

Abstract
Abstract
It is well established that contracting skeletal muscles produce free radicals. Given that radicals are known to play a prominent role in the pathogenesis of several diseases, the 1980s–90s dogma was that contraction-induced radical production was detrimental to muscle because of oxidative damage to macromolecules within the fibre. In contrast to this early outlook, it is now clear that both reactive oxygen species (ROS) and reactive nitrogen species (RNS) play important roles in cell signalling pathways involved in muscle adaptation to exercise and the remodelling that occurs in skeletal muscle during periods of prolonged inactivity. This review will highlight two important redox sensitive signalling pathways that contribute to ROS and RNS-induced skeletal muscle adaptation to endurance exercise. We begin with a historical overview of radical production in skeletal muscles followed by a discussion of the intracellular sites for ROS and RNS production in muscle fibres. We will then provide a synopsis of the redox-sensitive NF-κB and PGC-1α signalling pathways that contribute to skeletal muscle adaptation in response to exercise training. We will conclude with a discussion of unanswered questions in redox signalling in skeletal muscle in the hope of promoting additional research interest in this field.
Scott Powers (right), Erin Talbert (centre) and Peter Adhihetty (left) work in the department of Applied Physiology and Kinesiology at the University of Florida and collaborate on studies involving reactive oxygen species-linked signalling events in both skeletal and cardiac muscle. Their research backgrounds are in physiology and biochemistry. Scott Powers and Erin Talbert are currently collaborating on studies that focus on understanding the cell signalling pathways responsible for disuse muscle atrophy. Peter Adhihetty's research centres on investigating the role of mitochondrial dysfunction in both muscle and neural tissue in various diseases or disorders.
 |
Introduction
The first report that muscular exercise increases the production of reactive oxygen species (ROS) in humans appeared in 1978 (Dillard et al. 1978). This initial observation did not reveal the sources of ROS production during exercise, but a subsequent study demonstrated that contracting skeletal muscles are a prominent source of ROS production (Davies et al. 1982). It was later observed that contracting muscles also produce nitric oxide (NO) and other reactive nitrogen species (RNS) (Balon & Nadler, 1994). Since these early observations, many studies have confirmed that muscular exercise promotes the production of both ROS and RNS in skeletal muscle fibres (Powers & Jackson, 2008).
During the 1980s–90s it was widely believed that exercise-induced ROS production was damaging to skeletal muscle fibres and limited consideration was given to the possibility that contraction-induced ROS/RNS production could play an important signalling role in muscle adaptation to exercise. However, contemporary evidence indicates that increased ROS and RNS production plays a key role in the regulation of signalling pathways that are essential for muscle adaptation in response to endurance exercise training. The discovery that ROS/RNS plays a significant role in skeletal muscle adaptation to exercise is an exciting new area of research in exercise biology and is the focus of this review. Our report begins with an overview of the sources of contraction-induced ROS and RNS production in skeletal muscles followed by a discussion of the paradox that ROS plays a significant signalling role in muscle remodelling during both exercise training and disuse-induced muscle atrophy. We will then highlight two important redox-sensitive signalling molecules in skeletal muscle, nuclear factor-κB (NF-κB) and peroxisome proliferator-activated receptor-γ coactivator-1α (PCG-1α). This will be followed by a summary of the evidence that ROS and/or RNS contribute to skeletal muscle adaptation in response to endurance exercise training. We will conclude with a discussion of the gaps in our knowledge about redox control of muscle adaptation in the hope of stimulating additional research in this exciting area of exercise biology.
Sources of ROS/RNS production in contracting skeletal muscles
The chief parent radical species produced in muscle fibres are superoxide and NO, and both species can react with other molecules to form a wide range of ROS and RNS, respectively (Halliwell & Gutteridge, 2007). An overview of the principal sites of superoxide and NO production in cells follows.
Cellular sources of superoxide
Superoxide is produced by the addition of a single electron to ground state oxygen in a variety of intracellular locations. For example, superoxide production can occur in the mitochondrion, sarcoplasmic reticulum, transverse tubules, sarcolemma, and cytosol (Fig. 1). The main sites of superoxide production in the mitochondria are complexes I and III of the electron transport chain (Barja, 1999). Further, recent findings indicate that compared to mitochondria from slow type I muscle fibres, mitochondria from fast type II muscle fibres possess unique properties that promote higher levels of ROS production (Anderson & Neufer, 2006). The mechanism(s) to explain these differences remain unknown.
Figure 1. Illustration of the potential cellular sites for the production of superoxide in muscle fibres.
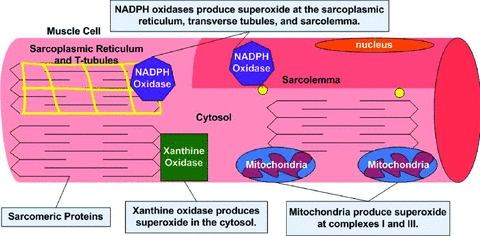
Note that primary sites for cellular superoxide production include mitochondria, NADPH oxidases (located within the sarcoplasmic reticulum, transverse tubules and the sarcolemma), and xanthine oxidase. See text for more details.
Mitochondria are commonly cited as the primary source of superoxide production in contracting muscle fibres because early reports suggested that 2–5% of the total oxygen consumed by mitochondria undergoes a one electron reduction to form superoxide (Boveris & Chance, 1973; Loschen et al. 1974). Based upon this assertion, authors have often assumed that the increased ROS generation that occurs in contracting muscles is directly linked to the increased oxygen consumption during exercise, implying a large increase (e.g. 50- to 100-fold) in superoxide generation by skeletal muscle during aerobic contractions (e.g. see Kanter, 1995; Urso & Clarkson, 2003). Nevertheless, increasing evidence argues against this assumption as emerging evidence reveals that the rate of superoxide production by mitochondria is much less that the early estimates that 2–5% of all molecular oxygen consumed by mitochondria is converted to superoxide. For example, Brand and colleagues calculates that less than 0.15% of oxygen consumed by the mitochondria is used to form superoxide (St-Pierre et al. 2002). Further, abundant data indicate that mitochondria produce more ROS in state 4 (basal) respiration compared to active state 3 (maximal ADP stimulated) respiration (Di Meo & Venditti, 2001; Adhihetty et al. 2005; Anderson & Neufer, 2006; Kavazis et al. 2009). Together, these results suggest that mitochondria are not the primary source of ROS production in muscle fibres during exercise.
