

Abstract
The impact of Nα-terminal acetylation on protein stability and protein function in general recently acquired renewed and increasing attention. Although the substrate specificity profile of the conserved enzymes responsible for Nα-terminal acetylation in yeast has been well documented, the lack of higher eukaryotic models has hampered the specificity profile determination of Nα-acetyltransferases (NATs) of higher eukaryotes. The fact that several types of protein N termini are acetylated by so far unknown NATs stresses the importance of developing tools for analyzing NAT specificities. Here, we report on a method that implies the use of natural, proteome-derived modified peptide libraries, which, when used in combination with two strong cation exchange separation steps, allows for the delineation of the in vitro specificity profiles of NATs. The human NatA complex, composed of the auxiliary hNaa15p (NATH/hNat1) subunit and the catalytic hNaa10p (hArd1) and hNaa50p (hNat5) subunits, cotranslationally acetylates protein N termini initiating with Ser, Ala, Thr, Val, and Gly following the removal of the initial Met. In our studies, purified hNaa50p preferred Met-Xaa starting N termini (Xaa mainly being a hydrophobic amino acid) in agreement with previous data. Surprisingly, purified hNaa10p preferred acidic N termini, representing a group of in vivo acetylated proteins for which there are currently no NAT(s) identified. The most prominent representatives of the group of acidic N termini are γ- and β-actin. Indeed, by using an independent quantitative assay, hNaa10p strongly acetylated peptides representing the N termini of both γ- and β-actin, and only to a lesser extent, its previously characterized substrate motifs. The immunoprecipitated NatA complex also acetylated the actin N termini efficiently, though displaying a strong shift in specificity toward its known Ser-starting type of substrates. Thus, complex formation of NatA might alter the substrate specificity profile as compared with its isolated catalytic subunits, and, furthermore, NatA or hNaa10p may function as a post-translational actin Nα-acetyltransferase.
The multisubunit and ribosome-associated protein Nα-acetyltransferases (NATs)1 are omnipresent enzyme complexes that catalyze the transfer of the acetyl moiety from acetyl-CoA to the primary α-amines of N termini of nascent proteins (1–3). As up to 50 to 60% of yeast proteins and 80 to 90% of human proteins are modified in this manner, Nα-acetylation is a widespread protein modification in eukaryotes (4–7), and the pattern of modification has remained largely conserved throughout evolution (4, 8). NATs belong to a subfamily of the Gcn5-related N-acetyltransferase superfamily of N-acetyltransferases, additionally encompassing the well-studied histone acetyltransferases that are implicated in epigenetic imprinting.
In yeast and humans, three main NAT complexes, NatA, NatB, and NatC were found to be responsible for the majority of Nα-terminal acetylations (1). The NatA complex, responsible for cotranslational Nα-terminal acetylation of proteins with Ser, Ala, Thr, Gly, and Val N termini, is composed of two main subunits, the catalytic subunit Naa10p (previously known as Ard1p) and the auxiliary subunit Naa15p (previously known as Nat1p/NATH) (9–11). Furthermore, a third catalytic subunit Naa50p (previously known as Nat5)—an acetyltransferase shown to function in chromosome cohesion and segregation (12–14)—was found to physically interact with the NatA complex of yeast (2), fruit fly (12), and human (15). Recently, human Naa50p (hNaa50p) was reported to display lysine or Nε-acetyltransferase as well as NAT activity (16), the latter was defined as NatE activity (16). Interestingly, the chaperone-like, Huntingtin interacting protein HYPK, identified as a novel stable interactor of human NatA, was functionally implicated in the N-terminal acetylation of an in vivo NatA substrate, demonstrating that NAT complex formation and composition may have an overall influence on the observed (degree of) Nα-acetylation (17). Further, subunits of the human NatA complex have been coupled to cancer-related processes and differentiation, with altered subunit expression reported in papillary thyroid carcinoma, neuroblastoma, and retinoic acid induced differentiation. Furthermore, the NatA catalytic subunit was found to be implicated in processes such as hypoxia-response and the β-catenin pathway (18, 19). Of note is that in line with the differential localization patterns of the individual NatA subunits (9, 13, 20, 21), other data indicate that these subunits might well exert NatA-independent enzymatic functions (13, 22, 23). Given that a significant fraction of hNaa10p and hNaa15p are nonribosomal (9), and given the multitude of postulated post-translational in vivo N-acetylation events recently reported (24–26), these observations argue in favor of the existence of NAT complexes and/or catalytic NAT-subunits acting post-translationally.
Similar to NatA, the NatB and NatC complexes, composed of the catalytic subunit Naa20p or Naa30p and the auxiliary subunits Naa25p or Naa35p and Naa38p respectively, are conserved from yeast to higher eukaryotes concerning their subunit composition as well as their substrate specificity. Both these complexes display activity toward methionine-starting N termini, with NatB preferring acidic residues as well as Asn and Gln at P2′-sites2, whereas NatC prefers hydrophobic amino acid residues at substrate P2′-sites (1, 27, 28).
Nα-acetylation affects various protein functions such as localization, activity, association, and stability (29, 30). Only recently a more generalized function of protein Nα-acetylation in generating so-called N-terminal degrons marking proteins for removal was put forward (31). The lack of mouse models in addition to the fact that (combined) knockdown of individual components of Nα-acetyltransferases only marginally affect the overall Nα-acetylation status (4) have so far hampered the molecular characterization of the substrate specificity profile of (yet uncharacterized) NATs. To date, all eukaryote Nα-acetylation events are assumed to be catalyzed by the five known NATs (32). However, an additional level of complexity is imposed by the fact that in contrast to yeast, higher eukaryotes express multiple splice variants of various NAT subunits as well as paralogs thereof (33, 34), further implicating that a specific NAT's substrate specificity might be altered in this way, in addition to the possible existence of substrate redundancy. Moreover, regulation of substrate specificity and stability of NAT activity can be imposed by differential complex formation and post-translational modifications including phosphorylation, auto-acetylation, and specific proteolytic cleavage of the catalytic subunits (9, 16, 17). As such, a detailed understanding of the substrate specificity of NATs, and the regulation thereof, could help unravel the physiological substrate repertoires as well as the associated physiological roles of NATs in the normal and the disease state.
The specificity of Nα-acetyltransferases and their endogenous substrates were originally studied by two-dimensional-PAGE: Nα-acetylation neutralizes the N-terminal positive charge, resulting in an altered electrophoretic protein migration during isoelectric focusing (35–38). Recently, this altered biophysical property was also exploited to enrich for protein N-termini using low pH strong cation exchange (SCX) chromatography (24, 39). As an example, SCX prefractionation combined with N-terminal combined fractional diagonal chromatography, a targeted proteomics technology negatively selecting for protein N-terminal peptides, stable isotope labeling of amino acids in cell culture, and amino-directed modifiers (40), was used to study the in vivo substrate repertoires of human as well as yeast NatA (4).
Nevertheless, the various methods reported today to study in detail Nα-terminal acetylation and thus the specificities of different NATs make use of a limited and therefore somewhat biased set of synthesized peptide substrates and comprise the rather laborious detection of radioactive acetylated products as well as enzyme-coupled methods quantifying acetyl-CoA conversion. Because (proteome-derived) peptide libraries have been used extensively to study epitope mapping (41), protein-protein interactions (42), protein modifications such as phosphorylation (43), and proteolysis (44, 45), as well as for determining the substrate specificity of the Nα-deblocking peptide deformylase (46), we reckoned that the development of an oligopeptide-based acetylation assay should allow for more comprehensive screening of NAT-like activities. We here report on the development of a peptide-based method to systematically screen for the in vitro sequence specificity profile of individual NATs as well as endogenous NAT complexes. In summary, SCX enriched, Nα-free peptide libraries, derived from natural proteomes build up the peptide substrate pool. And, upon incubation, NAT Nα-acetylated peptides are enriched by a second SCX fractionation step, resulting in a positive selection of NAT-specific peptide substrates. By use of this proteome-derived peptide library approach, we here delineated (differences in) the specificity profiles of hNaa50p and hNaa10p as isolated hNatA components, as well as of assayed their combined activity when in their native hNatA complex.
EXPERIMENTAL PROCEDURES
Cell Culture
Human K-562 cells were from the ATCC (CCL-243, American Type Culture Collection, Manassas, VA, USA) and were grown in RPMI 1640 medium supplemented with 10% fetal calf serum (Invitrogen, Carlsbad, CA), 100 units/ml penicillin (Invitrogen), and 100 μg/ml streptomycin (Invitrogen). Human A-431 cells (ATCC, CLR-1555) were grown in Dulbecco's modified Eagle's medium supplemented with with 2 mm l-glutamine, 50 mg/ml gentamicin, and 10% newborn calf serum (Lonza Group, Basel, Switzerland). Cells were cultured at 37 °C and in 5% CO2.
