

Abstract
Shotgun proteomics data analysis usually relies on database search. However, commonly used protein sequence databases do not contain information on protein variants and thus prevent variant peptides and proteins from been identified. Including known coding variations into protein sequence databases could help alleviate this problem. Based on our recently published human Cancer Proteome Variation Database, we have created a protein sequence database that comprehensively annotates thousands of cancer-related coding variants collected in the Cancer Proteome Variation Database as well as noncancer-specific ones from the Single Nucleotide Polymorphism Database (dbSNP). Using this database, we then developed a data analysis workflow for variant peptide identification in shotgun proteomics. The high risk of false positive variant identifications was addressed by a modified false discovery rate estimation method. Analysis of colorectal cancer cell lines SW480, RKO, and HCT-116 revealed a total of 81 peptides that contain either noncancer-specific or cancer-related variations. Twenty-three out of 26 variants randomly selected from the 81 were confirmed by genomic sequencing. We further applied the workflow on data sets from three individual colorectal tumor specimens. A total of 204 distinct variant peptides were detected, and five carried known cancer-related mutations. Each individual showed a specific pattern of cancer-related mutations, suggesting potential use of this type of information for personalized medicine. Compatibility of the workflow has been tested with four popular database search engines including Sequest, Mascot, X!Tandem, and MyriMatch. In summary, we have developed a workflow that effectively uses existing genomic data to enable variant peptide detection in proteomics.
DNA sequence variation is associated with diseases and differential drug response. As a paradigmatic example, cancers are diseases of clonal proliferations caused by mutations in oncogenes and tumor suppressor genes (1). After several decades of searching through traditional biology approaches, many mutant genes have been causally implicated in oncogenesis (2). Facilitated by the new genomic techniques such as SNP (single nucleotide polymorphism) arrays and deep-sequencing, the identification of cancer genes has made enormous progress over the past several years (3–7). The genomic abnormalities of cancer are expressed through aberrant proteins and proteomes and their altered functions. Although proteins reflecting the genomic changes in cancer have the potential to become clinically meaningful biomarkers, their discovery and validation has proven to be challenging. As a result, few biomarker candidates have translated into clinical use.
Over the past decade, mass spectrometry (MS)-based shotgun proteomics has emerged as a high-throughput, unbiased method for the identification of proteins in complex samples (8, 9). Its application to tumor specimens holds great potential in identifying mutant proteins in human cancers. However, because shotgun proteomics data analysis usually relies on database search and because commonly employed protein sequence databases do not contain protein variation information, the application of shotgun proteomics to the detection of protein sequence variants remains a big challenge.
Several research groups have made valuable efforts on enabling the identification of variant peptides based on the exhaustive search of all possible sequence variants. A modified version of Sequest provides automated search of human hemoglobin gene variants through dynamically generating all possible single-nucleotide variations and then constructing a database that translates these sequences to peptides (10). Roth et al. (11) developed a human protein database tailored for the “top-down” MS approach by combinatorial consideration of protein variability in a search. Similarly, the error-tolerant search in Mascot (12) and the refinement search in X!Tandem (13) allow exhaustive test of all amino acid substitutions that can arise from single-base nucleotide substitutions in each protein. Because of the greatly expanded search space, it is difficult to apply meaningful measure of statistical significance for the variant identifications and the results require careful interpretation (12).
An effective approach to limit the search space of protein variants is to consider only those derived from known coding SNPs. A SNP annotation method was presented by Bunger et al. in which MS/MS spectra were searched against reference protein databases and a separate SNP database created from peptides from the National Center for Biotechnology Information (NCBI) dbSNP database (14). Schandorff et al. established the MSIPI protein sequence database through elongating the original International Protein Index sequences with coding-SNPs from dbSNP, sequence conflicts, and N-terminal peptides (15). More recently, a web-based platform SysPIMP was created for identifying human disease-related mutant sequences based on the X!Tandem search of shotgun proteomics data (16). SysPIMP collects human disease-related mutant sequences from the Online Mendelian Inheritance in Man(17), Protein Mutant Database (18), and SwissProt database (19).
Despite these exciting developments, the problem of applying shotgun proteomics to the identification of protein variants in human cancers has not been addressed adequately. First, mutations, especially cancer-specific ones, are not specifically considered in existing approaches. NCBI's dbSNP database provides a general catalog of genome variation to address large-scale sampling designs required by association studies. It has been an invaluable resource for applying genetic approaches to understanding the etiology of different cancers (20). However, cancer somatic mutations are collected in the Catalogue of Somatic Mutations in Cancer (http://www.sanger.ac.uk/genetics/CGP/cosmic) (21) and other cancer specific databases (22) rather than dbSNP. As a result, most cancer-specific mutations have been omitted from previous studies. Recently, we developed a human Cancer Proteome Variation database (CanProVar1, http://bioinfo.vanderbilt.edu/canprovar/) (23) that comprehensively integrates proteome variation data from a variety of cancer specific variation data sources including HPI (24, 25), COSMIC, OMIM, and large-scale mutational profiling studies on cancer genes and cancer genomes (6, 7). Confirmed coding variations in NCBI's dbSNP are also included in CanProVar. This cancer-centric proteome variation repository provides an opportunity to create a protein sequence database that can facilitate protein variant detection in shotgun proteomics analysis of human cancer samples.
Second, although limiting protein variants to known coding SNPs and mutations could effectively reduce the search space as compared with the exhaustive test of all possible amino acid substitutions, this method still significantly increases the number of entries in a protein sequence database, which in turn increases the risk of false positive identifications. Many previous reports failed to address this critical problem (14). In the study by Bunger et al. (14), a peptide is assigned as an “alternative allele” SNP if the search score for its match against the dbSNP is at least 15% higher than the score for corresponding reference hit. The threshold of 15% was chosen based on manual examination to provide the best balance between false positives and false negatives (14). Although it was proven successful in this study, selection of the score threshold requires manual examination by experienced researchers and cannot be generalized and automated. Other problems introduced by adding variations to sequence databases include (1) efficient storage of variation information in the database, (2) compatibility of the database with different search engines, and (3) interpretability of reports that include both variant and wild-type peptides.