In addition to mitochondria, superoxide can be produced by numerous other cellular locations including NADPH oxidases located within the sarcoplasmic reticulum, transverse tubules and sarcolemma (Fig. 1) (Powers & Jackson, 2008). Unfortunately, limited information currently exists about the regulation of these systems in muscle during exercise.
Evidence also indicates that xanthine oxidase produces superoxide in the cytosol of contracting rat skeletal muscles (Gomez-Cabrera et al. 2005). However, compared to rats, human skeletal muscles contain lower levels of xanthine oxidase and debate continues as to whether xanthine oxidase plays an important role in superoxide production in human skeletal muscle (Linder et al. 1999; Gomez-Cabrera et al. 2003).
The dismutation of superoxide in cells produces hydrogen peroxide (H2O2) and this process can occur spontaneously or by action of the superoxide dismutases (SOD) (Halliwell & Gutteridge, 2007). H2O2 is a non-radical and a weak oxidant with a relatively long half-life, which permits its diffusion within cells and across cell membranes (Halliwell & Gutteridge, 2007). Further, H2O2 reacts with many different cellular molecules and can activate a variety of signalling pathways. Collectively, these properties make H2O2 an important ROS signalling molecule in cells (Veal et al. 2007).
Nitric oxide production in contracting muscles
Nitric oxide is synthesized from the amino acid l-arginine using three different isoforms of nitric oxide synthase (NOS1, NOS2 and NOS3). Further, a fourth mitochondrial nitric oxide synthase may also exist (Ghafourifar & Cadenas, 2005). Normally skeletal muscle expresses two of these isoforms (i.e. NOS1 and NOS3) (Moylan & Reid, 2007). However, NOS2 can also be expressed in skeletal muscle during inflammatory states (Moylan & Reid, 2007). Nitric oxide is known to have many signalling functions and can readily react with superoxide to form the strong oxidizing agent peroxynitrite leading to the depletion of thiol groups in cells (Moylan & Reid, 2007). This modification of cellular thiol groups could alter redox signalling and may play an important role in numerous cell signalling pathways (Jones, 2006). Peroxynitrite formation also reduces the bioavailability of both superoxide and NO, which could also influence cell signalling events (Halliwell & Gutteridge, 2007; Powers & Jackson, 2008).
ROS as signalling molecule in skeletal muscle remodelling: the ROS paradox
Many studies have concluded that inactivity-induced ROS production in skeletal muscle contributes to disuse muscle atrophy (Powers et al. 2005; Powers et al. 2007). Paradoxically, growing evidence also suggests that intracelluar ROS production is a required signal for the normal remodelling that occurs in skeletal muscle in response to repeated bouts of endurance exercise (Hamilton et al. 2003; Gomez-Cabrera et al. 2008; Ristow et al. 2009). Therefore, how does the ROS production that occurs in muscle fibres during exercise avoid resulting in muscle atrophy? Unfortunately, a definitive answer to this question is not currently available. Nonetheless, our knowledge about the biological implications of exercise-induced ROS has expanded rapidly in recent years and has provided some clues to this apparent mystery. Based upon current evidence, it appears that at least two potential explanations for this apparent ROS paradox exist. First, while continuous and high rates of free radical production can damage cellular components, depress protein synthesis, and activate proteases, an acute bout of muscular exercise that results in acute production of low-to-moderate levels of oxidants does not generate this response. On the contrary, an acute and small increase in ROS production during a bout of muscular exercise appears to play an important role in the regulation of cell signalling pathways that promote gene expression leading to an increased oxidative phenotype of skeletal muscle (Droge, 2002; Jackson, 2008).
A second potential factor that may contribute to the ‘ROS paradox’ is that the site(s) of ROS production in muscle may differ between contracting fibres and fibres exposed to prolonged periods of inactivity. For example, a recent study indicates that prolonged muscle inactivity results in a large increase in mitochondrial ROS production (Kavazis et al. 2009). In contrast, it seems unlikely that mitochondria are the primary source of ROS production in contracting muscle fibres (Powers & Jackson, 2008). Therefore, it is feasible that the different sites of muscle ROS production in these two conditions may also influence redox-sensitive signalling and contribute to the ROS paradox in skeletal muscle.
Regardless of the explanation for the ROS paradox, the remainder of this report will provide a summary of two key redox sensitive signalling pathways in skeletal muscle and will highlight the evidence that ROS/RNS is required for normal adaptation of skeletal muscle to endurance exercise training.
Redox sensitive signalling pathways in skeletal muscle
Skeletal muscle is a malleable tissue that can undergo significant phenotypic changes in response to repeated bouts of exercise. Indeed, as few as five consecutive days of endurance exercise results in significant improvements in both the oxidative and antioxidant capacity of skeletal muscle fibres (Vincent et al. 1999, 2000). During the past decade, much has been learned about the exercise-induced cell signalling pathways that mediate these changes. Interestingly, many of these pathways appear to be initiated, or at least potentiated, by ROS and RNS signals. Several of these redox-sensitive pathways result in changes in transcription factor activity, either increasing or decreasing the transcription of target genes. The next segment will highlight the regulation of two important redox-sensitive transcription factors that are involved in muscle adaptation in response to endurance exercise training. Specifically, we will discuss the role that ROS/RNS play in the activation of exercise-induced signalling via nuclear factor-κB (NF-κB) and PGC-1α in skeletal muscle fibres.
Redox control of NF-κB activation
Again, it is well known that repeated bouts of endurance exercise result in adaptive changes in skeletal muscle fibres resulting in an oxidative phenotype with improved antioxidant capacity (Hammeren et al. 1992; Powers et al. 1992; Criswell et al. 1993; Powers et al. 1994). Understanding the cell signalling pathways responsible for exercise-induced muscle adaptation remains an important topic for research in skeletal muscle biology. In this regard, robust evidence reveals that redox-sensitive pathways use ROS or RNS to transfer signals from the cytoplasm to the nucleus to promote gene expression (Droge, 2002; Ji et al. 2006; Upham & Trosko, 2009). An important signalling link between contraction-induced ROS production and skeletal muscle remodelling involves the redox regulation of the NF-κB family of transcriptional activators. NF-κB transcription factors are evolutionary conserved signalling molecules that control the expression of numerous genes involved in a large number of cell processes such as inflammation, cell growth, stress responses, and apoptosis (Kramer & Goodyear, 2007). For example, several antioxidant enzymes including copper-zinc superoxide dismutase, manganese superoxide dismutase, and γ-glumatylcysteine synthetase contain NF-κB binding sites in the 5′-flanking region of their promoter (Allen & Tresini, 2000). Therefore, these genes are potential targets for ROS-mediated signalling via activation of NF-κB. A brief summary of NF-κB regulation and evidence that active NF-κB plays an important role in exercise-induced muscle adaptation follows.
