

Abstract
Ubiquitin (Ub) is a small and highly conserved protein that can covalently modify protein substrates. Ubiquitination is one of the major post-translational modifications that regulate a broad spectrum of cellular functions. The advancement of mass spectrometers as well as the development of new affinity purification tools has greatly expedited proteome-wide analysis of several post-translational modifications (e.g. phosphorylation, glycosylation, and acetylation). In contrast, large-scale profiling of lysine ubiquitination remains a challenge. Most recently, new Ub affinity reagents such as Ub remnant antibody and tandem Ub binding domains have been developed, allowing for relatively large-scale detection of several hundreds of lysine ubiquitination events in human cells. Here we review different strategies for the identification of ubiquitination site and discuss several issues associated with data analysis. We suggest that careful interpretation and orthogonal confirmation of MS spectra is necessary to minimize false positive assignments by automatic searching algorithms.
Ub (Ub)1 is a small protein modifier that can be covalently attached to protein substrates to regulate their stability, localization, and/or activity. In a canonical reaction, ubiquitination is catalyzed by an enzymatic cascade composed of Ub activating enzyme (E1), Ub conjugate enzyme (E2), and Ub E3 ligase (1–4). Substrate specificity is primarily determined by the E3 ligase (5), although the E2 enzyme may also have its influence (6, 7). Ubiquitination reaction will generate an isopeptide bond between the carboxyl terminus of Ub and the ε-amino group of lysine in the substrate (8), and in rare cases, the N-terminal amine or Cys residue might also be modified (9, 10). Ubiquitination is reversible by deubiquitination enzymes (DUBs), which can cleave off the Ub moieties from the substrate (11, 12). In the human proteome, greater than 500 Ub E3 ligases and around 100 DUBs are present to modify potentially thousands of substrates (13–15). In addition, cell evolves with proteins with Ub binding domains (UBD) for Ub binding, further expanding the functional diversity of the Ub proteasome system signaling network (16, 17).
Substrates can be modified by different forms of Ub. They can be mono-ubiquitinated at one or multiple lysine residues or poly-ubiquitinated by poly-Ub chains. Ub contains seven internal lysine residues and all can serve as conjugation sites to build up poly-Ub chains that may impart different functions (18, 19). Poly-Ub can regulate either proteolysis (such as K11 and K48 linkage) or nonproteolysis (K63 linkage) of the substrates (20–22). Other poly-Ub linkages may also regulate proteasome-mediated protein degradation (18, 23–25). Furthermore, the existence of the N-terminal head-to-tail linear poly-Ub chain has been proposed, which may directly activate protein kinases in immune response (26, 27). However, direct chemical evidence by mass spectrometry that supports such poly-Ub chain is still lacking. In addition to these homogenous poly-Ub chains, heterogeneous poly-Ub chains that form fork structures with mixed linkages have also been detected in vivo (28–30). It is possible that some of these uncommon structures may be resistant to proteasome mediated degradation (31, 32).
Pinpointing the lysine residue(s) for Ub conjugation is essential for the molecular understanding of ubiquitination. The identification of ubiquitination sites provides the ultimate proof that the putative substrate is indeed ubiquitinated. Large-scale analysis of ubiquitination sites has been a daunting task with only a few successful attempts in the past (28–30, 33–38). As the abundance of ubiquitinated species is generally too low to be directly detected by mass spectrometry, strategies that can enrich the substrates are required. A conventional way for ubiquitination enrichment is to utilize epitope-tagged Ub. Recently, several affinity reagents were developed for ubiquitinated substrate and peptide enrichment, including Ub-chain specific antibodies, the ubiquitination remnant antibody, and tandem Ub binding domains (29, 30, 39–41).
The ubiquitinated species, once enriched and purified, will be identified by mass spectrometry (MS). Application of fast speed, high resolution and high mass accuracy mass spectrometers for “shotgun” proteomics has greatly increased peptide identification and potentially eliminated false-positive assignments (42–45). Powered by technology advancement, we and others have identified several hundreds of lysine ubiquitination sites in human cells. Concurrently, the large amount of spectra collected at high precursor mass accuracy now pose new challenges for the accurate assignment of ubiquitination sites using automatic search algorithms.
Here we review different affinity purification strategies for ubiquitination enrichment, and discuss technical issues that one may encounter during data analysis. We present examples of mis-assignment by automatic search and offer practical solutions to these problems. We suggest that careful manual inspection of individual MS/MS spectrum is still an important final step in eliminating false positive identifications from automatic search.
Different Strategies to Isolate Ubiquitinated Proteins
Because of the relatively low cellular abundance, ubiquitinated substrates have to be enriched by Ub affinity reagents for large-scale identification of ubiquitination sites. Following enrichment, the isolated samples will then be separated to reduce the complexity before MS analysis. Currently there are at least three different strategies for large-scale ubiquitination profiling (Figs. 1A–1C): (A) Ub epitope-tag expressing systems, (B) Ub binding domains, especially tandem UBDs that offer high poly-Ub binding affinity, and (C) Ub antibodies (either poly-Ub linkage-specificity antibody or ubiquitination remnant-recognizing antibody).
Fig. 1.

