

Abstract
Common germline genetic variation in the population is associated with susceptibility to epithelial ovarian cancer. Microcell-mediated chromosome transfer and expression microarray analysis identified nine genes associated with functional suppression of tumorogenicity in ovarian cancer cell lines; AIFM2, AKTIP, AXIN2, CASP5, FILIP1L, RBBP8, RGC32, RUVBL1 and STAG3. Sixty-three tagging single nucleotide polymorphisms (tSNPs) in these genes were genotyped in 1,799 invasive ovarian cancer cases and 3,045 controls to look for associations with disease risk. Two SNPs in RUVBL1, rs13063604 and rs7650365, were associated with increased risk of serous ovarian cancer [HetOR = 1.42 (1.15–1.74) and the HomOR = 1.63 (1.10–1.42), p-trend = 0.0002] and [HetOR = 0.97 (0.80–1.17), HomOR = 0.74 (0.58–0.93), p-trend = 0.009], respectively. We genotyped rs13063604 and rs7650365 in an additional 4,590 cases and 6,031 controls from ten sites from the United States, Europe and Australia; however, neither SNP was significant in Stage 2. We also evaluated the potential role of tSNPs in these nine genes in ovarian cancer development by testing for allele-specific loss of heterozygosity (LOH) in 286 primary ovarian tumours. We found frequent LOH for tSNPs in AXIN2, AKTIP and RGC32 (64, 46 and 34%, respectively) and one SNP, rs1637001, in STAG3 showed significant allele-specific LOH with loss of the common allele in 94% of informative tumours (p = 0.015). Array comparative genomic hybridisation indicated that this nonrandom allelic imbalance was due to amplification of the rare allele. In conclusion, we show evidence for the involvement of a common allele of STAG3 in the development of epithelial ovarian cancer.
Keywords: risk of ovarian cancer, polymorphism, association studies
BRCA1 and BRCA2 mutations are responsible for the majority of families containing more than two cases of ovarian cancer.1-3 However, BRCA1 and BRCA2 mutation carriers are rare in the population, and these genes are thought to explain <40% of familial ovarian cancer risk.4 It has been proposed that low-penetrance susceptibility alleles that are more common in the population contribute to a significant fraction of the excess familial risk.5
The most widely used study design for identifying common low-penetrance susceptibility alleles for disease is the genetic association study, in which the frequency of single nucleotide polymorphisms (SNPs) is compared between individuals with the disease and unaffected controls. Studies have used either a candidate gene approach, in which SNPs in genes hypothesised to have a functional role in disease development are analysed for their disease association, or a genome wide association study (GWAS) design, which is an empirical approach that evaluates hundreds of thousands of SNPs distributed throughout the genome without any a priori functional role in the disease being studied.
During the last 3 years, there have been numerous reports describing common SNPs conferring susceptibility to several common diseases, including several cancers (reviewed in Refs. 6,7). Most published genetic association studies for ovarian cancer have used a candidate gene approach with genes selected from pathways including steroid hormone metabolism, DNA repair and cell cycle control, as well as known oncogenes and tumour suppressor genes.8-13 Many of the studies reporting statistically significant associations were performed using small sample sizes. Recently, a multicentre international consortium [Ovarian Cancer Association Consortium (OCAC)] has enabled replication analysis of many of these initial findings in samples sizes of up to 9,000 ovarian cancer cases and 11,500 controls. These studies have shown that the majority of genetic associations so far reported are likely to be either weak effects or false-positive associations.14,15
One possible explanation for the failure of candidate gene studies to identify true genetic associations could be that the strategies used for candidate gene selection are inadequate. Often, gene selection is based on predicted rather than a known role for genes in ovarian cancer development; selecting genes for which there is experimentally demonstrable evidence of functional involvement in ovarian cancer may prove a more successful strategy for gene selection. For example, a recently published study in which an in vitro model of ovarian cancer suppression was used to identify genes that might be associated with ovarian cancer prognosis, identified common genetic variants in a gene (RBBP8) that showed evidence of association with ovarian cancer survival.16
Few genetic association studies have provided a functional rationale for the susceptibility variants that have been identified. There are several possible reasons for this. One reason is that confirmed susceptibility SNPs are rarely nonsynonymous coding variants within genes. The associated SNP may not be the disease-causing variant but instead is probably in linkage disequilibrium with the true causal variant. A substantial proportion of disease associated SNPs are not located within or near known genes suggesting that noncoding DNA variation may impart functional effects; however, our understanding of the biological function of noncoding DNA is rudimentary, making functional analysis of these susceptibility alleles challenging.
In our study, we have evaluated the effects on ovarian cancer risk of common variants in nine candidate susceptibility genes (AIFM2, AKTIP, AXIN2, CASP5, FILIP1L, RBBP8, RGC32, RUVBL1 and STAG3) identified after performing microcell-mediated chromosome transfer (MMCT) of chromosome 18 into ovarian cancer cell lines16,17 as described below. These genes were first genotyped in 1,799 invasive ovarian cancer cases and 3,045 unaffected controls from three studies; significant associations were genotyped in an additional 4,590 cases and 6,031 controls from ten studies that are part of the OCAC, resulting in a total of 6,389 invasive ovarian cancer cases and 9,076 controls from 13 studies. We also tested the functional effects of SNPs in these candidate genes in primary ovarian tumours by evaluating allele-specific loss of heterozygosity (LOH) for nondisease associated alleles and the frequency of somatic alterations at putative susceptibility loci using array comparative genomic hybridisation (aCGH) analysis.
Material and Methods
Identifying functional candidate genes using MMCT
MMCT of normal human Chromosome 18 was achieved by polyethylene glycol fusion of the epithelial ovarian cancer cell lines TOV112D and TOV21G (American Tissue Culture Collection, LGC Standards, Middlesex, United Kingdom)18 and mouse (A9): human monochromosome hybrid donor cell lines carrying a selectable fusion gene marker, hygromycin phosphotransferase.17 In vitro phenotypic analysis was performed by assaying anchorage independent growth in soft agar and invasion through matrigel as described previously.17 For MMCT hybrids displaying significant neoplastic suppression, a combination of cytogenetic analysis, DNA microarray analysis and microsatellite genotyping confirmed the uptake of a complete or partial human chromosome 18 in MMCT hybrids. Expression microarray analysis was performed on the parental cell lines and four chromosome 18 MMCT hybrids, two generated from each of the parental cell lines as described previously.16 All samples were performed in triplicate. The microarray (Applied Biosystems version 2) contained 32,878 probes for the interrogation of 29,098 genes. An analysis of variance test was used to generate p values for statistical differences between groups. The p values were adjusted for multiple comparisons.19
Candidate gene selection was based on genes that showed significant differential expression between hybrid and parental cell lines.16 Lists of genes that were up or down regulated in hybrids from TOV21G, TOV112D, or both cancer cell lines were generated. The top 30 ranked genes in each list, based on p value and expression fold change, were compiled into a single master list. The functions of these genes were obtained from Gene Cards (http://www.genecards.org) and NCBI Entrez Gene (http://www.ncbi.nlm.nih.gov/sites/entrez). Tagged SNPs for each gene were identified from HapMap data release 22/phase II, April 2007, including putative regulatory regions up and down stream of each gene (within 5kb). Common SNPs (minor allele frequency ≥ 0.05) from each gene with a minimum correlation coefficient (r2) of 0.8 were selected and tagged with Haploview and coworkers20 and Tagger21 using the aggressive tagging option. Candidate genes were selected if the function was known or predicted and had a plausible role in cancer; if there was at least a common SNP (MAF ≥ 0.05) for every 2 kb of the gene; and if the number of tSNPs for a gene was between 3 and 20 tSNPs (to be sufficiently tagged and fit into an iplex multiplex). Nine genes AIFM2, AKTIP, AXIN2, CASP5, FILIP1L, RBBP8, RGC32, RUVBL1 and STAG3 were selected for genotyping from which there were 68 tagging SNPs (tSNPs).