In this paper, we present an integrated workflow to address the above problems. First, we created a variation-containing protein sequence database based on the CanProVar database. Next, we developed a workflow for identifying both wild-type and variant peptides simultaneously from shotgun proteomics data. We used data sets from colorectal cancer cell lines and human patient samples to demonstrate our workflow. Identified variants were validated through genomic sequencing. Moreover, we tested the compatibility of the workflow with popular search engines including MyriMatch(26), Sequest(27), Mascot(28), and X!Tandem(13). A postprocessing tool was also developed to generate easily interpretable reports based on the output from different search engines. Finally, we benchmarked our workflow against the exhaustive search-based methods.
EXPERIMENTAL PROCEDURES
Proteomics Data Sets
The human proteomics datasets from colorectal adenocarcinoma cell lines (RKO, SW480, and HCT-116) and three colorectal tumor specimens were generated in the Ayers Institute at Vanderbilt. The cell lines were obtained from American Type Culture Collection (ATCC, Manassas, VA) and grown and harvested within 6 months of date of purchase, or grown from frozen stocks that had been made within 6 months of original purchase. They were grown in 10% fetal bovine serum and penicillin and streptomycin supplemented medium at 37 °C with 5% CO2. SW480 was grown in RPMI 1640 medium, whereas HCT-116 and RKO were grown in McCoy's5A medium. Cells were grown to 80% confluency, the growth medium was aspirated, cells were washed once in 1× phosphate-buffered saline and collected in 1× phosphate-buffered saline. Cells were centrifuged at 300 × g for 5 min and supernatant was discarded. Cell pellets were stored at −80 °C until cell lysis could be carried out. Biological replicates were harvested ∼1 week apart from the identical cell culture. These replicates were processed separately and independently through the complete analysis procedure. Colorectal tumor specimens were obtained from the Vanderbilt colorectal cancer repository under an IRB-approved protocol that included informed consent from the patients. We obtained three Stage III sigmoid carcinoma specimens based on availability of the biological material and confirmed for the presence of more than 70% tumor cells by a certified pathologist (Dr M.K. Washington). A total of 60 μm thickness for each of the frozen specimens was sectioned and collected into microcentrifuge tubes.
Mass spectrometry methods have been described in detail (29, 30). In summary, proteins from cell line or tissue samples were reduced, alkylated with iodoacetamide, and digested with trypsin. The resulting peptides were separated on isoelectric focusing strips that were cut into 15 (for cell lines) or 20 (for human tissues) separate fractions. Each of these fractions was analyzed by a second separation on a liquid chromatography column, followed by MS/MS analysis on an LTQ-Orbitrap. Binary spectral data present in the raw files were converted to the mzML format using the msConvert tool in the ProteoWizard library (v2.0.1757, 01/27/2010) (31).
Variations and Protein Reference Database
Protein variation data were downloaded from the CanProVar database on 9/26/2009, which included 41,541 nonsynonymous SNPs in 30,322 proteins and 8570 cancer-related variations in 2921 proteins (23). A corresponding normal protein database was downloaded from Ensembl (human, v53) at ftp://ftp.ensembl.org/pub/current_fasta/homo_sapiens/pep/.
Search Parameters
We tested our workflow against four popular database search engines, including MyriMatch(26) (v1.5.6), Sequest(27) (TurboSEQUEST v27), Mascot(28) (v2.2.04), and X!Tandem(13) (X!TANDEM TORNADO v2008.02.01.3). MyriMatch was used as the primary search engine in this study. All cysteines were assumed to be carbamidomethylated, and methionines were allowed to be oxidized. A precursor error of up to 0.007 m/z was permitted, whereas fragment ions were required to fall within 0.5 m/z of their expected locations. Ambiguous identifications that mapped to three or more peptide sequences with equal scores were excluded. One missed cleavage was permitted and no nonspecific cleavage was allowed. The configurations for all search engines are provided in supplemental File S1.
Genomic Sequence Verification
Genomic DNA from cell lines RKO, SW480, and HCT-116 was isolated using a DNeasy® kit (Qiagen). After identification of putative variant peptides by shotgun proteomics, the corresponding exons encoding the protein sequences were amplified using a HotStarTaq® Master Mix Kit (Qiagen). The following polymerase chain reaction (PCR) conditions were used: 96 °C × 15 min, followed by 40 cycles of 95 °C × 30 s, 60 °C × 30 s, 72 °C × 60 s, and a final extension of 72 °C × 10 min. A list of all the primers used for the PCR amplifications is provided in supplemental File S2. Excess primers and nucleotides were digested using ExoSAP (USB). Sequencing reactions were performed by using Applied Biosystems Version 3.1 Big Dye Terminator chemistry and then analyzed on an Applied Biosystems 3730XL Sequencer. All sequence chromatograms were read in both forward (F) and reverse (R) directions.
RESULTS
Setup of the Workflow
As illustrated in Fig. 1, our workflow for identifying wild-type and variant peptides based on shotgun proteomics data includes three steps: database creation, peptide identification, and post-processing.
Fig. 1.

Workflow for identifying variant peptides from shotgun proteomics data. A, MS/MS data in one of the standard formats is searched using a selected search engine against an integrated database including both a regular protein sequence database and the MS-CanProVar. B, For each pepXML file generated from an experimental run, the false discovery rates (FDR) for wild-type and variant identifications are estimated separately. C, Both wild-type and variant peptides are identified from MS/MS dataset based on a selected FDR cutoff and reported.
The variation-containing protein sequence database was created based on the Ensembl protein database (human, v53) and the CanProVar database (23). Missense variations, nonsense variations and single amino acid deletions and insertions were included in the database. After the naming convention in dbSNP, each cancer-related variation in CanProVar was given an identifier prefixed with “cs.” For each single amino acid alteration, the sequence covering the enclosing tryptic peptide and the two flanking tryptic peptides was taken as an independent entry in the FASTA format. Peptide entries with less than 4 residues were excluded because they cannot be confidently identified in shotgun proteomics. Adding the flanking peptides allows for the identification of peptides with missed enzyme cleavage (15). This database construction approach shares the same space-saving advantage as appending sequence variants to the original protein sequence, which was adopted in the study of Schandorff et al. (15). We chose to keep these peptides as independent entries because related variation information can be easily recorded in the sequence header, which includes corresponding protein ID, the start and end positions of the peptide in the protein, and the identifier of the variation in database dbSNP or CanProVar. These new peptide entries resulted in an increase of about 3.4% in the tryptic peptide database size. Our protein sequence database comprised the complete Ensembl protein database (v53, 47,509 entries) and an additional 97,637 peptide entries with variations from 29,873 Ensembl proteins. Among these, 10,254 peptide entries carried cancer-related variations. We named this protein sequence database MS-CanProVar. Reverse sequences were appended as decoy sequences for false discovery rate (FDR) estimation (32). MS-CanProVar can be downloaded at http://bioinfo.vanderbilt.edu/canprovar.
