∼152 Da had been assigned to carbamidomethylated cysteines in all cases, and we therefore knew that it was in fact a modification of ∼209 Da.
Calcium and Sodium Adducts
The most dominant modification detected in this data set turned out to be from the formation of calcium-peptide adducts. The estimated mass shift caused by this modification is 37.94689 Da, differing by only 0.00005 Da from the theoretical value of 37.94694 Da (one calcium mass minus two hydrogen masses). Potassium adduct formation would cause a very similar mass shift of 37.95588 Da, but the detected mass shift is nearly 0.01 Da away from this theoretical value, well beyond the mass accuracy of LTQ-FT instruments as well as the average mass accuracy of the detected modifications for this data set. Fig. 4 shows the scatter-histogram of delta vectors in the mass interval containing this modification mass. We can see that the Δm values associated with this modification are so concentrated that the standard deviation is only 0.0026 Da. Also, this modification has almost no effect on peptide retention times, which is a common characteristic of metal adducts, because they are usually formed upon ionization and thus will “coelute” with the unmodified peptides. Moreover, no molecular formula composed of the C, H, N, O, and S elements exists such that its molecular mass matches the observed mass shift within a tolerance of 0.01 Da. Therefore, we strongly believe that this modification is because of calcium adduct formation, although it was rarely reported before. As an example, supplementary Fig. S1 illustrates three spectra that were generated from a peptide with different numbers of calcium adduct formations. The calcium in this data set might have come from the CaCl2 added to the sample to enhance the activity of trypsin (59). Another kind of metal adduct abundantly present in this data set was sodium adduct, which is common in LC-MS/MS experiments. By incorporating the calcium/sodium adduct into database search as variable modifications (at aspartic acid, glutamic acid, and the peptide C terminus), 142 spectra and 205 spectra were identified as calcium-peptide and sodium-peptide adducts, respectively. On the other hand, 488 spectra were identified as calcium-peptide adducts through peptide propagation among the single and double-calcium-related spectral pairs with PEP ≤ 0.02. For sodium, this number was 372. The overlapping spectra (134 for calcium and 170 for sodium) identified by database search and peptide propagation had identical peptide sequence assignments, indicating the reliability of these identifications.
Fig. 4.

Scatter-histogram of observed Δ data points around the nominal mass value of 38 Da for the ISB standard protein mix data set. The dense data cluster in the small square, which was automatically located by the DeltAMT algorithm, was induced by the calcium adduct formation.
Deamidation
Deamidation (0.98433 Da) was another abundant modification observed in this data set. It slightly increased the peptide retention time (0.8 min on average). By incorporating it as a variable modification (on asparagine and glutamine) into sequence database search, 165 spectra of deamidated peptides were identified. On the other hand, through peptide propagation among the spectral pairs detected with PEP ≤ 0.02 for this modification, 98 spectra were identified as deamidated peptides. Of these spectra, 87 were also identified by database search and had identical peptide sequence assignments.
Carbamidomethyl DTT
A modification with a mass shift of 151.99699 Da was reported by DeltAMT. Peptide propagation and modification site location results showed that this modification occurred on the cysteine residues. Because we set the cysteine carbamidomethylation (57.021464 Da) as a fixed modification in the basic database search, the actual mass shift caused by this modification is in fact 209.01845 (151.99699 + 57.021464) Da. This is exactly the mass of carbamidomethylated DTT modification of cysteine that was discovered by Chalkley et al. on the same data set (Mixture 2, QTOF instrument) (22). Our result confirms this discovery and further shows that peptides with this modification tend to elute significantly earlier (1.842 min on average) than the ordinary carbamidomethylated peptides. By adding this modification into sequence database search, 82 spectra with this modification were identified.
Oxidation and Dehydration
Oxidation (15.99421 Da) was detected as expected. It significantly decreased the retention time (-4.101 min on average). Oxidation had already been specified as a variable modification (on methionine) in the basic database search, and 99 spectra of oxidized peptides were successfully identified. The mass shift of 18.00828 Da corresponds to the neutral mass of a water molecule. In addition to chemical modifications, such as pyroglutamic acid formation from N-terminal glutamic acids, it could also result from in-source fragmentation. Incorporating it into database search (dehydration of threonine, serine and aspartic acid, formation of pyroglutamic acid at the peptide N terminus) identified 14 spectra of dehydrated peptides.
