

Abstract
Abstract
Duchenne muscular dystrophy (DMD) is the most devastating type of muscular dystrophy, leading to progressive weakness of respiratory (e.g. diaphragm) and locomotor muscles (e.g. gastrocnemius). DMD is caused by X-linked defects in the gene that encodes for dystrophin, a key scaffolding protein of the dystroglycan complex (DCG) within the sarcolemmal cytoskeleton. As a result of a compromised dystroglycan complex, mechanical integrity is impaired and important signalling proteins (e.g. nNOS, caveolin-3) and pathways are disrupted. Disruption of the dystroglycan complex leads to high susceptibility to injury with repeated, eccentric contractions as well as inflammation, resulting in significant damage and necrosis. Chronic damage and repair cycling leads to fibrosis and weakness. While the link between inflammation with damage and weakness in the DMD diaphragm is unresolved, elevated oxidative stress may contribute to damage, weakness and possibly fibrosis. While utilization of non-specific antioxidant interventions has yielded inconsistent results, recent data suggest that NAD(P)H oxidase could play a pivotal role in elevating oxidative stress via integrated changes in caveolin-3 and stretch-activated channels (SACs). Oxidative stress may act as an amplifier, exacerbating disruption of the dystroglycan complex, upregulation of the inflammatory transcription factor NF-κB, and thus functional impairment of force-generating capacity.
Dr. John Lawler is a Professor in Health & Kinesiology and Biomedical Engineering at Texas A&M University. Dr. Lawler earned a BSE in Biomedical Engineering from Duke University and a PhD in Exercise Physiology and Physiology from the University of Florida. His laboratory currently focuses on redox regulation of the interface between the myocyte and extracellular matrix in (a) skeletal muscle wasting (e.g., disuse, aging, muscular dystrophy) and (b) remodeling and fibrosis in the aging heart. Dr. Lawler has 71 peer-review publications to his credit. He lives in College Station, TX with his wife Lynn, their children Amanda and Matthew, Daisy the blonde Labrador retriever, and an orange tabby who responds to name Baxter.
Pathophysiological significance
The subsarcolemmal cytoskeleton ensures mechanical resilience, includes proteins that serve as sensors of mechanical stress and strain, regulates satellite cell activation, and governs protein turnover (Rando, 2001). The initial triggers for dozens of muscular dystrophies stem from a mutation of a gene encoding for proteins normally located in the subsarcolemmal cytoskeleton (e.g. dystrophin, ß-dystroglycan, caveolin-3), including the dystroglycan complex (DGC) (Rando, 2001; Tidball & Wehling-Henricks, 2007). Duchenne muscular dystrophy (DMD) is the most common and devastating type of muscular dystrophy with an incidence of 1 in every 3500 males, as a result in a mutation of the dystrophin gene (Nakamura & Takeda, 2011; Spencer & Tidball, 2001). Symptoms of Duchenne muscular dystrophy usually appear between 3 years of age, and by the age of 12 patients are typically no longer able to breathe or walk unassisted (Escolar & Scacheri, 2001). Muscle wasting and weakness are especially critical for muscles of locomotion and respiratory muscles (e.g. diaphragm), with fast-twitch fibres being particularly susceptible to myopathy with DMD (Tkatchenko et al. 2000). Decline of diaphragm muscle function is directly related to the clinical need for mechanical ventilation in more advanced stages of DMD, and respiratory muscle failure remains a leading cause of death before the age of 30 (Escolar & Scacheri, 2001). The mdx mouse model, which also has a mutation of the dystrophin gene, is a common analogue for human Duchenne muscular dystrophy. The diaphragm muscle in the mdx mouse is highly susceptible to oxidative stress and experiences a disease progression similar to human DMD (Tkatchenko et al. 2000; Hartel et al. 2001).
The etiology of Duchenne muscular dystrophy is characterized by progressive damage, inflammation, fibrosis, and weakness of respiratory and limb skeletal muscles. Muscle damage and pathology with DMD are proposed to result from (a) high susceptibility to material fatigue injury and (b) chronic inflammation (Tidball & Wehling-Henricks, 2007). Material fatigue injuries occur in biological tissues, including skeletal muscle with repeated mechanical strain. Repeated lengthening or eccentric contractions of sufficient load and frequency induce a multifocal material fatigue injury in skeletal muscle (Warren et al. 1993). The load magnitude is often moderate, much lower than a maximal contraction, and material fatigue is believed to cause the initial damage phase associated with delayed-onset muscle soreness (Warren et al. 1993). Susceptibility to repetitive, eccentric-contraction damage and thus material fatigue injury is enhanced with DMD (Rousseau et al. 2010). Thus, a lower load magnitude in DMD muscles is required to induce damage with repeated eccentric contractions, possibly related to an impaired ability to transfer transverse or lateral forces (Ramaswamy et al. 2011).
Anti-inflammatory, corticosteroid drugs reduce oxidative stress, damage, apoptosis and disease progression with DMD (Lim et al. 2004), but have numerous side effects that limit their long-term use. While the link between inflammation with muscle damage and weakness in Duchenne muscular dystrophy is unresolved, elevated ‘oxidative stress’ has been proposed as a contributing mechanism (Tidball & Wehling-Henricks, 2007). Muscle damage, wasting and weakness have been noted with deficiencies in vitamin E and the Cu–Zn isoform of superoxide dismutase (Binder et al. 1965; Muller et al. 2006). Reduction in oxidative stress indeed correlates with slowed muscle wasting and relief of clinical symptoms with DMD, including respiratory muscle distress (Anderson et al. 2000; Bonafati et al. 2000; Carter & McDonald 2000). However, cause and effect were not established and the mechanisms by which glucocorticoids provide relief in DMD models remain uncertain. Perhaps oxidative stress may be upstream of nuclear factor-kappaB (NF-κB) and pro-inflammatory targets.