A schematic presentation of three different strategies to isolate ubiquitinated substrates. A, The affinity purification strategy using Ub tag that allows purification under proteome denaturing condition. B, Tandem Ub binding domain for endogenous substrate enrichment. C, Poly-Ub chain specific antibody or ubiquitination signature remnant antibody. D, Three different contaminants of either protein precipitation during incubation, nonspecific binders to the matrix, or specific binding proteins to the affinity bait and poly-Ub conjugates exist during purification process.
The Ub epitope-tagging strategy was initially applied for profiling yeast ubiquitome. Using an engineered yeast strain where all Ub genes are null and the expression level of exogenous Ub is controlled, Peng et al. identified an impressive number of ubiquitination sites, totaling 110, using a LCQ mass spectrometer (28). A similar approach was adapted in the mammalian system, in which a tandem His-biotin tagged Ub was stably expressed in HeLa cells for efficient substrate isolation. In this experiment, ∼50 ubiquitination sites were identified with a linear trap quadrupole (LTQ)-Orbitrap mass spectrometer (38). Most recently, Danielsen et al. reported the identification of ∼750 lysine ubiquitination sites from two human cell lines that stably express HA-Ub, which stands by far the largest collection of ubiquitination sites (46). Remarkably, all the MS/MS spectra from this study were collected by LTQ Orbitrap Velos with higher-energy collisional dissociation method where both precursor and fragment peptides were analyzed in the Orbitrap with high mass accuracy, and with the inherent large mass-range of fragment peptides that together improve the overall spectra quality.
One of the major advantages of Ub epitope-tagging strategy is that substrates can be purified under protein denaturing conditions, thereby to a large extent eliminating nonsubstrate proteins. However, tagging Ub in animal tissues or pathological specimens is difficult with limited success (47–49). In addition, it remains uncertain to which extent Ub overexpression may interfere with normal cellular functions. Although it may not be a major concern for the engineered yeast strain, in which the endogenous Ub protein is eliminated and the level of exogenous Ub can be properly controlled, expression of the tagged Ub in mammalian systems entails the possibility of its influence on the physiology of the cell by changing the kinetics of the ubiquitination enzymatic reaction, interfering with the cell cycle, and/or by altering the subcellular localization of exogenous Ub (e.g. sequester the exogenous Ub in the nucleus). It is much less optimal to investigate linkage specific substrates by expression of a tagged lysine mutant Ub. Competition between the endogenous Ub with the exogenous mutant for conjugation (mixed poly-Ub chains) will render the data difficult to interpret. The presence of four Ub loci in the mammalian genome makes it a daunting task to analyze the substrates in a clear Ub gene knockout background. Recently, however, a strategy using tetracycline-inducible RNAi that eliminates endogenous Ub and expression of an exogenous K63R mutant Ub was developed that may partially circumvent this problem (50). This method holds the potential to profile linkage specific ubiquitination substrates.
UBDs can be used to isolate endogenous poly-Ub proteins. UBDs are small structural entities that have affinity to Ub. Over 20 different UBDs have been discovered (16, 17, 51, 52). The existence of a variety of UBDs implies their functional diversities in the Ub proteasome system signaling network. Although all UBDs bind Ub, the binding affinity varies greatly with the Kd value in the range of several hundred millimoles to a few micromoles (16). However, the inherent low affinity of a natural UBD is generally not sufficient for large-scale purification of substrates.
The development of engineered tandem UBD with antibody-like affinity makes it an attractive reagent for substrate purification. We have used such quadruple-UBD (qUBD) to detect close to 300 lysine ubiquitination sites from human cells that were not treated with proteasome inhibitors using a high speed LTQ Velos Orbitrap mass spectrometry. A complication for this approach is that purification cannot be performed under denaturing conditions; thus, “contaminant proteins” (especially true for UBD proteins that bind poly-Ub chains) will be inevitably copurified and sequenced by MS, which may mask the identification of low abundant substrates. Moreover, UBDs are more favorable for poly-Ub chains than mono-Ub and this strategy could be biased toward substrates that are polyubiquitininated. One intriguing question about UBDs is whether they have preference toward specific type of poly-Ub linkage or the length of the chain, which remains to be fully investigated. A better understanding of UBD and Ub interaction may facilitate the design of poly-Ub linkage-specific reagents with higher affinity and selectivity.
High-affinity Ub antibodies are powerful tools for ubiquitination profiling. Previous attempts to generate high affinity Ub antibody from mice and rabbits were not successful. Recently, several linkage-specific monoclonal antibodies selected from phage display libraries were developed and have been shown to be useful for immunoprecipitation and Western blot (39, 40). Whether or not these antibodies can efficiently profile linkage-specific substrates remains unclear, but it is expected that future experiments will reveal their utility soon.
Although tandem UBDs and presumably Ub antibodies are effective tools for substrate purification, ideally, comprehensive ubiquitination site mapping requires extra analytical work to separate ubiquitinated peptides from the unmodified ones. An excellent reagent for this purpose has been recently introduced by Xu et al. The Gly-Gly remnant antibody was generated against the Gly-Gly signature peptides (29). Because this antibody recognizes specifically the modified peptides, it is more efficient for ubiquitination site profiling than any other method that isolates the entire substrates. The authors have further demonstrated the power of this reagent by profiling hundreds of ubiquitination sites in human cells. It is noteworthy that as Gly-Gly remnant antibody does not distinguish ubiquitination from certain Ub-like modifications such as ISG15 or NEDD8, as a result, it may be necessary to couple with other purification method in order to obtain a homogenous population of ubiquitinated species for peptide IP.