Acetaldehyde
The last interesting modification reported had an estimated mass shift of 26.01532 Da. This is probably the acetaldehyde (+26) modification, which was also detected by Chalkley et al. (22). Here, we further show that this modification significantly delayed the elution of peptides by about 2.6 min on average. Through peptide propagation, we found that this modification mostly occurred on peptide N termini. Incorporating this modification (on histidine, lysine, and peptide N terminus) into database search, 38 spectra of peptides with this modification were identified, 35 of which had this modification assigned to the N termini of peptides.
Nonspecific Digestion
Mass shifts corresponding to amino acid residues or their combinations were also reported, including isoleucine (I) or leucine (L), arginine (R), lysine (K), valine (V), glycine (G)—isoleucine (I) or leucine (L), and alanine (A)—valine (V). All of them occurred at the termini of tryptic peptides, suggesting that they might represent nonspecific or semispecific digestion of proteins by trypsin. However, the small retention time differences caused by the I/L and GI/L or AV removal also indicated in-source fragmentation. When overlapping tryptic and semi-tryptic peptides were detected by LC-MS/MS, DeltAMT could report their differences as modifications. A database search was carried out with the same search parameters as used in the basic database search but allowing semi-specific digestion, and 218 spectra of semitryptic peptides were identified. One the other hand, through peptide propagation among the spectral pairs detected with PEP ≤ 0.02 for nonspecific digestion, 14 spectra were identified as semi-tryptic peptides. Again, the nine spectra identified by both methods had identical peptide sequence assignments.
False Positives
The only uninterpreted mass shift detected by DeltAMT was 104.09558 Da. Six spectral pairs were detected for this mass shift with PEP ≤ 0.02. However, three of them were conflicting pairs. That is, the two spectra in each of the three pairs were identified as different peptides by database search. For the other three pairs, none of the spectra in them was identified by database search. Moreover, the retention time shift was extraordinarily large (-13 min on average). Therefore, we consider this mass shift a false positive.
Comparison with MS-Alignment
We also analyzed the ISB data set with the MS-Alignment algorithm for comparison and validation purposes. The 4085 spectra in this data set were searched by MS-Alignment against the same database as was searched by pFind. The whole database, including target and decoy sequences, contained less than 6 m amino acids in total. The search parameters were set as follows: instrument, FT-Hybrid; protease, Trypsin; mods, 2; blind, 1; mod, +57, C, fix. It took about 9 h for MS-Alignment to complete the search. The search results were filtered by their p values and the FDR was estimated using the target-decoy strategy. At 2% FDR, a total of 1034 spectra were identified, including 961 spectra matched to peptides from the standard proteins, 55 from contaminant proteins, 10 from decoy sequences, and eight from background yeast proteins. The matches to decoy and yeast sequences were false positives and were discarded. The remaining 1016 identifications were accepted, among which 367 were peptides with positive modification masses, 30 were peptides with negative modification masses, and 619 were unmodified peptides. Fig. 5 illustrates the histogram of modification mass shifts detected by MS-Alignment. It turned out that almost all of the abundant modifications (> 3 identified spectra) detected by MS-Alignment were also detected by DeltAMT. The only one missed by DeltAMT had a mass shift corresponding to ammonia loss, but MS-Alignment failed to detect deamidation that was discovered by DeltAMT. Overall, the two algorithms largely confirmed each other's findings. Although MS-Alignment detected many low-abundance modifications, this was achieved at the cost of a long search time. Because ammonia loss, e.g. N-terminal cyclization of glutamine or carbamidomethylated cysteine, is so common and was abundantly detected by MS-Alignment, we examined the reason why DeltAMT failed to find it. We decreased the d-score cut-off value from 10 to 3 and re-analyzed the data. As a result, a putative modification with d-score of 5.0 was reported with a mass shift of 17.02595 Da (-0.0006 Da from the theoretical mass of NH3). The estimated standard deviation of the mass dimension was 0.002 Da, as small as those estimated for other putative modifications. However, the estimated standard deviation of the retention time dimension was 4.05 min, much larger than those estimated for other putative modifications. It seemed that the ammonia loss had a less consistent effect on the retention times of peptides than other modifications we found. In fact, this mass shift may have three different sources: N-terminal cyclization—then the retention times should be significantly different, and ammonium adduct formation or in source fragmentation—then the retention times should be the same. This explained why this modification was so poorly scored that it was missed by the original analysis of DeltAMT.
Fig. 5.

Modification mass shifts detected by the MS-Alignment algorithm and the corresponding numbers of identified spectra. Those mass shifts with more than three identified spectra are annotated with the nominal mass shift values and modification names.