NF-κB comprises a family of five transcription factors (p65, Rel B, c-Rel, p52 and p50). To act as a transcriptional factor, two of these proteins must dimerize; this dimerization facilitates nuclear translocation and the subsequent binding of NF-κB to the kB consensus sequence of the target genes (Bakkar & Guttridge, 2010). Active NF-κB transcription factors can promote a wide range of cellular outcomes depending upon the cell type (Jackman & Kandarian, 2004). All five of the NF-κB family members are expressed in skeletal muscle but evidence indicates that the p50–p65 heterodimer is responsible for the majority of NF-κB activity in muscle (Jackman & Kandarian, 2004).
In unstressed cells, the nuclear localization sequence of NF-κB is bound to inhibitory IkB proteins in the cytosol and these inhibitory proteins prevent the dimerization of p50 to p65. However, increased levels of ROS in the cytosol can activate IkB-α kinase (IKK) resulting in the phosphorylation of IkB proteins; this initiates ubiquitination and subsequent IkB degradation via the proteasome (Kabe et al. 2005). Degradation of IkB removes the inhibition and liberates NF-κB complexes so that dimerization and nuclear translocation can occur (Kabe et al. 2005) (Fig. 2).
Figure 2. Steps leading to NF-κB activation and signalling in cells.
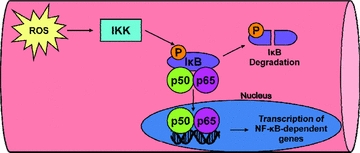
In unstressed cells, the nuclear localization sequence of NF-κB is bound to inhibitory IkB proteins in the cytosol and these inhibitory proteins inhibit the dimerization of p50 to p65. During periods of oxidant stress, increased levels of ROS in the cytosol can activate IkB-α kinase (IKK), which results in phosphorylation of IkB proteins. This phosphorylation initiates ubiquitination and subsequent IkB degradation via the proteasome. Degradation of IkB removes the inhibition and releases NF-κB complexes so that dimerization and nuclear translocation can occur. See text for more details.
Many studies have reported that an acute bout of endurance exercise results in NF-κB activation in skeletal muscle (Ji et al. 2006; Ji, 2007). In this regard, time course studies reveal that a decrease in cytosolic IkB and an increase in phosphorylated IkB proteins occurs immediately post-exercise (Ji et al. 2004). Current evidence indicates that NF-κB binding to DNA reaches a peak at ∼2 h following an acute bout of exercise (Ji et al. 2004). Similarly, exposure of C2C12 muscle cells to 1–2 mmol l−1 H2O2 results in NF-κB activation with maximal NF-κB/DNA binding occurring at 2 h following exposure to ROS (Zhou et al. 2001).
Although ROS can promote NF-κB activation and subsequent gene expression, the DNA binding activity of oxidized NF-κB is diminished suggesting that ROS may also inhibit NF-κB transcriptional activity (Kabe et al. 2005). Hence, although NF-κB was once considered to be a prototypic redox sensitive transcription factor, the observation that ROS can both promote and inhibit NF-κB transcriptional activation has led to debate regarding the redox control of NF-κB signalling (Pantano et al. 2006). Nonetheless, a growing body of evidence suggests that exercise-induced ROS production promotes contraction-induced NF-κB activation in skeletal muscle (Ji et al. 2007; Ji, 2008). Indeed, several studies reveal that exercise-induced NF-κB activation in skeletal muscle requires ROS as an upstream signal and that NF-κB activation is essential for exercise training-induced expression of antioxidant enzymes (Hollander et al. 2001; Ji et al. 2004; Gomez-Cabrera et al. 2005). For example, Gomez-Cabrera and colleagues have demonstrated that antioxidant-mediated suppression of exercise-induced ROS emission in contracting skeletal muscles results in blunted NF-κB activation as evidenced by reduced nuclear binding of NF-κB (Gomez-Cabrera et al. 2005). Further, the decrease of contraction-induced ROS production and NF-κB activation in these experiments also prevented the exercise-induced increase in muscle levels of manganese SOD (MnSOD) mRNA and MnSOD protein.
To summarize, ROS generated by contracting muscles plays an important role in the activation of NF-κB in skeletal muscle in response to exercise. Moreover, exercise-induced activation of NF-κB appears to be a requirement for exercise-induced expression of MnSOD and perhaps many other cellular proteins that are critical to training-induced muscle adaptation. Identifying other important gene targets of NF-κB in skeletal muscle is likely to remain an important topic for future studies of exercise-induced cell signalling.
Redox control of PGC-1α
An important skeletal muscle adaptation that occurs with endurance training is the increase in mitochondrial content in the muscle fibre due to mitochondrial biogenesis. Exercise-induced increases in mitochondria in skeletal muscle are associated with a variety of health-related benefits including improvements in tissue oxidative capacity, exercise tolerance and insulin resistance (Holloszy, 1967; Holloszy & Coyle, 1984; Irrcher et al. 2003a, 2008; Hawley & Zierath, 2004). Mitochondrial biogenesis is a unique and complicated process since mitochondria are composed of gene products from both the nuclear and mitochondrial genomes. Thus, mitochondrial biogenesis requires the coordinated response of the nuclear and mitochondrial genomes to maintain the correct stoichiometric arrangement of proteins during organelle synthesis. This coordinated response is accomplished by a vast array of transcription factors and transciptional coactivators (Hood, 2001; Adhihetty et al. 2003).