Oxidative stress and upregulation of the inflammatory transcription factor NF-κB are believed to contribute to myopathy during disuse, cachexia, chronic heart failure, chronic obstructive pulmonary disease, AIDS and cancer (Buck & Chojkier 1996; Adams et al. 1999; Dalla Libera et al. 2001; Lawler et al. 2003; Abrogast et al. 2007; Powers et al. 2007). While oxidative stress is elevated and integrated with inflammatory cell (macrophages, T-cells), cause and effect have remained uncertain with Duchenne muscular dystrophy (Tidball & Wehling-Henricks, 2007). Unfortunately, our understanding of the importance of redox signalling in DMD pathology has been partially obscured by the use of non-specific scavenger approaches and scientific designs that have yielded inconsistent results. For example, green tea extract (Buetler et al. 2002), purified epigallocatechin-3-gallate from green tea (Nakae, 2008), and low-iron diet (Bomman et al. 1998) reduced oxidative stress and muscle damage with DMD. In contrast, vitamin E, vitamin C and superoxide dismutase treatments have resulted in no clinical improvements (Walton & Nattrass, 1954; Roelofs et al. 1979; Fenichel et al. 1988; Stern et al. 1998). Inconsistency of non-specific antioxidant interventions in ameliorating Duchenne muscular dystrophy may lie within the nature of redox microenvironments of the sarcolemma (Fisher, 2009; Ushio-Fukai, 2009). For example, the reactive oxygen species (ROS)-generating NAD(P)H oxidase complex (Nox2) is elevated in the sarcolemma with both Duchenne muscular dystrophy and disuse (Williams & Allen, 2007).
Recent publications suggest that oxidative stress may play a more important role than previously understood. The antioxidant N-acetylcysteine was found to protect against extensor digitorum longus muscle (EDL) muscle damage, internal nuclei and weakness in locomotor muscles of mdx mice (Whitehead et al. 2008). NAD(P)H oxidase is upregulated in the mdx mouse, and thus a potential contributor to oxidative stress and DMD pathology (Williams & Allen, 2007; Spurney et al. 2008; Whitehead et al. 2008). Inhibition of NAD(P)H oxidase protected against delayed muscle development and apoptosis (Whitehead et al. 2008). It is possible that Nox2 may hyper-respond to stretch in DMD muscles, possibly as a result of upregulation of stretch-activated Ca2+ channels (SACs), including transient receptor potential channel 1 (TRPC1) and caveolin-3 (Whitehead et al. 2006; Gervásio et al. 2008). Indeed, new evidence indicates that Nox2 is activated by stretch and contributes to muscle damage in the tibialis anterior of mdx mice (Whitehead et al. 2010). Downstream of oxidative stress, NF-κB may exacerbate DGC disruption and muscle damage through matrix metalloproteinase-9 (MMP-9) (Li et al. 2009). A model is proposed that connects ROS to exacerbation of pathology of DMD in diaphragm and limb muscles via NAD(P)H oxidase activation of caveolin-3 and NF-κB (Fig. 1).
Figure 1. Redox signalling model for Duchenne muscular dystrophy (DMD), proposing oxidative stress as an amplifier of dystroglycan complex (DGC) disruption, muscle damage and pathology.
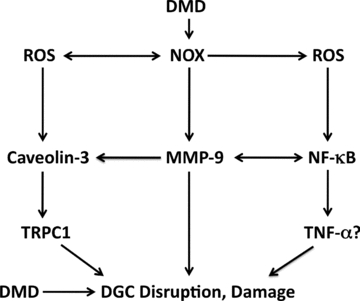
In this model, NAD(P)H oxidase and NOX contribute to upregulation of caveolin-3 and transient receptor potential channel 1 (TRPC1), as well as activation of nuclear factor kappaB (NF-κB). NF-kB may contribute to damage via tumour-necrosis factor-alpha (TNF-α). ROS, reactive oxygen species.
The subsarcolemmal cytoskeleton
Skeletal muscle is a highly specialized and adaptive mesodermic tissue capable of rapid remodelling in response to changes in loading and stretch (i.e. mechanotransduction) (Smith et al. 2002). The subsarcolemmal cytoskeleton ensures mechanical integrity of skeletal muscle and initiates cell signalling in response to alterations in mechanical stress (i.e. mechanotransduction) (Rando, 2001). The subsarcolemmal cytoskeleton is anchored to the sarcolemma where it (a) transfers forces between the muscle cells and extracellular matrix (ECM) proteins, and (b) initiates cell signalling responses to mechanical strain, endocrine influences and the chemical environment (Disatnik & Rando, 1999). The cytoskeleton forms two aggregates in skeletal muscle secured to extracellular proteins such as laminin: (a) the dystroglycan complex and (b) the focal adhesion complex (Yang et al. 1995; Rando, 2001). The dystroglycan complex includes α-,ß-dystroglycan, α-,ß-,γ-,∂-sarcoglycan subunits, biglycan, the scaffolding proteins dystrophin and dystrobrevin, utrophin, syncoilin, Grb2, α-,ß-syntrophin and neuronal nitric oxide synthase (nNOS), and is associated with caveolin-3. The dystroglycan complex plays a major role in linking the actin cytoskeleton to the extracellular matrix, stabilizing the myocyte during contraction and relaxation, and transmitting force generated in the muscle sarcomeres to the ECM. DGC proteins regulate cell signalling involved in mechanical strain, protein turnover, growth, blood flow and adhesion proteins, including satellite cell activation and protein turnover (Rando, 2001; Kosek & Bamman, 2008).
The dystrophin protein is a large (427 kD), rod-like scaffolding protein co-localized with α-syntrophin, nNOS and calmodulin in the DGC (Jarrett & Foster 1999; Rando, 2001; Tidball & Wehling-Henricks, 2007; Kosek & Bamman, 2008). Dystrophin appears be important in cytoskeleton-dependent stiffness and the elastic properties of skeletal muscle (Puttini et al. 2009). The μ-splice variant of nNOS is a key signalling protein integral to the DGC (Percival et al. 2008). nNOS is anchored to the sarcolemmal cytoskeleton by binding of a PDX motif at its N-terminus to α- or ß-syntrophin (Rando 2001; Compton et al. 2005; Fanin et al. 2009). Both α- and ß-syntrophin bind nNOS, which in turn attachs to dystrophin and dystrobrevin (Kameya et al. 1999; Rando, 2001; Jones et al. 2003; Compton et al. 2005). Caveolin-3 is a small muscle-specific sarcolemmal protein localized in caveolae, invaginations central to growth and insulin signalling (Fecchi et al. 2006). Caveolin-3 also binds to dystroglycan complex proteins including ß-dystroglycan and nNOS, possibly regulating DGC assembly (Venema et al. 1997; Galbiati et al. 2001; Allen et al. 2010). Caveolin-3 inhibits nNOS via distinct scaffolding domains (Venema et al. 1997; Sunada et al. 2001; Whitehead et al. 2008), and also binds to transient receptor potential channel-1 (TRPC1), a stretch-activated Ca2+ channel (Gervásio et al. 2008). Either upregulation of caveolin-3 or suppression via genetic ablation exacerbates Duchenne muscular dystrophy or causes limb-girdle muscular dystrophy (LGMD-1C) (Galbiati et al. 2001).