Results for the MaxQuant HeLa Proteome Data Set
Overview
A direct application of the DeltAMT algorithm to the MaxQuant HeLa proteome data set generated a long list of putative modifications (supplementary Table S3). The dominant modifications were as expected, i.e. SILAC labels (8.01400 Da and 10.00844 Da) and oxidation (15.99477 Da), because this data set was from a SILAC-treated sample. However, many other modifications were reported, most of them being additive pseudo-modifications. Their masses were characterized by fixed mass ladders, e.g. 44.026 Da, suggesting that they were not peptide modifications but were actually derived from nonpeptide polymers. Retention time distribution of the spectra detected as polymers suggested that the majority of polymers were eluted in the beginning and the end of chromatography. A plot of retention times versus precursor masses of all MS/MS spectra uncovered two clusters of polymers, one in the first 30 min and the other in the last 10 min (shown in supplementary Fig. S2). These two clusters contain a total of 1811 spectra, none of which was identified by database search. Therefore, all of the components eluted during these two stages were probably polymers. Following removal of these polymer spectra, we reanalyzed the remaining spectra using DeltAMT. As expected, all modification masses corresponding to polymers disappeared, and only SILAC labels and oxidation were reported with d-scores above 10. When the d-score cut-off value was relaxed to three, potential low-abundance modifications were revealed.
Nonpeptide Polymers
Three series of polymers were recognized, each with a mass ladder of 44.02616, 43.04219, or 72.02107 Da. The first one is probably the well-known polyethylene glycol (PEG) with the structure -(CH2-CH2-O-)n, in which CH2-CH2-O corresponds to 44.02621 Da, differing by 0.00005 Da from the detected mass value. PEG is widely used in biomedical research (e.g. in protein purification) and is often unavoidable in mass spectrometry (60). The second polymer is likely the polyethyleneimine (PEI) with the structure -(CH2-CH2-NH-)n, in which CH2-CH2-NH corresponds to 43.04220 Da, differing by 0.00001 Da from the detected mass value. The third polymer possibly has the formula (C3H4O2)n, where C3H4O2 corresponds to 72.02113 Da, differing by 0.00006 Da from the detected mass value. This may be the polymer of acrylic acid -[CH2-CH(COOH)-]n. The second and the third polymers seemed to have formed some kind of copolymers because several combinations of them were detected.
Low-abundance Modifications
To examine if lower-abundance modifications were present, we decreased the d-score cut-off from ten to three. As a result, several potential modifications were revealed, including methylation, carbamylation, deamidation, etc. (listed in Table IV). As observed in previous studies (21, 61), methylation increased the retention times of peptides, whereas carbamylation decreased the retention times. Their combinations were consistent in terms of retention time shifts. For example, the retention time shift caused by dimethylation was approximately twice as much as that by methylation. The mass shift of about 44.026 Da was still detected, indicating that PEG might still exist in the retention time interval considered (from the 31st minute to the 130th minute). However, the corresponding retention time shift was very different from the one detected from the whole retention time range. Therefore, it might also be a modification on peptides, possibly hydroxyethylation, which would result in the same mass shift as observed. No reliable spectral pairs were detected for those possible low-abundance modifications, and we did not make further efforts to validate them.
Table IV. Detected modifications for the MaxQuant HeLa proteome data set (within retention time ranging from the 31st minute to the 130th minute).
|
Mono-modifications | |||||
|---|---|---|---|---|---|
| Δm (Da) | Δt (min) | d-score | Pairs (PEP) | Interpretation | Mass deviation (Da) |
| 8.01398 | 0.0299 | 813.0 | 4194 (0.02) | SILAC label | −0.00022 |
| 10.00844 | 0.0306 | 516.7 | 2297 (0.02) | SILAC label | 0.00017 |
| 15.99474 | 0.0545 | 163.8 | 1395 (0.02) | Oxidation | −0.00018 |
| 14.01558 | 1.577 | 8.3 | 0 | Methylation | −0.00007 |
| 43.00604 | −2.106 | 8.3 | 0 | Carbamylation | 0.00023 |
| 41.02658 | −1.208 | 4.5 | 0 | Acetonitrile adduct or amidination with methyl acetimidate | 0.00003 |
| 0.98466 | −0.067 | 3.9 | 0 | Deamidation | 0.00064 |
|
Additive pseudo-modifications | |||||
|---|---|---|---|---|---|
| Δm (Da) | Δt (min) | d-score | Pairs (PEP) | Interpretation | Mass deviation (Da) |
| 31.98956 | 0.0715 | 116.1 | 898 (0.02) | Double oxidations or di-oxidation | −0.00027 |
| 114.04269 | −0.536 | 10.3 | 35 (0.10) | Dimethylation + di-carbamylation or di-carbamidomethylation or GlyGly ubiquitination | −0.00024 |
| 28.03128 | 3.638 | 9.1 | 0 | Dimethylation | −0.00002 |
| 44.02608 | 4.571 | 7.6 | 0 | Polymer (PEG) or hydroxyethylation | −0.00014 |