Preparation of Proteome-derived Peptide Libraries
Proteome-derived peptide libraries were generated from human K-562 cells. Cells were repeatedly (3×) washed in d-phosphate-buffered saline and then re-suspended at 7 × 106 cells per ml in lysis buffer (50 mm sodium phosphate buffer pH 7.5, 100 mm NaCl, 1% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonic acid (CHAPS), and 0.5 mm EDTA) in the presence of protease inhibitors (Complete protease inhibitor mixture tablet (Roche Diagnostics, Mannheim, Germany)). Following lysis for 10 min on ice, the lysate was cleared by centrifugation for 10 min at 16,000 × g and solid guanidinum hydrochloride was added to the supernatant to a final concentration of 4 m. The protein samples were reduced and S-alkylated, followed by tri-deuteroacetylation of primary amines and digestion with trypsin as described previously (39, 47). The resulting peptide mixtures were vacuum dried. The dried peptides were redissolved in 500 μl 50% acetonitrile. The sample was acidified to pH 3.0 using a stock solution of 1% trifluoroacetic acid (TFA) in 50% acetonitrile and further diluted with 10 mm sodium phosphate in 50% acetonitrile to a final volume of 1 ml. This peptide mixture was then loaded onto an AccuBONDII SCX SPE cartridge (Agilent Technologies, Waldbronn, Germany) and SCX separation (SCX fractionation 1) of Nα-blocked N-terminal peptides and C-terminal peptides from Nα-free peptides was performed as described previously (39, 48). The flow-through containing the Nα-blocked N-terminal peptides and C-terminal peptides was discarded and the SCX-bound fraction (containing the Nα-free peptides) was collected by elution with 4 ml of 400 mm NaCl and 10 mm sodium phosphate in 40% of acetonitrile (pH 3.0). Eluted peptides were vacuum dried and redissolved in 1 ml of high performance liquid chromatography (HPLC) solvent A (10 mm ammonium acetate in 2% acetonitrile, pH 5.5). C18 solid-phase extraction (SPE desalting step) of the Nα-free peptides was performed by loading the peptide mixture onto a AccuBONDII ODS-C18 SPE cartridge (1 ml tube, 100 mg, Agilent Technologies, Santa Clara, CA). This cartridge has a binding capacity of 1 mg of peptides and thus for each milligram of material, a separate cartridge was used. Prior to sample loading, the cartridges were washed with 2 ml of 50% acetonitrile and then washed with 5 ml of HPLC solvent A. Sample loading was followed by washing the C18 cartridge with 5 ml of 2% acetonitrile. Peptides were eluted with 3 ml of 70% acetonitrile and subsequently vacuum dried.
In Vitro Peptide Library-based NAT Assay
One hundred nanomols of the desalted Nα-free peptide pool was reconstituted in acetylation buffer (50 mm Tris-HCl (pH 8.5), 1 mm dithiotreitol, 800 μm EDTA, 10% glycerol) and equimolar amounts of a stable isotope encoded variant of acetyl-CoA, 13C2-acetyl CoA, (99% 13C2-acetyl CoA, ISOTEC-Sigma (lithium salt)) and the indicated amount of enzyme was added to a final reaction volume of 1 ml. The reaction was allowed to proceed for 1 h (for the recombinant enzymes) or 2 h (for the immunoprecipitated complexes) at 37 °C and stopped by addition of acetic acid to a 5% final concentration. SPE was then performed as described above.
NAT Oligopeptide-substrate Recovery and Reverse Phase (RP)-HPLC-based Separation
Peptides starting with pyroglutamate were unblocked prior to the second SCX fractionation step. Here, 25 μl of pGAPase (25 U/ml) (TAGZymeTM kit, Qiagen, Hilden, Germany) was activated for 10 min at 37 °C by addition of 1 μl of 50 mm EDTA (pH 8.0), 1 μl of 800 mm NaCl, and 11 μl of freshly prepared 50 mm cysteamine-HCl. Twenty-five microliters of Qcyclase (50 U/ml, TAGZymeTM) was then added to the pGAPase solution. The dried peptides were re-dissolved in 212 μl of buffer containing 16 mm NaCl, 0.5 mm EDTA, 3 mm cysteamine, and 50 μm aprotinin. The activated pGAPase and Q-cyclase mixture was added to the peptide sample and the mixture (275 μl total volume) was incubated for 60 min at 37 °C. Two hundred and seventy-five microliters acetonitrile was then added and the sample was acidified to pH 3 using a 1% TFA stock solution in 50% acetonitrile. The sample was further diluted with 10 mm sodium phosphate in 50% acetonitrile to a final volume of 1 ml. SCX enrichment of Nα-blocked N-terminal peptides was performed as described (39) (SCX fractionation 2). The SCX fraction containing the newly blocked N-terminal peptides was vacuum dried and re-dissolved in 100 μl of HPLC solvent A. To prevent oxidation of methionine between the primary and secondary RP-HPLC separations (and thus unwanted segregation of methionyl peptides (49)), methionines were uniformly oxidized to sulfoxides prior to the primary RP-HPLC run by adding 2 μl of 30% (w/v) H2O2 (final concentration of 0.06%) for 30 min at 30 °C. This peptide mixture was injected onto a RP-column (Zorbax® 300SB-C18 Narrowbore, 2.1 mm (internal diameter) x 150 mm length, 5 μm particles, Agilent Technologies) and the RP-HPLC separation was performed as described previously (39). Fractions of 30 s wide were collected from 20 to 80 min following sample injection (120 fractions). To reduce liquid chromatography (LC)-tandem MS (MS/MS) analysis time, fractions eluting 12 min apart were pooled, vacuum dried and re-dissolved in 40 μl of 2% acetonitrile. In total, 24 pooled fractions per setup were subjected to LC-MS/MS analysis (see below).
LC-MS/MS Analysis
LC-MS/MS analysis was performed using an Ultimate 3000 HPLC system (Dionex, Amsterdam, The Netherlands) in-line connected to a LTQ Orbitrap XL mass spectrometer (Thermo Electron, Bremen, Germany) and, per LC-MS/MS analysis, 2 μl of sample was consumed. LC-MS/MS analysis and generation of MS/MS peak lists were performed as described (50). These MS/MS peak lists were then searched with Mascot using the Mascot Daemon interface (version 2.2.0, Matrix Science). The Mascot search parameters were set as follows. Searches were performed in the Swiss-Prot database with taxonomy set to human (UniProtKB/SwissProt database version 2010_05 of 20-Apr-2010 containing 20,286 human protein sequences). Trideutero-acetylation at lysines, carbamidomethylation of cysteine and methionine oxidation to methionine-sulfoxide were set as fixed modifications. Variable modifications were trideutero-acetylation, acetylation, and 13C2-acetylation of protein N termini and pyroglutamate formation of N-terminal glutamine. Endoproteinase Arg-C/P (Arg-C specificity with arginine-proline cleavage allowed) was set as enzyme allowing no missed cleavages. The mass tolerance on the precursor ion was set to 10 ppm and on fragment ions to 0.5 Da. Only MS/MS-spectra and corresponding identifications that exceeded the corresponding MASCOT's threshold score of identity (at 95% confidence level) and that were ranked one, were withheld. The false discovery rate was estimated according to the method described by Käll et al. (51), and was found <2% at the spectrum level and <3% at the peptide level. Identified MS/MS spectra are made publicly available in the Proteomics Identification Database (PRIDE) (74) under the accession codes 13643, 13644, and 13645.
Plasmid Construction, Protein Expression, and Purification
The cDNA encoding hNaa10p and hNaa50p ORFs were cloned into pETM-41 [MBP(Maltose Binding Protein)/His-fusion] and pETM-30 (GST/His- fusion, generously provided by G. Stier, EMBL, Heidelberg, Germany) for expression in E. coli. Correct cloning was verified by DNA sequencing and the plasmids were transformed into E. coli BL21 Star™ (DE3) cells (Invitrogen) by heat shock. Two hundred milliliter cell cultures were grown in LB (Luria Bertani) medium to an OD600 nm of 0.6 at 37 °C and subsequently transferred to 20 °C. Following 30 min of incubation, protein expression was induced by IPTG (1 mm). Following 17 h of incubation, the cultures were harvested by centrifugation and the pellets stored at −20 °C. E. coli pellets containing recombinant proteins were thawed at 4 °C and the cells lysed by sonication and french press in lysis buffer (1 mm dithiotreitol, 50 mm Tris-HCl (pH 7.4), 300 mm NaCl, 1 tablet EDTA-free protease Inhibitor mixture per 50 ml (Roche)). The cell extracts were applied on a metal affinity FPLC column (HisTrap HP, GE Healthcare, Uppsala, Sweden). Appropriate fractions containing recombinant protein were pooled and further purified using size exclusion chromatography (Superdex™ 75, GE Healthcare). The purity of the fractions corresponding to purified monomeric recombinant proteins were analyzed on Coomassie stained SDS-PAGE gels and the protein concentrations determined by OD280 nm measurements.