After creation of the database, shotgun proteomics data from a cancer sample can be searched against the database using a database search engine (Fig. 1A). The next important step is the confidence evaluation of the peptide identifications, i.e. FDR estimation. It has been suggested that a higher risk of false positives could be associated with variant peptide identifications as compared with that for wild-type ones (14). In order to systematically investigate this problem, we searched the SW480 dataset against MS-CanProVar with MyriMatch and used the standard FDR estimation method (32) with no special treatment to variant identifications. Peptides with an FDR <0.05 were separated into a wild-type group and a variant group, and the score distributions of these two groups were plotted (Fig. 2A). The score distribution for the variant group showed a significant shift toward the low-score end. Similar results were observed in data from other cancer cell lines (data not shown). These results suggest that although the two groups of peptides were identified using the same FDR cutoff, the variant group does have a higher risk of false positive identifications. Follow-up genomic analysis further confirmed this concern. As shown in Table I, among the 11 putative variant peptides randomly chosen with FDR < 0.1, only six were confirmed with genomic sequencing. With a threshold of FDR < 0.05, the confirmation rate was six of nine.
Fig. 2.

Search score distributions for the variant (red) and wildtype (green) peptides identified with FDR < 0. 05 in the SW480 data set. A, Under regular FDR estimation, an apparent shift to the low-score end was observed in the distribution for variant identifications as compared with that for the wild-type identifications. B, Naïve separate estimation reduced the bias to a certain degree. C,The new refined separate FDR estimation approach proposed in this study further improved the quality of variant peptide identifications.
Table I. Genomic sequencing verification for the variant peptides identified in SW480. NS_FDR and RS_FDR, respectively, refer to naïve separate FDR estimation and the refined separate FDR estimation proposed in this study. Peptides with FDRs higher than the given FDR cutoffs are marked with dashes. Stars and cross marks represent successful or failed genomic sequencing verification, respectively.
| No. | Peptide | Gene | Variation | FDR .1 | FDR .05 | NS_FDR .05 | RS_FDR .05 |
|---|---|---|---|---|---|---|---|
| 1 | LAAETGEGEGEPLSR | DIDO1 | T1568A | * | * | * | * |
| 2 | DPAEPMSPGEATQSGARPADR | MYBBP1A | Q8E | * | * | * | * |
| 3 | ASSSILINESEPTTNIQIR | NSFL1C | D179N | X | X | X | - |
| 4 | AGTDSPVSCASITEER | CDCA2 | V717I | * | * | * | * |
| 5 | GTETFEPEDK | CD3EAP | K259T | * | * | * | * |
| 6 | LDSTDFTSTIK | TFRC | G142S | * | * | * | * |
| 7 | FAALDDEEEDKEEEIIK | ABCF1 | N198D | X | X | X | X |
| 8 | ELFQTPGPSEESMSDEK | MKI67 | T760S | X | - | - | - |
| 9 | SDSELNNEVAAR | CYBRD1 | S266N | * | * | * | * |
| 10 | QLVNMCMNPDPEK | NEK7 | I275M | X | X | X | - |
| 11 | EILDEAYAMAGVGSPYVSR | ERBB2 | V773A | X | - | - | - |
For FDR estimation, all forward sequences are considered as expressed and present. Nevertheless, for a specific sample, only some of the forward sequences are expressed. Moreover, the proportion of expressed sequences among all variant sequences in the database is expected to be significantly lower than that among all wild-type sequences, i.e. variant sequences are expected to have a lower prior probability of being present in a specific sample. However, the FDR estimation is for all matches above the selected score threshold without discriminating wild-type from variant sequences. Therefore, the combined FDR estimation will lead to a higher false negative rate for wild-type peptide identifications and a higher false positive rate for variant peptide identifications. This might not be a big problem for wild-type identifications because variant sequences only comprise a small fraction of the database. Nevertheless, when we consider only variant peptide identifications, the real FDR for the subgroup could be much higher than the combined estimation.
To address this problem, we first estimated the FDRs for wildtype and variant peptides separately. Specifically, only variant peptides and corresponding decoys were considered for the FDR estimation of variant peptide identifications. With this naïve separate FDR estimation, more stringent score cutoffs were set for the variant peptides than for the wild-type ones in most of the experimental runs, and the risk of high false positives for the variant group was reduced according to the score distribution plot (Fig. 2B). However, in some experimental runs, a lower search score cutoff was set for the variant peptides than that for the wild-type ones. Indeed, for variant identifications, we found that the search score cutoff corresponding to a preselected FDR level (e.g. 0.05) varied dramatically across the experiment runs, a phenomenon we did not see in the wildtype searches. This may be because of the small number of matches found in the variant peptides and variant decoys. In the target-decoy search strategy for FDR estimation, one can estimate the total number of false positives that meet a specific score threshold by doubling the number of selected decoy matches. This represents the number of observed incorrect decoy matches, combined with the hidden incorrect target matches. When the number of total matches is very low for a given subset of peptides, the estimate of false positives becomes highly variable. As a result, no improvement on the genomic confirmation rate was observed (Table I).
To achieve a more robust estimation of the total number of false positives, we proposed to combine information based on decoys from both variant and wild-type sequences and calculate FDR for variant identifications based on the following formula:
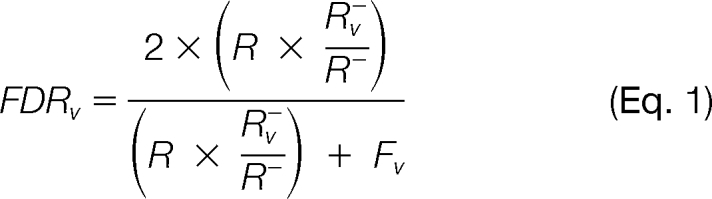 |
Here, R and Fv are the numbers of reverse matches and forward variant matches above the score threshold, respectively. R− and R− are the numbers of reverse matches and variant reverse matches falling below the score threshold, respectively. The Rv−/R− ratio provides an estimate of the proportion of variant sequences in the database.