Etiology of Duchenne muscular dystophy
Disturbances in the dystrglycan complex, including loss of dystrophin and attached proteins (e.g. ß-dystroglycan, α-syntrophin, nNOS), reduce the mechanical integrity of skeletal muscle eliciting a lower threshold to injuries with repeated stretch or eccentric contractions (Warren et al. 1993; Childers et al. 2002). DMD compromises the mechanical resilience of the cytoskeleton and cellular membrane to repeated mechanical strain resulting in material fatigue injury to muscles (Warren et al. 1993; Rando, 2001). Thus, muscles in DMD patients and animal models display a lower threshold for injury with repeated stretch or eccentric contractions (Childers et al. 2002; Rousseau et al. 2010). Importantly, Ramaswamy et al. (2011) recently published exciting new data that suggested that the dystroglycan complex is the weak link in transfer of lateral or transverse forces from the contractile apparatus with Duchenne muscular dystrophy, in a manner shared with the ageing process.
Loss of nNOS from the sarcolemma appears central to damage and myopathy with DMD as well (Tidball & Wehling-Henricks, 2004; Kim, 2009). Canonical thought is that damage leads to necrosis, apoptosis and weakness in Duchenne muscular dystrophy (Rando, 2001; Tidball & Wehling-Henricks, 2007). Apoptosis and necrosis may lead to loss of satellite cells, myonuclei and muscle fibres with a profound effect on muscle weakness, wasting, impaired muscle regeneration and etiology of the disease (Sandri et al. 2001; Mikhailov et al. 2002; Sandri & Carraro, 2002). An alternative hypothesis suggests that disruption of the dystroglycan complex elevates stretch-activated channels such as TRPC1, and elicits a Ca2+-driven cascade (Allen et al. 2010). In addition, caveolin-3 may exacerbate loss of DGC proteins and disease pathogensis, possibly via TRPC1-regulated Ca2+ release (Gervásio et al. 2008).
Recent evidence suggests that matrix metalloproteinase-9 (MMP-9) cleaves ß-dystroglycan and exacerbates loss of DGC proteins from the sarcolemma, enhancing muscle damage with DMD (Li et al. 2009). Resultant muscle damage invites infiltration of inflammatory cells, primarily macrophages and T-cells, and initiates necrosis and apoptosis (Spencer & Tidball, 2001). Necrosis and apoptosis may remove satellite cells, myonuclei and muscle fibres, with a profound effect on muscle contractility and regeneration (Sandri et al. 2001; Mikhailov et al. 2002; Sandri & Carraro, 2002). Thus, alterations in the DCG also may impair regeneration and healing (Tidball & Wehling-Henricks, 2007). With repeated chronic cycling of damage and repair, fibrotic tissue fills in lost sarcomeres and muscle fibres over time (Gosselin et al. 1994; Gosselin & Martinez, 2004). As damage–repair cycling continues, a catenation of Wg (wingless), from wing development in Drosophila and Int, a homologous breast tumor gene (wnt) signalling and satellite cell function are impaired and satellite cells display shortened telomeres, and may differentiate to adipocytes, fibroblasts or cease to divide (Alexakis et al. 2007; Lund et al. 2007; Pescatori et al. 2007). Differentiation and reduction of satellite cell activation may exacerbate weakening and wasting of muscle fibres, further impairing function and thus respiratory muscle failure.
DMD and inflammation
Increasing evidence indicates inflammatory processes are highly integrated into muscle wasting with DMD (Spencer & Tidball, 2001). Autoreactive immune cells including T-cells and macrophages invade skeletal muscle in DMD (Spencer & Tidball, 2001). Indeed, CD4+ and CD8+ T-cell depletion decreases histopathology of mdx mouse muscle (Spencer & Tidball, 2000). Circulating inflammatory cytokines may be a 1000 times higher in DMD human patients than healthy controls (Watanabe, 2001). Stretch, damage, cytokines, inflammation and oxidative stress activate the transcription factor NF-κB through the phosphorylation and release of the inhibitor protein I-κB (inhibitory kappaB) (Gius et al. 1999). Indeed, the inflammatory cytokines tumour necrosis factor-α (TNF-α), transforming growth factor-beta (TGF-ß) and interleukin 1ß as well as the inflammatory transcription factor NF-κB are significantly elevated with DMD and in the mdx mouse (Kumar & Boriek, 2003; Monici et al. 2003). NF-κB translocates to the nucleus where it binds with DNA (i.e. activation), and then amplifies release of ROS and pro-inflammatory proteins and peptides (Crépieux et al. 1997; Kumar et al. 2004). Indeed, Monici et al. (2003) and Boriek and colleagues (Kumar & Boriek, 2003, Kumar et al. 2004) reported that NF-κB activity was higher in limb muscle and the diaphragm of mdx mice compared with wild-types. As a consequence, interventions that reduce NF-κB in DMD models significantly reduce damage and pathology (Nakae et al. 2001; Lim et al. 2004; Messina et al. 2006; Peterson et al. 2011). While the mechanisms remain unclear, NF-κB is linked with necrosis, activation of ubiquitin ligases and muscle wasting (Messina et al. 2006; Senf et al. 2008).
Cyclic damage and inflammation can also promote fibrosis or accumulation of connective tissue proteins such as collagen (Gosselin & Martinez, 2004). Collagen accumulation with repeated damage and repair is exacerbated by inflammatory cytokines (e.g. TNF-α, TGF-ß) and oxidative stress (Gosselin & Martinez, 2004). Collagen turnover is regulated by MMPs and by upstream tissue inhibitors of MMPs (TIMPs) (Kassiri & Khokha, 2005).