Peroxisome proliferator-activated receptor-γ (PPAR-γ) coactivator-1α (PGC-1α) has been shown to be an integral regulator of mitochondrial biogenesis. PGC-1α interacts with and coactivates a variety of transcription factors and nuclear receptors involved in the upregulation of both nuclear- and mitochondrial-encoded genes involved in organelle synthesis, which include oestrogen-related receptor-α (ERR-α; Huss et al. 2002), nuclear respiratory factors (NRF-1 and NRF-2; Scarpulla, 2006), thyroid hormone receptor (TR; Irrcher et al. 2003b) and myocyte enhancer factor (MEF; Lin et al. 2002). PGC-1α has been shown to regulate a variety of cellular processes such as adaptive thermogenesis, glucose metabolism, muscle fibre-type specialization and oxidative phosphorylation in many tissues (Puigserver et al. 1998; Wu et al. 1999; Lin et al. 2002). Within skeletal muscle, numerous studies have shown that PGC-1α is capable of dictating mitochondrial content and function, and potently activating mitochondrial biogenesis (Ljubicic et al.; Wu et al. 1999; Vega et al. 2000; Huss & Kelly, 2004). Thus, PGC-1α has emerged as a key protein that can regulate mitochondrial content in tissues and has been referred to as the ‘master regulator’ of mitochondrial biogenesis.
PGC-1α expression in muscle can be induced by a variety of stimuli associated with muscular exercise (Goto et al. 2000; Baar et al. 2002; Irrcher et al. 2003a,b; Pilegaard et al. 2003; Russell et al. 2003). The exercise-induced signalling mechanisms leading to PGC-1α induction have been primarily attributed to contractile activity-mediated increases in AMPK and p38 MAPK activation. While many studies have confirmed that both acute and chronic exercise can activate pathways leading to PGC-1α induction, much less is known about the specific role that ROS plays in mitochondrial biogenesis. Nonetheless, emerging studies reveal that exercise increased ROS and RNS production in muscle occurs coincident with enhanced mitochondrial biogenesis signalling markers. However, these observations are correlative and do not demonstrate cause-and-effect. Thus, two important questions emerge: (1) is PGC-1α transcriptional activity influenced by redox control? and (2) are the exercise inductions of PGC-1α expression in skeletal muscle dependent upon ROS and/or RNS production?
PGC-1α appears to be sensitive to the redox status of the cell because treatment of cultured muscle myotubes with exogenous hydrogen peroxide causes induction of PGC-1α, and the antioxidant N-acetylcysteine inhibits this upregulation (Irrcher et al. 2009). In this regard, Irrcher et al. (2009) predict that elevated cellular levels of ROS induce PGC-1α transcription indirectly, via AMPK activation (Fig. 3). Thus, PGC-1α appears to be part of a redox-sensitive pathway similar to the ROS-sensitive transcription factor NF-κB. In fact, the human PGC-1α promoter contains an NF-κB binding site, which suggests NF-κB may also regulate the expression of PGC-1α (Irrcher et al. 2008). Analysis of the human PGC-1α promoter has revealed a variety of consensus transcription binding sites to the following transcription factors: specificity protein 1 (SP1), cAMP response element binding protein (CREB), CREB related family member, activating transcription factor (ATF2), forkhead transcription factor (FKHR), p53, EBox binding proteins, GATA and MEF2 (Irrcher et al. 2008). Many of these transcription factors have been shown to be ROS-sensitive which indicates numerous potential possibilities for redox control of PGC-1α expression. However, these are only theoretical possibilities which have not been confirmed and further studies are necessary. Additionally, RNS, particularly NO, may also be involved in the regulation of PGC-1α (Lira et al. 2010). Specifically, Lira et al. (2010) report that NO production promotes PGC-1α expression via NO-mediated activation of AMPK (i.e. AMPK α1 isoform) (Fig. 3). Therefore, the current evidence suggests that both ROS and RNS can contribute to PGC-1α expression via a common signalling pathway (i.e. AMPK activation).
Figure 3. Schematic diagram of the key steps involved in PGC-1α expression and activation in skeletal muscle fibres.
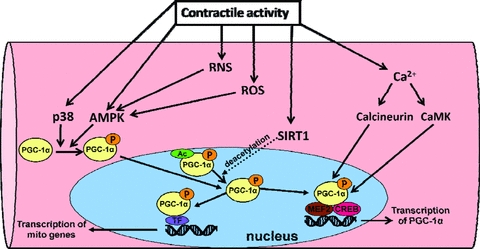
Note that the dotted arrow leading from SIRT1 and directed toward the arrow leading from PCG-1α is driving this reaction forward resulting in deacetylation of PGC-1α and increasing the pool of non-acetylated PGC-1α in the nucleus. See text for more details. Legend: Ac, acetylation; p, phosphorylation.
It has recently been shown that PGC-1α activity is also regulated by a variety of post-translational modifications including phosphsphorylation, acetylation, methylation and ubiquination (Jager et al. 2007; Rodgers et al. 2008; Dominy et al. 2010). Studies have demonstrated that p38 MAPK and AMPK are capable of phosphorylating PGC-1α at a variety of amino acid residues, which results in a more stable and active PGC-1α protein. The acetylase transferase (GCN5) acetylates PGC-1α at several lysine residues to inactivate PGC-1α while the NAD+-dependent deacetylase Sirt1 removes acetyl groups leading to activation of PGC-1α (Fig. 3). These post-translational modifications have been proposed to activate and/or deactivate PGC-1α located in the nucleus or within the cytosol of the cell. Recently, Wright et al. (2007) reported that acute exercise causes the activation a cytosolic pool of PGC-1α, which then translocates to the nucleus to initiate mitochondrial gene expression prior to increases in overall PGC-1α expression. Thus, post-translational modifications appear to represent an immediate mechanism to activate PGC-1α and initiate PGC-1α-dependent gene expression. To date, it is unclear whether these post-translational modifications are under redox control but this certainly represents another potential complexity that may be involved in ROS-mediated induction of PGC-1α.
Is contraction-induced ROS production required for PGC-1α induction in skeletal muscle?