Study individuals
In the first stage of our study, we genotyped 1,799 invasive ovarian cancer cases and 3,045 unaffected controls from three populations; MALOVA, Denmark (446 cases; 1,221 controls) and two UK studies; SEARCH (847 cases; 1,229 controls) and UKOPS (506 cases; 595 controls). Of the 1,799 cases, 849 were serous histology; 279 endometrioid; 196 mucinous; 166 clear cell and 309 with mixed/other histology or undifferentiated.
In the second stage, two putative positive associations were followed up in ten additional case control studies as follows: (1) USC, USA (391 cases; 546 controls); (2) DOV, USA (530 cases; 716 controls); (3) HOP, USA (280 case; 603 controls); (4) GEO, USA (327 cases; 429 controls); (5) NCO, USA (622; 747 controls); (6) HAW, USA (70 cases; 158 controls) (7) POC, Poland (456 cases; 460 controls); (8) BAV, Germany (228 cases; 234 controls); (9) GER, Germany (218 cases; 416 controls) and (10) AUS, Australia (768 cases; 1,122 controls). An additional 553 cases and 467 controls from UKOPS and SEARCH called UKO (B) were also included in Stage 2. A total of 4,590 cases and 6,031 controls were genotyped in Stage 2. Only the nonHispanic White individuals from these studies were included in the analysis to avoid heterogeneity due to ethnicity. Details for several of these studies have been published previously15,22 and are summarised in Supporting Information Table 1. Local ethics committee approval was given for the collections and genotyping in all individuals.
SNP genotyping
Of the 68 tSNPs, three SNPs (AIFM2 rs2271695, AXIN2 rs4128941 and rs2240308) failed manufacture and could not be efficiently tagged by any other SNP. Therefore, 65 tSNPs were genotyped in Stage 1 using a combination of iPLEX Gold (Sequenom Inc., Hamburg, Germany) and TaqMan ABI 7900HT Sequence Detection System (Applied Biosystems, Cheshire, United Kingdom) as previously described.12 Two iPlex multiplex experiments were designed to analyse 50 SNPs, and the remaining 13 SNPs were analysed by Taqman (Table 1). TaqMan was used to genotype the two SNPs in the Stage 2 studies, apart from the AUS samples which were genotyped for rs7650365 by iplex. All genotyping included duplicate samples and no DNA template controls for quality control purposes as described previously.15 DNA plates with <90% call rates and studies with <95% call rates were excluded. Studies deviating from Hardy-Weinberg equilibrium (HWE) p < 0.05 in controls were excluded. This is reflected in the variable numbers of cases and/or controls that were successfully genotyped for each tSNP. Two SNPs (RGC32 rs3783197 and RUVBL1 rs13091198) failed on genotyping quality control criteria.
Table 1.
Candidate genes selected from differential gene expression between parental cancer cells lines and MMCT-18 hybrids
| Gene | Expression: parental vs hybrid lines |
Cytoband | Function | Gene size (bp) |
SNPs with MAF > 0.05 |
No. tSNPs |
tSNPs analysed |
SNPs captured r2 ≥ 0.8 |
SNPs captured r2 ≥ 0.8 (%) |
|---|---|---|---|---|---|---|---|---|---|
| AIFM2 | 3.4 fold increase in TOV112D and 1.8 fold increase in TOV21G hybrids |
10q22.1 | TP53-induced apoptosis; over expression induces apoptosis |
34,711 | 17 | 13 | 12 | 13/17 | 76 |
| AKTIP | 5.7 fold increase in TOV112D and 3 fold increase in TOV21G hybrids |
16q12.2 | Apoptosis; interacts with PKB/Akt; |
11,978 | 7 | 4 | 4 | 7/7 | 100 |
| AXIN2 | 5 fold increase in TOV112D hybrids |
17q23-q24 | Inhibitor of β-catenin in Wnt signalling pathway; LOH in breast and other cancers. |
33,084 | 14 | 12 | 10 | 12/14 | 86 |
| CASP5 | 7 fold increase in TOV21G hybrids |
11q22.2-q22.3 | Regulation of apoptosis. |
14,729 | 17 | 9 | 9 | 17/17 | 100 |
| FILIP1L | 5 fold increase in TOV112D hybrids |
3q12.1 | Down regulated in ovarian cancer. |
281,369 | 135 | 8 | 8 | 135/135 | 100 |
| RBBP8 | 7 fold increase in TOV112D hybrids |
18q11.2 | RB1 binding protein; transcriptional regulation of BRCA1; DNA repair; tumour suppressor |
93,155 | 39 | 4 | 4 | 39/39 | 100 |
| RGC32 | 3 fold decrease in TOV112D hybrids |
13q14.11 | Cell cycle progression regulation; induced by p53 in response to DNA damage. |
13,323 | 17 | 8 | 7 | 15/17 | 88 |
| RUVBL1 | 47 fold decrease in TOV112D and 2 fold decrease in TOV21G hybrids |
3q21 | Interacts with MYC; involved in cell growth |
42,857 | 29 | 7 | 6 | 22/29 | 76 |
| STAG3 | 9 fold increase in TOV21G hybrids |
7q22.1 | Component of cohesin complex; chromosome segregation |
43,764 | 28 | 3 | 3 | 28/28 | 100 |
| Total | 303 | 68 | 63 | 288/303 | 95 |
Captured SNPs refer to the number and proportion of criteria SNPs covered by the HapMap Data Release 22.
Abbreviations: MAF: minor allele frequencies; MMCT: microcell-mediated chromosome transfer; SNPs: single nucleotide polymorphisms.
Loss of heterozygosity analysis
DNA was extracted from 286 formalin-fixed paraffin-embedded (FFPE) tumour tissues from the Danish MALOVA study. Areas with >80% tumour cells were extracted using a Proteinase K DNA extraction method as previously described.23 Tumour DNA and matching germline DNA samples were genotyped for 50 tSNPs by iPlex. Additional quality control criteria were applied to genotypes in tumour tissues; FFPE samples that passed for <80% of the assays were excluded. SNPs with pass rates of <90% for the genomic DNA samples were excluded. After quality control, LOH data were available for 37 tSNPs.
The ratio of the allele peak heights between the tumour and the germline DNA for heterozygous individuals was used to determine LOH as described previously.23 L = (at2 × an1)/(at1 × an2), where at1,at2 and an1,an2 are the peak heights of the two alleles of the tumour and the germline DNA, respectively. A value of L < 0.6 and L > 1.67 was considered LOH.24
Genotyping of 95 of the tumour samples was repeated to confirm that the peak height ratios were reproducible. If a reaction showed LOH in one experiment and then there was a decrease in peak height for the same allele in the other experiment with peak height ratios close to the LOH limit values (<0.8 or >1.4), they were classed as concordant. If the concordance of LOH between the duplicate samples was <85%, the assay was excluded from the analysis. For duplicate samples that were concordant, the log average of the L value was used. If discordant both results were removed.