| 71.03701 | 0.446 | 7.5 | 0 | Dimethylation + carbamylation | −0.00010 |
| 85.05261 | 2.096 | 5.6 | 0 | Trimethylation + carbamylation | −0.00015 |
Database Search and Peptide Identification
The 13,572 tandem mass spectra were searched against the target-decoy International Protein Index human database using almost the same search parameters as those used for the ISB data set with the following exceptions. SILAC labels, i.e. Arginine-13C615N4 (10.008 Da) and Lysine-13C615N2 (8.014 Da), were set as variable modifications in addition to methionine oxidation. The precursor mass tolerance was set to ±7 ppm, as suggested by the authors of the data set. At 1% FDR, 6586 spectra were identified, 2894 of them having SILAC labels. Through peptide propagation on the detected 6491 SILAC pairs with PEP ≤ 0.02, 358 additional spectra were identified. During propagation, 48 edge conflicts and three node conflicts occurred, and involved spectral pairs were discarded. The increase in the number of identified spectra was relatively small because the SILAC labels had already been specified in database search. Besides SILAC labels, oxidation was the most abundant modification detected in this data set. Through peptide propagation on 2293 oxidation-related spectral pairs with PEP ≤ 0.02, 985 additional spectra were identified. During propagation, 33 edge conflicts and three node conflicts occurred, and involved spectral pairs were discarded. The total number of interpreted spectra increased by 48%, taking into account the1811 detected polymer spectra.
DISCUSSION
We present in this paper a novel algorithm named DeltAMT to detect the presence of abundant protein modifications from LC-MS/MS data. The unique feature of DeltAMT is its exclusive and complete use of peptide precursor information. Thus, it is extremely fast and insensitive to the quality of fragmentation spectra. Discovering abundantly present modifications in a sample can not only increase the spectral identification rate but also consequently increase the chance of identifying low-abundance proteins or modifications. In addition to modifications, DeltAMT can also effectively detect other events, such as isotopic labels, nonspecific digestion, and polymer contaminants. Therefore, DeltAMT can be used in many circumstances. For example, one may want to check if any unwanted chemical modifications or polymers have been heavily introduced during sample processing. To this end, it takes only several minutes for DeltAMT to analyze one LC-MS/MS run and report discoveries, based on which experimental protocols can be improved. In fact, we have successfully performed such data analysis and protocol improvement in experiments for core fucosylated glycoprotein identification using a preliminary version of the algorithm (62, 63). The analysis results of DeltAMT are also potentially beneficial for quantitation analysis of proteins. For example, one can avoid selecting peptides prone to chemical modifications for quantitation analysis. Furthermore, the increased spectral identification rate can potentially improve the accuracy of label-free quantitation.
One limitation of DeltAMT is that it works for abundant modifications only. However, several fragment information-based methods already exist that are able to detect low-abundance modifications. We consider DeltAMT a powerful complement to these methods. To detect lower-abundance modifications with DeltAMT, one may use lower d-score cut-offs. This will increase the level of false positives, but if anything of interest is detected, further analysis can be followed. Another issue worth mentioning is that because DeltAMT uses retention time information, its performance and analysis results depend on sample separation strategy and experimental configuration. For example, in experiments with offline fractionation, if the modified and unmodified versions of a peptide are separated into different fractions, then this modification will not be detected by DeltAMT. Moreover, retention time shifts caused by modifications vary with LC conditions. For instance, for acidic modifications, depending on the ion pairing agent used in the mobile phase, the modified peptides may elute earlier (e.g. when trifluoroacetic acid is the ion pairing agent) or later (e.g. when acetic acid or formic acid is used) than their unmodified counterparts. Finally, we point out that in the current version of DeltAMT, the peptide precursor data are from the MS/MS spectra exported by the instrument control software, and a simple strategy is used for data preprocessing. They are subject to imperfections, such as redundancy, incorrect mono-isotopic masses and coarse retention time values. In the future, we plan to extract more accurate precursor information directly from the MS spectra by recovering the retention profiles and recognizing isotopic clusters of peptides. Despite these limitations mentioned above, considering the trivial cost and informative outputs of DeltAMT, we expect that it will become a routine data analysis tool in most proteomics pipelines.