Precautions to be Taken in Affinity Purifications
One of the major challenges for large-scale substrate purification is to overcome the interference from contaminant proteins. There are mainly three types of protein contaminants for Ub affinity purification (Fig. 1D): (1) nonspecific binders to the solid matrix (e.g. agarose beads) for affinity reagents, (2) precipitated proteins that are accumulated during incubation, and (3) specific binders to poly-ubiquitinated proteins and/or Ub itself. Although it is very difficult to completely eliminate them all, the amount of contaminants can be significantly reduced by (1) carefully removing the insoluble precipitations following high-speed centrifugation, (2) shortening the affinity incubation time, (3) increasing protein solubility with stronger detergent such as 0.1% SDS, and (4) washing the beads with higher concentration of salt.
The second challenge during purification is to preserve the ubiquitination by restricting the DUB activity. There are ample DUBs in the cells that readily remove Ub from the conjugates. The use of DUB inhibitors of both Cys alkylation (such as iodoacetamide, choloroacetamide, or N-ethylmaleimide) and zinc chelating chemical (1, 10-σ-phenanthroline) can alleviate the loss of poly-Ub and preserve the intact modified substrates (53). In addition, the use of freshly prepared cell lysate might be helpful to preserve ubiquitinated species.
It has been shown that excessive Cys alkylation by iodoacetamide (IAA; 55 mm) can lead to chemically induced artifact with the same atomic composition of Gly-Gly remnant (C4H6N2O2; 114.043 Da) that mimics lysine ubiquitination (54). Use of an alternative alkylation chemical such as chloroacetamide might avoid the occurrence of such artifact. Further study suggests that the pseudo ubiquitination signature can be significantly reduced or eliminated at room temperature or at low dosage of IAA (such as at 1 mm) (18). When use IAA as the alkylation reagent for DUB inactivation, it is critical to monitor the conditions during sample preparation to avoid excessive alkylation and/or high temperature that induce ubiquitination artifacts.
Potential Caveats of Ubiquitination Site Assignments and False Discovery Rate Estimation
Ubiquitination can be identified by mass spectrometry with the detection of a mass shift of 114.043 Da—the ubiquitination signature that is derived from the di-glycine remnant of Ub following trypsin cleavage. Ubiquitination peptides (as well as sites) are identified by searching the protein sequence database to match a tryptic peptide sequence with the addition of the ubiquitination signature mass on the particular amino acid, typically on Lys residues. Such identifications are then evaluated with corresponding scores derived from various statistical models. In some cases, a scoring threshold is setup with an empirical filter followed by manual verifications (55–58). A more widely practiced strategy is the “target-decoy” searching in which the decoy hits are considered incorrect to filter the unreliable matches (59, 60). The target-decoy strategy is useful in estimating false discovery rate (FDR), which allows the application of identical criteria over various conditions. This has proven to be successful for peptide sequence identification, but becomes complicated when post-translational modifications (PTMs) are introduced. PTMs will certainly expand the search space; however, the impact of such expansion for different PTMs may not be identical. For instance, because of the discrete nature of amino acid mass, phosphorylation (monoisotopic mass of 79.966 Da) would not be confused with any amino acid and thus may have limited impact on the search space; such a scenario does not apply for the ubiquitination remnant (114.043 Da), which is di-glycine by nature and has indistinguishable mass with Asn. As a result, the search space is potentially expanded with the possibility of observing a 114.043 Da increase to a candidate peptide sequence assigned to either lysine ubiquitination or the extra amino acid Asn or Gly-Gly in the candidate sequence. The current implementation of FDR calculation has not taken such a situation into consideration. It is expected that the value of FDR allowing for modification is potentially inflated, and the extent of inflation is dependent on the nature of modification.
A considerable amount of false positive identification of ubiquitinated peptides was observed at 1% FDR. We searched 62 liquid chromatography-tandem MS (LC-MS/MS) runs to tentatively identify ubiquitinated peptides using two most popular search engines (Sequest and Mascot) (61, 62). Interestingly, at 1% FDR, Sequest (filtered with monotonic score of Xcorr) identified ∼500 unique ubiquitinated peptides, whereas Mascot (with ion score) identified 223 and is much more reliable. Manual verification, however, could only confidently assign 47% of the unique identifications (IDs) recovered by both algorithms combined at 1% FDR (data not shown).
It was also noticed that various algorithms may have different sensitivity and specificity for ubiquitination ID. For example, Xu, et al. (29) observed that the overlap of ubiquitinated peptide IDs from two different algorithms is less than 60%. A similar observation was made by us (Fig. 2). As expected, the common IDs made by the two algorithms tend to pass manual verification readily, but the unique ones contain more false positives, which is especially true for the assignments by Sequest (Xcorr). It is very risky to combine the search results for ubiquitination IDs by different algorithms, and the 1% FDR as defined for the search of unmodified peptides does not apply in the search of ubiquitinated peptides.
Fig. 2.