In this formula, the number of false positives in variant identifications is estimated by the total number of false positives and the proportion of variant sequences in the database. Our assumption is that there is no difference between the decoys from wild-type sequences and variant sequences for FDR estimation. This formula may provide a more accurate estimation of the number of false positives for variant peptide identifications because the estimate is based on data that are less subject to variation.
The score distribution plot showed that the new FDR estimation could significantly improve the confidence of variant peptide identifications (Fig. 2C). Genomic sequencing verification for the detected variations also showed that the new FDR estimation method significantly outperformed both combined and naïve separate FDR estimation. As shown in Table I, variant peptide identification based on the new method achieved a confirmation rate of six of seven, an improvement as compared with the rate of six of nine acquired based on combined or naïve separate FDR estimations. Moreover, although the seventh mutation ABCF1N198D was not confirmed at the genomic level, this change may actually happen after translation through the deamidation of the asparagine residue. On the basis of these verification results, the new refined FDR estimation approach for variant peptide identifications was employed in our workflow (Fig. 1B). In the last step of the workflow, both wild-type and variant peptides are identified based on the refined separate FDR estimation and an easily interpretable report is generated. (Fig. 1C).
Application on Human Cancer Data Sets
With the procedure described above, we performed database search and peptide identification for three data sets from colorectal cancer cell lines RKO, HCT-116, and SW480, respectively. MyriMatch was used as the search engine, and the FDR threshold was set to 0.05 for both wild-type and variant peptides. Thus, 6284, 9145, and 20,023 unique peptides were identified in SW480, RKO, and HCT-116 samples respectively, which were mapped to 1148, 1784, and 2927 indiscernible protein groups using IDPicker (33, 34). The number of variant peptides was 20, 27, and 34 for SW480, RKO, and HC-116 respectively (supplemental Files S3 and S4), corresponding to 0.3%, 0.3%, and 0.2% of all peptides identified in each cell line.
We randomly selected 10 and nine putative variant peptides from the RKO and HCT-116 data sets for genomic sequencing verification and the confirmation rate were 8 of 10 and nine of nine, respectively. Combining the genomic sequencing result for SW480, the overall confirmation rate for all three cell lines was 88% (23/26). A complete list of the variant peptides and associated information can be found in supplemental File S3. In the HCT-116 data set, we detected a variation G13D in KRAS (Fig. 3). KRAS was one of the first genes identified as a transforming gene (oncogene) capable of driving tumor formation in experimental model systems. The G13D variation is not only a known mutation in the HCT-116 cell line but has also been found in 21% of 335 colorectal tumors in a large-scale mutational profiling study (35).
Fig. 3.

Sequence validation of the KRASG13D identified in the HCT-116 data set. A tandem MS spectrum with m/z 507.304 was identified as peptide LVVVGAGDVGK. The peaks from y4 to y9 ions and b8 to b11 ions indicate a −58 Da mass shift corresponding to the substitution of glycine with aspartic acid. The inset on the top right corner shows genomic sequencing of PCR product from region surrounding the mutation with corresponding predicted amino-acids. Sequencing revealed a heterozygous point mutation. Consistently, the wildtype peptide LVVVGAGGVGK was also detected in the HCT-116 data set.
In addition to the cancer cell lines, we also applied the procedure on three data sets from clinical colorectal tumor specimens. A total of 37,827 unique peptides were identified in these data sets, which were mapped to 5581 indiscernible protein groups using IDPicker. The number of distinct variant peptides detected in these samples was 204 (supplemental Files S5 and S6), corresponding to 0.5% of all identified peptides. As shown in Fig. 4A, 101, 78, and 139 variant peptides were detected in each of the three patients, respectively. Five peptides carried known cancer-related mutations, and 4 of them were found in more than one patient (Fig. 4B). The mutation TP53P309S was found in two colorectal cancer patients in this study. Mutations in TP53 are the most commonly observed mutations in any cancer-associated genes with ∼50% of all human cancers harboring inactivating mutations in this tumor suppressor gene (36). The majority of TP53 mutations cause increased half-life of a functionally inactive p53 protein leading to loss of cell cycle control, resistance to programmed cell death (apoptosis) and the capacity of infinite growth (immortality) in cells harboring such mutations. The mutation TP53P309S has been reported in the SW480 cell line. Inhibiting mutant TP53(R273H/P309S) expression in SW480 reduces cell proliferation, in vitro and in vivo tumorigenicity, and resistance to anticancer drugs (37, 38). The mutation SMARCA4W764R was also observed in two patients. Although not reported in colorectal cancer previously, this mutation has been reported in lung cancer (39). A variety of other mutations within SMARCA4 (also named BRG1) were found in several cell lines derived from carcinomas of the breast, lung, pancreas, and prostate etc., and SMARCA4 has been suggested as a drug target for cancer treatment (40). As a subunit of mammalian SWI/SNF chromatin remodeling complexes, SMARCA4 is a critical regulator of TP53 and has been found to be necessary for the proliferation of malignant cells (41).
Fig. 4.

Variant peptides and known cancer-related mutations identified from clinical colorectal tumor specimens from three patients. A, Venn diagram shows the number of identified unique variant peptides and their overlaps among three patients. B, Five known cancer-related mutations were detected in patients, four of which were observed in two patients.
Test for Compatibility with Multiple Search Engines
To ensure compatibility between our workflow and popular proteomics search engines, we tested the procedure with Sequest, Mascot, X!Tandem as well as MyriMatch. The output files of these search engines are written in the pepXML format or can be transferred into the pepXML format via converters. The variation information for each variant peptide is included the pepXML files. Currently, there is no specific software available to extract this information. Therefore we created a tool CanProVar-Parser that can be used to estimate FDR, perform identification for both variant and wildtype peptides, and parse peptide information for reporting. Peptide-related information in a report includes protein mapping, variations, spectral count, FDR value, match rank and spectrum source. The CanProVar-Parser is written in Perl and can be downloaded from http://bioinfo.vanderbilt.edu/canprovar. Applying our procedure on the RKO data set identified 27, 22, 29, and 29 distinct variant peptides using MyriMatch, Sequest, Mascot, and X!Tandem, respectively. Twenty-five variant peptides were detected by two or more search engines (Fig. 5). It is not unusual to get moderately overlapping results from different search engines, and integrating results from multiple search engines has been proposed as a way to improve peptide identification (42–44). The ability to use our procedure with different search engines makes it possible to perform this type of integration.