Acknowledgments
We would like to thank the anonymous reviewers who pointed out the problems in the initial version of the paper and gave many informative and constructive suggestions. We gratefully appreciate the help of Dr. Meng-Qiu Dong in explaining some of the detected modifications and in improving the writing of the paper. We are also grateful to Wantao Ying, Zuofei Yuan, and Haipeng Wang for valuable discussions.
Footnotes
* This work was supported by the National Natural Science Foundation of China under Grant Nos. 30900262 and 20735005, the CAS Knowledge Innovation Program under Grant No. KGGX1-YW-13, the National High Technology Research and Development Program (863) of China under Grant Nos. 2007AA02Z315, 2008AA02Z309, and 2006AA02A308, and the National Key Basic Research and Development Program (973) of China under Grant No. 2010CB912701.
 This article contains supplemental Tables S1 to S3 and Figs. S1 and S2.
This article contains supplemental Tables S1 to S3 and Figs. S1 and S2.
1 The abbreviations used are:
- LC-MS/MS
- liquid chromatography-tandem MS
- AMT
- accurate mass and time
- DeltAMT
- delta accurate mass and time
- FDR
- false discovery rate
- ISB
- Institute for Systems Biology
- MS/MS
- tandem mass spectrometry
- PEP
- posterior error rate
- SILAC
- stable isotope labeling with amino acids in cell culture.
REFERENCES
- 1. Aebersold R., Mann M. (2003) Mass spectrometry-based proteomics. Nature 422, 198–207 [DOI] [PubMed] [Google Scholar]
- 2. Mann M., Jensen O. N. (2003) Proteomic analysis of post-translational modifications. Nat. Biotechnol. 21, 255–261 [DOI] [PubMed] [Google Scholar]
- 3. Witze E. S., Old W. M., Resing K. A., Ahn N. G. (2007) Mapping protein post-translational modifications with mass spectrometry. Nat. Methods 4, 798–806 [DOI] [PubMed] [Google Scholar]
- 4. Eng J. K., McCormack A. L., Yates J. R., III (1994) An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass. Spectrom. 5, 976–989 [DOI] [PubMed] [Google Scholar]
- 5. Perkins D. N., Pappin D. J., Creasy D. M., Cottrell J. S. (1999) Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20, 3551–3567 [DOI] [PubMed] [Google Scholar]
- 6. Craig R., Beavis R. C. (2004) TANDEM: matching proteins with tandem mass spectra. Bioinformatics 20, 1466–1467 [DOI] [PubMed] [Google Scholar]
- 7. Fu Y., Yang Q., Sun R., Li D., Zeng R., Ling C. X., Gao W. (2004) Exploiting the kernel trick to correlate fragment ions for peptide identification via tandem mass spectrometry. Bioinformatics 20, 1948–1954 [DOI] [PubMed] [Google Scholar]
- 8. Colinge J., Masselot A., Giron M., Dessingy T., Magnin J. (2003) OLAV: towards high-throughput tandem mass spectrometry data identification. Proteomics 3, 1454–1463 [DOI] [PubMed] [Google Scholar]
- 9. Geer L. Y., Markey S. P., Kowalak J. A., Wagner L., Xu M., Maynard D. M., Yang X., Shi W., Bryant S. H. (2004) Open mass spectrometry search algorithm. J. Proteome Res. 3, 958–964 [DOI] [PubMed] [Google Scholar]
- 10. Dancík V., Addona T. A., Clauser K. R., Vath J. E., Pevzner P. A. (1999) De novo peptide sequencing via tandem mass spectrometry. J. Comput. Biol. 6, 327–342 [DOI] [PubMed] [Google Scholar]
- 11. Frank A., Pevzner P. (2005) PepNovo: de novo peptide sequencing via probabilistic network modeling. Anal. Chem. 77, 964–973 [DOI] [PubMed] [Google Scholar]
- 12. Ma B., Zhang K., Hendrie C., Liang C., Li M., Doherty-Kirby A., Lajoie G. (2003) PEAKS: powerful software for peptide de novo sequencing by MS/MS. Rapid Commun. Mass Spectrom. 17, 2337–2342 [DOI] [PubMed] [Google Scholar]
- 13. Taylor J. A., Johnson R. S. (2001) Implementation and uses of automated de novo peptide sequencing by tandem mass spectrometry. Anal. Chem. 73, 2594–2604 [DOI] [PubMed] [Google Scholar]
- 14. Mann M., Wilm M. (1994) Error-tolerant identification of peptides in sequence databases by peptide sequence tags. Anal. Chem. 66, 4390–4399 [DOI] [PubMed] [Google Scholar]