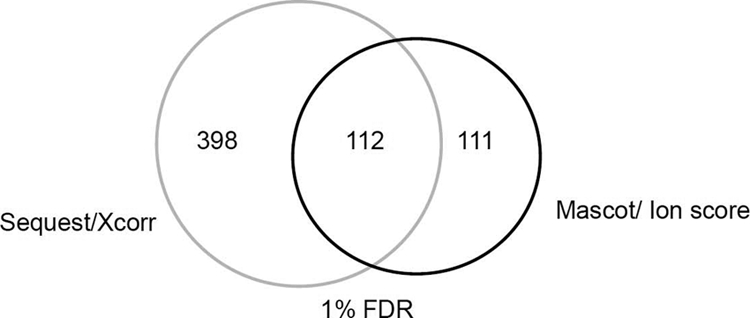
A comparison between Sequest and Mascot for ubiquitination identification at 1% FDR.
As single scores used for target-decoy search strategy might be less able to provide enough resolution to separate correct and incorrect assignments, use of composite scores that incorporate multiple factors (e.g. mass accuracy, peptide charge and length distribution, and enzymatic specificity) may increase the sensitivity and presumably the specificity of the peptide identifications (63, 64). In addition, it is also worth knowing that a handful of factors such as the type of mass spectrometer, precursor ion fragmentation pattern, or the target-decoy strategy used for search (separated, concatenated, or shuffled sequence database) may also impact the final results (65–67). A comprehensive evaluation of different parameters for ubiquitination search will certainly help for more accurate estimation of FDR.
When low resolution traps such as LTQ (where mass accuracy of precursor mass is generally less than 1 Da) is used, false-positive assignments may occur to peptides that contain internal lysine residues adjacent to certain amino acids (such as Leu, Asn, or Asp that has similar mass to that of Gly-Gly) (68). Most of such ambiguous assignments could be distinguished when the parent ions are analyzed by a high resolution mass spectrometer with a mass accuracy of several parts per million (ppm). Use of high mass accuracy mass spectrometers for proteome-wide PTM mapping (45) is always preferable. Measuring fragmentation ions with high mass accuracy using the Orbitrap as a detector might also help for ubiquitination site ID, although compromised sensitivity is generally expected.
It is noteworthy that even for high mass accuracy data of 1% FDR with great probability scores, false positive identifications occur by mislocalized modification sites or ambiguous fragmentation pattern. For example, as shown In Fig. 3A, as Asn has near-identical mass (114.043 Da) to that of the Gly-Gly remnant, miscleavage of Lys-Asn at either every carboxyl terminus of a protein or the Lys-Asn-Lys (Arg) motif can be mis-assigned to the adjacent lysine residue as ubiquitination. Moreover, when iodoacetamide is used for Cys alkylation, which requires the addition of 57.021 Da (the mass of carbamidomethyl) as a dynamic modification, two unmodified Cys with the exact ubiquitination signature mass may confuse the program for lysine ubiquitination assignment (Fig. 3B). Indeed, based on our observation, virtually all C-terminal ubiquitination candidates are improperly assigned. In addition, a significant proportion of ambiguous or weak assignments (Fig. 3C) happens in the 1% FDR data (>30%) from the Sequest search, although to a much less extent when Mascot is used. Fig. 3D shows a relatively challenging scenario that is difficult to judge by the MS/MS patterns alone. However, two other important parameters suggest it to be more likely a false-positive assignment. First, the full mass accuracy (ΔM) of the spectrum is >10 ppm, which is considered the outlier of the mass accuracy distribution (which follows the Gaussian distribution with σ <2ppm). Second, the peptide coverage for the particular protein is so too low (<3%) that only this “modified” form, but not any other peptide derived from this protein, is identified. In this regard, the peptide coverage is informative and should be used as an additional constraint for modification assignment.
Fig. 3.

Four sample MS/MS spectra misassigned for ubiquitination filtered at 1% FDR using Mascot. Note all the spectra have good ion score (25–120). A, A peptide spectrum with C-terminal Asn (N) results in false-positive ubiquitination assignment as C-terminal lysine. B, Two Cys (C) alkylation (+57.021 Da) leads to false positive ubiquitination identification. C, An ambiguous ubiquitination spectrum where major fragmentation peaks do not match. D, A false postive ubiquitination assignment rejected by precursor mass deviation and low peptide coverage.
Although it has been reported that trypsin is capable of cleaving the C-terminal ubiquitinated lysine of synthetic Ub peptides (69), we have not observed such spectra, consistent with a previous report using synthetic Gly-Gly peptides (68). We did detect a larger ubiquitination remnant of LRGG (383.228 Da) on a small proportion of poly-Ub peptides (Figs. 4A–4C). The LRGG-modified poly-Ub forms tend to be slightly more hydrophilic and eluted earlier than the typical Ub-linkage peptides (data not shown). Adding another ubiquitination parameter of LRGG-modified lysine (+383.228Da) for the database search might increase the chance for ubiquitination identification.
Fig. 4.

A–C, MS/MS spectra of K11, K48 and K63 ubiquitination tryptic peptides conjugated with larger remnant (LRGG).