Immunoprecipitation of NAT Complexes
An aliquot of 5 × 106 A-431 cells (per sample) were harvested and lysed in 500 μl of lysis buffer (50 mm Tris (pH 8), 50 mm NaCl, 0.5% Nonidet P-40, 5 mm EDTA, 1 mm Pefabloc (Roche)). A total of 50 μl of protein A/G Agarose (Santa Cruz) was added to the lysates and incubated for 1 h at 4 °C. Following centrifugation at 2000 × g for 2 min, the supernatants were collected and incubated for another 2 h at 4 °C with 3 μg anti-hNaa15p (anti-NATH in (9)) or custom-made unspecific rabbit IgG (Biogenes). The samples were centrifuged as above and 50 μl of Protein A/G Agarose was added to the supernatants. Following incubation for 16 h, three repetitive rounds of centrifugation and washing (two times in lysis buffer and once in acetylation buffer), the samples were used for an in vitro peptide acetylation assay.
In Vitro N-Acetyltransferase Assay Using Synthetic Peptides
Purified MBP-hNaa10p (40 nm) or GST-hNaa50p (50 nm) was mixed with selected oligopeptide substrates (200 μm) and 500 μm of acetyl-CoA in a total volume of 60 μl acetylation buffer. For MBP-hNaa10p, the samples were incubated at 37 °C for 10 min, whereas for GST-hNaa50p, the samples were incubated at 37 °C for 30 min. The enzyme activities were quenched by adding 5 μl of 10% TFA. The acetylation reactions were quantified using RP-HPLC as described previously (16). When making use of immunoprecipitated material as enzyme input, the assay was carried out the same way using a 60 min incubation time. In order to determine the concentration of the NatA complex used in the assays, we analyzed the immunoprecipated NatA complex by SDS-PAGE and SYPRO Ruby (BioRad) staining. The relative intensities of hNaa10p, hNaa15p, and hNaa50p were quantified and compared with purified recombinant proteins and each other using Image Gauge V 4.0 and further corrected for the staining intensities contributed by the different protein species based on amino acid composition (52). This semiquantitative analysis suggested that the complex is present in a 1:1:1 (1:0.96:0.87) stoichiometry and furthermore that one sample of immunoprecipitated NatA complex contained 0.4 pmol NatA (hNaa10p). Time course analysis of the different enzymes was performed to investigate initial velocity conditions (supplemental Fig. S1).
Synthetic Peptide Sequences
Peptides (supplemental Table 1) were custom-made (Biogenes) to a purity of 80–95%. All peptides contain seven unique amino acids at their N terminus, as these are the major determinants influencing N-terminal acetylation. The next 17 amino acids are essentially identical to the adrenocortictropic hormone peptide sequence (RWGRPVGRRRRPVRVYP) however, lysines were replaced by arginines to minimize any potential interference by Nε-acetylation.
Gel Filtration Assay
A cell pellet from ∼2 × 107 A-431 cells was resuspended in homogenization buffer (0.25 m Sucrose, 140 mm NaCl, 1 mm EDTA, 1 mm Pefabloc (Roche), 20 mm Tris-HCl pH 8.0) and homogenized using a cell-cracker (EMBL Germany) using six strokes with a 4 μm gap. Following centrifugation, at 17,000 × g for 20 min, 500 μl of clear cell lysate (supernatant) was applied on a Superdex 200 10/300 GL (GE Healthcare) gel filtration column. The column was pre-equilibrated with 50 mm Tris-HCl (pH 8.0), 200 mm NaCl and 10% glycerol using settings as recommended by the manufacturer. Two hundred and fifty microliter fractions were collected and analyzed by SDS-PAGE and Western blotting using anti-hNaa10p and anti-hNaa15p.
Ribosome Isolation
Approximately 2 × 107 HEK293 cells were used per experiment. Prior to harvesting, cells were treated with 10 μg/ml cycloheximide for 5 min at 37 °C. Cells were harvested, lysed with KCl-containing ribosome lysis buffer (1.1% (w/v) KCl, 0.15% (w/v) triethanolamine, 0.1% (w/v) magnesium acetate, 8.6% (w/v) sucrose, 0.05% (w/v) sodium deoxycholate, 0.5% (v/v) Triton-X100, 0.25% (v/v) Pefabloc) and incubated on ice for 15 min. Following removing the nuclear and membrane containing fraction by centrifugation at 400 × g for 10 min, 700 μl cell lysate was ultra-centrifuged at 436,000 × g for 25 min on a 0.4 ml cushion of 25% sucrose in KCl ribosome lysis buffer using a MLA-130 rotor (Beckman, Geneva, Switzerland). Pellets were resuspended in ribosome lysis buffer with the indicated KCl concentrations, followed by ultracentrifugation as described above. Pellets were resuspended in KCl ribosome lysis buffer, and prepared for analysis by SDS-PAGE and Western blotting.
RESULTS
Proteome-derived Peptide Library Generation and Isolation of NAT-specific Peptide Substrates
The proteome-derived peptide library generation workflow and NAT-substrate isolation assay is shown in Fig. 1. At the protein level, cysteine sulfhydryls are protected by carbamidomethylation and primary amines are modified by trideutero-acetylation. This primary amine protection step, besides being a prerequisite for the applied selection strategy (see below), additionally excludes nonenzymatic acetyl-CoA modification, which was reported to occur spontaneously on Nε-amines of lysine side chains (53). Subsequently, the modified protein mixture is digested with trypsin, resulting in the formation of endoproteinase Arg-C type of peptides (because trypsin will not cleave at trideutero-acetylated lysines). To create a peptide library that is mainly composed of Nα-free peptides and thus to generate a large substrate pool for NATs, the naturally acetylated and the in vitro blocked (trideutero-acetylated) N-terminal peptides are removed from this modified peptide pool by a first round of SCX separation at pH 3.0. Here, the flow-through, or SCX nonretained fraction composed of peptides with an overall net charge of zero and thus containing the Nα-blocked peptides (39), is discarded and a high-salt elution step is applied to recover the SCX-resin bound, Nα-free peptide fraction. Before incubation with the NAT of interest, the recovered peptides are further purified by solid-phase extraction.
Fig. 1.

Proteome-derived peptide library generation and screen for Nα-acetylation of peptide substrates by purified NATs or NAT-complexes. A, Peptide library generation. At the protein level, cysteines are modified to carbamidomethylcysteine by iodoacetamide and primary amines are modified by trideutero-acetylation. Proteome-derived peptide libraries are generated by digesting the modified proteome with trypsin, yielding Arg-C specific peptides. In order to enrich for Nα-free peptides that are introduced upon digestion, SCX separation at pH 3.0 is performed. The flow-through fraction, composed of peptides with an overall net charge of zero and thus containing the in vivo and in vitro blocked peptides, is being discarded. The bound peptide fraction is then eluted and peptides are subsequently purified by desalting over a SPE cartridge. B, Peptide-substrate screen. Upon purification, the Nα-free-peptide library is dissolved in an NAT compatible buffer and incubated with a purified NAT of interest. Only peptides that become Nα-acetylated will elute in the flow-through fraction of second SCX-fractionation at pH 3.0, and these peptides are subsequently analyzed by MS/MS-analysis.
Following purification, the desalted Nα-free-peptide library is dissolved in a NAT compatible buffer and incubated with the acetyl donor acetyl-CoA and a purified NAT enzyme or NAT complex of interest. In order to distinguish between possibly copurified in vivo or in vitro Nα-acetylated/trideutero-acetylated background peptides and enzymatic in vitro NAT-Nα-acetylated peptides, an isotopic, and thus MS-distinguishable variant of acetyl-CoA, 1,2-13C2-acetyl-CoA, is used as acetyl donor. Enzymatically acetylated peptides are then isolated by a second SCX fractionation step as described above, but now remaining glutamine-starting peptides are removed prior to the second SCX fractionation step to avoid having pyroglutamate peptides co-enriched with acetylated peptides. We only implemented this step prior to the second SCX fractionation step as we wanted Gln-starting peptides to serve as potential oligopeptide substrates for the NATs assayed. In general, following completion of the isolation protocol, only 2% pyroglutamate peptides remained, all of which held proline as the second amino acid, which is expected given that this residue is refractory to pGAPase activity (39). As such, and because spontaneous Nα-acetylation is not observed, mainly enzymatically Nα-acetylated peptides are found in the flow-through fraction of this second low pH SCX fractionation step. Following LC-MS/MS analysis, the NAT substrate peptides identified were used to determine the sequence specificity of the studied NAT (see below).