- 15. Tabb D. L., Saraf A., Yates J. R., 3rd (2003) GutenTag: high-throughput sequence tagging via an empirically derived fragmentation model. Anal. Chem. 75, 6415–6421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tanner S., Shu H., Frank A., Wang L. C., Zandi E., Mumby M., Pevzner P. A., Bafna V. (2005) InsPecT: identification of posttranslationally modified peptides from tandem mass spectra. Anal. Chem. 77, 4626–4639 [DOI] [PubMed] [Google Scholar]
- 17. Martens L., Apweiler R. (2009) Algorithms and Databases. In: Reinders J., Sickmann A. eds. Proteomics Methods and Protocols, 245–260, Humana Press, New York: [DOI] [PubMed] [Google Scholar]
- 18. Hunyadi-Gulyás É., Medzihradszky K. F. (2004) Factors that contribute to the complexity of protein digests. Drug Discovery Today: TARGETS 3, 3–10 [Google Scholar]
- 19. Nesvizhskii A. I., Roos F. F., Grossmann J., Vogelzang M., Eddes J. S., Gruissem W., Baginsky S., Aebersold R. (2006) Dynamic spectrum quality assessment and iterative computational analysis of shotgun proteomic data: toward more efficient identification of post-translational modifications, sequence polymorphisms, and novel peptides. Mol. Cell Proteomics 5, 652–670 [DOI] [PubMed] [Google Scholar]
- 20. Tsur D., Tanner S., Zandi E., Bafna V., Pevzner P. A. (2005) Identification of post-translational modifications by blind search of mass spectra. Nat. Biotechnol. 23, 1562–1567 [DOI] [PubMed] [Google Scholar]
- 21. Savitski M. M., Nielsen M. L., Zubarev R. A. (2006) ModifiComb, a new proteomic tool for mapping substoichiometric post-translational modifications, finding novel types of modifications, and fingerprinting complex protein mixtures. Mol. Cell Proteomics 5, 935–948 [DOI] [PubMed] [Google Scholar]
- 22. Chalkley R. J., Baker P. R., Medzihradszky K. F., Lynn A. J., Burlingame A. L. (2008) In-depth analysis of tandem mass spectrometry data from disparate instrument types. Mol. Cell Proteomics 7, 2386–2398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nielsen M. L., Savitski M. M., Zubarev R. A. (2006) Extent of modifications in human proteome samples and their effect on dynamic range of analysis in shotgun proteomics. Mol. Cell Proteomics 5, 2384–2391 [DOI] [PubMed] [Google Scholar]
- 24. Yates J. R., 3rd, Eng J. K., McCormack A. L., Schieltz D. (1995) Method to correlate tandem mass spectra of modified peptides to amino acid sequences in the protein database. Anal. Chem. 67, 1426–1436 [DOI] [PubMed] [Google Scholar]
- 25. Yates J. R., 3rd, Eng J. K., McCormack A. L. (1995) Mining genomes: correlating tandem mass spectra of modified and unmodified peptides to sequences in nucleotide databases. Anal. Chem. 67, 3202–3210 [DOI] [PubMed] [Google Scholar]
- 26. Creasy D. M., Cottrell J. S. (2004) Unimod: Protein modifications for mass spectrometry. Proteomics 4, 1534–1536 [DOI] [PubMed] [Google Scholar]
- 27. Creasy D. M., Cottrell J. S. (2002) Error tolerant searching of uninterpreted tandem mass spectrometry data. Proteomics 2, 1426–1434 [DOI] [PubMed] [Google Scholar]
- 28. Craig R., Beavis R. C. (2003) A method for reducing the time required to match protein sequences with tandem mass spectra. Rapid Commun. Mass Spectrom. 17, 2310–2316 [DOI] [PubMed] [Google Scholar]
- 29. Pevzner P. A., Dancík V., Tang C. L. (2000) Mutation-tolerant protein identification by mass-spectrometry. J. Comput. Biol. 7, 777–787 [DOI] [PubMed] [Google Scholar]
- 30. Tanner S., Pevzner P. A., Bafna V. (2006) Unrestrictive identification of post-translational modifications through peptide mass spectrometry. Nat. Protoc. 1, 67–72 [DOI] [PubMed] [Google Scholar]
- 31. Chalkley R. J., Baker P. R., Huang L., Hansen K. C., Allen N. P., Rexach M., Burlingame A. L. (2005) Comprehensive analysis of a multidimensional liquid chromatography mass spectrometry data set acquired on a quadrupole selecting, quadrupole collision cell, time-of-flight mass spectrometer: II. New developments in Protein Prospector allow for reliable and comprehensive automatic analysis of large datasets. Mol. Cell Proteomics 4, 1194–1204 [DOI] [PubMed] [Google Scholar]
- 32. Hansen B. T., Davey S. W., Ham A. J., Liebler D. C. (2005) P-Mod: an algorithm and software to map modifications to peptide sequences using tandem MS data. J. Proteome Res. 4, 358–368 [DOI] [PubMed] [Google Scholar]