Concluding Remarks
The availability of the methods described in this review has opened a door for profiling of ubiquitinated substrates. However, very few published studies focused on the quantitative aspects of ubiquitination analysis, such as ubiquitination substrates, Ub E3 ligase complex, or linkage quantification (70–73). The greatest challenge is to systematically identify E3 ligase or DUB substrates by quantitative proteomics so that one can pair the substrates with the enzymes. To achieve such a goal, the first step is to comprehensively identify ubiquitinated substrates under different conditions. It seems likely that most of the ubiquitinated species remain undetected, as suggested by the rather small overlap of ubiquitination data sets reported by three groups recently (29, 30, 46), although part of the reason can be attributed to different purification strategies or data quality. A genetic approach based on high-throughput fluorescence measurement called Global Protein Stability Profiling has been successfully developed to correlate protein level and stability to potential Ub E3 ligases (74). This, however, is an indirect method, and would preclude the detection of substrates whose ubiquitination does not directly control protein turnover. A direct method would require quantitative profiling of the global “substrate ubiquitination status,” which will reveal all classes of substrates.
With the advancement of Ub affinity purification and high resolution protein and peptide separation, paralleled with the improvement of mass spectrometers, the day for ubiquitination profiling at proteome-level is fast approaching. One of the major challenges to Ub proteomics is matching substrates to the corresponding Ub enzymes and studying the dynamics of ubiquitinated proteomes under different biological conditions such as different genetic backgrounds, or with specific drug treatment. As more ubiquitination data accumulates, it is equally important to improve the current statistical methods for more accurate estimation of FDR. Although careful verification of each single MS/MS spectra that tentatively identifies the ubiquitination site is the most reliable methods, it requires experience and will become a bottleneck for global profiling. Ub proteomics is now at the stage of “exploding” with numerous exciting opportunities and new challenges.
Acknowledgments
The authors thank Dr. Yi Wang for critical readings of the manuscript and helpful comments.
Footnotes
* This work is supported in part by NIH CA84199 and Welch Foundation (J. Q.).
1 The abbreviations used are:
- Ub
- ubiquitin
- DUB
- deubiquitination enzyme
- UBD
- Ub binding domains
- MS/MS
- tandem mass spectrometry.
REFERENCES
- 1. Hershko A., Ciechanover A. (1998) The ubiquitin system. Annu. Rev. Biochem. 67, 425–479 [DOI] [PubMed] [Google Scholar]
- 2. Pickart C. M. (2001) Mechanisms underlying ubiquitination. Annu. Rev. Biochem. 70, 503–533 [DOI] [PubMed] [Google Scholar]
- 3. Hochstrasser M. (2006) Lingering mysteries of ubiquitin-chain assembly. Cell 124, 27–34 [DOI] [PubMed] [Google Scholar]
- 4. Deshaies R. J., Joazeiro C. A. (2009) RING domain E3 ubiquitin ligases. Annu. Rev. Biochem. 78, 399–434 [DOI] [PubMed] [Google Scholar]
- 5. Pickart C. M., Fushman D. (2004) Polyubiquitin chains: polymeric protein signals. Curr. Opin. Chem. Biol. 8, 610–616 [DOI] [PubMed] [Google Scholar]
- 6. Nagy V., Dikic I. (2010) Ubiquitin ligase complexes: from substrate selectivity to conjugational specificity. Biol. Chem. 391, 163–169 [DOI] [PubMed] [Google Scholar]
- 7. Ye Y., Rape M. (2009) Building ubiquitin chains: E2 enzymes at work. Nat. Rev. Mol. Cell Biol. 10, 755–764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Weissman A. M. (2001) Themes and variations on ubiquitylation. Nat. Rev. Mol. Cell Biol. 2, 169–178 [DOI] [PubMed] [Google Scholar]
- 9. Ciechanover A., Ben-Saadon R. (2004) N-terminal ubiquitination: more protein substrates join in. Trends Cell Biol. 14, 103–106 [DOI] [PubMed] [Google Scholar]
- 10. Cadwell K., Coscoy L. (2005) Ubiquitination on nonlysine residues by a viral E3 ubiquitin ligase. Science 309, 127–130 [DOI] [PubMed] [Google Scholar]
- 11. Komander D., Clague M. J., Urbé S. (2009) Breaking the chains: structure and function of the deubiquitinases. Nat. Rev. Mol. Cell Biol. 10, 550–563 [DOI] [PubMed] [Google Scholar]