Numerous training studies have provided in vivo evidence that ROS are important signalling molecules mediating many of the exercise-induced adaptations in skeletal muscle. The majority of these studies utilize dietary antioxidant supplementation to suppress exercise-induced redox signalling in skeletal muscles. For example, Hamilton et al. (2003) illustrated that a diet rich in antioxidants prevents training-induced increases in heat shock protein 72 (HSP72) in the heart. This study provided the first evidence that exercise-induced ROS production was critical for the training-induced adaptive responses of HSP72. These findings were confirmed and extended by Jackson et al. (2004) who demonstrated that training-induced increases in HSP60, HSP72 and HSP73 in human skeletal muscle were blunted by a diet high in vitamin E and β-carotene (Jackson et al. 2004). Thus, antioxidant treatments were initially shown to blunt the HSP72 responses to exercise, which clearly indicates the importance of ROS as signalling molecules. However, the link between exercise-induced ROS production and exercise-induced elevations in muscle PGC-1α was only recently identified and a brief summary of this work follows.
Gomez-Cabrera et al. (2008) reported that exercising rats supplemented with vitamin C do not exhibit the normal phenotypic changes observed in skeletal muscle following a program of endurance training. Specifically, vitamin C supplementation diminished the exercise-induced increase in maximal oxygen consumption and also prevented the training-mediated rise in muscle antioxidant enzymes and PGC-1α protein levels in rat skeletal muscle. These investigators also supplemented human subjects with vitamin C and found the results to be in agreement with their findings in rodents (Gomez-Cabrera et al. 2008). Recent work has further confirmed that supplementing human subjects with vitamins C and E leads to diminished training-induced improvements in insulin sensitivity, as well as the expression of antioxidant defence enzymes and PGC-1α (Ristow et al. 2009). Taken together, these results support the notion that contraction-induced ROS molecules are critical signalling molecules for exercise-induced adaptations in vivo and that PGC-1α is under redox control.
Conclusions and future directions
A growing body of literature reveals that ROS and RNS are important signalling molecules for exercise-training induced adaptations in skeletal muscles. Indeed, both human and animal studies confirm that prevention of exercise-induced redox signalling via antioxidant supplementation results in a blunted training response in skeletal muscles.
Although our understanding of redox signalling pathways in skeletal muscle has grown in recent years, many unanswered questions remain. For example, the primary sites of superoxide production in contracting muscle remain controversial and additional work is required to determine the main sites of ROS production in contracting muscle and how this varies with exercise conditions (i.e. low intensity exercise versus high intensity exercise). Further, the inability to perform quantitative measures of ROS production in muscle cells is a significant limitation to investigators. Indeed, significant technological advances are required to provide investigators with the necessary tools to better understand the sites of ROS production and redox compartmentalization in muscle fibres. This is a critical area for future work because improving our understanding of ROS production and redox compartmentalization in muscle fibres will likely provide important insight into specific redox signalling pathways.
Also, whilst it is established that activation of NF-κB contributes to exercise-induced signalling in skeletal muscle, the identification of unknown gene targets of NF-κB in skeletal muscle remains an important topic for future research. Moreover, details regarding the specific signalling role that NF-κB plays in promotion of exercise-induced expression of PGC-1α should receive additional experimental attention.
Finally, emerging evidence indicates that ROS are required for contraction-induced expression of PGC-1α in the active muscle fibres. However, limited information exists regarding the explicit redox controlled signalling pathways that regulate PGC-1α activation and mitochondrial biogenesis in skeletal muscle. Clearly, there is much more to be learned about the redox control of skeletal muscle adaptation to exercise.
References
- Adhihetty PJ, Irrcher I, Joseph AM, Ljubicic V, Hood DA. Plasticity of skeletal muscle mitochondria in response to contractile activity. Exp Physiol. 2003;88:99–107. doi: 10.1113/eph8802505. [DOI] [PubMed] [Google Scholar]
- Adhihetty PJ, Ljubicic V, Menzies KJ, Hood DA. Differential susceptibility of subsarcolemmal and intermyofibrillar mitochondria to apoptotic stimuli. Am J Physiol Cell Physiol. 2005;289:C994–C1001. doi: 10.1152/ajpcell.00031.2005. [DOI] [PubMed] [Google Scholar]
- Allen RG, Tresini M. Oxidative stress and gene regulation. Free Radic Biol Med. 2000;28:463–499. doi: 10.1016/s0891-5849(99)00242-7. [DOI] [PubMed] [Google Scholar]
- Anderson EJ, Neufer PD. Type II skeletal myofibres possess unique properties that potentiate mitochondrial H2O2 generation. Am J Physiol Cell Physiol. 2006;290:C844–851. doi: 10.1152/ajpcell.00402.2005. [DOI] [PubMed] [Google Scholar]
- Baar K, Wende AR, Jones TE, Marison M, Nolte LA, Chen M, Kelly DP, Holloszy JO. Adaptations of skeletal muscle to exercise: rapid increase in the transcriptional coactivator PGC-1. FASEB J. 2002;16:1879–1886. doi: 10.1096/fj.02-0367com. [DOI] [PubMed] [Google Scholar]