- 33. Tang W. H., Halpern B. R., Shilov I. V., Seymour S. L., Keating S. P., Loboda A., Patel A. A., Schaeffer D. A., Nuwaysir L. M. (2005) Discovering known and unanticipated protein modifications using MS/MS database searching. Anal. Chem. 77, 3931–3946 [DOI] [PubMed] [Google Scholar]
- 34. Havilio M., Wool A. (2007) Large-scale unrestricted identification of post-translation modifications using tandem mass spectrometry. Anal. Chem. 79, 1362–1368 [DOI] [PubMed] [Google Scholar]
- 35. Baumgartner C., Rejtar T., Kullolli M., Akella L. M., Karger B. L. (2008) SeMoP: a new computational strategy for the unrestricted search for modified peptides using LC-MS/MS data. J. Proteome Res. 7, 4199–4208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chen Y., Chen W., Cobb M. H., Zhao Y. (2009) PTMap–a sequence alignment software for unrestricted, accurate, and full-spectrum identification of post-translational modification sites. Proc. Natl. Acad. Sci. U.S.A. 106, 761–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Searle B. C., Dasari S., Turner M., Reddy A. P., Choi D., Wilmarth P. A., McCormack A. L., David L. L., Nagalla S. R. (2004) High-throughput identification of proteins and unanticipated sequence modifications using a mass-based alignment algorithm for MS/MS de novo sequencing results. Anal. Chem. 76, 2220–2230 [DOI] [PubMed] [Google Scholar]
- 38. Han Y., Ma B., Zhang K. (2005) SPIDER: software for protein identification from sequence tags with de novo sequencing error. J. Bioinform. Comput. Biol. 3, 697–716 [DOI] [PubMed] [Google Scholar]
- 39. Kim S., Na S., Sim J. W., Park H., Jeong J., Kim H., Seo Y., Seo J., Lee K. J., Paek E. (2006) MODi: a powerful and convenient web server for identifying multiple post-translational peptide modifications from tandem mass spectra. Nucleic Acids Res. 34, W258–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Shen Y., Tolić N., Hixson K. K., Purvine S. O., Anderson G. A., Smith R. D. (2008) De novo sequencing of unique sequence tags for discovery of post-translational modifications of proteins. Anal. Chem. 80, 7742–7754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Liu C., Yan B., Song Y., Xu Y., Cai L. (2006) Peptide sequence tag-based blind identification of post-translational modifications with point process model. Bioinformatics 22, e307–313 [DOI] [PubMed] [Google Scholar]
- 42. Bandeira N., Tsur D., Frank A., Pevzner P. (2006) A new approach to protein identification, in 10th Annual International Conference on Research in Computational Molecular Biology, Venice, Italy, April 2–5, 2006, Springer-Verlag, Berlin Heidelberg [Google Scholar]
- 43. Bandeira N. (2007) Spectral networks: a new approach to de novo discovery of protein sequences and posttranslational modifications. BioTechniques 42, 687–695 [DOI] [PubMed] [Google Scholar]
- 44. Falkner J. A., Falkner J. W., Yocum A. K., Andrews P. C. (2008) A spectral clustering approach to MS/MS identification of post-translational modifications. J. Proteome Res. 7, 4614–4622 [DOI] [PubMed] [Google Scholar]
- 45. Ahrné E., Masselot A., Binz P. A., Müller M., Lisacek F. (2009) A simple workflow to increase MS2 identification rate by subsequent spectral library search. Proteomics 9, 1731–1736 [DOI] [PubMed] [Google Scholar]
- 46. Lam H., Deutsch E. W., Eddes J. S., Eng J. K., King N., Stein S. E., Aebersold R. (2007) Development and validation of a spectral library searching method for peptide identification from MS/MS. Proteomics 7, 655–667 [DOI] [PubMed] [Google Scholar]
- 47. Ye D., Fu Y., Sun R. X., Wang H. P., Yuan Z. F., Chi H., He S. M. (2010) Open MS/MS spectral library search to identify unanticipated post-translational modifications and increase spectral identification rate. Bioinformatics 26, i399–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Smith R. D., Anderson G. A., Lipton M. S., Pasa-Tolic L., Shen Y., Conrads T. P., Veenstra T. D., Udseth H. R. (2002) An accurate mass tag strategy for quantitative and high-throughput proteome measurements. Proteomics 2, 513–523 [DOI] [PubMed] [Google Scholar]