Anti-inflammatory and immunosuppressant drugs such as glucocorticoids (e.g. prednisone and deflazacort) consistently reduce skeletal muscle weakness, damage and progression of DMD in patients (Anderson et al. 2000; Bonafati et al. 2000; Carter & McDonald 2000; Sussman 2002). Immunosuppressants such as cyclosporine A reduce muscle damage in mdx mice (De Luca et al. 2005). Corticosteroids also reduce oxidative stress (Tarnopolsky et al. 2004). Unfortunately, steroidal anti-inflammatories cause serious side-effects including impaired growth and maturation, weight gain, osteopaenia, immunosuppression and susceptibility to infection (Carter & McDonald 2000; Skrabek & Anderson, 2001).
Oxidative stress and DMD
Sources of oxidative stress in respiratory and locomotor muscles with DMD are thought to include infiltration of inflammatory cells (e.g. myeloperoxidase), NAD(P)H oxidase, mitochondria and decoupling of inducible nitric oxide synthase (iNOS) (Adams et al. 1999; Williams & Allen 2007); Spurney et al. 2008; Tidball & Wehling-Henricks, 2007; Whitehead et al. 2008. Furthermore, oxidative stress may also be exacerbated in muscle wasting disease by insufficient stress response including heat shock proteins and insulin-like growth factor (Bouchentouf et al. 2004; Lawler et al. 2006; Senf et al. 2008). Elevated oxidative stress may promote inflammatory cell invasion, exacerbate damage and interfere with cell signalling that can promote repair (Tidball & Wehling-Henricks, 2007). Markers of oxidative stress and lipid peroxidation are elevated with DMD (Grinio et al. 1984; Tidball & Wehling-Henricks, 2007). Muscles in Duchenne muscular dystrophy patients may also be more susceptible to oxidative stress. For example, Rando et al. (1998) showed that mdx myotubes are killed more easily by oxidants. He proposed a ‘two-hit’ hypothesis where the combination of oxidative stress plus disturbances in the dystroglycan complex lead to pathology with DMD.
Downregulation and dislocation of nNOS from the dystroglycan complex may also elevate oxidative stress and lead to muscle damage (Wehling et al. 2001; Nguyen & Tidball, 2003; Shiao et al. 2004; Tidball & Wehling-Henricks, 2004). nNOS in skeletal muscle appears to be attached to dystroglycan complex scaffolding proteins dystrophin and dystrobrevin via α- and ß-syntrophin (Rando, 2001). Dislocation of nNOS from the dystroglycan complex could (a) increase NAD(P)H oxidase activity, (b) increase inflammation, (c) increase protein degradation via activation of ubiquitin ligases, (d) impair satellite cell activation, and (e) increase the risk of material fatigue injury (Nguyen & Tidball, 2003; Tidball & Wehling-Henricks, 2004; Tidball & Wehling-Henricks, 2007; Suzuki et al. 2008). Further, impairment of mitochondrial function with DMD can also lead to oxidative stress, apoptosis and necrosis (Bernardi, 1999).
The Nox2 isoform of NAD(P)H oxidase contains membrane-bound gp91phox, p22phox and the cytosolic subunits p47phox and p67phox (Nguyen & Tidball, 2003; Whitehead et al. 2008). NAD(P)H oxidase is a source of oxidative stress, localized in cell membranes and inflammatory cells (Nguyen & Tidball, 2003). While NAD(P)H oxidase releases superoxide anions (O2•−) into the interstitial space, O2•− may enter a cell easily via chloride channel-3 (ClC3) while H2O2, following dismutation, diffuses across cell membranes facilitated by aquaporin channels (Fisher 2009). Mounting data suggest that NAD(P)H oxidase is an important contributor of skeletal muscle pathology and muscle wasting including mechanical ventilation (McClung et al. 2007) and ageing (Vasilaki et al. 2006). Further, knockout of the NAD(P)H oxidase regulatory subunit gp91phox reduces membrane and fibre damage during reloading following hindlimb unloading (Nguyen & Tidball, 2003). Importantly, elevations of NAD(P)H oxidase have recently been identified in the heart and muscles of mdx mice that indeed may play a significant role in muscle damage, weakness and wasting (Williams & Allen, 2007; Spurney et al. 2008).
Recent evidence suggests that oxidative stress may sensitise stretch-activated Ca2+ channels, including TRPC1, thus upregulating caveolin-3 (Allen et al. 2010). Now evidence suggests NAD(P)H oxidase is involved in response to stretch and damage in mdx mice (Whitehead et al. 2010). Upregulation of caveolin-3 is believed to contribute to enhanced dislocation of DGC proteins with DMD, including nNOS dislocation from the DGC (Gervásio et al. 2008). MMP-9 cleaves ß-dystroglycan from α-dystroglycan, and is potentially under redox modulation via NF-κB (Li et al. 2009).
Data from Suzuki et al. (2007) implicated disruption in DGC integrity and nNOS dissociation with activation of muscle wasting. DGC disruption may lead to muscle wasting with other models. Ageing results in dissociation of dystrophin (Rice et al. 2006) from the sarcolemmal and reduces nNOS (Song et al. 2009), linked with sarcopenia and weakness (Rice et al. 2006; Marzetti et al. 2007, Kim 2009). Indeed, a very new publication reported that the dystroglycan is the mechanical weak link in the transfer of transverse forces linking the contractile apparatus and z-disc with ageing (Ramaswamy et al. 2011). Therefore, disruption of the DGC and subsarcolemmal cytoskeleton may be a shared signalling mechanism that elicits myopathy across a number of pathologies.
The diaphragm muscle in particular suffers from significant fibrosis, damage and weakness with DMD, and is also susceptible to oxidative stress (Tkatchenko et al. 2000; Van Gammeren et al. 2004). Oxidant production may be higher in the mdx diaphragm than limb muscles, which could contribute to more profound fibrosis, weakness and fatigue in that muscle (Stevens & Faulkner, 2000; Hartel et al. 2001). In addition, the diaphragm muscle in the mdx genetic mouse model experiences muscle damage, disease progression and gene expression profiles similar to human DMD pathology (Tkatchenko et al. 2000; Tidball & Wehling-Henricks, 2007). Therefore, the study of the mechanisms by which oxidative stress causes pathology in the mdx diaphragm, and development and testing of targeted, antioxidant therapeutics is vital in translation to human health and pathology with DMD.