- 12. Reyes-Turcu F. E., Wilkinson K. D. (2009) Polyubiquitin binding and disassembly by deubiquitinating enzymes. Chem. Rev. 109, 1495–1508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Semple C. A. (2003) The comparative proteomics of ubiquitination in mouse. Genome Res. 13, 1389–1394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Reyes-Turcu F. E., Ventii K. H., Wilkinson K. D. (2009) Regulation and cellular roles of ubiquitin-specific deubiquitinating enzymes. Annu. Rev. Biochem. 78, 363–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nijman S. M., Luna-Vargas M. P., Velds A., Brummelkamp T. R., Dirac A. M., Sixma T. K., Bernards R. (2005) A genomic and functional inventory of deubiquitinating enzymes. Cell 123, 773–786 [DOI] [PubMed] [Google Scholar]
- 16. Hicke L., Schubert H. L., Hill C. P. (2005) Ubiquitin-binding domains. Nat. Rev. Mol. Cell Biol. 6, 610–621 [DOI] [PubMed] [Google Scholar]
- 17. Dikic I., Wakatsuki S., Walters K. J. (2009) Ubiquitin-binding domains - from structures to functions. Nat. Rev. Mol. Cell Biol. 10, 659–671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Xu P., Duong D. M., Seyfried N. T., Cheng D., Xie Y., Robert J., Rush J., Hochstrasser M., Finley D., Peng J. (2009) Quantitative proteomics reveals the function of unconventional ubiquitin chains in proteasomal degradation. Cell 137, 133–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kirkpatrick D. S., Hathaway N. A., Hanna J., Elsasser S., Rush J., Finley D., King R. W., Gygi S. P. (2006) Quantitative analysis of in vitro ubiquitinated cyclin B1 reveals complex chain topology. Nat. Cell Biol. 8, 700–710 [DOI] [PubMed] [Google Scholar]
- 20. Chau V., Tobias J. W., Bachmair A., Marriott D., Ecker D. J., Gonda D. K., Varshavsky A. (1989) A multiubiquitin chain is confined to specific lysine in a targeted short-lived protein. Science 243, 1576–1583 [DOI] [PubMed] [Google Scholar]
- 21. Deng L., Wang C., Spencer E., Yang L., Braun A., You J., Slaughter C., Pickart C., Chen Z. J. (2000) Activation of the IkappaB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell 103, 351–361 [DOI] [PubMed] [Google Scholar]
- 22. Jin L., Williamson A., Banerjee S., Philipp I., Rape M. (2008) Mechanism of ubiquitin-chain formation by the human anaphase-promoting complex. Cell 133, 653–665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chastagner P., Israël A., Brou C. (2006) Itch/AIP4 mediates Deltex degradation through the formation of K29-linked polyubiquitin chains. EMBO Rep. 7, 1147–1153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Johnson E. S., Ma P. C., Ota I. M., Varshavsky A. (1995) A proteolytic pathway that recognizes ubiquitin as a degradation signal. J. Biol. Chem. 270, 17442–17456 [DOI] [PubMed] [Google Scholar]
- 25. Koegl M., Hoppe T., Schlenker S., Ulrich H. D., Mayer T. U., Jentsch S. (1999) A novel ubiquitination factor, E4, is involved in multiubiquitin chain assembly. Cell 96, 635–644 [DOI] [PubMed] [Google Scholar]
- 26. Kirisako T., Kamei K., Murata S., Kato M., Fukumoto H., Kanie M., Sano S., Tokunaga F., Tanaka K., Iwai K. (2006) A ubiquitin ligase complex assembles linear polyubiquitin chains. EMBO J. 25, 4877–4887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tokunaga F., Sakata S., Saeki Y., Satomi Y., Kirisako T., Kamei K., Nakagawa T., Kato M., Murata S., Yamaoka S., Yamamoto M., Akira S., Takao T., Tanaka K., Iwai K. (2009) Involvement of linear polyubiquitylation of NEMO in NF-kappaB activation. Nat. Cell Biol. 11, 123–132 [DOI] [PubMed] [Google Scholar]
- 28. Peng J., Schwartz D., Elias J. E., Thoreen C. C., Cheng D., Marsischky G., Roelofs J., Finley D., Gygi S. P. (2003) A proteomics approach to understanding protein ubiquitination. Nat. Biotechnol. 21, 921–926 [DOI] [PubMed] [Google Scholar]
- 29. Xu G., Paige J. S., Jaffrey S. R. (2010) Global analysis of lysine ubiquitination by ubiquitin remnant immunoaffinity profiling. Nat. Biotechnol. 28, 868–873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shi Y., Chan D. W., Jung S. Y., Malovannaya A., Wang Y., Qin J. (2010) A dataset of human endogenous ubiquitination sites. Mol. Cell Proteomics [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kim H. T., Kim K. P., Lledias F., Kisselev A. F., Scaglione K. M., Skowyra D., Gygi S. P., Goldberg A. L. (2007) Certain pairs of ubiquitin-conjugating enzymes (E2s) and ubiquitin-protein ligases (E3s) synthesize nondegradable forked ubiquitin chains containing all possible isopeptide linkages. J. Biol. Chem. 282, 17375–17386 [DOI] [PubMed] [Google Scholar]
- 32. Kim H. T., Kim K. P., Uchiki T., Gygi S. P., Goldberg A. L. (2009) S5a promotes protein degradation by blocking synthesis of nondegradable forked ubiquitin chains. EMBO J. 28, 1867–1877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Vasilescu J., Smith J. C., Ethier M., Figeys D. (2005) Proteomic analysis of ubiquitinated proteins from human MCF-7 breast cancer cells by immunoaffinity purification and mass spectrometry. J. Proteome. Res. 4, 2192–2200 [DOI] [PubMed] [Google Scholar]