The Specificity Profile of Recombinant hNaa50p
Because Nα-acetylation activity of recombinant hNaa50p has been shown to target various Met-starting peptides (16), our approach was first challenged using recombinant hNaa50p. hNaa50p N-terminally fused to maltose-binding protein was expressed in E. coli, purified, and incubated with a proteome-derived peptide library from human cells. The enzyme-treated peptide library was then processed as described above. Following LC-MS/MS analysis of the second SCX flow-through fraction, 3602 MS/MS spectra linked to 1860 peptides were identified. Of these, 1342 peptides (72%) were enzymatically, in vitro Nα-13C2-acetylated. Other peptides were either incompletely retained Nα-free peptides (444 peptides) or otherwise Nα-blocked peptides, including the pGAPase refractory pyroglutamate-proline starting peptides. The hNaa50p specificity profile, delineated following multiple sequence alignment analysis using iceLogo (54) (Fig. 2A) revealed that hNaa50p was predominantly selective for peptides starting with leucine (697) or methionine (341); the P1 specificity accounting for 55 and 27% (or 82% in total) of all unique hNaa50p oligopeptide substrates identified. Besides being an N-end rule destabilizing residue (31, 55), leucine belongs to the class of residues of which the gyration radii precludes initiator Met-removal by methionine aminopeptidases (MAPs) (56, 57), and thus represents a physiologically irrelevant P1′-residue for hNaa50p cotranslational acetylation. The unexpected high selectivity for leucine-starting peptides however might be explained by the fact that leucine and methionine share similar physicochemical characteristics (58, 59). And, given their similar side-chain hydrophobic nature, as well as preservation of favorable van der Waals interactions, leucine has been the preferred (if not exclusive) residue of choice for the substitution of methionine in proteins (60–62). Furthermore, given that N-terminal prolines inhibit Nα-acetylation (8), the complete lack at position P1′ and P2′ and the very limited occurrence of Pro at P3′, indeed indicates the highly inhibitory potential of N-terminally positioned prolines on hNaa50p-activity. Medium-sized hydrophobic residues Leu (184 or 14%), Lys (Lys can be here considered hydrophobic, because its side-chain was blocked by acetylation) (183 or 14%), Ala (151 or 12%), and Met (131 or 10%) show an increased frequency of occurrence at P2′. Because the Nα-acetyltransferase(s) responsible for Met-Lys, Met-Ala, and Met-Met starting N termini are not yet characterized, our results hint to hNaa50p as a potential candidate.
Fig. 2.

Sequence logos of the NAT-specific substrate peptide sequences from a Nα-free-peptide library derived from human K-562 cells. IceLogos (54) of the 1271 unique recombinant hNaa50p substrate peptides (A) and of the unique 2399 recombinant hNaa10p substrate sequences identified (B) are shown. C1, Heat map and icelogo showing enriched and depleted residues present in the 52 peptides found to be acetylated by the immunoprecipitated hNatA complex. C2 and C3, heat maps and iceLogos as in C1 but only for the 22 Ser- and Ala-starting hNatA substrate peptides and the 22 Met- or Leu-starting hNatA substrate peptides identified respectively. In all iceLogos, peptide substrate motifs are given as 1, 2, 3, … with Nα-acetylation occurring at position 1. Further, only statistically significant residues (p < 0.05) are shown, with amino acids heights indicating the degree of conservation of an amino acid at the indicated position. The frequency of amino acid occurrence at each position in each sequence set was compared with amino acid frequencies in the human subsection of the Swiss-Prot 56.0 database. Residues with statistically significant 0% frequency of occurrence as compared with Swiss-Prot are shown in pink. SPE, solid phase extraction; SCX, strong cation exchange.
The Specificity Profile of Recombinant hNaa10p
Similarly to hNaa50p, an maltose binding protein (MBP)-hNaa10p fusion protein was expressed in E. coli and the purified enzyme was incubated with a proteome-derived library of Nα-free peptides. Overall, 6,169 MS/MS spectra (65% of all spectra identified) belonged to 2,399 unique Nα-13C2-acetylated peptides. Surprisingly, the observed substrate specificity of the individual catalytic subunit hNaa10p does not correlate with the proposed specificity of the protein Nα-terminal acetyltransferase complex hNatA in which it predominantly resides. Isolated hNaa10p appears to favor peptides starting with acidic residues and more specifically Glu-starting N termini (Fig. 2B): 777 (32%) of all identified hNaa10p-acetylated substrate peptides start with Glu, whereas 287 (12%) and 250 (10%) carried an Asp or Glu at their P2′-position. Combined, these peptides account for 1250 (52%) of all peptides identified. Of further note is that Asp at P2′ (Asp is generally underrepresented at P1′ as only 71 (3%) of all unique peptides identified are Asp-starting peptides) is enriched in peptides not holding Glu at P1′ (84% of all P2′-Asp containing hNaa10p substrate peptides), indicating that having at least one acidic residue at P1′ (Glu) or P2′ (Asp) is highly preferred for isolated recombinant hNaa10p-mediated acetylation. In this respect, it is also noteworthy that 84 (3.5%) of the identified peptides start with a Glu-Glu motif.
As for the remaining peptides, only 344 (14%) point to the proposed NatA-type specificity; i.e. starting with Ala (184), Cys (44), Gly (18), Ser (49), Val (37), or Thr (12). Of particular note is that Ser and Thr, being the preferred P1′ substrate of the human NatA complex, with a frequency of in vivo (partial) Nα-acetylation of over 99 and 92% (4), respectively, are here highly underrepresented (Fig. 2B). In general, Ala has a higher frequency of occurrence, especially at P2′ and P3′ (Fig. 2B). This observation hints to the fact that residues beyond P1′ might steer hNaa10p/substrate-interaction. As observed for the hNaa50p study, proline is also inhibitory for Nα-acetylation by hNaa10p.
The Specificity Profile of hNatA
To assess the N-acetyltransferase activity of an endogenous NAT complex, and more in particular hNaa50p and hNaa10p in their native context, the activity of the immunoprecipitated endogenous hNaa15p-hNaa10p-hNaa50p complex was assayed. Because this NatA complex was immunoprecipitated by anti-hNaa15p, which was shown to effectively co-immunoprecipitate hNaa10p and hNaa50p (9, 15, 17), all the hNaa10p and hNaa50p present was stably bound to hNaa15p, and thus only genuine NatA complexes were assayed for as described above. In contrast to the high number of substrates identified when using recombinant enzymes, only 52 unique Nα-13C2-acetylated peptides were identified as hNatA substrates (Fig. 2C1). These results are probably indicative for the fact that, as compared with the recombinant enzyme preparation, far less enzyme was used (i.e. endogenous NAT complexes). Interestingly, the delineated NatA substrate specificity was mainly directed toward Ser-starting peptides (22 peptides, 43% of total) (Fig. 2C2) and Met- and Leu-starting peptides (22 or 43%) (Fig. 2C3), whereas only one Asp- and one Glu-starting peptide, i.e. the preferred recognition motif of recombinant hNaa10p (Fig. 2B), were identified. Taken together, our results let us discern two different types of Nα-acetylation specificities in the immunoprecipitated NatA complex, hinting to the fact that activities of both catalytic subunits present in the hNatA complex, hNaa10p and hNaa50p, were assayed for during our experiment. Additionally, the extended specificity profile of hNaa50p within the hNatA complex agrees very well with that of individual, recombinant hNaa50p because, besides the similar preference at P2′, acidic residues were found enriched from P3′ to P5′ (Figs. 2A and 2C3). Of further note is that 22 out of the 26 (85%) P2′-residues in Ser- or Ala-starting peptides are occupied by Ser (11), Ala (6), or Thr (5), representative for optimal NatA-type substrate N termini in vivo (4) (Fig. 2C2), and thus demonstrating that the in vitro specificity profile observed, can be extrapolated to the in vivo NatA-type substrate specificity (4).