- 49. Zimmer J. S., Monroe M. E., Qian W. J., Smith R. D. (2006) Advances in proteomics data analysis and display using an accurate mass and time tag approach. Mass Spectrom. Rev. 25, 450–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Potthast F., Gerrits B., Häkkinen J., Rutishauser D., Ahrens C. H., Roschitzki B., Baerenfaller K., Munton R. P., Walther P., Gehrig P., Seif P., Seeberger P. H., Schlapbach R. (2007) The Mass Distance Fingerprint: a statistical framework for de novo detection of predominant modifications using high-accuracy mass spectrometry. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 854, 173–182 [DOI] [PubMed] [Google Scholar]
- 51. Griffiths S. W., Cooney C. L. (2002) Development of a peptide mapping procedure to identify and quantify methionine oxidation in recombinant human alpha1-antitrypsin. J. Chromatogr. A 942, 133–143 [DOI] [PubMed] [Google Scholar]
- 52. Hsu Y. R., Narhi L. O., Spahr C., Langley K. E., Lu H. S. (1996) In vitro methionine oxidation of Escherichia coli-derived human stem cell factor: effects on the molecular structure, biological activity, and dimerization. Protein Sci. 5, 1165–1173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Dasari S., Wilmarth P. A., Rustvold D. L., Riviere M. A., Nagalla S. R., David L. L. (2007) Reliable detection of deamidated peptides from lens crystallin proteins using changes in reversed-phase elution times and parent ion masses. J. Proteome Res. 6, 3819–3826 [DOI] [PubMed] [Google Scholar]
- 54. Cox J., Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 [DOI] [PubMed] [Google Scholar]
- 55. Klimek J., Eddes J. S., Hohmann L., Jackson J., Peterson A., Letarte S., Gafken P. R., Katz J. E., Mallick P., Lee H., Schmidt A., Ossola R., Eng J. K., Aebersold R., Martin D. B. (2008) The standard protein mix database: a diverse data set to assist in the production of improved Peptide and protein identification software tools. J. Proteome Res. 7, 96–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Cox J., Mann M. (2007) Is proteomics the new genomics? Cell 130, 395–398 [DOI] [PubMed] [Google Scholar]
- 57. Wang L. H., Li D. Q., Fu Y., Wang H. P., Zhang J. F., Yuan Z. F., Sun R. X., Zeng R., He S. M., Gao W. (2007) pFind 2.0: a software package for peptide and protein identification via tandem mass spectrometry. Rapid Commun. Mass Spectrom. 21, 2985–2991 [DOI] [PubMed] [Google Scholar]
- 58. Elias J. E., Gygi S. P. (2007) Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 4, 207–214 [DOI] [PubMed] [Google Scholar]
- 59. Green N. M., Neurath H. (1953) The effects of divalent cations on trypsin. J. Biol. Chem. 204, 379–390 [PubMed] [Google Scholar]
- 60. Shen J., Buko A. (2002) Rapid identification of proteins in polyethylene glycol-containing samples using capillary electrophoresis electrospray mass spectrometry. Anal. Biochem. 311, 80–83 [DOI] [PubMed] [Google Scholar]
- 61. Wilmarth P. A., Tanner S., Dasari S., Nagalla S. R., Riviere M. A., Bafna V., Pevzner P. A., David L. L. (2006) Age-related changes in human crystallins determined from comparative analysis of post-translational modifications in young and aged lens: does deamidation contribute to crystallin insolubility? J. Proteome Res. 5, 2554–2566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Fu Y., Jia W., Lu Z., Wang H., Yuan Z., Chi H., Li Y., Xiu L., Wang W., Liu C., Wang L., Sun R., Gao W., Qian X., He S. M. (2009) Efficient discovery of abundant post-translational modifications and spectral pairs using peptide mass and retention time differences. BMC Bioinformatics 10 Suppl 1, S50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Jia W., Lu Z., Fu Y., Wang H. P., Wang L. H., Chi H., Yuan Z. F., Zheng Z. B., Song L. N., Han H. H., Liang Y. M., Wang J. L., Cai Y., Zhang Y. K., Deng Y. L., Ying W. T., He S. M., Qian X. H. (2009) A strategy for precise and large scale identification of core fucosylated glycoproteins. Mol. Cell Proteomics 8, 913–923 [DOI] [PMC free article] [PubMed] [Google Scholar]