- 34. Matsumoto M., Hatakeyama S., Oyamada K., Oda Y., Nishimura T., Nakayama K. I. (2005) Large-scale analysis of the human ubiquitin-related proteome. Proteomics. 5, 4145–4151 [DOI] [PubMed] [Google Scholar]
- 35. Maor R., Jones A., Nuhse T. S., Studholme D. J., Peck S. C., Shirasu K. (2007) Multidimensional protein identification technology (MudPIT) analysis of ubiquitinated proteins in plants. Mol. Cell Proteomics. 6, 601–610 [DOI] [PubMed] [Google Scholar]
- 36. Tagwerker C., Flick K., Cui M., Guerrero C., Dou Y., Auer B., Baldi P., Huang L., Kaiser P. (2006) A tandem affinity tag for two-step purification under fully denaturing conditions: application in ubiquitin profiling and protein complex identification combined with in vivo cross-linking. Mol. Cell Proteomics. 5, 737–748 [DOI] [PubMed] [Google Scholar]
- 37. Mayor T., Lipford J. R., Graumann J., Smith G. T., Deshaies R. J. (2005) Analysis of polyubiquitin conjugates reveals that the Rpn10 substrate receptor contributes to the turnover of multiple proteasome targets. Mol. Cell Proteomics. 4, 741–751 [DOI] [PubMed] [Google Scholar]
- 38. Meierhofer D., Wang X., Huang L., Kaiser P. (2008) Quantitative analysis of global ubiquitination in HeLa cells by mass spectrometry. J. Proteome. Res. 7, 4566–4576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Matsumoto M. L., Wickliffe K. E., Dong K. C., Yu C., Bosanac I., Bustos D., Phu L., Kirkpatrick D. S., Hymowitz S. G., Rape M., Kelley R. F., Dixit V. M. (2010) K11-linked polyubiquitination in cell cycle control revealed by a K11 linkage-specific antibody. Mol. Cell 39, 477–484 [DOI] [PubMed] [Google Scholar]
- 40. Newton K., Matsumoto M. L., Wertz I. E., Kirkpatrick D. S., Lill J. R., Tan J., Dugger D., Gordon N., Sidhu S. S., Fellouse F. A., Komuves L., French D. M., Ferrando R. E., Lam C., Compaan D., Yu C., Bosanac I., Hymowitz S. G., Kelley R. F., Dixit V. M. (2008) Ubiquitin chain editing revealed by polyubiquitin linkage-specific antibodies. Cell 134, 668–678 [DOI] [PubMed] [Google Scholar]
- 41. Hjerpe R., Aillet F., Lopitz-Otsoa F., Lang V., England P., Rodriguez M. S. (2009) Efficient protection and isolation of ubiquitylated proteins using tandem ubiquitin-binding entities. EMBO Rep. 10, 1250–1258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Nesvizhskii A. I., Vitek O., Aebersold R. (2007) Analysis and validation of proteomic data generated by tandem mass spectrometry. Nat. Methods 4, 787–797 [DOI] [PubMed] [Google Scholar]
- 43. Makarov A., Denisov E., Kholomeev A., Balschun W., Lange O., Strupat K., Horning S. (2006) Performance evaluation of a hybrid linear ion trap/orbitrap mass spectrometer. Anal. Chem. 78, 2113–2120 [DOI] [PubMed] [Google Scholar]
- 44. Marshall A. G., Hendrickson C. L. (2008) High-resolution mass spectrometers. Annu. Rev. Anal. Chem. 1, 579–599 [DOI] [PubMed] [Google Scholar]
- 45. Zubarev R., Mann M. (2007) On the proper use of mass accuracy in proteomics. Mol. Cell Proteomics. 6, 377–381 [DOI] [PubMed] [Google Scholar]
- 46. Danielsen J. M., Sylvestersen K. B., Bekker-Jensen S., Szklarczyk D., Poulsen J. W., Horn H., Jensen L. J., Mailand N., Nielsen M. L. (2011) Mass spectrometric analysis of lysine ubiquitylation reveals promiscuity at site level. Mol. Cell Proteomics. 10, M110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Franco M., Seyfried N. T., Brand A. H., Peng J., Mayor U. (2010) A novel strategy to isolate ubiquitin conjugates reveals wide role of ubiquitination during neural development. Mol. Cell Proteomics [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tsirigotis M., et al. (2001)Analysis of ubiquitination in vivo using a transgenic mouse model. BioTechniques 31, 120–126, 128,, 130 [DOI] [PubMed] [Google Scholar]
- 49. Jeon H. B., Choi E. S., Yoon J. H., Hwang J. H., Chang J. W., Lee E. K., Choi H. W., Park Z. Y., Yoo Y. J. (2007) A proteomics approach to identify the ubiquitinated proteins in mouse heart. Biochem. Biophys. Res. Commun. 357, 731–736 [DOI] [PubMed] [Google Scholar]
- 50. Xu M., Skaug B., Zeng W., Chen Z. J. (2009) A ubiquitin replacement strategy in human cells reveals distinct mechanisms of IKK activation by TNFalpha and IL-1beta. Mol. Cell 36, 302–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Young P., Deveraux Q., Beal R. E., Pickart C. M., Rechsteiner M. (1998) Characterization of two polyubiquitin binding sites in the 26 S protease subunit 5a. J. Biol. Chem. 273, 5461–5467 [DOI] [PubMed] [Google Scholar]
- 52. Wilkinson C. R., Seeger M., Hartmann-Petersen R., Stone M., Wallace M., Semple C., Gordon C. (2001) Proteins containing the UBA domain are able to bind to multi-ubiquitin chains. Nat. Cell Biol. 3, 939–943 [DOI] [PubMed] [Google Scholar]