Targeted antioxidant therapeutic development and DMD
Oxidative stress is a potential upstream contributor to inflammatory signalling and DMD pathology, and thus a therapeutic target (Tidball & Wehling-Henricks, 2007; Whitehead et al. 2008). Reduction in oxidative stress indeed correlates with slowed muscle wasting and relief of clinical symptoms with DMD including respiratory muscle distress (Anderson et al. 2000; Bonafati et al. 2000; Carter & McDonald 2000). Sources of oxidative stress with DMD include inflammatory cells (e.g. myeloperoxidase), NAD(P)H oxidase, mitochondria and decoupling of nitric oxide synthases (Adams et al. 1999; Tidball & Wehling-Henricks, 2007; Williams & Allen, 2007; Spurney et al. 2008; Whitehead et al. 2008). Dislocation of nNOS from the dystroglycan complex could also (a) increase NAD(P)H oxidase activity, (b) increase inflammation, (c) increase protein degradation via activation of ubiquitin ligases, (d) impair satellite cell activation, and (e) increase the risk of material fatigue injury (Nguyen & Tidball, 2003; Tidball & Wehling-Henricks, 2004; Suzuki et al. 2007; Tidball & Wehling-Henricks, 2007).
Unfortunately, non-specific scavenger approaches have yielded mixed and confounding results in ameliorating DMD pathology. Green tea extracts including epigallocatechin-3-gallate (Buetler et al. 2002; Nakae et al. 2008), low-iron diet (Bomman et al. 1998) and N-acetylcysteine (Whitehead et al. 2008) protected against muscle damage, while tocopherols, ascorbate and penicillamine treatments (Walton & Nattrass 1954; Roelofs et al. 1979; Stern et al. 1982; Fenichel et al. 1988) elicited no improvements. Only more recently has the tie between oxidative stress and pathology in muscles in Duchenne muscular dystrophy moved beyond association.
Loss of nNOS from the sarcolemma may have a profound effect on oxidative stress, mechanical integrity and capability for satellite cell activation, growth and repair (Suzuki et al. 2007; Tidball & Wehling-Henricks, 2007). Tidball and colleagues (Wehling et al. 2001; Tidball & Wehling-Henricks, 2004) reported that overexpression of nNOS increased nNOS localization at the sarcolemma, and partially rescued ß-dystroglycan, α-sarcoglycan and ß-sarcoglycan, concomitant with reduced oxidative stress and muscle damage in mdx mice. Thus, interventions that stabilize sarcolemmal localization of nNOS and other DGC proteins may prove effective in ameliorating DMD.
Whitehead et al. (2008) demonstrated that 6 week treatment of the antioxidant N-acetylcysteine was found to protect against damage, incidence of internal nuclei and weakness in the EDL muscle of 8-week-old mdx mice. In addition, N-acetylcysteine also increased protein levels of the ß-dystroglycan and utrophin while caveolin-3 was reduced by N-acetylcysteine. N-acetylcysteine also reduced dihydroethidine oxidation (marker of oxidative stress) and protein expression of the p65 subunit of NF-κB (Whitehead et al. 2008), an inflammatory transcription factor that also stimulates proteolysis via the ubiquitin ligase (muscle ring finger-1: MuRF1, atrogin-1) activation. N-acetylcysteine treatment also reduced macrophage invasion and collagen accumulation in the hearts of mdx mice (Williams & Allen, 2007). N-acetylecysteine also attenuated the elevation of caveolin-3 protein expression noted in the EDL of mdx mice.
New evidence indicates that NAD(P)H oxidase is upregulated in the mdx mouse, and is thus a potential source of oxidative stress and pathology (Williams & Allen, 2007; Spurney et al. 2008; Whitehead et al. 2008). Inhibition of NAD(P)H oxidase appears to reduce markers of apoptosis in the diaphragm of mdx mice by elevating Bcl-2 (B-cell lymphoma 2) and BAG-4 (Bcl-2-associated athanogene-4) are mitochondrial-expressed stress proteins that are anti-apoptotic. in the diaphragm (Kwak et al. 2008). Nox may be an important regulator of DMD pathology; however, additional studies are required.
Inhibition of TRPC1 by streptomycin reduces muscle damage and depresses muscle contractility in mdx mice (Whitehead et al. 2006; Gervásio et al. 2008). TRPC1 appears to co-localize with caveolin-3, and both are upregulated in DMD (Gervásio et al. 2008). Indeed, TRPC1 upregulation was dependent on caveolin-3. H2O2 increased TRPC1 protein expression in myotubes, suggesting that upregulation of TPRC1 via caveolin-3 may be redox dependent (Gervásio et al. 2008).
Li et al. (2009) reported that matrix MMP-9 appears to be upstream of NF-κB activation. Inhibition or genetic ablation of MMP-9 reduced muscle membrane damage and necrosis. Furthermore, MMP-9 knockout rescued ß-dystroglycan protein expression and reduced caveolin-3 with a small increase in nNOS levels. Although uncertain, MMP-9 may be a nexus of control in the regulation of caveolin-3 and amplification of DMD-induced disruption of DGC integrity, inflammation and myopathy.
Green tea extract, which contains antioxidant compounds such as epigallocatechin-3-gallate (EGCG), reduced oxidative stress in a dose-dependent manner and attenuated the number of necrotic and regenerating fibres in the EDL muscle (Buetler et al. 2002). Injection of EGCG extract reduced oxidative stress, lipofuscin, membrane damage (creatine kinase leakage) and necrosis (Nakae et al. 2008). Long-term EGCG treatment also blunts NF-κB activity (Evans et al. 2010). EGCG is a powerful antioxidant and inhibitor of NF-κB (Aggarwal & Shishodia, 2004, Ichiyanagi et al. 2004), and thus offers a more targeted therapeutic strategy than many nutraceuticals.