The calculated frequency of overall LOH for a specific gene was based on the combined analysis of multiple tSNPs within the gene. LOH was recorded if any informative SNP in a gene showed LOH, even if other informative tSNPs did not show LOH. Tumour DNA samples from 12 cases were also analysed by array CGH analysis as previously described.17
Statistical methods
Deviation from HWE was assessed in controls within study populations using standard χ2 test. Unconditional logistic regression was used to assess the association between each tSNP and risk of ovarian cancer for each study and pooled across studies (stratified by study) with the primary test of association being a test for trend (p-trend) as described previously.12 A log-additive co-dominant model was used, and odds ratios for the heterozygote and rare homozygote relative to the common homozygote were estimated by stratified logistic regression. The programme TagSNPs25 was used to model the relevant multi-marker haplotypes resulting from aggressive SNP tagging. Analysis to test for association with the variants was first performed on all invasive cases and then restricted to the serous histological subtype as this was the most frequent subtype. There were insufficient numbers of the other subtypes for analysis.
We also conducted analysis to determine if haplotype effects were present as described previously.12 Haplotype blocks were defined using the confidence interval option of Haploview and coworkers,20 with minor adjustments to include adjacent SNPs but maintaining the cumulative frequency of the common haplotypes to [mt 90%. AKTIP, RBBP8, RGC32, RUVBL1 and STAG3 had one haplotype block. The other genes (AIFM2, AXIN2, CASP5 and FILIP1L) had two haplotype blocks. Unconditional logistic regression was used to test the association between each haplotype relative to the most common haplotype.25-27 Haplotypes that occurred with a frequency of 2% or greater in the combined data were considered “common”, and those with <2% frequency were pooled as rare haplotypes. To assess the existence of allele-specific LOH, the Fishers exact two tailed test was used to determine if there was a significant deviation from random LOH.
Results
Evaluating ovarian cancer risk associated with common variants in nine candidate genes
We have previously shown that microcell-mediated transfer of normal human chromosome 18 in ovarian cancer cell lines induces functional suppression of the neoplastic phenotype in vitro and in vivo.17 Subsequently, gene expression microarray analysis was used to compare the transcriptome of parental cancer cell lines with Chromosome 18 cell line hybrids; this identified nine candidate genes (AIFM2, AKTIP, AXIN2, CASP5, FILIP1L, RBBP8, RGC32, RUVBL1 and STAG3) that were differentially expressed in chromosome 18 hybrids suggesting they are associated with neoplastic suppression in this model (Table 1).
We selected tagging SNPs (tSNPs) with minor allele frequencies (MAF) ≥ 0.05 in each of these genes and evaluated their association with ovarian cancer risk after genotyping up to 1,799 invasive epithelial ovarian cancers and 3,045 healthy controls from three different population-based ovarian cancer case control studies. Sixty-eight tSNPs were identified in these genes but five could not be genotyped because they failed manufacture or quality control, and therefore, data were available for 63 tSNPs. We found evidence of risk association for tSNPs in AXIN2, CASP5 and RUVBL1 when the data for all histological subtypes were combined. The rs11079571 in AXIN2 showed evidence of an increased ovarian cancer risk [heterozygous odds ratio (HetOR) with 95% confidence intervals (CI) 1.23 (1.00–1.51); homozygous OR (HomOR) = 1.73 (0.99–3.01), p-trend = 0.038]; the minor alleles of rs518604 in CASP5 and rs13063604 in RUVBL1 were also associated with an increased risk [HetOR = 1.39 (1.06–1.81), HomOR = 1.44(1.05–1.97), p-trend = 0.0124; and HetOR = 1.14 (0.97–1.34), HomOR = 1.39 [1.02–1.89], p-trend = 0.0192, respectively). We found no evidence of association for any tSNPs in the AIFM2, AKTIP, FILIP1L, RBBP8, RGC32 and STAG3 genes. These data are summarised in Table 2 and Supporting Information Tables 2 and 3.
Table 2.
Genotype specific risks from stage 1 (SEA, UKO and MAL)
| All subtypes |
Serous |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Gene | tSNP | Controls1 | Cases1 | Serous cases |
HetOR2 (95% CI) | HomOR2 (95% CI) | p-trend | HetOR2 (95% CI) | HomOR2 (95% CI) | p-trend |
| AXIN2 | rs11079571 | 1,206 | 839 | 326 | 1.23 (1.00–1.51) | 1.73 (0.99–3.01) | 0.038 | 1.22 (0.92–1.63) | 1.74 (0.84–3.63) | 0.11 |
| CASP5 | rs518604 | 1,195 | 438 | 270 | 1.39 (1.06–1.81) | 1.44 (1.05–1.97) | 0.012 | 1.36 (0.98–1.88) | 1.45 (0.99–2.11) | 0.031 |
| RBBP8 | rs4474794 | 2,895 | 1,764 | 829 | 0.94 (0.82–1.07) | 0.88 (0.72–1.06) | 0.21 | 0.83 (0.70–0.98) | 0.80 (0.63–1.03) | 0.032 |
| RUVBL1 | rs13063604 | 1,724 | 1,266 | 537 | 1.14 (0.97–1.34) | 1.39 (1.02–1.89) | 0.019 | 1.42 (1.15–1.74) | 1.63 (1.10–2.42) | 0.0002 |
| RUVBL1 | rs7650365 | 2,672 | 1,645 | 769 | 1.08 (0.93–1.26) | 0.86 (0.72–1.03) | 0.11 | 0.97 (0.80–1.17) | 0.74 (0.58–0.93) | 0.009 |
| STAG3 | rs1637001 | 2,967 | 1,784 | 843 | 0.86 (0.76–0.98) | 0.92 (0.73–1.16) | 0.07 | 0.84 (0.71–0.99) | 0.77 (0.56––1.05) | 0.018 |
Bold indicates significant p values <0.05.
The differences in the numbers of cases and controls between tSNPs reflect the exclusion of data from some studies that did not pass genotyping quality control criteria.
OR and CI compared to common homozygote.
Abbreviations: OR: odds ratio; CI: confidence interval.
When the analysis of these 63 SNPs was restricted to serous ovarian cancer cases, the most common histological subtype, additional ovarian cancer risk associations were identified (Table 2). These included rs13063604 and rs7650365 in RUVBL1 [HetOR = 1.42 (1.15–1.74) HomOR = 1.63 (1.10–1.42), p-trend = 0.0002; and HetOR = 0.97 (0.80–1.17), HomOR = 0.74 (0.58–0.93) p-trend = 0.009); rs518604 in CASP5 [HetOR = 1.36 (0.98–1.88), HomOR = 1.45 (0.99–2.11), p-trend = 0.031]; rs4474794 in RBBP8 [HetOR = 0.83 (0.70–0.98), HomOR = 0.80 (0.63–1.03), p-trend = 0.032]; and rs1637001 in STAG3 [HetOR = 0.84 (0.71–0.99), HomOR = 0.77 (0.56–1.05), p-trend = 0.018].
The two most significant of these associations (rs13063604 and rs7650365 in RUVBL1) were further investigated after genotyping up to 4,437 additional cases (of which 2,534 were serous) and 5,885 controls from the OCAC. The rs13063604 was genotyped in fewer samples as a second batch of Taqman assay failed manufacture. Neither tSNP was significant in the additional genotyping data alone, and rs7650365 was not significant in a combined analysis (Table 3). However, for rs13063604, the association remained significant in the combined analysis [HetOR = 1.13 (1.00–1.27), HomOR =1.22 (0.96–1.56), p-trend = 0.019; Supporting Information Figures 1 and 2]. These results appear to be driven by the Stage 1 data. In a combined analysis for all histological subtypes, the association for rs13063604 was also marginally significant [HetOR = 1.08 (0.98–1.19), HomOR =1.19 (0.98–1.45), p-trend = 0.033].
Table 3.