Fig. 5.

Overlap of variant peptides identified by different search engines in the RKO data set.
Comparison with Exhaustive Search-based Methods
Relying on exhaustive search for all amino acid substitutions that can arise from single base nucleotide substitutions in each protein, the error tolerant search in Mascot (12) and the refinement search in X!Tandem (13) allow the detection of variant peptides without using existing information on genomic sequence variations. To benchmark our method, we performed analysis on the RKO data set using these exhaustive search-based methods and the Ensembl protein database. We controlled the FDR at a 5% level for the wild-type peptide identifications. For variant peptide identifications, we followed the suggestion from Mascot (http://www.matrixscience.com/help/error_tolerant_help.html): (a) they must have scores of at least the identity threshold for wild-type identifications; and (b) they must have scores in excess of the highest scoring match to the wild-type sequences. Accordingly, the error-tolerant search in Mascot identified 10,344 wildtype peptides and 1298 variant peptides, whereas the refinement search in X!Tandem identified 9705 wildtype peptides and 595 variant peptides.
First, we compared the variant peptides identified by exhaustive search using the two search engines and found very limited overlap (Fig. 6). Specifically, 95 and 90% of the identifications were unique to Mascot and X!Tandem, respectively. In contrast, the overlap between the two search engines using our method was much higher, with only 27% nonoverlapping identifications for both search engines. The extremely small percentage of overlap between exhaustive search results from the two search engines raises a concern of potentially high false positive rates (i.e. low specificity). Nevertheless, exhaustive search-based methods identified many more variant peptides than our method even if we only considered common identifications from the two search engines, suggesting a possibly higher sensitivity of these methods.
Fig. 6.

Comparison between exhaustive search-based methods and MS-CanProVar-based methods. A, Overlap of variant peptides identified by Mascot and X!Tandem based on exhaustive search. B, Overlap of variant peptides identified by Mascot and X!Tandem based on MS-CanProVar. Bold numbers represent the count of variant peptides and corresponding percentages in each section of the Venn Diagrams. Numbers in the parentheses represent the count of overlap with the 8 confirmed variant peptides in each section of the Venn Diagrams.
To gain some insight into the sensitivity of the exhaustive search-based methods, we compared their variant identifications against the eight variant peptides detected by MyriMatch in the RKO cell line and confirmed by genomic sequencing (supplemental Files S3). Surprisingly, despite the large numbers of variant peptide identifications, for each search engine, only three out of the eight peptides were identified (Fig. 6). In contrast, with our method, seven out of the eight were identified by both Mascot and X!Tandem. Although a conclusion cannot be made based on the limited number of true positives, these results do not provide evidence for a superior sensitivity of the exhaustive search-based methods.
DISCUSSION
We have created a protein variation-containing database MS-CanProVar and a workflow for the simultaneous identification of wild-type and variant peptides based on the database. A novel FDR estimation method was introduced in the workflow to ensure high reliability of the variant identifications. Hundreds of variant peptides were identified from three colorectal cancer cell lines and three tumor specimens used in this study. Most of the variants were derived from the dbSNP database and are likely to represent polymorphisms. Whether these polymorphisms are associated with cancer will require large-scale association studies. Some known cancer-related mutations have been identified, including those associated with cell proliferation, tumorigenesis, and drug resistance.
A major concern on the use of variation-containing databases for shotgun proteomics data searching is the high risk of false positive identifications (14). In this study, we systematically investigated this risk by comparing the search score distributions of wild-type and variant peptide identifications and proposed a modified FDR estimation method to automatically handle this issue. By contrast, existing studies require manual selection of a more stringent threshold for variant peptide identifications (14). It is also worth mentioning that in our workflow, although FDR estimations were carried out separately for variant and wild-type peptide identifications, the database search was done at the same time, similar to Schandorff et al. (15). In Bunger et al. study (14), separate searches were performed for the reference and variant databases. When a variant database is searched separately, a best match to a variant peptide may be because of the absence of the competition from the truly presenting wild-type protein.
Genomic sequencing was used to provide an objective evaluation of the reliability of the peptide variants identified using our workflow and confirmation rates of around 90% were achieved. Besides false discoveries generated by the workflow, inconsistency between proteomics identifications and genomic sequencing results can also be explained by mass shifts because of various peptide modifications (14). For example, the alteration ABCF1N198D detected in the SW480 data set might be because of deamidation as this alteration was not confirmed by genomic sequencing. Oxidation (+16), formylation (+28), and acetylation (+42) are other common modifications on peptides (14). Discerning whether a mass shift has resulted from a sequence variation or post-translational modification may require sequencing for confirmation. For example, although the mass shift in MYBBP1AQ8E could be explained by the deamidation of the glutamine residue, this alteration was confirmed at the genomic level (Table I).
As pointed out by Schandorff et al. (15), searching against variation-containing protein databases should provide a new dimension to clinical proteomics projects. In cancer care, detection of expressed mutant peptides and proteins of individual patients by proteomics techniques may have an important impact on the development of personalized medicine. Although only five known cancer-related mutations were detected in the tumor specimens from three colorectal cancer patients, each patient showed a specific mutation pattern (Fig. 4B). These mutation patterns provide both germline and somatic mutation information at a proteome level that could potentially facilitate personalized cancer care.