- 53. Cooper E. M., Cutcliffe C., Kristiansen T. Z., Pandey A., Pickart C. M., Cohen R. E. (2009) K63-specific deubiquitination by two JAMM/MPN+ complexes: BRISC-associated Brcc36 and proteasomal Poh1. EMBO J. 28, 621–631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Nielsen M. L., et al. (2008) Iodoacetamide-induced artifact mimics ubiquitination in mass spectrometry. Nat. Methods 5, 459–460 [DOI] [PubMed] [Google Scholar]
- 55. Chen Y., Kwon S. W., Kim S. C., Zhao Y. (2005) Integrated approach for manual evaluation of peptides identified by searching protein sequence databases with tandem mass spectra. J. Proteome Res. 4, 998–1005 [DOI] [PubMed] [Google Scholar]
- 56. Chen Y., Zhang J., Xing G., Zhao Y. (2009) Mascot-derived false positive peptide identifications revealed by manual analysis of tandem mass spectra. J. Proteome. Res. 8, 3141–3147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Vasilescu J., Smith J. C., Zweitzig D. R., Denis N. J., Haines D. S., Figeys D. (2008) Systematic determination of ion score cutoffs based on calculated false positive rates: application for identifying ubiquitinated proteins by tandem mass spectrometry. J. Mass Spectrom. 43, 296–304 [DOI] [PubMed] [Google Scholar]
- 58. Washburn M. P., Wolters D., Yates J. R., 3rd (2001) Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat. Biotechnol. 19, 242–247 [DOI] [PubMed] [Google Scholar]
- 59. Elias J. E., Gygi S. P. (2007) Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 4, 207–214 [DOI] [PubMed] [Google Scholar]
- 60. Moore R. E., Young M. K., Lee T. D. (2002) Qscore: an algorithm for evaluating SEQUEST database search results. J. Am. Soc. Mass Spectrom. 13, 378–386 [DOI] [PubMed] [Google Scholar]
- 61. Perkins D. N., Pappin D. J., Creasy D. M., Cottrell J. S. (1999) Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20, 3551–3567 [DOI] [PubMed] [Google Scholar]
- 62. Eng J. K., et al. (1994) An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 5, 957–989 [DOI] [PubMed] [Google Scholar]
- 63. Elias J. E., Gygi S. P. (2010) Target-decoy search strategy for mass spectrometry-based proteomics. Methods Mol. Biol. 604, 55–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Käll L., Canterbury J. D., Weston J., Noble W. S., MacCoss M. J. (2007) Semi-supervised learning for peptide identification from shotgun proteomics datasets. Nat. Methods 4, 923–925 [DOI] [PubMed] [Google Scholar]
- 65. Jiang X., Jiang X., Han G., Ye M., Zou H. (2007) Optimization of filtering criterion for SEQUEST database searching to improve proteome coverage in shotgun proteomics. BMC. Bioinformatics 8, 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Higdon R., Hogan J. M., van B. G., Kolker E. (2005) Randomized sequence databases for tandem mass spectrometry peptide and protein identification. OMICS. 9, 364–379 [DOI] [PubMed] [Google Scholar]
- 67. Käll L., Storey J. D., MacCoss M. J., Noble W. S. (2008) Assigning significance to peptides identified by tandem mass spectrometry using decoy databases. J. Proteome. Res. 7, 29–34 [DOI] [PubMed] [Google Scholar]
- 68. Seyfried N. T., Xu P., Duong D. M., Cheng D., Hanfelt J., Peng J. (2008) Systematic approach for validating the ubiquitinated proteome. Anal. Chem. 80, 4161–4169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Denis N. J., Vasilescu J., Lambert J. P., Smith J. C., Figeys D. (2007) Tryptic digestion of ubiquitin standards reveals an improved strategy for identifying ubiquitinated proteins by mass spectrometry. Proteomics. 7, 868–874 [DOI] [PubMed] [Google Scholar]
- 70. Hitchcock A. L. (2003) A subset of membrane-associated proteins is ubiquitinated in response to mutations in the endoplasmic reticulum degradation machinery. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Bennett E. J., Shaler T. A., Woodman B., Ryu K. Y., Zaitseva T. S., Becker C. H., Bates G. P., Schulman H., Kopito R. R. (2007) Global changes to the ubiquitin system in Huntington's disease. Nature 448, 704–708 [DOI] [PubMed] [Google Scholar]
- 72. Phu L., Izrael-Tomasevic A., Matsumoto M. L., Bustos D. J., Dynek J. N., Fedorova A. V., Bakalarski C. E., Arnott D., Deshayes K., Dixit V. M., Kelley R. F., Vucic D., Kirkpatrick D. S. (2010) Improved quantitative mass spectrometry methods for characterizing complex ubiquitin signals. Mol. Cell Proteomics [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Lee J. E., Sweredoski M. J., Graham R. L., Kolawa N. J., Smith G. T., Hess S., Deshaies R. J. (2010) The steady-state repertoire of human SCF Ubiquitin ligase complexes does not require ongoing Nedd8 conjugation. Mol. Cell Proteomics [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Yen H. C., Elledge S. J. (2008) Identification of SCF ubiquitin ligase substrates by global protein stability profiling. Science 322, 923–929 [DOI] [PubMed] [Google Scholar]