In conclusion, emerging evidence is suggesting that oxidative stress may be an important amplifier of pathology in respiratory and limb muscles with Duchenne muscular dystrophy. Based upon recent evidence, a cascade involving TRPC1, NAD(P)H oxidase, caveolin-3 and NF-κB may contribute to damage, inflammation and impaired contractile function in skeletal muscles of patients suffering from DMD. Pharmaceutical developments that target specific mechanisms elevating oxidative stress with DMD could attenuate myopathy and provide relief for patients.
Acknowledgments
Support has been provided by NIH (AR054084), and the Sydney and J.L. Huffines Institute for Sports Medicine.
Glossary
Abbreviations
- DGC
dystroglycan complex
- DMD
Duchenne muscular dystrophy
- ECM
extracellular matrix
- MMP
matrix metalloproteinase
- ROS
reactive oxygen species
- SAC
stretch-activated channel
- TGF
transforming growth factor
- TNF
tumour necrosis factor
- TRPC1
transient receptor potential canonical 1
References
- Abrogast S, Smith J, Matuszczak JY, Hardin BJ, Moylan JS, Smith JD, et al. Bowman-Birk inhibitor concentrate prevents atrophy, weakness, and oxidative stress in soleus muscle of hindlimb-unloaded mice. J Appl Physiol. 2007;102:956–964. doi: 10.1152/japplphysiol.00538.2006. [DOI] [PubMed] [Google Scholar]
- Adams V, Jiang H, Yu J, Möbius-Winkler S, Fiehn E, Linke A, et al. Apoptosis in skeletal muscle myocytes of patients with chronic heart failure is associated with exercise intolerance. J Am Coll Cardiol. 1999;33:959–965. doi: 10.1016/s0735-1097(98)00626-3. [DOI] [PubMed] [Google Scholar]
- Aggarwal BB, Shishodia S. Suppression of the nuclear factor-κB activation pathway by spice-derived phytochemicals: reasoning for seasoning. Ann N Y Acad Sci. 2004;1030:434–441. doi: 10.1196/annals.1329.054. [DOI] [PubMed] [Google Scholar]
- Alexakis C, Partridge T, Bou-Gharios G. Implication of the satellite cell in dystrophic muscle fibrosis: a self-perpetuating mechanism of collagen overproduction. Am J Physiol Cell Physiol. 2007;293:C661–C669. doi: 10.1152/ajpcell.00061.2007. [DOI] [PubMed] [Google Scholar]
- Allen DG, Gervasio OL, Yeung EW, Whitehead NP. Calcium and the damage pathways in muscular dystrophy. Can J Physiol Pharmacol. 2010;88:83–91. doi: 10.1139/Y09-058. [DOI] [PubMed] [Google Scholar]
- Anderson JE, Weber M, Vargas C. Deflazacort increases laminin expression and myogenic repair, and induces early persistent functional gain in mdx mouse muscular dystrophy. Cell Transplant. 2000;9:551–564. doi: 10.1177/096368970000900411. [DOI] [PubMed] [Google Scholar]
- Bernardi P. Mitochondria in muscle cell death. Ital J Neurol Sci. 1999;20:395–400. doi: 10.1007/s100720050057. [DOI] [PubMed] [Google Scholar]
- Binder HJ, Herting DC, Hurst V, Finch SC, Sprio HM. Tocopherol deficiency in man. N Engl J Med. 1965;273:1289–1297. doi: 10.1056/NEJM196512092732401. [DOI] [PubMed] [Google Scholar]
- Bomman L, Rossouw H, Gericke GS, Polla BS. Effects of iron deprivation on the pathology and stress protein expression in murine X-linked muscular dystrophy. Biochem Pharmacol. 1998;56:751–757. doi: 10.1016/s0006-2952(98)00055-0. [DOI] [PubMed] [Google Scholar]
- Bonafati MD, Ruzza G, Bonometto P, Bernardinelli A, Gorni K, Orcesi S, Lanzi G, Angelini C. A multicenter, double-blind, randomized trial of deflazacort versus prednisone in Duchenne muscular dystrophy. Muscle Nerve. 2000;23:1344–1347. doi: 10.1002/1097-4598(200009)23:9<1344::aid-mus4>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- Bouchentouf M, Benabdallah BF, Tremblay JP. Myoblast survival enhancement and transplantation success improvement by heat-shock treatment in mdx mice. Transplantation. 2004;77:1349–1356. doi: 10.1097/01.tp.0000121503.01535.f5. [DOI] [PubMed] [Google Scholar]
- Buck M, Chojkier M. Muscle wasting and dedifferentiation induced by oxidative stress in a model of cachexia is prevented by inhibitors of nitric oxide synthesis and antioxidants. EMBO J. 1996;15:1753–1765. [PMC free article] [PubMed] [Google Scholar]
- Buetler TM, Renard M, Offord EA, Schneider H, Ruegg UT. Green tea extract decreases muscle necrosis in mdx mice and protects against reactive oxygen species. Am J Clin Nutr. 2002;75:749–753. doi: 10.1093/ajcn/75.4.749. [DOI] [PubMed] [Google Scholar]
- Carter GT, McDonald CM. Preserving function in Duchenne dystrophy with long-term pulse prednisone therapy. Am J Phys Med Rehabil. 2000;79:455–458. doi: 10.1097/00002060-200009000-00009. [DOI] [PubMed] [Google Scholar]
- Childers MK, Okamura CS, Bogan DJ, Bogan JR, Petroski GF, McDonald K, Kornegay JN. Eccentric contraction injury in dystrophic canine muscle. Arch Phys Med Rehabil. 2002;83:1572–1578. doi: 10.1053/apmr.2002.35109. [DOI] [PubMed] [Google Scholar]
- Compton AG, Cooper ST, Hill PM, Yang N, Froehner SC, North KN. The syntrophin-dystrobrevin subcomplex in human neuromuscular disorders. J Neuropathol Exp Neurol. 2005;64:350–361. doi: 10.1093/jnen/64.4.350. [DOI] [PubMed] [Google Scholar]