Genotype specific risks for Stage 2 and all samples combined for the two most significant serous RUVBL1 tSNPs
| Serous cases |
All subtypes |
Serous |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Gene | tSNP | Controls1 | Cases1 | HetOR2 (95% CI) | HomOR2 (95% CI) | p-trend | HetOR2 (95% CI) | HomOR2 (95% CI) | p-trend | |
| rs13063604 | POC | 427 | 432 | 203 | 0.99 (0.75,1.32) | 1.6 (0.88,2.91) | 0.38 | 1.01 (0.71,1.43) | 1.59 (0.77,3.29) | 0.46 |
| DOV | 706 | 526 | 301 | 0.95 (0.75,1.21) | 0.74 (0.45,1.22) | 0.34 | 0.96 (0.72,1.27) | 0.67 (0.36,1.25) | 0.35 | |
| HOP | 559 | 263 | 151 | 0.94 (0.69,1.28) | 1.44 (0.78,2.66) | 0.67 | 0.93 (0.63,1.37) | 1.45 (0.69,3.03) | 0.76 | |
| STA | 418 | 315 | 213 | 1.25 (0.92,1.70) | 1.21 (0.60,2.47) | 0.20 | 1.19 (0.84,1.68) | 1.29 (0.59,2.83) | 0.32 | |
| USC | 529 | 379 | 237 | 1.15 (0.86,1.52) | 0.97 (0.58,1.65) | 0.65 | 0.96 (0.69,1.33) | 0.9 (0.48,1.66) | 0.74 | |
| All | 2,639 | 1,915 | 1218 | 1.04 (0.92,1.18) | 1.09 (0.85,1.41) | 0.40 | 1 (0.86,1.16) | 1.05 (0.77,1.42) | 0.83 | |
| Summary | Stage 1 | 1,724 | 1,266 | 537 | 1.14 (0.97–1.34) | 1.39 (1.02–1.89) | 0.019 | 1.42 (1.15–1.74) | 1.63 (1.10–2.42) | 0.0002 |
| Stage 2 | 2,639 | 1,915 | 1,218 | 1.04 (0.92–1.18) | 1.09 (0.85–1.41) | 0.40 | 1.00 (0.86–1.16) | 1.05 (0.77–1.42) | 0.83 | |
| Combined | 4,363 | 3,181 | 1,755 | 1.08 (0.98–1.19) | 1.19 (0.98–1.45) | 0.033 | 1.13 (1.00–1.27) | 1.22 (0.96–1.56) | 0.019 | |
| rs7650365 | POC | 562 | 543 | 343 | 0.87 (0.66,1.15) | 0.8 (0.58,1.12) | 0.20 | 0.89 (0.65,1.21) | 0.8 (0.56,1.14) | 0.23 |
| DOV | 706 | 524 | 300 | 1 (0.76,1.30) | 1.05 (0.77,1.45) | 0.79 | 0.97 (0.70,1.33) | 0.93 (0.64,1.36) | 0.75 | |
| HOP | 579 | 266 | 151 | 0.69 (0.49,0.97) | 0.88 (0.59,1.31) | 0.43 | 0.65 (0.43,0.98) | 0.75 (0.46,1.24) | 0.21 | |
| STA | 405 | 313 | 208 | 0.86 (0.61,1.21) | 0.81 (0.53,1.24) | 0.33 | 0.83 (0.56,1.24) | 1.02 (0.64,1.62) | 0.97 | |
| USC | 533 | 382 | 237 | 0.97 (0.72,1.32) | 1.03 (0.71,1.49) | 0.96 | 1 (0.69,1.43) | 1.22 (0.79,1.87) | 0.43 | |
| AUS | 1,121 | 766 | 462 | 1 (0.80,1.25) | 1.23 (0.94,1.59) | 0.15 | 0.96 (0.74,1.25) | 1.27 (0.94,1.72) | 0.14 | |
| BAV | 229 | 215 | 124 | 1.03 (0.66,1.59) | 1.25 (0.74,2.12) | 0.46 | 0.89 (0.53,1.49) | 1.17 (0.64,2.14) | 0.72 | |
| GER | 410 | 215 | 112 | 1.38 (0.93,2.05) | 0.98 (0.60,1.59) | 0.98 | 1.19 (0.73,1.95) | 0.89 (0.49,1.64) | 0.86 | |
| HAW | 157 | 70 | 36 | 0.82 (0.42,1.59) | 0.64 (0.30,1.36) | 0.29 | 1.03 (0.43,2.47) | 0.82 (0.30,2.22) | 0.79 | |
| NCO | 725 | 600 | 349 | 1 (0.77,1.29) | 0.92 (0.68,1.24) | 0.63 | 0.9 (0.67,1.22) | 0.84 (0.59,1.19) | 0.34 | |
| UKO (B) | 458 | 543 | 212 | 1.1 (0.82,1.48) | 0.91 (0.64,1.29) | 0.68 | 1.22 (0.82,1.82) | 1.22 (0.77,1.92) | 0.43 | |
| All | 5,885 | 4,437 | 2534 | 0.97 (0.88,1.06) | 0.97 (0.87,1.09) | 0.62 | 0.94 (0.84,1.05) | 0.99 (0.87,1.13) | 0.86 | |
| Summary | Stage 1 | 2,672 | 1,645 | 769 | 1.08 (0.93–1.26) | 0.86 (0.72–1.03) | 0.11 | 0.97 (0.80–1.17) | 0.74 (0.58–0.93) | 0.009 |
| Stage 2 | 6,010 | 4,437 | 2,534 | 0.97 (0.88–1.06) | 0.97 (0.87–1.09) | 0.62 | 0.94 (0.84–1.05) | 0.99 (0.87–1.13) | 0.86 | |
| Combined | 8,682 | 6,129 | 3,303 | 1.02 (0.94–1.1) | 0.96 (0.87–1.05) | 0.40 | 0.94 (0.86–1.04) | 0.92 (0.82–1.03) | 0.142 | |
Bold indicates significant p values <0.05.
The p value heterogeneity between studies for Stage 2 for all subtypes was 0.482 and 0.547 and for serous only was 0.624 and 0.579 for rs13063604 and rs7650365, respectively. Not all studies could be genotyped for rs13063604 as a second batch of Taqman assay failed manufacture.
The difference in the numbers of cases and controls between tSNPs reflects the exclusion of data from some studies that did not pass genotyping quality control criteria.
OR and CI compared to common homozygote.
Abbreviations: OR: odds ratio; CI: confidence interval.
From the Stage 1 data, we also found evidence of association with ovarian cancer risk for four haplotypes (Supporting Information Table 4). Haplotypes h0011 and h1111 in AXIN2 Block 2 were associated with decreased and increased risk respectively [OR = 0.57 (0.34–0.95) p = 0.031; and OR = 1.21 (1.03–1.42) p = 0.023]. This is consistent with the presence or absence of the rare allele of rs11079571 in the second position of the block that was shown to have an increased risk (described above). Haplotypes h000 and h100 in Block 1 of CASP5 were associated with a decreased risk [OR = 0.72 (0.56–0.94) p = 0.015], and increased risk [OR = 1.13 (1.03–1.24) p = 0.012], respectively. These haplotypes contain the common and minor allele, respectively, of rs518604 in the first position, which was associated with an increased risk in the analysis described earlier. A global haplotype analysis showed that CASP5 block 1 and RUVBL1 were significant (p = 8.4 × 10−6 and p = 0.0016, respectively).