- Bakkar N, Guttridge DC. NF-κB signaling: a tale of two pathways in skeletal myogenesis. Physiol Rev. 2010;90:495–511. doi: 10.1152/physrev.00040.2009. [DOI] [PubMed] [Google Scholar]
- Balon TW, Nadler JL. Nitric oxide release is present from incubated skeletal muscle preparations. J Appl Physiol. 1994;77:2519–2521. doi: 10.1152/jappl.1994.77.6.2519. [DOI] [PubMed] [Google Scholar]
- Barja G. Mitochondrial oxygen radical generation and leak: sites of production in states 4 and 3, organ specificity, and relation to aging and longevity. J Bioenerg Biomembr. 1999;31:347–366. doi: 10.1023/a:1005427919188. [DOI] [PubMed] [Google Scholar]
- Boveris A, Chance B. The mitochondrial generation of hydrogen peroxide. General properties and effect of hyperbaric oxygen. Biochem J. 1973;134:707–716. doi: 10.1042/bj1340707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Criswell D, Powers S, Dodd S, Lawler J, Edwards W, Renshler K, Grinton S. High intensity training-induced changes in skeletal muscle antioxidant enzyme activity. Med Sci Sports Exerc. 1993;25:1135–1140. [PubMed] [Google Scholar]
- Davies KJ, Quintanilha AT, Brooks GA, Packer L. Free radicals and tissue damage produced by exercise. Biochem Biophys Res Commun. 1982;107:1198–1205. doi: 10.1016/s0006-291x(82)80124-1. [DOI] [PubMed] [Google Scholar]
- Di Meo S, Venditti P. Mitochondria in exercise-induced oxidative stress. Biol Signals Recept. 2001;10:125–140. doi: 10.1159/000046880. [DOI] [PubMed] [Google Scholar]
- Dillard CJ, Litov RE, Savin WM, Dumelin EE, Tappel AL. Effects of exercise, vitamin E, and ozone on pulmonary function and lipid peroxidation. J Appl Physiol. 1978;45:927–932. doi: 10.1152/jappl.1978.45.6.927. [DOI] [PubMed] [Google Scholar]
- Dominy JE, Jr, Lee Y, Gerhart-Hines Z, Puigserver P. Nutrient-dependent regulation of PGC-1α's acetylation state and metabolic function through the enzymatic activities of Sirt1/GCN5. Biochim Biophys Acta. 2010;1804:1676–1683. doi: 10.1016/j.bbapap.2009.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Droge W. Free radicals in the physiological control of cell function. Physiol Rev. 2002;82:47–95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- Ghafourifar P, Cadenas E. Mitochondrial nitric oxide synthase. Trends Pharmacol Sci. 2005;26:190–195. doi: 10.1016/j.tips.2005.02.005. [DOI] [PubMed] [Google Scholar]
- Gomez-Cabrera MC, Borras C, Pallardo FV, Sastre J, Ji LL, Vina J. Decreasing xanthine oxidase-mediated oxidative stress prevents useful cellular adaptations to exercise in rats. J Physiol. 2005;567:113–120. doi: 10.1113/jphysiol.2004.080564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Cabrera MC, Domenech E, Romagnoli M, Arduini A, Borras C, Pallardo FV, Sastre J, Vina J. Oral administration of vitamin C decreases muscle mitochondrial biogenesis and hampers training-induced adaptations in endurance performance. Am J Clin Nutr. 2008;87:142–149. doi: 10.1093/ajcn/87.1.142. [DOI] [PubMed] [Google Scholar]
- Gomez-Cabrera MC, Pallardo FV, Sastre J, Vina J, Garcia-del-Moral L. Allopurinol and markers of muscle damage among participants in the Tour de France. JAMA. 2003;289:2503–2504. doi: 10.1001/jama.289.19.2503-b. [DOI] [PubMed] [Google Scholar]
- Goto M, Terada S, Kato M, Katoh M, Yokozeki T, Tabata I, Shimokawa T. cDNA Cloning and mRNA analysis of PGC-1 in epitrochlearis muscle in swimming-exercised rats. Biochem Biophys Res Commun. 2000;274:350–354. doi: 10.1006/bbrc.2000.3134. [DOI] [PubMed] [Google Scholar]
- Halliwell B, Gutteridge J. Free Radicals in Biology and Medicine. Oxford: Oxford University Press; 2007. [Google Scholar]
- Hamilton KL, Staib JL, Phillips T, Hess A, Lennon SL, Powers SK. Exercise, antioxidants, and HSP72: protection against myocardial ischemia/reperfusion. Free Radic Biol Med. 2003;34:800–809. doi: 10.1016/s0891-5849(02)01431-4. [DOI] [PubMed] [Google Scholar]
- Hammeren J, Powers S, Lawler J, Criswell D, Martin D, Lowenthal D, Pollock M. Exercise training-induced alterations in skeletal muscle oxidative and antioxidant enzyme activity in senescent rats. Int J Sports Med. 1992;13:412–416. doi: 10.1055/s-2007-1021290. [DOI] [PubMed] [Google Scholar]
- Hawley JA, Zierath JR. Integration of metabolic and mitogenic signal transduction in skeletal muscle. Exerc Sport Sci Rev. 2004;32:4–8. doi: 10.1097/00003677-200401000-00002. [DOI] [PubMed] [Google Scholar]
- Hollander J, Fiebig R, Gore M, Ookawara T, Ohno H, Ji LL. Superoxide dismutase gene expression is activated by a single bout of exercise in rat skeletal muscle. Pflugers Arch. 2001;442:426–434. doi: 10.1007/s004240100539. [DOI] [PubMed] [Google Scholar]
- Holloszy JO. Biochemical adaptations in muscleEffects of exercise on mitochondrial oxygen uptake and respiratory enzyme activity in skeletal muscle. J Biol Chem. 1967;242:2278–2282. [PubMed] [Google Scholar]
- Holloszy JO, Coyle EF. Adaptations of skeletal muscle to endurance exercise and their metabolic consequences. J Appl Physiol. 1984;56:831–838. doi: 10.1152/jappl.1984.56.4.831. [DOI] [PubMed] [Google Scholar]
- Hood DA. Invited Review: contractile activity-induced mitochondrial biogenesis in skeletal muscle. J Appl Physiol. 2001;90:1137–1157. doi: 10.1152/jappl.2001.90.3.1137. [DOI] [PubMed] [Google Scholar]
- Huss JM, Kelly DP. Nuclear receptor signaling and cardiac energetics. Circ Res. 2004;95:568–578. doi: 10.1161/01.RES.0000141774.29937.e3. [DOI] [PubMed] [Google Scholar]