hNaa10p and hNatA Acetylate Actin N Termini
We further wanted to pursue whether the observed activity of the hNaa10p catalytic subunit of NatA toward acidic N termini could be of physiological relevance. Of note is that very exceptionally, mature protein N termini start with an acidic amino acid, as acidic residues normally retain their initiator methionine. Actins from most eukaryotic species however undergo a unique N-terminal protein modification process, which has been shown to play important roles in the interaction of actin with several actin-binding proteins (63–68). N-terminal actin modifications, include the post-translational removal of the acetylated initiator methionine, in the case of β- and γ-actin by an unconventional MAP, and the cotranslational removal of the initiator methionine and post-translational removal of the N-terminal acetylated cysteine in the case of α-actin, leaving all of them with an acidic residue at their newly exposed N terminus (69–71). Final maturation of these N termini requires Nα-acetylation. However, this unconventional post-translational Nα-acetylating activity has so far remained uncharacterized. Therefore, a previously developed method based on RP-HPLC peptide separation, enabling the determination of kinetic constants and efficiency of Nα-acetylation was here used for studying the degree of actin peptide Nα-acetylation (16). Purified hNaa10p was incubated with various actin-derived, N-terminal-like peptides and acetyl-CoA, and product formation was followed for each of these peptides (Fig. 3A). In these experiments, the efficiency of Nα-acetylation correlated with the degree of in vivo Nα-acetylation states of the respective substrate N termini given that the AVFAD motif represents a semioptimal substrate (derived from a partially acetylated in vivo substrate), whereas the SESSS motif is representative of a fully Nα-acetylated protein. Further, the SPTPP-starting peptide representative of an in vivo unacetylated protein N terminus was here also found to remain unacetylated following incubation with purified hNaa10p (4). However, and in line with the substrate specificity determined from the hNaa10p peptide library screen, hNaa10p strongly preferred the acidic actin N termini over these well-characterized in vitro and in vivo substrates as actin N termini appear to be Nα-acetylated about a 1000-fold better by hNaa10p. As such, these actin-derived N-terminal peptides represent by far the best hNaa10p in vitro substrate peptides identified to date. Another important issue is whether hNaa10p in the context of the endogenous NatA complex is capable of acetylating these actin N termini. As demonstrated above, the immunoprecipitated NatA complex preferred Ser besides Met and Leu-starting N termini. To this end, we analyzed the capacity of the immunoprecipitated hNatA complex to acetylate the actin-derived N-terminal peptides. The immunoprecipitated hNatA complex acetylated the acidic γ-actin N terminus (EEEIA) two- to threefold better as compared with the established in vivo acetylated peptide SESSS (fully Nα-acetylated N terminus in vivo), whereas on the other hand, SESSS is twofold more efficiently acetylated by the NatA complex as compared with the β-actin N terminus (DDDIA) (Fig. 3B). Consequently, both purified hNaa10p and the hNatA complex prefer the γ-actin N terminus over the known Ser-type of substrates. However, there is a dramatic specificity shift between purified hNaa10p and the hNatA complex as hNatA prefers Ser-starting peptides, whereas recombinant hNaa10p prefers acidic actin N termini. Further analyses demonstrated that isolated hNaa50p is unable to significantly acetylate the actin N termini (Fig. 3C). Thus, the NAT activity of the NatA complex toward actin N termini is solely a consequence of hNaa10p activity, and not hNaa50p and we therefore propose hNaa10p and/or hNatA as the post-translational actin Nα-acetyltransferase.
Fig. 3.

NAT-activity of recombinant hNaa10p, recombinant hNaa50p and immunoprecipitated hNatA toward synthetic N-terminal peptides. A, Purified MBP-hNaa10p (40 nm) was mixed with oligopeptide substrates (200 μm) and saturated levels of acetyl-CoA (500 μm) and incubated in acetylation buffer at 37 °C for 10 min. In the assays where the AVFAD, SESSS, MLGPE, and SPTP based oligopeptides were tested, the concentration of MBP-hNaa10p was increased to 150 nm, mixed with 200 μm of the indicated oligopeptides in acetylation buffer and incubated for 25 min at 37 °C. The acetylation activity was determined by RP-HPLC peptide separation. Error bars indicate the standard deviation based on three independent experiments. White bars indicate peptides representative of the β- and γ-actin N termini. The black bar and gray bar respectively indicates the peptide representative of a fully and partially in vivo Nα-acetylated NatA substrate N terminus (4). The first five amino acids in the peptides are indicated, for further details see supplementary Table S1. B, Human A431 cells were harvested, lysed and subjected to immunoprecipitation, using anti-hNaa15p or control antibodies. The beads containing hNatA complexes were mixed with the indicated oligopeptide substrates (200 μm) at saturated levels of acetyl-CoA (500 μm) and incubated for 60 min at 37 °C in acetylation buffer. Values from negative control immunoprecipitations were subtracted from the NatA values to obtain specific activity values. C, Purified GST-hNaa50p (50 nm) was assayed as in A, but with an incubation time of 30 min.
A Major Fraction of NatA is not Associated With Ribosomes and a Minor Fraction of Cytosolic hNaa10p Exists Independent of the NatA Complex
The substrate specificity results (Fig. 3) indicate that hNaa10p may act alone on acidic actin N termini. To address whether some cellular hNaa10p may exist independently of the NatA complex, we performed gel filtration analyses of A-431 cell homogenates. Both hNaa15p and hNaa10p co-eluted in fractions corresponding to the predicted NatA complex (Fig. 4). Interestingly, some hNaa10p, but not hNaa15p, additionally eluted in fractions corresponding to predicted hNaa10p monomers (i.e. around the carbonic anhydrase standard protein of 29 kDa), indicating that a minor fraction of hNaa10p exists independently of hNaa15p in cells, and is capable of carrying out post-translational acetylation of actins and other substrates. Another not mutually exclusive possibility however is that the NatA complex acetylates acidic N termini post-translationally. In yeast, all NatA is associated with ribosomes (2) making such an option unlikely. To this end, we isolated polysomes and profiled hNaa10p, hNaa15p, and hNaa50p by SDS-PAGE and Western blotting. In contrast to yeast, all subunits of the human NatA complex are mostly nonribosomal, whereas only a small but significant fraction is associated with ribosomes (Fig. 5). In conclusion, our results are in agreement with a role of both hNaa10p and the NatA complex in post-translational acetylation of processed actins.
Fig. 4.

A minor fraction of hNaa10p does not co-fractionate with hNaa15p. Human A-431 cells were homogenized and subjected to gel filtration using a Superdex-200 column. Fractions were collected and analyzed by SDS-PAGE and Western blotting using anti-hNaa10p and anti-hNaa15p. Fraction numbers are indicated above the lanes and standard proteins used to calibrate the gel filtration column are indicated below the lanes. Standard proteins include ferritin (440 kDa), aldolase (158 kDa), conalbumin (75 kDa), and carbonic anhydrase (29 kDa). “Ctr” indicates internal total A-431 lysate control used to track the proteins of interest and to verify equal exposure of different gels run in parallel. Results shown are representative of three independent experiments.
Fig. 5.
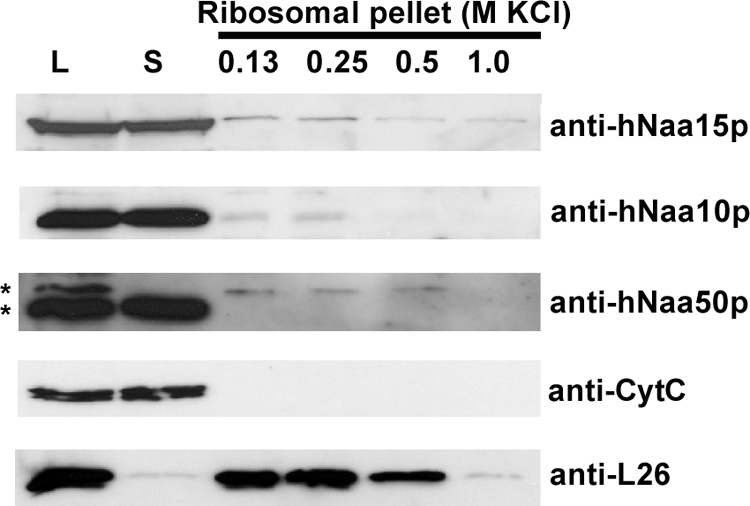
A major fraction of hNatA subunits does not cosediment with polysomes. Polysomal pellets from HEK-293 cells were resuspended in buffer containing increasing concentrations of KCl. Cell lysate (L), supernatant post first ultracentrifugation (S) and polysomal pellets following KCl treatment were analyzed by SDS-PAGE and Western blotting. The membrane was incubated with anti-hNaa15p, anti-hNaa10p, anti-hNaa50p, anti-L26 (ribosomal protein), and anti-CytC antibodies. Results shown are representative of three independent experiments. *Indicates two distinct hNaa50p specific bands (verified by sihNAA50 treatment in an independent analysis, data not shown) of which the slower migrating variant specifically associates with ribosomes.
DISCUSSION
In this study, we report on a novel method that allows systematic analysis of proteome-derived peptidic substrates of NATs. In principle, our method can also be exploited to study the substrate specificity of other Nα-modifying enzymes introducing either neutral or negatively charged groups at the Nα-free N termini, or when using a peptide library enriched for Nα-modified peptides, to study the action of Nα-deblocking enzymes such as deformylases. When using the Nα-free peptide libraries reported here, typically up to 65 to 75% of all identified MS/MS spectra, converging in more than thousand unique peptides, originate from enzymatically tagged, Nα-13C2-acetylated peptides. This high yield points to an excessive positive enrichment of oligopeptide-substrates of the NATs assayed. Given that the capture efficiency for Nα-free peptides by the strong cation resin was calculated to be around 98% at pH 3.0 (39), not surprisingly, the remaining peptides were categorized as incompletely retained Nα-free, or incompletely depleted Nα-blocked peptides including pGAPase refractory pyroglutamate-starting peptides. However, the use of SCX imposes some inherent drawbacks because His-containing peptides, given that they carry one additional positive charge, are retained by the SCX resin and can consequently not be assayed. In addition, the side-chains of lysine and cysteine are blocked by acetylation and carbamidomethylation respectively, which might alter NAT substrate recognition. Of note, as compared with Swiss-Prot, the general occurrence of modified cyteines after performing an LC-MS/MS-based quality control of the proteome derived peptide libraries revealed a general underrepresentation (data not shown). Further, because no information on the efficiency of Nα-acetylation can be obtained by our method, a previously developed method based on RP-HPLC was used to determine efficiencies of Nα-acetylation (16). However, in this respect it is noteworthy that in an independent peptide library screen for hNaa10p substrates, which made use of half of the peptide input material, although less substrate peptides were identified, the percentage of Glu- and, to a much lower extent, Asp-starting peptides increased to 70%, most likely reflecting the fact that Glu-starting peptides are much more efficiently Nα-acetyated as compared with the other identified peptidic substrates (data not shown).