Evaluating a functional role for SNPs and candidate genes in primary ovarian tumours
We simultaneously evaluated whether or not the candidate genes and/or tSNPs described above were involved in the somatic genetic development of primary ovarian cancers by studying loss of heterozygosity (LOH) in 286 tumours for which germline genotype data were also available. We hypothesised that low-penetrance susceptibility genes behave like tumour suppressor genes according to Knudson’s hypothesis28 and undergo nonrandom loss of the wild-type (nonrisk) allele during tumour development.
Genotyping was performed concurrently in germline and matching tumour DNA for 50 tSNPs from the nine genes performed by iPlex; 37 tSNPs passed the quality control criteria we established for genotyping of FFPE tumour samples (Table 4; Supporting Information Table 5). We observed frequent LOH for tSNPs in the AXIN2, AKTIP and RGC32 (64, 46 and 34% LOH, respectively; Table 4) suggesting a role for these genes (or regions) in ovarian tumour development. We found evidence of allele-specific LOH for one SNP, rs1637001 in STAG3; in 16 tumours informative for LOH for this tSNP, the common A allele showed significant preferential loss over the G allele in 94% of cases (p = 0.015; Figure 1).
Table 4.
Frequency of LOH in tumours for each candidate gene
| Gene | Chromosomal location | No. tSNPs per gene | No. tumours heterozygous | No. tumours with LOH | Percentage of LOH |
|---|---|---|---|---|---|
| AIFM2 | 10q22.1 | 7 | 202 | 55 | 27 |
| AKTIP | 16q12.2 | 4 | 137 | 63 | 46 |
| AXIN2 | 17q23-q24 | 5 | 235 | 150 | 64 |
| CASP5 | 11q22.2-q22.3 | 5 | 214 | 70 | 33 |
| FILIP1L | 3q12.1 | 5 | 196 | 63 | 32 |
| RBBP8 | 18q11.2 | 3 | 148 | 34 | 23 |
| RGC32 | 13q14.11 | 3 | 185 | 63 | 34 |
| RUVBL1 | 3q21 | 3 | 167 | 51 | 31 |
| STAG3 | 7q22.1 | 2 | 162 | 30 | 19 |
A tumour was considered to be heterozygous if heterozygous at any tSNP within the gene and to have LOH if LOH in at least one of the tSNPs. Abbreviations: LOH: loss of heterozygosity; tSNPs: tagging single nucleotide polymorphisms.
Figure 1.
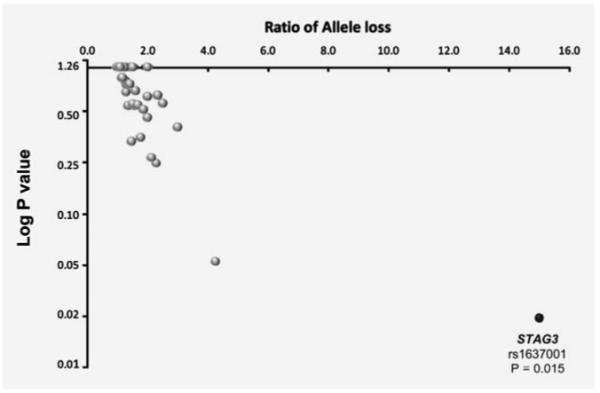
Allele-specific LOH for 37 tSNPs. For each of the 37 SNPs the log of the p-value of difference from random loss of alleles is plotted against the ratio of allele loss. The grey spots indicate SNPs showing random LOH; the black spot is rs1637001 in STAG3, which shows significant deviation from random LOH (p = 0.015).
We then used array comparative genomic hydridisation (aCGH) analysis of ovarian tumours to establish whether the allelic imbalances we observed for rs1637001 in STAG3 were the result of deletion of the common A allele or amplification of the rare G allele. The aCGH was performed for 12 ovarian tumours that showed allelic imbalance for rs1637001. Nine of these tumours showed copy number gain for STAG3, and three tumours were copy number neutral (Figure 2); none of the tumours showed deletion of STAG3. These data suggest that, for the majority of tumours, there was amplification of the rare G allele rather than deletion of the common A allele. For tumours that were copy number neutral for STAG3, it is possible that the somatic alterations that were revealed by LOH analysis were not detected by aCGH due to subsequent amplification of the remaining allele.
Figure 2.

The aCGH profiles of tumours showing copy number variation for rs1637001 in STAG3. (a) Array CGH profiles are presented in linear chromosome order, from the telomere of the p-arm to the telomere of the q-arm for each chromosome. Yellow spots indicate no change in copy number between normal and tumour DNA; green spots indicate an increase in DNA copy number in the tumour; red spots indicates loss of DNA copy number. (b) aCGH profiles for Chromosome 7 only in three tumours. The position of STAG3 is indicated by the blue line and arrow. (i) A tumour showing normal copy number at STAG3 and LOH with loss of allele A; thus the predicted genotype is GG. (ii) A tumour showing three copies of STAG3 and LOH with loss of allele A; thus the predicted genotype is AGG. (iii) A tumour with LOH and loss of allele G, with four copies of STAG3; thus, the predicted genotype is AAAG.
Discussion
We have used a functional approach to identify candidate genes from an in vitro model of ovarian cancer suppression, and evaluated whether common germline genetic variation for 63 SNPs in these genes is associated with low-penetrance susceptibility to the disease. Of the nine genes identified, we found statistically significant evidence of association with disease risk at the 5% level for three SNPs (in genes AXIN1, CASP5 and RUVBL1) when all ovarian cancer histological subtypes were analysed, and for an additional three SNPs (in genes RUVLB1, RBBP8 and STAG3) when the analysis was restricted to the serous subtype.
The strongest associations identified were for two SNPs in RUVLB1. However, these associations may still be false positives; more stringent significance levels are required to ensure that an identified association is a true positive. It has been proposed that the significance level of candidate gene studies should be p < 10−4.29 Therefore, we further investigated the associations for rs13063604 and rs7650365 in RUVBL1 as part of an international multicentre case control study by the OCAC. This analysis failed to replicate independently the association for either of these SNPs, although the combined analysis of all genotyping remained marginally significant for rs13063604. This variant is located in Intron 9 of the gene and does not appear to be functionally significant per se; however, it tags nine other SNPs in the gene, two of which are located in the 3′untranslated region. Using the bioinformatics tool Pupasuite (http://pupasuite.bioinfo.cipf.es/), none of these SNPs were predicted to be potentially functionally significant. There is evidence to suggest that RUVBL1 plays an important role in the development of ovarian cancer and other cancer types; RUVBL1 expression is elevated in ovarian, breast, colon, bladder and lung cancers [Oncomine(Compendia Bioscience, Ann Arbor, MI)].
The data for rs13063604 and rs7650365 in RUVBL1 were stronger in serous ovarian cancer cases than in all histological subtypes combined. If different germline genetic variants confer susceptibility to different disease subtypes, then disease heterogeneity may be one of the reasons for a lack of success in identifying genetic susceptibility alleles for ovarian cancer. There is increasing evidence from high-penetrance gene studies that the underlying genetic basis of ovarian cancer can contribute to disease heterogeneity. For example, BRCA1 and BRCA2 appear to confer susceptibility mainly to the development of the serous subtype.30 Some studies have also suggested that disease heterogeneity can be influenced by common alleles of low penetrance. Studies by the OCAC that are sufficiently large to enable stratification by subtype have identified genetic variants that may be associated with specific subtypes of the disease.22,31
From the data presented, we cannot rule out the possibility that risk associations exist for other germline genetic variants in these genes (AIFM2, AKTIP, AXIN2, CASP5, FILIP1L, RBBP8, RGC32, RUVBL1 and STAG3). The combined sample size from the three studies provides 98% power at the 5% significance level to detect a co-dominant allele with a frequency of 0.3 that confers a relative risk of 1.2, and 95% power to detect a dominant allele with a frequency of 0.1 that confers a relative risk of 1.3. Of the 303 common variants, 288 were tagged with r2 ≥ 0.8 and 290 with r2 ≥ 0.5; however, some unidentified common as yet SNPs may not have been tagged efficiently, and we did not evaluate rarer variants with MAFs <0.05. Risk associations for these genes may also exist for some of the rarer histological subtypes of ovarian cancer, but we did not have sufficient statistical power to detect such associations.