- Huss JM, Kopp RP, Kelly DP. Peroxisome proliferator-activated receptor coactivator-1α (PGC-1α) coactivates the cardiac-enriched nuclear receptors estrogen-related receptor-α and -γ. Identification of novel leucine-rich interaction motif within PGC-1α. J Biol Chem. 2002;277:40265–40274. doi: 10.1074/jbc.M206324200. [DOI] [PubMed] [Google Scholar]
- Irrcher I, Adhihetty PJ, Joseph AM, Ljubicic V, Hood DA. Regulation of mitochondrial biogenesis in muscle by endurance exercise. Sports Med. 2003a;33:783–793. doi: 10.2165/00007256-200333110-00001. [DOI] [PubMed] [Google Scholar]
- Irrcher I, Adhihetty PJ, Sheehan T, Joseph AM, Hood DA. PPARγ coactivator-1α expression during thyroid hormone- and contractile activity-induced mitochondrial adaptations. Am J Physiol Cell Physiol. 2003b;284:C1669–1677. doi: 10.1152/ajpcell.00409.2002. [DOI] [PubMed] [Google Scholar]
- Irrcher I, Ljubicic V, Hood DA. Interactions between ROS and AMP kinase activity in the regulation of PGC-1α transcription in skeletal muscle cells. Am J Physiol Cell Physiol. 2009;296:C116–123. doi: 10.1152/ajpcell.00267.2007. [DOI] [PubMed] [Google Scholar]
- Irrcher I, Ljubicic V, Kirwan AF, Hood DA. AMP-activated protein kinase-regulated activation of the PGC-1α promoter in skeletal muscle cells. PLoS One. 2008;3:e3614. doi: 10.1371/journal.pone.0003614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackman RW, Kandarian SC. The molecular basis of skeletal muscle atrophy. Am J Physiol Cell Physiol. 2004;287:C834–843. doi: 10.1152/ajpcell.00579.2003. [DOI] [PubMed] [Google Scholar]
- Jackson MJ. Free radicals generated by contracting muscle: by-products of metabolism or key regulators of muscle function? Free Radic Biol Med. 2008;44:132–141. doi: 10.1016/j.freeradbiomed.2007.06.003. [DOI] [PubMed] [Google Scholar]
- Jackson MJ, Khassaf M, Vasilaki A, McArdle F, McArdle A. Vitamin E and the oxidative stress of exercise. Ann N Y Acad Sci. 2004;1031:158–168. doi: 10.1196/annals.1331.015. [DOI] [PubMed] [Google Scholar]
- Jager S, Handschin C, St-Pierre J, Spiegelman BM. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1α. Proc Natl Acad Sci U S A. 2007;104:12017–12022. doi: 10.1073/pnas.0705070104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji LL. Antioxidant signaling in skeletal muscle: A brief review. Exp Gerontol. 2007;42:582–593. doi: 10.1016/j.exger.2007.03.002. [DOI] [PubMed] [Google Scholar]
- Ji LL. Modulation of skeletal muscle antioxidant defense by exercise: Role of redox signaling. Free Radic Biol Med. 2008;44:142–152. doi: 10.1016/j.freeradbiomed.2007.02.031. [DOI] [PubMed] [Google Scholar]
- Ji LL, Gomez-Cabrera MC, Steinhafel N, Vina J. Acute exercise activates nuclear factor (NF)-kappaB signaling pathway in rat skeletal muscle. FASEB J. 2004;18:1499–1506. doi: 10.1096/fj.04-1846com. [DOI] [PubMed] [Google Scholar]
- Ji LL, Gomez-Cabrera MC, Vina J. Exercise and hormesis: activation of cellular antioxidant signaling pathway. Ann N Y Acad Sci. 2006;1067:425–435. doi: 10.1196/annals.1354.061. [DOI] [PubMed] [Google Scholar]
- Ji LL, Gomez-Cabrera MC, Vina J. Role of nuclear factor κB and mitogen-activated protein kinase signaling in exercise-induced antioxidant enzyme adaptation. Appl Physiol Nutr Metab. 2007;32:930–935. doi: 10.1139/H07-098. [DOI] [PubMed] [Google Scholar]
- Jones DP. Redefining oxidative stress. Antioxid Redox Signal. 2006;8:1865–1879. doi: 10.1089/ars.2006.8.1865. [DOI] [PubMed] [Google Scholar]
- Kabe Y, Ando K, Hirao S, Yoshida M, Handa H. Redox regulation of NF-κB activation: distinct redox regulation between the cytoplasm and the nucleus. Antioxid Redox Signal. 2005;7:395–403. doi: 10.1089/ars.2005.7.395. [DOI] [PubMed] [Google Scholar]
- Kanter M. Free radicals and exercise: effects of nutritional antioxidant supplementation. Exerc Sport Sci Rev. 1995;23:375–397. [PubMed] [Google Scholar]
- Kavazis AN, Talbert EE, Smuder AJ, Hudson MB, Nelson WB, Powers SK. Mechanical ventilation induces diaphragmatic mitochondrial dysfunction and increased oxidant production. Free Radic Biol Med. 2009;46:842–850. doi: 10.1016/j.freeradbiomed.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer HF, Goodyear LJ. Exercise, MAPK, and NF-κB signaling in skeletal muscle. J Appl Physiol. 2007;103:388–395. doi: 10.1152/japplphysiol.00085.2007. [DOI] [PubMed] [Google Scholar]
- Lin J, Wu H, Tarr PT, Zhang CY, Wu Z, Boss O, Michael LF, Puigserver P, Isotani E, Olson EN, Lowell BB, Bassel-Duby R, Spiegelman BM. Transcriptional co-activator PGC-1α drives the formation of slow-twitch muscle fibres. Nature. 2002;418:797–801. doi: 10.1038/nature00904. [DOI] [PubMed] [Google Scholar]
- Linder N, Rapola J, Raivio KO. Cellular expression of xanthine oxidoreductase protein in normal human tissues. Lab Invest. 1999;79:967–974. [PubMed] [Google Scholar]
- Lira VA, Brown DL, Lira AK, Kavazis AN, Soltow QA, Zeanah EH, Criswell DS. Nitric oxide and AMPK cooperatively regulate PGC-1 in skeletal muscle cells. J Physiol. 2010;588:3551–3566. doi: 10.1113/jphysiol.2010.194035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ljubicic V, Joseph AM, Saleem A, Uguccioni G, Collu-Marchese M, Lai RY, Nguyen LM, Hood DA. Transcriptional and post-transcriptional regulation of mitochondrial biogenesis in skeletal muscle: effects of exercise and aging. Biochim Biophys Acta. 1800:223–234. doi: 10.1016/j.bbagen.2009.07.031. [DOI] [PubMed] [Google Scholar]
- Loschen G, Azzi A, Richter C, Flohe L. Superoxide radicals as precursors of mitochondrial hydrogen peroxide. FEBS Lett. 1974;42:68–72. doi: 10.1016/0014-5793(74)80281-4. [DOI] [PubMed] [Google Scholar]