- Crépieux P, Kwon H, Leclerc N, Spencer W, Richard S, Lin R, Hiscott J. IκBα physically interacts with a cytoskeleton-associated protein through its signal response domain. Mol Cell Biol. 1997;17:7375–7385. doi: 10.1128/mcb.17.12.7375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Criswell DS. Trolox attenuates mechanical ventilation-induced contractile Suppl. 2004. [DOI] [PubMed]
- Dalla-Libera L, Sabbadini R, Renken C, Ravara B, Sandri M, Betto R, et al. Apoptosis in the skeletal muscle of rats with heart failure is associated with increased serum levels of TNF-α and sphingosine. J Mol Cell Cardiol. 2001;33:1871–1878. doi: 10.1006/jmcc.2001.1453. [DOI] [PubMed] [Google Scholar]
- DeLuca A, Nico B, Liatonio A, Didonna MP, Fraysse B, Pierno S, Burdi R, Mangieri D, Rolland JF, Camerino C, Zallone A, Confalonieri P, Andreetta F, Arnoldi E, Courdier-Fruh I, Magyar JP, Frigeri A, Pisoni M, Svelto M, Camerino DC. A mulitidisciplinary evaluation of the effectiveness of cyclosporine A in dystrophic mdx mice. Am J Pathol. 2005;166:477–489. doi: 10.1016/S0002-9440(10)62270-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Disatnik M-H, Rando TA. Integrin-mediated muscle cell spreading. J Biol Chem. 1999;274:32486–32492. doi: 10.1074/jbc.274.45.32486. [DOI] [PubMed] [Google Scholar]
- Escolar DM, Scacheri CG. Pharmacologic and genetic therapy for childhood muscular dystrophies. Curr Neurol Neurosci Rep. 2001;1:168–174. doi: 10.1007/s11910-001-0013-y. [DOI] [PubMed] [Google Scholar]
- Evans NP, Misyak SA, Robertson JL, Bassaganya-Riera J, Robert W, Grange RW. Immune-mediated mechanisms potentially regulate the disease time course of Duchenne muscular dystrophy and provide targets for therapeutic intervention. PM R. 2010;1:755–768. doi: 10.1016/j.pmrj.2009.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fecchi K, Volonte D, Hezel MP, Schmeck K, Galbiati F. Spatial and temporal regulation of GLUT4 translocation by flotillin-1 and caveolin-3 in skeletal muscle cells. FASEB J. 2006;20:705–707. doi: 10.1096/fj.05-4661fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenichel GM, Brooke MH, Griggs RC, Mendell JR, Miller JP, Moxley RT, 3rd, et al. Clinical investigation in Duchenne muscular dystrophy: penicillamine and vitamin E. Muscle Nerve. 1988;11:1164–1168. doi: 10.1002/mus.880111110. [DOI] [PubMed] [Google Scholar]
- Fanin M, Tasca E, Nascimbeni AC, Angelini C. Sarcolemmal neuronal nitric oxide synthase defect in limb-girdle muscular dystrophy: an adverse modulating factor in the disease course? J Neuropathol Exp Neurol. 2009;68:383–390. doi: 10.1097/NEN.0b013e31819cd612. [DOI] [PubMed] [Google Scholar]
- Fisher AB. Redox signaling across cell membranes. Antioxid Redox Signal. 2009;11:1349–1356. doi: 10.1089/ars.2008.2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galbiati F, Razani B, Lisanti MP. Caveolae and caveolin-3 in muscular dystrophy. Trends Mol Med. 2001;7:435–441. doi: 10.1016/s1471-4914(01)02105-0. [DOI] [PubMed] [Google Scholar]
- Gervásio OL, Whitehead NP, Yeung EW, Phillips WD, Allen DG. TRPC1 binds to caveolin-3 and is regulated by Src kinase – role in Duchenne muscular dystrophy. J Cell Sci. 2008;121:2246–2255. doi: 10.1242/jcs.032003. [DOI] [PubMed] [Google Scholar]
- Girgenrath M, Dominov JA, Kostek CA, Miller JB. Inhibition of apoptosis improves outcome in a model of congential muscular dystrophy. J Clin Invest. 2004;114:1635–1639. doi: 10.1172/JCI22928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gius D, Botero A, Shah S, Curry HA. Intracellular oxidation/reduction status in the regulation of transcription factors NF-kB and AP-1. Toxicol Lett. 1999;106:93–106. doi: 10.1016/s0378-4274(99)00024-7. [DOI] [PubMed] [Google Scholar]
- Gosselin LE, Martinez DA. Impact of TNF-α blockade on TGF-β1 and type I collagen mRNA expression in dystrophic muscle. Muscle Nerve. 2004;30:244–246. doi: 10.1002/mus.20056. [DOI] [PubMed] [Google Scholar]
- Gosselin LE, Martinez DA, Vailas AC, Sieck GC. Passive length-force properties of senescent diaphragm: relationship with collagen characteristics. J Appl Physiol. 1994;76:2680–2685. doi: 10.1152/jappl.1994.76.6.2680. [DOI] [PubMed] [Google Scholar]
- Grinio LP, Orlov ON, Prilipko LL, Kagan VE. Lipid peroxidation in children with Duchenne's hereditary myopathy (in Russian) Biull Eksp Biol Med. 1984;98:423–425. [PubMed] [Google Scholar]
- Hartel JV, Granchelli JA, Hudecki MS, Pollina CM, Gosselin LE. Impact of prednisone on TGF-β1 and collagen in diaphragm muscle from mdx mice. Muscle Nerve. 2001;24:428–432. doi: 10.1002/1097-4598(200103)24:3<428::aid-mus1018>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- Ichiyanagi T, Hatano Y, Matsuo S, Konishi T. Simultaneous comparison of relative reactivities of twelve major anthocyanins in bilberry towards reactive nitrogen species. Chem Pharm Bull. 2004;52:1312–1315. doi: 10.1248/cpb.52.1312. [DOI] [PubMed] [Google Scholar]
- Jarrett BW, Foster JL. Alternative binding of actin and calmodulin to multiple sites on dystrophin. J Biol Chem. 1999;270:5578–5586. doi: 10.1074/jbc.270.10.5578. [DOI] [PubMed] [Google Scholar]
- Jones KJ, Compton AG, Yang N, Mills MA, Peters MF, Mowat D, et al. Deficiency of the syntrophins and a-dystrobrevin in patients with inherited myopathy. Neuromuscul Disord. 2003;13:456–467. doi: 10.1016/s0960-8966(03)00066-x. [DOI] [PubMed] [Google Scholar]