During the course of this study, a GWAS for ovarian cancer was completed and published.22 This GWAS study analysed a partly overlapping set of patients as used in this study, with cases from UKOPS and SEARCH as well as other UK studies. With access to the data from this GWAS (which analysed 1,819 cases and 2,343 controls for 507,094 genotyped SNPs and ~2 million imputed SNPs) we were able to reevaluate all tSNPs identified in these nine genes including the five SNPs that were not analysed; we found no evidence of association with disease risk in the GWAS data for all histologies combined (Supporting Information Table 6). Three SNPs were significant in the serous only analysis; two in CASP5 rs518604 and rs523104 and one in FILIP1L rs12494994. CASP5 rs518604 is the only SNP significant in both studies, and it is currently being investigated further in additional cases and controls.
One of the features of our study is that candidate genes were selected on the basis of having a possible functional role in an in vitro model of ovarian cancer suppression. To our knowledge, none of the nine candidate genes investigated have been previously assessed for their association with ovarian cancer risk. For some of the genes, we identified, there are previously reported evidence of a role in ovarian cancer development, in addition to that described for RUVBL1: For example, we have previously shown that germline variants in the BRCA1 interacting gene RBBP8 may be associated with survival after a diagnosis of ovarian cancer,16 and another study has shown downregulation of FILIP1L in primary ovarian tumours suggesting it may be an ovarian cancer tumour suppressor gene [Oncomine (Compendia Bioscience, Ann Arbor, MI)].
Although numerous common genetic variants have now been shown to be associated with the risks of several cancer types, rarely has any functional rationale been established for their risk association. Therefore, we investigated whether SNPs within all nine genes were somatically altered in primary ovarian cancers. The SNP rs1637001 in the STAG3 gene shows significant nonrandom allelic imbalance by LOH analysis in tumours. This was shown by array CGH to be the likely result of amplification of the minor G allele; amplification rather than deletion of an allele has previously been suggested as a potential mechanism to explain detected LOH at a SNP.32 The same SNP in this gene also showed evidence of association with disease risk in serous ovarian cancer cases, although this was not confirmed in the imputed data from the GWAS. The potential synergy between a putative risk association for a germline genetic variant and the preferential somatic amplification of one of the alleles during tumour development is intriguing, and further investigation is worthwhile.
Although there are several examples in the published literature of allele-specific imbalance of polymorphic markers in primary tumours (reviewed in Refs. 33-36), synergy between genetic risk alleles and somatic alterations has not been reported before. For example, in one study, analysis of the DAL1 gene in breast cancer found that 94% of tumours showing LOH retained the C allele of the C2166T SNP; in another study, 73% of lung tumours with LOH involving the P34 gene retained the G allele of the A106G SNP; and a third study reported 83% of breast tumours with loss of the pro allele of Arg72Pro in the P53 gene. What is unclear for the rs1637001 variant located in the 3′UTR of STAG3 is whether or not the preferential allelic amplification targets rs1637001 or STAG3 specifically. The amplified region detected by aCGH in tumours extended across several genes including STAG3; neither is it clear that the amplification is functionally relevant to ovarian cancer development. More detailed in vitro cell biology studies will be needed to address this. STAG3 is a component of the meiosis specific cohesion complex.37,38 It is not expressed in embryonic stem cells that form follicle like ovarian structures, and this is thought to contribute to the inability of those cells to progress through meiosis.39 The gene is activated in lymphoma cells after mutant p53 has been induced by irradiation.40 Other studies have implicated STAG3 mutations with chromosomal instability in colorectal cancer,41 and the gene is associated with chromosome segregation and downregulation in testicular cancer.42
In conclusion, we have used a functional model of ovarian cancer suppression to identify and test candidate susceptibility genes for ovarian cancer. We have identified functional evidence suggesting allele-specific imbalance for somatic genetic alterations in primary ovarian tumours for a SNP in the STAG3 gene. These studies highlight the importance of international consortia like the OCAC to validate putative genetic risk associations; but they also emphasise some of the limitations and challenges that face the scientific community in trying to elucidate the functional rationale underlying the numerous genetic associations that have been identified for multiple complex disease traits.
Acknowledgements
This work was undertaken at UCLH/UCL who received a proportion of funding from the Department of Health’s NIHR Biomedical Research Centres funding scheme. M.N. and L.Q. are funded by the MRC. Genotyping was funded by a Cancer Research UK project Grant (no. C8804/A7058). The UKOPS study is funded by the OAK Foundation and the Eve Appeal. SEARCH is funded through a programme grant from Cancer Research UK. The authors thank the family and friends of Kathryn Sladek Smith for their generous support of OCAC through their donations to the Ovarian Cancer Research Fund. We also thank the SEARCH team, the local general practices and nurses, the Eastern Cancer Registration and Information Centre, the EPIC-Norfolk investigators and Craig Luccarini and Don Conroy. The UKOPS team also thanks all members of the research team, including research nurses, research scientists, data entry personnel and consultant gynaecological oncologists for their help in establishing the UKOPS case control collection and particularly Andy Ryan Jeremy Ford, Nyaladzi Balogun and Martin Widschwendter. Debby Bass, Shari Hutchison, Carlynn Jackson, Jessica Kopsic and Mary Hartley for the HOPE project. The AOCS Management Group (D. Bowtell, G. Chenevix-Trench, A. deFazio, D. Gertig, A. Green, P. Webb) gratefully acknowledges the contribution of all the clinical and scientific collaborators (see http://www.aocstudy.org/). The AOCS and ACS Management Group (A. Green, P. Parsons, N. Hayward, P. Webb, D. Whiteman) thank all of the project staff and collaborating institutions. GCT is a Senior Principal Research Fellow of the NHMRC. Finally, the authors thank all the study participants who contributed to this research.
Grant sponsor: Cancer Research UK project; Grant number: C8804/A7058; Grant sponsor: National Cancer Institute, the Danish Cancer Society (MALOVA); Grant number: RO1 CA61107; Grant Sponsor: The Roswell Park Alliance and the National Cancer Institute (GEOCS study); Grant numbers: CA71766, CA16056; Grant sponsor: NIH (the DOVE study); Grant number: RO1 CA87538; Grant sponsor: NCI; Grant numbers: K07-CA80668, R01CA095023; Grant sponsor: Department of Defence (The HOPE project); Grant number: DAMD17-02-1-0669; Grant sponsor: The California Cancer Research Program; Grant numbers: 00-01389V-20170, 2110200; Grant sponsor: US Public Health Service and California Department of Health Services (Cancer Reporting Program; University of Southern California); Grant numbers: CA14089, CA17054, CA61132, CA63464, N01-PC-67010, R03-CA113148, 050-E8709; Grant sponsor: US Public Health Service; Grant number: R01-CA-58598; Grant sponsor: Department of Health and Human Services, National Institutes of Health (HAW study); Grant numbers: N01-CN67001, NO1-PC-035137.