Previous reports and the present study show that individual catalytic subunits inherently hold enzymatic activity, demonstrating that the substrate specificity observed for intact NAT complexes is at least partially contained when only studying their catalytic subunits (28). In this regard, hNaa50p was reported to display Met-Leu Nα-acetylating activity, whereas additionally allowing small, hydrophobic as well as negatively charged residues at P3′ substrate positions (16). In this study, the Met-Leu substrate preference was verified, and additionally, specificities directed toward Met-Lys, Met-Met, and Met-Ala starting N termini were observed. Because only about 0.12% of all Arg-C generated peptides are estimated to start with a methionine, the nearly 27% of Met-starting hNaa50p substrate sequences identified here, implicate a more than 200-fold selective enrichment. Therefore, in vitro, Met might very well be preferred over Leu at P1′, especially because Leu at P1′-positions (55% of all peptides identified) only represents a 30-fold enrichment given that 1.6% of all Arg-C generated peptides start with a Leu. In contrast to the results previously reported (16), we could not observe a preference of Pro at P4′. Nevertheless, and in contrast to the zero tolerance at P1′ and P2′ and very limited tolerance at P3′, nearly 6% of the P4′-positions were occupied by a proline in the hNaa50p peptide substrates, indicating that this residue is tolerated at P4′, but not preferred. The lack of significant activity of the NatA-complex toward the MLGP-peptide (as was shown for purified hNaa50p) might be explained by the observation that subtle changes in P2′-substrate specificity were observed when assaying hNaa50p alone opposed to when it is present in its endogenous complex (Fig. 2A versus Fig. 2C3). Nevertheless, the extended specificity profile from P3′ to P5′ was nearly indistinguishable. Because the Nα-acetyltransferase(s) responsible for Met-Lys, Met-Ala, and Met-Met starting N termini, N termini often found to be Nα-acetylated in vivo in human cells, are not yet characterized, our results indicate hNaa50p to be a likely candidate for acetylating these protein N termini. In fact, its activity toward Met-Leu starting peptides is shared with the catalytic subunit Naa30p of NatC, and thus points to substrate redundancy among NATs.
Interestingly, the specificity shift between purified hNaa10p and the hNatA complex toward a relative preference of hNatA for Ser besides Met-starting peptides, and of the hNaa10p for acidic (actin) N termini, may reflect an in vivo activity of hNaa10p to post-translationally acetylate actin N termini independent of the NatA complex. In support of this, our gel filtration assays demonstrated that a fraction of cellular hNaa10p exists independently of hNaa15p and the NatA complex (Fig. 4). In this respect it is also noteworthy that a significant portion of the NAT subunits, hNaa10p, hNaa50p, and hNaa15p, can be detected in the nonpolysomal bound fraction, indicative for the fact that they may additionally serve other purposes, alone or in complex, or allow for different substrate preferences of the enzyme (Fig. 5). An interesting observation is that hNaa15p (named Tubedown in this study) colocalizes with actin fibers and specifically interacts with the actin-binding protein cortactin in endothelial cells (72). This places hNaa15p and possibly the hNatA complex in close proximity to actins which may facilitate post-translational N-terminal acetylation of actin. Because functional roles for actin Nα-acetylation have been demonstrated in yeast and fruit fly, identifying the actin-NAT is important. The in vitro data presented here strongly imply that hNaa10p/hNatA is a very likely actin-NAT candidate, especially bearing in mind that none of the other known human NAT significantly acetylate the actin N termini in vitro (Arnesen et al., unpublished data). However, because of the lack of KO models and viewing the fact that we previously demonstrated that despite a significant hNatA knockdown (i.e. 95% reduction of hNaa10p expression), solely a partial loss in N-terminal acetylation was observed for proteins displaying suboptimal hNatA substrate sequences in vivo (4), the in vivo role of NatA/hNaa10p in actin function of higher eukaryotic species awaits functional analyses. An overview illustrating the known and postulated Nα-acetylating activities of the hNatA-complex, hNatE and individual catalytic subunits is shown in Fig. 6.
Fig. 6.

Overview of the hNatA, hNatE, and individual hNaa10p and hNaa50p enzymatic activities. The ribosome-bound subunits hNaa10p and hNaa15p combine to form the functional hNatA complex and cotranslationally acetylate nascent polypeptides (e.g. polypeptide with an N-terminal serine). hNaa50p, responsible for the observed NatE activity, may physically associate with hNaa10p and hNaa15p of the hNatA complex. NatE activity is mainly directed toward MK, MM, MA, and MI starting N termini. Nonpolysomal bound hNaa10p and hNaa50p might be responsible for the post-translationally Nα-acetylation of acidic actin N termini and other hNaa50p-type substrate N termini respectively, independent of the NatA complex. HYPK is stably associated with the NatA complex and might contact with nascent polypeptides. ACTG, γ-actin; ACTB, β-actin; SS, small ribosomal subunit; LG, large ribosomal subunit.
Acknowledgments
P.V.D. is a Postdoctoral Fellow of the Research Foundation - Flanders (FWO-Vlaanderen). Nina Glomnes is thanked for technical assistance.
Footnotes
* We further acknowledge support of research grants from the Fund for Scientific Research - Flanders (Belgium) (project numbers G.0042.07 and G.0440.10), the Concerted Research Actions (project BOF07/GOA/012) from the Ghent University, the Inter University Attraction Poles (IUAP06), the Norwegian Research Council (Grant 197136 to TA), and the Norwegian Cancer Society (to JRL and TA).
 This article contains supplemental Fig. S1 and Table S1.
This article contains supplemental Fig. S1 and Table S1.
2 In analogy with the nomenclature used for the description of protease subsites (73), we introduce a nomenclature where the substrate amino acid residues of NATs are called P-sites (for peptide sites). The Nα-acetylated amino acid residue is labeled P1′, and amino acids downstream of this residue are labeled p2′, P3′, etc.
1 The abbreviations used are:
- NAT
- Nα-acetyltransferase
- Ac(D3)
- (trideutero)-acetyl
- Ard1
- arrest-defective 1 protein
- CoA
- Coenzyme A
- NAA#
- N-alpha acetyltransferase # (gene/protein)
- NATH
- N-acetyl transferase human
- pGAPase
- pyroglutamyl aminopeptidase
- SCX
- strong cation exchange
- FPLC
- fast protein liquid chromatography.