As compared with exhaustive search-based methods, limiting protein variants to those derived from known coding SNPs and mutations could effectively reduce the search space and thus lead to more reliable identifications. However, this advantage also simultaneously imposes a major limitation of dependence on known genomic sequence variations. The number of known cancer-related mutations detected in this study was moderate. Although this can be partially explained by the potentially low stability of mutated proteins, an obvious explanation is the limited database coverage. Cancer-related mutations in the current CanProVar database distribute highly unevenly in human proteins. Most proteins have very few cancer-related mutations whereas some well-known cancer genes have reported mutations in many positions in their protein sequences, such as TP53, CTNNB1, and PIK3CA. More than a hundred different cancer-related mutations have been reported in these proteins. This bias might be explained by the extreme instability of these important cancer genes, but it may also reflect lack of study of other genes. Ongoing large-scale cancer genome projects, such as the Cancer Genome Project of the Sanger Institute, The Cancer Genome Atlas project of the National Cancer Institute, and the National Human Genome Research Institute, will rapidly expand our knowledge on mutations in human cancers (21, 39). We will continuously incorporate results generated from these studies into CanProVar and MS-CanProVar to improve the sensitivity of our analysis workflow.
Although exhaustive search-based methods identified many more variant peptides, evaluation in this study based on limited true positives did not provide evidence for a superior sensitivity of these methods over our workflow. Recently, a sequence tagging-based “de novo” algorithm has been proposed as an attractive alternative for variant peptide identification (45). It will be interesting to perform a thorough comparison of these complementary approaches in order to highlight their distinct values. Moreover, only missense variations, nonsense variations and single amino acid deletions and insertions were included in MS-CanProVar. Other protein sequence variations such as splice variants and post-translational modifications are also critical in cancer studies and have been detected by shotgun proteomics (46, 47). Future work is required to improve our database and workflow for the inclusion of existing knowledge on these variations.
In summary, we have developed a workflow for variant peptide detection in shotgun proteomics studies. The workflow achieves a good balance between reliable variation detection and overall sensitivity of peptide identification. Compatibility of the workflow with popular database search engines has been extensively tested. Reliability of the identifications has been confirmed by genomic sequencing. Applying this workflow on human cancer proteomics studies should provide novel insight into cancer predisposition and potential personalized therapy.
Acknowledgments
We would like to thank Dr. Surendra Dasari and Mr. Mathew C. Chambers for their assistance in the preparation of the supplementary files.
Footnotes
* This work was conducted, in part, using the resources of the Advanced Computing Center for Research and Education at Vanderbilt University, Nashville, TN. This work was supported by the National Institutes of Health (NIH)/National Cancer Institute (NCI) through grant R01 CA126218, the NIH/National Institute of General Medical Sciences (NIGMS) through grant R01 GM088822, the NCI Clinical Proteomic Technologies Assessment for Cancer (CPTAC) program through 1U24CA126479, the NIH/National Center for Research Resources (NCRR) Clinical and Translational Science Awards (CTSA) program through UL1 RR024975, and a generous gift from the Jim Ayers Foundation. WP acknowledges funding from the NIH/NCI (VICC Cancer Center Core Grant (CA68485)) and an anonymous donor. JL acknowledges funding from the National Natural Science Foundation of China (Grant No. 31000582).
 This article contains supplemental Files S1 to S6.
This article contains supplemental Files S1 to S6.
1 The abbreviations used are:
- CanProVar
- the human Cancer Proteome Variation Database
- SNP
- single nucleotide polymorphism.
REFERENCES
- 1. Vogelstein B., Kinzler K. W. (2004) Cancer genes and the pathways they control. Nat. Med. 10, 789–799 [DOI] [PubMed] [Google Scholar]
- 2. Futreal P. A., Coin L., Marshall M., Down T., Hubbard T., Wooster R., Rahman N., Stratton M. R. (2004) A census of human cancer genes. Nat. Rev. Cancer 4, 177–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wood L. D., Parsons D. W., Jones S., Lin J., Sjöblom T., Leary R. J., Shen D., Boca S. M., Barber T., Ptak J., Silliman N., Szabo S., Dezso Z., Ustyanksky V., Nikolskaya T., Nikolsky Y., Karchin R., Wilson P. A., Kaminker J. S., Zhang Z., Croshaw R., Willis J., Dawson D., Shipitsin M., Willson J. K., Sukumar S., Polyak K., Park B. H., Pethiyagoda C. L., Pant P. V., Ballinger D. G., Sparks A. B., Hartigan J., Smith D. R., Suh E., Papadopoulos N., Buckhaults P., Markowitz S. D., Parmigiani G., Kinzler K. W., Velculescu V. E., Vogelstein B. (2007) The genomic landscapes of human breast and colorectal cancers. Science 318, 1108–1113 [DOI] [PubMed] [Google Scholar]