Abbreviations:
- GWAS
genome wide association study
- HetOR
heterozygous odds ratio
- HomOR
homozygous odds ratio
- HWE
Hardy-Weinberg equilibrium
- LOH
loss of heterozygosity
- MMCT
microcell-mediated chromosome transfer
- SNP
single nucleotide polymorphisms
- tSNP
tagging single nucleotide polymorphisms
Footnotes
Additional Supporting Information may be found in the online version of this article.
References
- 1.Stratton JF, Gayther SA, Russell P, Dearden J, Gore M, Blake P, Easton D, Ponder BA. Contribution of BRCA1 mutations to ovarian cancer. N Engl J Med. 1997;336:1125–30. doi: 10.1056/NEJM199704173361602. [DOI] [PubMed] [Google Scholar]
- 2.Ramus SJ, Harrington PA, Pye C, DiCioccio RA, Cox MJ, Garlinghouse-Jones K, Oakley-Girvan I, Jacobs IJ, Hardy RM, Whittemore AS, Ponder BA, Piver MS, et al. Screening for the. BRCA1-ins6kb. Ex13 mutation: potential for misdiagnosis. Mutation in brief #964. Online. Hum Mutat. 2007;28:525–6. doi: 10.1002/humu.9493. [DOI] [PubMed] [Google Scholar]
- 3.Gayther SA, Russell P, Harrington P, Antoniou AC, Easton DF, Ponder BA. The contribution of germline BRCA1 and BRCA2 mutations to familial ovarian cancer: no evidence for other ovarian cancer-susceptibility genes. Am J Hum Genet. 1999;65:1021–9. doi: 10.1086/302583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Antoniou AC, Easton DF. Risk prediction models for familial breast cancer. Future Oncol. 2006;2:257–74. doi: 10.2217/14796694.2.2.257. [DOI] [PubMed] [Google Scholar]
- 5.Pharoah PD, Antoniou A, Bobrow M, Zimmern RL, Easton DF, Ponder BA. Polygenic susceptibility to breast cancer and implications for prevention. Nat Genet. 2002;31:33–36. doi: 10.1038/ng853. [DOI] [PubMed] [Google Scholar]
- 6.Easton DF, Eeles RA. Genome-wide association studies in cancer. Hum Mol Genet. 2008;17:R109–15. doi: 10.1093/hmg/ddn287. [DOI] [PubMed] [Google Scholar]
- 7.McCarthy MI, Hirschhorn JN. Genome-wide association studies: past, present and future. Hum Mol Genet. 2008;17:R100–1. doi: 10.1093/hmg/ddn298. [DOI] [PubMed] [Google Scholar]
- 8.Pearce CL, Hirschhorn JN, Wu AH, Burtt NP, Stram DO, Young S, Kolonel LN, Henderson BE, Altshuler D, Pike MC. Clarifying the PROGINS allele association in ovarian and breast cancer risk: a haplotype-based analysis. J Natl Cancer Inst. 2005;97:51–9. doi: 10.1093/jnci/dji007. [DOI] [PubMed] [Google Scholar]
- 9.Song H, Ramus SJ, Shadforth D, Quaye L, Kjaer SK, Dicioccio RA, Dunning AM, Hogdall E, Hogdall C, Whittemore AS, McGuire V, Lesueur F, et al. Common variants in RB1 gene and risk of invasive ovarian cancer. Cancer Res. 2006;66:10220–6. doi: 10.1158/0008-5472.CAN-06-2222. [DOI] [PubMed] [Google Scholar]
- 10.Song H, Ramus SJ, Kjaer SK, Hogdall E, Dicioccio RA, Whittemore AS, McGuire V, Hogdall C, Jacobs IJ, Easton DF, Ponder BA, Dunning AM, et al. Tagging single nucleotide polymorphisms in the BRIP1 gene and susceptibility to breast and ovarian cancer. PLoS ONE. 2007;2:e268. doi: 10.1371/journal.pone.0000268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Song H, Ramus SJ, Quaye L, DiCioccio RA, Tyrer J, Lomas E, Shadforth D, Hogdall E, Hogdall C, McGuire V, Whittemore AS, Easton DF, et al. Common variants in mismatch repair genes and risk of invasive ovarian cancer. Carcinogenesis. 2006;27:2235–42. doi: 10.1093/carcin/bgl089. [DOI] [PubMed] [Google Scholar]
- 12.Quaye L, Song H, Ramus SJ, Di Cioccio RA, McGuire V, Hogdall E, Hogdall C, Blaakr J, Easton DF, Ponder BA, Jacobs I, Kjaer SK, et al. Tagging single nucleotide polymorphisms in candidate oncogenes and susceptibility to ovarian cancer. Br J Cancer. 2009;100:993–1001. doi: 10.1038/sj.bjc.6604947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fasching PA, Gayther SA, Pearce CL, Schildkraut JM, Goode E, Thiel F, Chenevix-Trench G, Chang-Claude J, Wang-Gohrke S, Ramus SJ, Pharoah PDP, Berchuck A, et al. Role of genetic polymorphisms and ovarian cancer susceptibility. Mol Oncol. 2009;3:171–81. doi: 10.1016/j.molonc.2009.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gayther SA, Song H, Ramus SJ, Kjaer SK, Whittemore AS, Quaye L, Tyrer J, Shadforth D, Hogdall E, Hogdall C, Blaeker J, DiCioccio R, et al. Tagging single nucleotide polymorphisms in cell cycle control genes and susceptibility to invasive epithelial ovarian cancer. Cancer Res. 2007;67:3027–35. doi: 10.1158/0008-5472.CAN-06-3261. [DOI] [PubMed] [Google Scholar]
- 15.Ramus SJ, Vierkant RA, Johnatty SE, Pike MC, Van Den Berg DJ, Wu AH, Pearce CL, Menon U, Gentry-Maharaj A, Gayther SA, Dicioccio RA, McGuire V, et al. Consortium analysis of seven candidate snps for ovarian cancer. Int J Cancer. 2008;123:380–8. doi: 10.1002/ijc.23448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Quaye L, Dafou D, Ramus SJ, Song H, Gentry-Maharaj A, Notaridou M, Hogdall E, Kjaer SK, Christensen L, Hogdall C, Easton DF, Jacobs I, et al. Functional complementation studies identify candidate genes and common genetic variants associated with ovarian cancer survival. Hum Mol Genet. 2009;18:1869–78. doi: 10.1093/hmg/ddp107. [DOI] [PubMed] [Google Scholar]
- 17.Dafou D, Ramus SJ, Choi K, Grun B, Trott DA, Newbold RF, Jacobs IJ, Jones C, Gayther SA. Neoplastic suppression in epithelial ovarian cancer cells lines by microcell-mediated transfer of chromosomes 5, 6 and 18. Int J Cancer. 2009;124:1037–44. doi: 10.1002/ijc.24058. [DOI] [PubMed] [Google Scholar]
- 18.Provencher DM, Lounis H, Champoux L, Tétrault M, Manderson EN, Wang JC, Eydoux P, Savoie R, Tonin PN, Mes-Masson AM. Characterization of four novel epithelial ovarian cancer cell lines. In Vitro Cell Dev Biol Anim. 2000;36:357–61. doi: 10.1290/1071-2690(2000)036<0357:COFNEO>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 19.Benjamini Y, Yekutieli D. Quantitative trait loci analysis using the false discovery rate. Genetics. 2005;171:783–90. doi: 10.1534/genetics.104.036699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–5. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- 21.de Bakker PI, Yelensky R, Pe’er I, Gabriel SB, Daly MJ, Altshuler D. Efficiency and power in genetic association studies. Nat Genet. 2005;37:1217–23. doi: 10.1038/ng1669. [DOI] [PubMed] [Google Scholar]