REFERENCES
- 1. Polevoda B., Sherman F. (2003) N-terminal acetyltransferases and sequence requirements for N-terminal acetylation of eukaryotic proteins. J. Mol. Biol. 325, 595–622 [DOI] [PubMed] [Google Scholar]
- 2. Gautschi M., Just S., Mun A., Ross S., Rücknagel P., Dubaquié Y., Ehrenhofer-Murray A., Rospert S. (2003) The yeast N(alpha)-acetyltransferase NatA is quantitatively anchored to the ribosome and interacts with nascent polypeptides. Mol. Cell. Biol. 23, 7403–7414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Helbig A. O., Gauci S., Raijmakers R., van Breukelen B., Slijper M., Mohammed S., Heck A. J. (2010) Profiling of N-acetylated protein termini provides in-depth insights into the N-terminal nature of the proteome. Mol. Cell. Proteomics 9, 928–939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Arnesen T., Van Damme P., Polevoda B., Helsens K., Evjenth R., Colaert N., Varhaug J. E., Vandekerckhove J., Lillehaug J. R., Sherman F., Gevaert K. (2009) Proteomics analyses reveal the evolutionary conservation and divergence of N-terminal acetyltransferases from yeast and humans. Proc. Natl. Acad. Sci. U.S.A. 106, 8157–8162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lee F. J., Lin L. W., Smith J. A. (1989) N alpha-acetyltransferase deficiency alters protein synthesis in Saccharomyces cerevisiae. FEBS. Lett. 256, 139–142 [DOI] [PubMed] [Google Scholar]
- 6. Polevoda B., Sherman F. (2000) Nalpha -terminal acetylation of eukaryotic proteins. J. Biol. Chem. 275, 36479–36482 [DOI] [PubMed] [Google Scholar]
- 7. Driessen H. P., de Jong W. W., Tesser G. I., Bloemendal H. (1985) The mechanism of N-terminal acetylation of proteins. CRC. Crit. Rev. Biochem. 18, 281–325 [DOI] [PubMed] [Google Scholar]
- 8. Goetze S., Qeli E., Mosimann C., Staes A., Gerrits B., Roschitzki B., Mohanty S., Niederer E., Laczko E., Timmerman E., Lange V., Hafen E., Aebersold R., Vandekerckhove J., Basler K., Ahrens C., Gevaert K., Brunner E. (2009) Identification and functional characterization of N-terminal protein acetylations in Drosophila melanogaster. PLoS Biol. 7, e1000236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Arnesen T., Anderson D., Baldersheim C., Lanotte M., Varhaug J. E., Lillehaug J. R. (2005) Identification and characterization of the human ARD1-NATH protein acetyltransferase complex. Biochem. J. 386, 433–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mullen J. R., Kayne P. S., Moerschell R. P., Tsunasawa S., Gribskov M., Colavito-Shepanski M., Grunstein M., Sherman F., Sternglanz R. (1989) Identification and characterization of genes and mutants for an N-terminal acetyltransferase from yeast. EMBO. J. 8, 2067–2075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Park E. C., Szostak J. W. (1992) ARD1 and NAT1 proteins form a complex that has N-terminal acetyltransferase activity. EMBO. J. 11, 2087–2093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Williams B. C., Garrett-Engele C. M., Li Z., Williams E. V., Rosenman E. D., Goldberg M. L. (2003) Two putative acetyltransferases, san and deco, are required for establishing sister chromatid cohesion in Drosophila. Curr. Biol. 13, 2025–2036 [DOI] [PubMed] [Google Scholar]
- 13. Hou F., Chu C. W., Kong X., Yokomori K., Zou H. (2007) The acetyltransferase activity of San stabilizes the mitotic cohesin at the centromeres in a shugoshin-independent manner. J. Cell. Biol. 177, 587–597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pimenta-Marques A., Tostões R., Marty T., Barbosa V., Lehmann R., Martinho R. G. (2008) Differential requirements of a mitotic acetyltransferase in somatic and germ line cells. Dev. Biol. 323, 197–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Arnesen T., Anderson D., Torsvik J., Halseth H. B., Varhaug J. E., Lillehaug J. R. (2006) Cloning and characterization of hNAT5/hSAN: an evolutionarily conserved component of the NatA protein N-alpha-acetyltransferase complex. Gene 371, 291–295 [DOI] [PubMed] [Google Scholar]
- 16. Evjenth R., Hole K., Karlsen O. A., Ziegler M., Arnesen T., Lillehaug J. R. (2009) Human Naa50p (Nat5/San) displays both protein N alpha- and N epsilon-acetyltransferase activity. J. Biol. Chem. 284, 31122–31129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Arnesen T., Starheim K. K., Van Damme P., Evjenth R., Dinh H., Betts M. J., Ryningen A., Vandekerckhove J., Gevaert K., Anderson D. (2010) The chaperone-like protein HYPK acts together with NatA in cotranslational N-terminal acetylation and prevention of Huntingtin aggregation. Mol. Cell. Biol. 30, 1898–1909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Starheim K. K., Gromyko D., Velde R., Varhaug J. E., Arnesen T. (2009) Composition and biological significance of the human Nalpha-terminal acetyltransferases. BMC Proc. 3, S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Arnesen T., Thompson P. R., Varhaug J. E., Lillehaug J. R. (2008) The protein acetyltransferase ARD1: a novel cancer drug target? Curr. Cancer Drug Targets. 8, 545–553 [DOI] [PubMed] [Google Scholar]
- 20. Chun K. H., Cho S. J., Choi J. S., Kim S. H., Kim K. W., Lee S. K. (2007) Differential regulation of splicing, localization and stability of mammalian ARD1235 and ARD1225 isoforms. Biochem. Biophys. Res. Commun. 353, 18–25 [DOI] [PubMed] [Google Scholar]
- 21. Lee C. F., Ou D. S., Lee S. B., Chang L. H., Lin R. K., Li Y. S., Upadhyay A. K., Cheng X., Wang Y. C., Hsu H. S., Hsiao M., Wu C. W., Juan L. J. (2010) hNaa10p contributes to tumorigenesis by facilitating DNMT1-mediated tumor suppressor gene silencing. J. Clin. Invest. 120, 2920–2930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chu C. W., Hou F., Zhang J., Phu L., Loktev A. V., Kirkpatrick D. S., Jackson P. K., Zhao Y., Zou H. (2010) A novel acetylation of {beta}-tubulin by San modulates microtubule polymerization via down-regulating tubulin incorporation. Mol. Biol. Cell. 22, 448–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lim J. H., Park J. W., Chun Y. S. (2006) Human arrest defective 1 acetylates and activates beta-catenin, promoting lung cancer cell proliferation. Cancer Res. 66, 10677–10682 [DOI] [PubMed] [Google Scholar]
- 24. Dormeyer W., Mohammed S., Breukelen B. V., Krijgsveld J., Heck A. J. (2007) Targeted Analysis of Protein Termini. J. Proteome Res. 6, 4634–4465 [DOI] [PubMed] [Google Scholar]
- 25. Helbig A. O., Gauci S., Raijmakers R., van Breukelen B., Slijper M., Mohammed S., Heck A. J. (2010) Profiling of N-acetylated protein termini provides in-depth insights into the N-terminal nature of the proteome. Mol. Cell. Proteomics. 9, 928–939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Helbig A. O., Rosati S., Pijnappel P. W., van Breukelen B., Timmers M. H., Mohammed S., Slijper M., Heck A. J. (2010) Perturbation of the yeast N-acetyltransferase NatB induces elevation of protein phosphorylation levels. BMC Genomics 11, 685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Starheim K. K., Arnesen T., Gromyko D., Ryningen A., Varhaug J. E., Lillehaug J. R. (2008) Identification of the human N(alpha)-acetyltransferase complex B (hNatB): a complex important for cell-cycle progression. Biochem. J. 415, 325–331 [DOI] [PubMed] [Google Scholar]
- 28. Starheim K. K., Gromyko D., Evjenth R., Ryningen A., Varhaug J. E., Lillehaug J. R., Arnesen T. (2009) Knockdown of human N alpha-terminal acetyltransferase complex C leads to p53-dependent apoptosis and aberrant human Arl8b localization. Mol. Cell. Biol. 29, 3569–3581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Urbancikova M., Hitchcock-DeGregori S. E. (1994) Requirement of amino-terminal modification for striated muscle alpha-tropomyosin function. J. Biol. Chem. 269, 24310–24315 [PubMed] [Google Scholar]
- 30. Behnia R., Panic B., Whyte J. R., Munro S. (2004) Targeting of the Arf-like GTPase Arl3p to the Golgi requires N-terminal acetylation and the membrane protein Sys1p. Nat. Cell. Biol. 6, 405–413 [DOI] [PubMed] [Google Scholar]
- 31. Hwang C. S., Shemorry A., Varshavsky A. (2010) N-terminal acetylation of cellular proteins creates specific degradation signals. Science 327, 973–977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Polevoda B., Arnesen T., Sherman F. (2009) A synopsis of eukaryotic Nalpha-terminal acetyltransferases: nomenclature, subunits and substrates. BMC Proc. 3, S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Arnesen T., Betts M. J., Pendino F., Liberles D. A., Anderson D., Caro J., Kong X., Varhaug J. E., Lillehaug J. R. (2006) Characterization of hARD2, a processed hARD1 gene duplicate, encoding a human protein N-alpha-acetyltransferase. BMC Biochem. 7, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Arnesen T., Gromyko D., Kagabo D., Betts M. J., Starheim K. K., Varhaug J. E., Anderson D., Lillehaug J. R. (2009) A novel human NatA Nalpha-terminal acetyltransferase complex: hNaa16p-hNaa10p (hNat2-hArd1). BMC Biochem. 10, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Arnold R. J., Polevoda B., Reilly J. P., Sherman F. (1999) The action of N-terminal acetyltransferases on yeast ribosomal proteins. J. Biol. Chem. 274, 37035–37040 [DOI] [PubMed] [Google Scholar]