- Moylan JS, Reid MB. Oxidative stress, chronic disease, and muscle wasting. Muscle Nerve. 2007;35:411–429. doi: 10.1002/mus.20743. [DOI] [PubMed] [Google Scholar]
- Pantano C, Reynaert NL, Van Der Vliet A, Janssen-Heininger YM. Redox-sensitive kinases of the nuclear factor-κB signaling pathway. Antioxid Redox Signal. 2006;8:1791–1806. doi: 10.1089/ars.2006.8.1791. [DOI] [PubMed] [Google Scholar]
- Pilegaard H, Saltin B, Neufer PD. Exercise induces transient transcriptional activation of the PGC-1α gene in human skeletal muscle. J Physiol. 2003;546:851–858. doi: 10.1113/jphysiol.2002.034850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers SK, Criswell D, Lawler J, Ji LL, Martin D, Herb RA, Dudley G. Influence of exercise and fibre type on antioxidant enzyme activity in rat skeletal muscle. Am J Physiol Regul Integr Comp Physiol. 1994;266:R375–380. doi: 10.1152/ajpregu.1994.266.2.R375. [DOI] [PubMed] [Google Scholar]
- Powers SK, Criswell D, Lieu FK, Dodd S, Silverman H. Exercise-induced cellular alterations in the diaphragm. Am J Physiol Regul Integr Comp Physiol. 1992;263:R1093–1098. doi: 10.1152/ajpregu.1992.263.5.R1093. [DOI] [PubMed] [Google Scholar]
- Powers SK, Jackson MJ. Exercise-induced oxidative stress: cellular mechanisms and impact on muscle force production. Physiol Rev. 2008;88:1243–1276. doi: 10.1152/physrev.00031.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers SK, Kavazis AN, DeRuisseau KC. Mechanisms of disuse muscle atrophy: role of oxidative stress. Am J Physiol Regul Integr Comp Physiol. 2005;288:R337–344. doi: 10.1152/ajpregu.00469.2004. [DOI] [PubMed] [Google Scholar]
- Powers SK, Kavazis AN, McClung JM. Oxidative stress and disuse muscle atrophy. J Appl Physiol. 2007;102:2389–2397. doi: 10.1152/japplphysiol.01202.2006. [DOI] [PubMed] [Google Scholar]
- Puigserver P, Wu Z, Park CW, Graves R, Wright M, Spiegelman BM. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell. 1998;92:829–839. doi: 10.1016/s0092-8674(00)81410-5. [DOI] [PubMed] [Google Scholar]
- Ristow M, Zarse K, Oberbach A, Kloting N, Birringer M, Kiehntopf M, Stumvoll M, Kahn CR, Bluher M. Antioxidants prevent health-promoting effects of physical exercise in humans. Proc Natl Acad Sci U S A. 2009;106:8665–8670. doi: 10.1073/pnas.0903485106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers JT, Lerin C, Gerhart-Hines Z, Puigserver P. Metabolic adaptations through the PGC-1α and SIRT1 pathways. FEBS Lett. 2008;582:46–53. doi: 10.1016/j.febslet.2007.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell AP, Feilchenfeldt J, Schreiber S, Praz M, Crettenand A, Gobelet C, Meier CA, Bell DR, Kralli A, Giacobino JP, Deriaz O. Endurance training in humans leads to fibre type-specific increases in levels of peroxisome proliferator-activated receptor-γ coactivator-1 and peroxisome proliferator-activated receptor-α in skeletal muscle. Diabetes. 2003;52:2874–2881. doi: 10.2337/diabetes.52.12.2874. [DOI] [PubMed] [Google Scholar]
- Scarpulla RC. Nuclear control of respiratory gene expression in mammalian cells. J Cell Biochem. 2006;97:673–683. doi: 10.1002/jcb.20743. [DOI] [PubMed] [Google Scholar]
- St-Pierre J, Buckingham JA, Roebuck SJ, Brand MD. Topology of superoxide production from different sites in the mitochondrial electron transport chain. J Biol Chem. 2002;277:44784–44790. doi: 10.1074/jbc.M207217200. [DOI] [PubMed] [Google Scholar]
- Upham BL, Trosko JE. Oxidative-dependent integration of signal transduction with intercellular gap junctional communication in the control of gene expression. Antioxid Redox Signal. 2009;11:297–307. doi: 10.1089/ars.2008.2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urso ML, Clarkson PM. Oxidative stress, exercise, and antioxidant supplementation. Toxicology. 2003;189:41–54. doi: 10.1016/s0300-483x(03)00151-3. [DOI] [PubMed] [Google Scholar]
- Veal EA, Day AM, Morgan BA. Hydrogen peroxide sensing and signaling. Mol Cell. 2007;26:1–14. doi: 10.1016/j.molcel.2007.03.016. [DOI] [PubMed] [Google Scholar]
- Vega RB, Huss JM, Kelly DP. The coactivator PGC-1 cooperates with peroxisome proliferator-activated receptor α in transcriptional control of nuclear genes encoding mitochondrial fatty acid oxidation enzymes. Mol Cell Biol. 2000;20:1868–1876. doi: 10.1128/mcb.20.5.1868-1876.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent HK, Powers SK, Demirel HA, Coombes JS, Naito H. Exercise training protects against contraction-induced lipid peroxidation in the diaphragm. Eur J Appl Physiol Occup Physiol. 1999;79:268–273. doi: 10.1007/s004210050505. [DOI] [PubMed] [Google Scholar]
- Vincent HK, Powers SK, Stewart DJ, Demirel HA, Shanely RA, Naito H. Short-term exercise training improves diaphragm antioxidant capacity and endurance. Eur J Appl Physiol. 2000;81:67–74. doi: 10.1007/PL00013799. [DOI] [PubMed] [Google Scholar]
- Wright DC, Han DH, Garcia-Roves PM, Geiger PC, Jones TE, Holloszy JO. Exercise-induced mitochondrial biogenesis begins before the increase in muscle PGC-1α expression. J Biol Chem. 2007;282:194–199. doi: 10.1074/jbc.M606116200. [DOI] [PubMed] [Google Scholar]
- Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, Troy A, Cinti S, Lowell B, Scarpulla RC, Spiegelman BM. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell. 1999;98:115–124. doi: 10.1016/S0092-8674(00)80611-X. [DOI] [PubMed] [Google Scholar]
- Zhou LZ, Johnson AP, Rando TA. NF κB and AP-1 mediate transcriptional responses to oxidative stress in skeletal muscle cells. Free Radic Biol Med. 2001;31:1405–1416. doi: 10.1016/s0891-5849(01)00719-5. [DOI] [PubMed] [Google Scholar]