- 22.Song H, Ramus SJ, Tyrer J, Bolton K, Gentry-Maharaj A, Wozniak E, Anton-Culver H, Chang-Claude J, Cramer DW, DiCioccio R, Dörk T, Goode EL, et al. Genome-wide association study identifies a novel ovarian cancer susceptibility locus on 9p22.2. Nat Genet. 2009;41:996–1000. doi: 10.1038/ng.424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Khalique L, Ayhan A, Weale M, Jacobs IJ, Ramus SJ, Gayther SA. Genetic intra tumour heterogeneity in epithelial ovarian cancer: the implications for clinical diagnostics. J Pathol. 2007;211:286–95. doi: 10.1002/path.2112. [DOI] [PubMed] [Google Scholar]
- 24.Canzian F, Salovaara R, Hemminki A, Kristo P, Chadwick RB, Aaltonen LA, de la Chapelle A. Semiautomated assessment of loss of heterozygosity and replication error in tumors. Cancer Res. 1996;56:3331–7. [PubMed] [Google Scholar]
- 25.Stram DO, Haiman CA, Hirschhorn JN, Altshuler D, Kolonel LN, Henderson BE, Pike MC. Choosing haplotype-tagging SNPS based on unphased genotype data using a preliminary sample of unrelated subjects with an example from the Multiethnic Cohort Study. Hum Hered. 2003;55:27–36. doi: 10.1159/000071807. [DOI] [PubMed] [Google Scholar]
- 26.Gabriel SB, Schaffner SF, Nguyen H, Moore JM, Roy J, Blumenstiel B, Higgins J, DeFelice M, Lochner A, Faggart M, Liu-Cordero SN, Rotimi C, et al. The structure of haplotype blocks in the human genome. Science. 2002;296:2225–9. doi: 10.1126/science.1069424. [DOI] [PubMed] [Google Scholar]
- 27.Zaykin DV, Westfall PH, Young SS, Karnoub MA, Wagner MJ, Ehm MG. Testing association of statistically inferred haplotypes with discrete and continuous traits in samples of unrelated individuals. Hum Hered. 2002;53:79–91. doi: 10.1159/000057986. [DOI] [PubMed] [Google Scholar]
- 28.Knudson AG. All in the (cancer) family. Nat Genet. 1993;5:103–104. doi: 10.1038/ng1093-103. [DOI] [PubMed] [Google Scholar]
- 29.Thomas DC, Haile RW, Duggan D. Recent developments in genomewide association scans: a workshop summary and review. Am J Hum Genet. 2005;77:337–45. doi: 10.1086/432962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lakhani SR, Manek S, Penault-Llorca F, Flanagan A, Arnout L, Merrett S, McGuffog L, Steele D, Devilee P, Klijn JG, Meijers-Heijboer H, Radice P, et al. Pathology of ovarian cancers in BRCA1 and BRCA2 carriers. Clin Cancer Res. 2004;10:2473–81. doi: 10.1158/1078-0432.ccr-1029-3. [DOI] [PubMed] [Google Scholar]
- 31.Pearce CL, Wu AH, Gayther SA, Bale AE, Australian Cancer Study (Ovarian Cancer) and Australian Cancer Study Group. Beck PA, Beesley J, Chanock S, Cramer DW, DiCioccio R, Edwards R, Fredericksen ZS, et al. Progesterone receptor variation and risk of ovarian cancer is limited to the invasive endometrioid subtype: results from the Ovarian Cancer Association Consortium pooled analysis. Br J Cancer. 2008;98:282–8. doi: 10.1038/sj.bjc.6604170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.LaFramboise T, Weir BA, Zhao X, Beroukhim R, Li C, Harrington D, Sellers WR, Meyerson M. Allele-specific amplification in cancer revealed by SNP array analysis. PLoS Comput Biol. 2005;1:e65. doi: 10.1371/journal.pcbi.0010065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mao X, Hamoudi RA, Talbot IC, Baudis M. Allele-specific loss of heterozygosity in multiple colorectal adenomas: toward an integrated molecular cytogenetic map II. Cancer Genet Cytogenet. 2006;167:1–14. doi: 10.1016/j.cancergencyto.2005.08.030. [DOI] [PubMed] [Google Scholar]
- 34.Kittiniyom K, Mastronardi M, Roemer M, Wells WA, Greenberg ER, Titus-Ernstoff L, Newsham IF. Allele-specific loss of heterozygosity at the DAL-1/4.1B (EPB41L3) tumor-suppressor gene locus in the absence of mutation. Genes Chromosomes Cancer. 2004;40:190–203. doi: 10.1002/gcc.20034. [DOI] [PubMed] [Google Scholar]
- 35.Wang M, Vikis HG, Wang Y, Jia D, Wang D, Bierut LJ, Bailey-Wilson JE, Amos CI, Pinney SM, Petersen GM, de Andrade M, Yang P, et al. Identification of a novel tumor suppressor gene p34 on human chromosome 6q25.1. Cancer Res. 2007;67:93–9. doi: 10.1158/0008-5472.CAN-06-2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wegman PP, Marcus NJ, Malakkaran BP, Wingren S. Biological significance of allele specific loss of the p53 gene in breast carcinomas. Breast Cancer Res Treat. 2009;118:15–20. doi: 10.1007/s10549-008-0212-1. [DOI] [PubMed] [Google Scholar]
- 37.Pezzi N, Prieto I, Kremer L, Pérez Jurado LA, Valero C, Del Mazo J, Martínez AC, Barbero JL. STAG3, a novel gene encoding a protein involved in meiotic chromosome pairing and location of STAG3-related genes flanking the Williams-Beuren syndrome deletion. FASEB J. 2000;14:581–92. doi: 10.1096/fasebj.14.3.581. [DOI] [PubMed] [Google Scholar]
- 38.Prieto I, Suja JA, Pezzi N, Kremer L, Martínez AC, Rufas JS, Barbero JL. Mammalian STAG3 is a cohesin specific to sister chromatid arms in meiosis I. Nat Cell Biol. 2001;3:761–6. doi: 10.1038/35087082. [DOI] [PubMed] [Google Scholar]
- 39.Novak I, Lightfoot DA, Wang H, Eriksson A, Mahdy E, Höög C. Mouse embryonic stem cells form follicle-like ovarian structures but do not progress through meiosis. Stem Cells. 2006;24:1931–6. doi: 10.1634/stemcells.2005-0520. [DOI] [PubMed] [Google Scholar]
- 40.Kalejs M, Ivanov A, Plakhins G, Cragg MS, Emzinsh D, Illidge TM, Erenpreisa J. Upregulation of meiosis-specific genes in lymphoma cell lines following genotoxic insult and induction of mitotic catastrophe. BMC Cancer. 2006;6:6. doi: 10.1186/1471-2407-6-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Barber TD, McManus K, Yuen KW, Reis M, Parmigiani G, Shen D, Barrett I, Nouhi Y, Spencer F, Markowitz S, Velculescu VE, Kinzler KW, et al. Chromatid cohesion defects may underlie chromosome instability in human colorectal cancers. Proc Natl Acad Sci USA. 2008;105:3443–8. doi: 10.1073/pnas.0712384105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Skotheim RI, Lind GE, Monni O, Nesland JM, Abeler VM, Fosså SD, Duale N, Brunborg G, Kallioniemi O, Andrews PW, Lothe RA. Differentiation of human embryonal carcinomas in vitro and in vivo reveals expression profiles relevant to normal development. Cancer Res. 2005;65:5588–98. doi: 10.1158/0008-5472.CAN-05-0153. [DOI] [PubMed] [Google Scholar]