- 4. Weir B. A., Woo M. S., Getz G., Perner S., Ding L., Beroukhim R., Lin W. M., Province M. A., Kraja A., Johnson L. A., Shah K., Sato M., Thomas R. K., Barletta J. A., Borecki I. B., Broderick S., Chang A. C., Chiang D. Y., Chirieac L. R., Cho J., Fujii Y., Gazdar A. F., Giordano T., Greulich H., Hanna M., Johnson B. E., Kris M. G., Lash A., Lin L., Lindeman N., Mardis E. R., McPherson J. D., Minna J. D., Morgan M. B., Nadel M., Orringer M. B., Osborne J. R., Ozenberger B., Ramos A. H., Robinson J., Roth J. A., Rusch V., Sasaki H., Shepherd F., Sougnez C., Spitz M. R., Tsao M. S., Twomey D., Verhaak R. G., Weinstock G. M., Wheeler D. A., Winckler W., Yoshizawa A., Yu S., Zakowski M. F., Zhang Q., Beer D. G., Wistuba II, Watson M. A., Garraway L. A., Ladanyi M., Travis W. D., Pao W., Rubin M. A., Gabriel S. B., Gibbs R. A., Varmus H. E., Wilson R. K., Lander E. S., Meyerson M. (2007) Characterizing the cancer genome in lung adenocarcinoma. Nature 450, 893–898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. TCGA (2008) Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455, 1061–1068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sjöblom T., Jones S., Wood L. D., Parsons D. W., Lin J., Barber T. D., Mandelker D., Leary R. J., Ptak J., Silliman N., Szabo S., Buckhaults P., Farrell C., Meeh P., Markowitz S. D., Willis J., Dawson D., Willson J. K., Gazdar A. F., Hartigan J., Wu L., Liu C., Parmigiani G., Park B. H., Bachman K. E., Papadopoulos N., Vogelstein B., Kinzler K. W., Velculescu V. E. (2006) The consensus coding sequences of human breast and colorectal cancers. Science 314, 268–274 [DOI] [PubMed] [Google Scholar]
- 7. Greenman C., Stephens P., Smith R., Dalgliesh G. L., Hunter C., Bignell G., Davies H., Teague J., Butler A., Stevens C., Edkins S., O'Meara S., Vastrik I., Schmidt E. E., Avis T., Barthorpe S., Bhamra G., Buck G., Choudhury B., Clements J., Cole J., Dicks E., Forbes S., Gray K., Halliday K., Harrison R., Hills K., Hinton J., Jenkinson A., Jones D., Menzies A., Mironenko T., Perry J., Raine K., Richardson D., Shepherd R., Small A., Tofts C., Varian J., Webb T., West S., Widaa S., Yates A., Cahill D. P., Louis D. N., Goldstraw P., Nicholson A. G., Brasseur F., Looijenga L., Weber B. L., Chiew Y. E., DeFazio A., Greaves M. F., Green A. R., Campbell P., Birney E., Easton D. F., Chenevix-Trench G., Tan M. H., Khoo S. K., Teh B. T., Yuen S. T., Leung S. Y., Wooster R., Futreal P. A., Stratton M. R. (2007) Patterns of somatic mutation in human cancer genomes. Nature 446, 153–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Foster L. J., de Hoog C. L., Zhang Y., Zhang Y., Xie X., Mootha V. K., Mann M. (2006) A mammalian organelle map by protein correlation profiling. Cell 125, 187–199 [DOI] [PubMed] [Google Scholar]
- 9. Kislinger T., Cox B., Kannan A., Chung C., Hu P., Ignatchenko A., Scott M. S., Gramolini A. O., Morris Q., Hallett M. T., Rossant J., Hughes T. R., Frey B., Emili A. (2006) Global survey of organ and organelle protein expression in mouse: combined proteomic and transcriptomic profiling. Cell 125, 173–186 [DOI] [PubMed] [Google Scholar]
- 10. Gatlin C. L., Eng J. K., Cross S. T., Detter J. C., Yates J. R., 3rd (2000) Automated identification of amino acid sequence variations in proteins by HPLC/microspray tandem mass spectrometry. Anal. Chem. 72, 757–763 [DOI] [PubMed] [Google Scholar]
- 11. Roth M. J., Forbes A. J., Boyne M. T., 2nd, Kim Y. B., Robinson D. E., Kelleher N. L. (2005) Precise and parallel characterization of coding polymorphisms, alternative splicing, and modifications in human proteins by mass spectrometry. Mol. Cell. Proteomics 4, 1002–1008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Creasy D. M., Cottrell J. S. (2002) Error tolerant searching of uninterpreted tandem mass spectrometry data. Proteomics 2, 1426–1434 [DOI] [PubMed] [Google Scholar]
- 13. Craig R., Beavis R. C. (2004) TANDEM: matching proteins with tandem mass spectra. Bioinformatics 20, 1466–1467 [DOI] [PubMed] [Google Scholar]
- 14. Bunger M. K., Cargile B. J., Sevinsky J. R., Deyanova E., Yates N. A., Hendrickson R. C., Stephenson J. L., Jr. (2007) Detection and validation of non-synonymous coding SNPs from orthogonal analysis of shotgun proteomics data. J. Proteome Res. 6, 2331–2340 [DOI] [PubMed] [Google Scholar]
- 15. Schandorff S., Olsen J. V., Bunkenborg J., Blagoev B., Zhang Y., Andersen J. S., Mann M. (2007) A mass spectrometry-friendly database for cSNP identification. Nat. Methods 4, 465–466 [DOI] [PubMed] [Google Scholar]
- 16. Xi H., Park J., Ding G., Lee Y. H., Li Y. (2009) SysPIMP: the web-based systematical platform for identifying human disease-related mutated sequences from mass spectrometry. Nucleic Acids Res. 37, D913–920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hamosh A., Scott A. F., Amberger J. S., Bocchini C. A., McKusick V. A. (2005) Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res. 33, D514–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kawabata T., Ota M., Nishikawa K. (1999) The Protein Mutant Database. Nucleic Acids Res. 27, 355–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Boeckmann B., Bairoch A., Apweiler R., Blatter M. C., Estreicher A., Gasteiger E., Martin M. J., Michoud K., O'Donovan C., Phan I., Pilbout S., Schneider M. (2003) The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 31, 365–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Packer B. R., Yeager M., Staats B., Welch R., Crenshaw A., Kiley M., Eckert A., Beerman M., Miller E., Bergen A., Rothman N., Strausberg R., Chanock S. J. (2004) SNP500Cancer: a public resource for sequence validation and assay development for genetic variation in candidate genes. Nucleic Acids Res. 32, D528–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bamford S., Dawson E., Forbes S., Clements J., Pettett R., Dogan A., Flanagan A., Teague J., Futreal P. A., Stratton M. R., Wooster R. (2004) The COSMIC (Catalogue of Somatic Mutations in Cancer) database and website. Br. J. Cancer 91, 355–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Olivier M., Petitjean A., Teague J., Forbes S., Dunnick J. K., den Dunnen J. T., Langerod A., Wilkinson J. M., Vihinen M., Cotton R. G., Hainaut P. (2009) Somatic mutation databases as tools for molecular epidemiology and molecular pathology of cancer: proposed guidelines for improving data collection, distribution, and integration. Hum. Mutat. 30, 275–282 [DOI] [PubMed] [Google Scholar]
- 23. Li J., Duncan D. T., Zhang B. (2010) CanProVar: a human cancer proteome variation database. Hum. Mutat. 31, 219–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Boeckmann B., Blatter M. C., Famiglietti L., Hinz U., Lane L., Roechert B., Bairoch A. (2005) Protein variety and functional diversity: Swiss-Prot annotation in its biological context. C. R. Biol. 328, 882–899 [DOI] [PubMed] [Google Scholar]
