

Landsberg and colleagues in the 1980s first showed that an increase in the level of circulating insulin results in an increase in sympathetic activity, both in humans and in rats, even under conditions where the level of plasma glucose is clamped (for review see Landsberg, 2001). Insulin levels increase as a consequence of insulin resistance, which is commonly associated with obesity and the metabolic syndrome (Landsberg, 2001; Lambert et al. 2010). These findings led to the hypothesis that the hyperinsulinaemia associated with obesity causes an increase in sympathetic activity, which could account, at least in part, for obesity-related hypertension (Landsberg, 2001). In support of this hypothesis, epidemiological studies carried out in the 1990s showed that obese subjects had higher levels of insulin and blood pressure. The correlation between hyperinsulinaemia and hypertension is likely to be causal, because an increase in insulin levels in obese subjects leads to an increase in sympathetic activity, whereas an acute reduction in insulin levels in patients with hyperinsulinaemia results in a small but significant decrease in both sympathetic activity and blood pressure (Landsberg, 2001). In these early studies sympathetic activity was assessed indirectly by measuring urinary excretion of noradrenaline, but more recently the findings have been confirmed using the methods of noradrenaline spillover and microneurography (Lambert et al. 2010). Thus, although several other factors such as increased levels of circulating leptin are also likely to be important, there is now very substantial evidence that hyperinsulinaemia is a causal factor in obesity-related hypertension, as a consequence of its stimulatory effect on sympathetic nerve activity.
Where and how does insulin act in the brain to increase sympathetic activity? A study by Cassaglia et al. in a recent issue of The Journal of Physiology provides convincing evidence that an increased level of circulating insulin acts on neurons in the arcuate nucleus in the ventromedial hypothalamus, leading to increased sympathetic activity and baroreflex sensitivity (see Fig. 1). The authors found that in anaesthetized rats intravenous infusion of insulin caused a large (over 100%) increase in lumbar sympathetic nerve activity (LSNA) and in the gain of the baroreflex control of LSNA. These effects were reversed after inhibition by microinjection of the GABA receptor agonist muscimol into either the hypothalamic paraventricular nucleus (PVN) or the arcuate nucleus. Both of these nuclei contain a high density of insulin receptors. Cassaglia et al. found, however, that insulin increased sympathetic activity and baroreflex gain when injected locally into the arcuate nucleus, but had no effect when injected into the PVN. As the authors note, confirmation that the arcuate nucleus is the site at which circulating insulin triggers sympathoexcitation will require future experiments in which the effects of specific blockade of insulin receptors within the arcuate nucleus are tested.
Figure 1. Postulated central pathways subserving the effect of insulin on sympathetic activity.
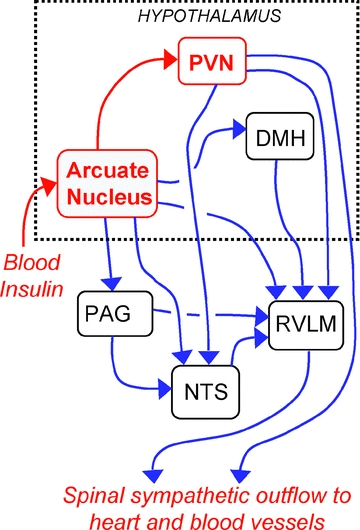
The components of these pathways that are postulated to be essential for this effect are in red, while other components that may also contribute are shown in blue. DMH, dorsomedial hypothalamus; NTS, nucleus of the solitary tract; PAG, periaqueductal grey; PVN, paraventricular nucleus; RVLM, rostral ventrolateral medulla.
If insulin acts on receptors in the arcuate nucleus, how is it transported from the blood? It does not readily cross the blood–brain barrier, but may access neurons within the arcuate nucleus via receptor-mediated endocytosis. On the other hand, the arcuate nucleus is unusual in that it contains highly permeable capillaries (Ciofi, 2011) like those in circumventricular organs, and so insulin may directly activate receptors in this nucleus without requiring a specific transport mechanism.
Although the results showed that the PVN is also an essential part of the neural pathway mediating the effects of insulin on the sympathetic outflow (see Fig. 1), insulin microinjection into the PVN had no effect on sympathetic activity even though the PVN contains a high density of insulin receptors, as mentioned above. As the authors point out, insulin has inhibitory effects on neurons, and so it is possible that the effects of insulin on PVN neurons may not be observable in anaesthesia, when the tonic activity of PVN-sympathetic neurons may be very low.
Apart from the PVN, the arcuate nucleus also projects to other brain regions that regulate the sympathetic outflow, including the dorsomedial hypothalamus, midbrain periaqueductal grey, rostral ventrolateral medulla and the nucleus of the solitary tract, (as shown in Fig. 1). These pathways may also subserve, at least in part, sympathoexcitatory responses evoked from the arcuate nucleus. Furthermore, neurons in the arcuate nucleus are also influenced by other hormones apart from insulin, such as leptin and ghrelin, which may also contribute to obesity-related hypertension (Rahmouni et al. 2005).
It is clear that much remains to be discovered about the central pathways and mechanisms by which circulating insulin can affect sympathetic outflow, both in the short and the long term. The present study, however, is an important step towards that goal.
References
- Cassaglia PA, Hermes SM, Aicher SA, Brooks VL. J Physiol. 2011;589:1643–1662. doi: 10.1113/jphysiol.2011.205575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciofi P. Neurosci Lett. 2011;487:187–190. doi: 10.1016/j.neulet.2010.10.019. [DOI] [PubMed] [Google Scholar]
- Lambert GW, Straznicky NE, Lambert EA, Dixon JB, Schlaich MP. Pharmacol Ther. 2010;126:159–172. doi: 10.1016/j.pharmthera.2010.02.002. [DOI] [PubMed] [Google Scholar]
- Landsberg L. J Hypertens. 2001;19:523–528. doi: 10.1097/00004872-200103001-00001. [DOI] [PubMed] [Google Scholar]
- Rahmouni K, Correia ML, Haynes WG, Mark AL. Hypertension. 2005;45:9–14. doi: 10.1161/01.HYP.0000151325.83008.b4. [DOI] [PubMed] [Google Scholar]