- 36. Polevoda B., Norbeck J., Takakura H., Blomberg A., Sherman F. (1999) Identification and specificities of N-terminal acetyltransferases from Saccharomyces cerevisiae. EMBO J. 18, 6155–6168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kimura Y., Takaoka M., Tanaka S., Sassa H., Tanaka K., Polevoda B., Sherman F., Hirano H. (2000) N(alpha)-acetylation and proteolytic activity of the yeast 20 S proteasome. J. Biol. Chem. 275, 4635–4639 [DOI] [PubMed] [Google Scholar]
- 38. Boucherie H., Sagliocco F., Joubert R., Maillet I., Labarre J., Perrot M. (1996) Two-dimensional gel protein database of Saccharomyces cerevisiae. Electrophoresis 17, 1683–1699 [DOI] [PubMed] [Google Scholar]
- 39. Staes A., Van Damme P., Helsens K., Demol H., Vandekerckhove J., Gevaert K. (2008) Improved recovery of proteome-informative, protein N-terminal peptides by combined fractional diagonal chromatography (COFRADIC). Proteomics 8, 1362–1370 [DOI] [PubMed] [Google Scholar]
- 40. Van Damme P., Van Damme J., Demol H., Staes A., Vandekerckhove J., Gevaert K. (2009) A review of COFRADIC techniques targeting protein N-terminal acetylation. BMC Proc. 3, S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Maier R. H., Maier C. J., Rid R., Hintner H., Bauer J. W., Onder K. (2010) Epitope mapping of antibodies using a cell array-based polypeptide library. J. Biomol Screen 15, 418–426 [DOI] [PubMed] [Google Scholar]
- 42. Beutling U., Städing K., Stradal T., Frank R. (2008) Large-scale analysis of protein-protein interactions using cellulose-bound peptide arrays. Adv Biochem. Eng. Biotechnol. 110, 115–152 [DOI] [PubMed] [Google Scholar]
- 43. Schutkowski M., Reineke U., Reimer U. (2005) Peptide arrays for kinase profiling. Chembiochem 6, 513–521 [DOI] [PubMed] [Google Scholar]
- 44. Diamond S. L. (2007) Methods for mapping protease specificity. Curr. Opin. Chem. Biol. 11, 46–51 [DOI] [PubMed] [Google Scholar]
- 45. Schilling O., Overall C. M. (2008) Proteome-derived, database-searchable peptide libraries for identifying protease cleavage sites. Nat. Biotechnol. 26, 685–694 [DOI] [PubMed] [Google Scholar]
- 46. Hu Y. J., Wei Y., Zhou Y., Rajagopalan P. T., Pei D. (1999) Determination of substrate specificity for peptide deformylase through the screening of a combinatorial peptide library. Biochemistry 38, 643–650 [DOI] [PubMed] [Google Scholar]
- 47. Van Damme P., Martens L., Van Damme J., Hugelier K., Staes A., Vandekerckhove J., Gevaert K. (2005) Caspase-specific and nonspecific in vivo protein processing during Fas-induced apoptosis. Nat. Methods 2, 771–777 [DOI] [PubMed] [Google Scholar]
- 48. Van Damme P., Staes A., Bronsoms S., Helsens K., Colaert N., Timmerman E., Aviles F. X., Vandekerckhove J., Gevaert K. Complementary positional proteomics for screening substrates of endo- and exoproteases. Nat. Methods 7, 512–515 [DOI] [PubMed] [Google Scholar]
- 49. Gevaert K., Van Damme J., Goethals M., Thomas G. R., Hoorelbeke B., Demol H., Martens L., Puype M., Staes A., Vandekerckhove J. (2002) Chromatographic isolation of methionine-containing peptides for gel-free proteome analysis: identification of more than 800 Escherichia coli proteins. Mol. Cell. Proteomics 1, 896–903 [DOI] [PubMed] [Google Scholar]
- 50. Ghesquière B., Colaert N., Helsens K., Dejager L., Vanhaute C., Verleysen K., Kas K., Timmerman E., Goethals M., Libert C., Vandekerckhove J., Gevaert K. (2009) In vitro and in vivo protein-bound tyrosine nitration characterized by diagonal chromatography. Mol. Cell. Proteomics. 8, 2642–2652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Käll L., Storey J. D., MacCoss M. J., Noble W. S. (2008) Assigning significance to peptides identified by tandem mass spectrometry using decoy databases. J. Proteome Res. 7, 29–34 [DOI] [PubMed] [Google Scholar]
- 52. Steinberg T. H., Chernokalskaya E., Berggren K., Lopez M. F., Diwu Z., Haugland R. P., Patton W. F. (2000) Ultrasensitive fluorescence protein detection in isoelectric focusing gels using a ruthenium metal chelate stain. Electrophoresis 21, 486–496 [DOI] [PubMed] [Google Scholar]
- 53. Berndsen C. E., Denu J. M. (2005) Assays for mechanistic investigations of protein/histone acetyltransferases. Methods 36, 321–331 [DOI] [PubMed] [Google Scholar]
- 54. Colaert N., Helsens K., Martens L., Vandekerckhove J., Gevaert K. (2009) Improved visualization of protein consensus sequences by iceLogo. Nat. Methods 6, 786–787 [DOI] [PubMed] [Google Scholar]
- 55. Varshavsky A. (1997) The N-end rule pathway of protein degradation. Genes Cells 2, 13–28 [DOI] [PubMed] [Google Scholar]
- 56. Tsunasawa S., Stewart J. W., Sherman F. (1985) Amino-terminal processing of mutant forms of yeast iso-1-cytochrome c. The specificities of methionine aminopeptidase and acetyltransferase. J. Biol. Chem. 260, 5382–5391 [PubMed] [Google Scholar]
- 57. Bradshaw R. A., Brickey W. W., Walker K. W. (1998) N-terminal processing: the methionine aminopeptidase and N alpha-acetyl transferase families. Trends Biochem. Sci. 23, 263–267 [DOI] [PubMed] [Google Scholar]
- 58. Finney J. L., Gellatly B. J., Golton I. C., Goodfellow J. (1980) Solvent effects and polar interactions in the structural stability and dynamics of globular proteins. Biophys J. 32, 17–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Guy H. R. (1985) Amino acid side-chain partition energies and distribution of residues in soluble proteins. Biophys. J. 47, 61–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Gustavsson N., Kokke B. P., Anzelius B., Boelens W. C., Sundby C. (2001) Substitution of conserved methionines by leucines in chloroplast small heat shock protein results in loss of redox-response but retained chaperone-like activity. Protein Sci. 10, 1785–1793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Matthews B. W. (1993) Structural and genetic analysis of protein stability. Annu. Rev. Biochem. 62, 139–160 [DOI] [PubMed] [Google Scholar]
- 62. Bordo D., Argos P. (1991) Suggestions for “safe” residue substitutions in site-directed mutagenesis. J. Mol. Biol. 217, 721–729 [DOI] [PubMed] [Google Scholar]
- 63. Sutoh K., Hatano S. (1986) Actin-fragmin interactions as revealed by chemical cross-linking. Biochemistry 25, 435–440 [DOI] [PubMed] [Google Scholar]
- 64. Sutoh K., Yin H. L. (1989) End-label fingerprintings show that the N- and C-termini of actin are in the contact site with gelsolin. Biochemistry 28, 5269–5275 [DOI] [PubMed] [Google Scholar]
- 65. Sutoh K., Mabuchi I. (1989) End-label fingerprintings show that an N-terminal segment of depactin participates in interaction with actin. Biochemistry 28, 102–106 [DOI] [PubMed] [Google Scholar]
- 66. Doi Y., Higashida M., Kido S. (1987) Plasma-gelsolin-binding sites on the actin sequence. Eur. J. Biochem. 164, 89–94 [DOI] [PubMed] [Google Scholar]
- 67. Adams S., DasGupta G., Chalovich J. M., Reisler E. (1990) Immunochemical evidence for the binding of caldesmon to the NH2-terminal segment of actin. J. Biol. Chem. 265, 19652–19657 [PubMed] [Google Scholar]
- 68. DasGupta G., Reisler E. (1991) Nucleotide-induced changes in the interaction of myosin subfragment 1 with actin: detection by antibodies against the N-terminal segment of actin. Biochemistry 30, 9961–9966 [DOI] [PubMed] [Google Scholar]
- 69. Cook R. K., Blake W. T., Rubenstein P. A. (1992) Removal of the amino-terminal acidic residues of yeast actin. Studies in vitro and in vivo. J. Biol. Chem. 267, 9430–9436 [PubMed] [Google Scholar]
- 70. Sheff D. R., Rubenstein P. A. (1992) Isolation and characterization of the rat liver actin N-acetylaminopeptidase. J. Biol. Chem. 267, 20217–20224 [PubMed] [Google Scholar]
- 71. Martin D. J., Rubenstein P. A. (1987) Alternate pathways for removal of the class II actin initiator methionine. J. Biol. Chem. 262, 6350–6356 [PubMed] [Google Scholar]
- 72. Paradis H., Islam T., Tucker S., Tao L., Koubi S., Gendron R. L. (2008) Tubedown associates with cortactin and controls permeability of retinal endothelial cells to albumin. J. Cell. Sci. 121, 1965–1972 [DOI] [PubMed] [Google Scholar]
- 73. Schechter I., Berger A. (1967) On the size of the active site in proteases. I. Papain. Biochem. Biophys. Res. Commun. 27(2), 157–162 [DOI] [PubMed] [Google Scholar]
- 74. Martens L., Hermjakob H., Jones P., Adamski M., Taylor C., States D., Gevaert K., Vandekerckhove J., Apweiler R. (2005) PRIDE: the proteomics identifications database. Proteomics 5, 3537–3545 [DOI] [PubMed] [Google Scholar]