- 25. O'Donovan C., Apweiler R., Bairoch A. (2001) The human proteomics initiative (HPI). Trends Biotechnol. 19, 178–181 [DOI] [PubMed] [Google Scholar]
- 26. Tabb D. L., Fernando C. G., Chambers M. C. (2007) MyriMatch: highly accurate tandem mass spectral peptide identification by multivariate hypergeometric analysis. J. Proteome Res. 6, 654–661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Eng J. K., MaCormack A. L., Yates J. R., 3rd (1994) An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database J. Am. Soc. Mass Spectrom. 5, 976–989 [DOI] [PubMed] [Google Scholar]
- 28. Perkins D. N., Pappin D. J., Creasy D. M., Cottrell J. S. (1999) Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20, 3551–3567 [DOI] [PubMed] [Google Scholar]
- 29. Slebos R. J., Brock J. W., Winters N. F., Stuart S. R., Martinez M. A., Li M., Chambers M. C., Zimmerman L. J., Ham A. J., Tabb D. L., Liebler D. C. (2008) Evaluation of strong cation exchange versus isoelectric focusing of peptides for multidimensional liquid chromatography-tandem mass spectrometry. J. Proteome Res. 7, 5286–5294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sprung R. W., Jr., Brock J. W., Tanksley J. P., Li M., Washington M. K., Slebos R. J., Liebler D. C. (2009) Equivalence of protein inventories obtained from formalin-fixed paraffin-embedded and frozen tissue in multidimensional liquid chromatography-tandem mass spectrometry shotgun proteomic analysis. Mol. Cell Proteomics 8, 1988–1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kessner D., Chambers M., Burke R., Agus D., Mallick P. (2008) ProteoWizard: open source software for rapid proteomics tools development. Bioinformatics 24, 2534–2536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Elias J. E., Gygi S. P. (2007) Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 4, 207–214 [DOI] [PubMed] [Google Scholar]
- 33. Zhang B., Chambers M. C., Tabb D. L. (2007) Proteomic parsimony through bipartite graph analysis improves accuracy and transparency. J. Proteome Res. 6, 3549–3557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ma Z. Q., Dasari S., Chambers M. C., Litton M. D., Sobecki S. M., Zimmerman L. J., Halvey P. J., Schilling B., Drake P. M., Gibson B. W., Tabb D. L. (2009) IDPicker 2.0: Improved protein assembly with high discrimination peptide identification filtering. J. Proteome Res. 8, 3872–3881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Oliveira C., Westra J. L., Arango D., Ollikainen M., Domingo E., Ferreira A., Velho S., Niessen R., Lagerstedt K., Alhopuro P., Laiho P., Veiga I., Teixeira M. R., Ligtenberg M., Kleibeuker J. H., Sijmons R. H., Plukker J. T., Imai K., Lage P., Hamelin R., Albuquerque C., Schwartz S., Jr., Lindblom A., Peltomaki P., Yamamoto H., Aaltonen L. A., Seruca R., Hofstra R. M. (2004) Distinct patterns of KRAS mutations in colorectal carcinomas according to germline mismatch repair defects and hMLH1 methylation status. Hum. Mol. Genet. 13, 2303–2311 [DOI] [PubMed] [Google Scholar]
- 36. Soussi T., Wiman K. G. (2007) Shaping genetic alterations in human cancer: the p53 mutation paradigm. Cancer Cell 12, 303–312 [DOI] [PubMed] [Google Scholar]
- 37. Bossi G., Lapi E., Strano S., Rinaldo C., Blandino G., Sacchi A. (2006) Mutant p53 gain of function: reduction of tumor malignancy of human cancer cell lines through abrogation of mutant p53 expression. Oncogene 25, 304–309 [DOI] [PubMed] [Google Scholar]
- 38. Yan W., Liu G., Scoumanne A., Chen X. (2008) Suppression of inhibitor of differentiation 2, a target of mutant p53, is required for gain-of-function mutations. Cancer Res. 68, 6789–6796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Forbes S. A., Bhamra G., Bamford S., Dawson E., Kok C., Clements J., Menzies A., Teague J. W., Futreal P. A., Stratton M. R. (2008) The Catalogue of Somatic Mutations in Cancer (COSMIC). Curr. Protoc. Hum. Genet. Chapter 10, Unit 10. 11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wong A. K., Shanahan F., Chen Y., Lian L., Ha P., Hendricks K., Ghaffari S., Iliev D., Penn B., Woodland A. M., Smith R., Salada G., Carillo A., Laity K., Gupte J., Swedlund B., Tavtigian S. V., Teng D. H., Lees E. (2000) BRG1, a component of the SWI-SNF complex, is mutated in multiple human tumor cell lines. Cancer Res. 60, 6171–6177 [PubMed] [Google Scholar]
- 41. Naidu S. R., Love I. M., Imbalzano A. N., Grossman S. R., Androphy E. J. (2009) The SWI/SNF chromatin remodeling subunit BRG1 is a critical regulator of p53 necessary for proliferation of malignant cells. Oncogene 28, 2492–2501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jones A. R., Siepen J. A., Hubbard S. J., Paton N. W. (2009) Improving sensitivity in proteome studies by analysis of false discovery rates for multiple search engines. Proteomics 9, 1220–1229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Searle B. C., Turner M., Nesvizhskii A. I. (2008) Improving sensitivity by probabilistically combining results from multiple MS/MS search methodologies. J. Proteome Res. 7, 245–253 [DOI] [PubMed] [Google Scholar]
- 44. Yu W., Taylor J. A., Davis M. T., Bonilla L. E., Lee K. A., Auger P. L., Farnsworth C. C., Welcher A. A., Patterson S. D. (2010) Maximizing the sensitivity and reliability of peptide identification in large-scale proteomic experiments by harnessing multiple search engines. Proteomics, 10, 1172–1189 [DOI] [PubMed] [Google Scholar]
- 45. Dasari S., Chambers M. C., Slebos R. J., Zimmerman L. J., Ham A. J., Tabb D. L. (2010) TagRecon: high-throughput mutation identification through sequence tagging. J. Proteome Res. 9, 1716–1726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Menon R., Omenn G. S. (2010) Proteomic characterization of novel alternative splice variant proteins in human epidermal growth factor receptor 2/neu-induced breast cancers. Cancer Res. 70, 3440–3449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Beausoleil S. A., Villén J., Gerber S. A., Rush J., Gygi S. P. (2006) A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat. Biotechnol. 24, 1285–1292 [DOI] [PubMed] [Google Scholar]