- Kameya S, Miyagoe Y, Nonaka I, Ikemoto T, Endo M, Hanaoka K, Nabeshima Y, Takeda S. α1-syntrophin gene disruption results in the absence of neuronal-type nitric-oxide synthase at the sarcolemma but does not induce muscle degeneration. J Biol Chem. 1999;274:2193–2200. doi: 10.1074/jbc.274.4.2193. [DOI] [PubMed] [Google Scholar]
- Kassiri Z, Khokha R. Myocardial extra-cellular matrix and its regulation by metalloproteinases and their inhibitors. Thromb Haemost. 2005;93:212–219. doi: 10.1160/TH04-08-0522. [DOI] [PubMed] [Google Scholar]
- Kim HB. Texas A&M University; 2009. Effect of the catalase/superoxide dismutase mimetic EUK-134 on damage, inflammation, and force generation on the diaphragm muscle of mdx mice. Doctoral Dissertation. [Google Scholar]
- Kosek D, Bamman M. Modulation of the dystrophin-associated protein complex in response to resistance training in young and older men. J Appl Physiol. 2008;104:1476–1484. doi: 10.1152/japplphysiol.00708.2007. [DOI] [PubMed] [Google Scholar]
- Kumar A, Boriek AM. Mechanical stress activates nuclear factor-κB pathway in skeletal muscle fibres: a possible role in Duchenne muscular dystrophy. FASEB J. 2003;17:386–396. doi: 10.1096/fj.02-0542com. [DOI] [PubMed] [Google Scholar]
- Kumar A, Takada Y, Boriek AM, Aggarwal BB. Nuclear factor-kB: its role in health and disease. J Mol Med. 2004;82:434–448. doi: 10.1007/s00109-004-0555-y. [DOI] [PubMed] [Google Scholar]
- Kwak HB, Kim JH, Lawler JM. NAD(P)H oxidase inhibition upregulates anti-apoptotic BAG-4 protein expression in the mdx diaphragm. FASEB J. 2008;22:959.8. abstract. [Google Scholar]
- Lawler JM, Song W, Demaree SR. Hindlimb unloading increases oxidative stress and disrupts antioxidant capacity in skeletal muscle. Free Rad Biol Med. 2003;35:9–16. doi: 10.1016/s0891-5849(03)00186-2. [DOI] [PubMed] [Google Scholar]
- Lawler JM, Song W, Kwak HB. Differential regulation of heat shock proteins by hindlimb unloading and reloading in the rat soleus. Muscle Nerve. 2006;33:200–207. doi: 10.1002/mus.20454. [DOI] [PubMed] [Google Scholar]
- Li H, Mittal A, Makonchuk DY, Bhatnagar S, Kumar A. Matrix metalloproteinase-9 inhibition ameliorates pathogenesis and improves skeletal muscle regeneration in muscular dystrophy. Hum Mol Genet. 2009;18:2584–2598. doi: 10.1093/hmg/ddp191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim JH, Kim DY, Bang MS. Effects of exercise and steroid on skeletal muscle apoptosis in the mdx mouse. Muscle Nerve. 2004;30:456–462. doi: 10.1002/mus.20139. [DOI] [PubMed] [Google Scholar]
- Lund TC, Grange RW, Lowe DA. Telomere shortening in diaphragm and tibialis anterior muscles of aged mdx mice. Muscle Nerve. 2007;36:387–390. doi: 10.1002/mus.20824. [DOI] [PubMed] [Google Scholar]
- Marzetti E, Privitera G, Simili V, Wohlgemuth SE, Aulisa L, Pahor M, Leeuwenburgh C. Multiple pathways to the same end: mechanisms of myonuclear apoptosis in sarcopenia of aging. Sci World J. 2010;10:340–349. doi: 10.1100/tsw.2010.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClung JM, Kavazis AN, DeRuisseau KC, Falk DJ, Deering MA, Lee Y, Sugiura T, Powers SK. Caspase-3 regulation of diaphragm myonuclear domain during mechanical ventilation-induced atrophy. Am J Respir. 2007;175:150–159. doi: 10.1164/rccm.200601-142OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messina S, Bitto A, Aguennouz M, Minutoli L, Monici MC, Altavilla D, Squadrito F, Vita G. Nuclear factor κ-B blockade reduces skeletal muscle degeneration and enhances muscle function in Mdx mice. Exp Neurol. 2006;198:234–241. doi: 10.1016/j.expneurol.2005.11.021. [DOI] [PubMed] [Google Scholar]
- Mikhailov YM, Kroptov AV, Zelenin AV, Krutilina RI, Kolesnikov VA, Zelenina IA, et al. The Bcl-xL and ACR-1 genes promote differentiation and reduce apoptosis in muscle fibers of mdx mice (in Russian) Genetika. 2002;38:1445–1450. [PubMed] [Google Scholar]
- Monici MC, Aguennouz M, Mazzeo A, Messina C, Vita G. Activation of nuclear factor-κB in inflammatory myopathies and Duchenne muscular dystrophy. Neurology. 2003;60:993–997. doi: 10.1212/01.wnl.0000049913.27181.51. [DOI] [PubMed] [Google Scholar]
- Muller FL, Song W, Liu Y, Chaudhuri A, Pieke-Dahl S, Strong R, et al. Absence of CuZn superoxide dismutase leads to elevated oxidative stress and acceleration of age-dependent skeletal muscle atrophy. Free Radic Biol Med. 2006;40:1993–2004. doi: 10.1016/j.freeradbiomed.2006.01.036. [DOI] [PubMed] [Google Scholar]
- Nakae Y, Stoward PJ, Shono M, Matsuzaki T. Most apoptotic cells in mdx diaphragm muscle contain accumulated lipofuscin. Histochem Cell Biol. 2001;115:205–214. doi: 10.1007/s004180100250. [DOI] [PubMed] [Google Scholar]
- Nakamura A, Takeda S. Mammalian models of duchenne muscular dystrophy: pathological characteristics and therapeutic applications. J Biomed Biotechnol. 2011;2011:184393. doi: 10.1155/2011/184393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakae Y, Stoward PJ, Shono M, Matsuzaki T. Most apoptotic cells in mdx diaphragm muscle contain accumulated lipofuscin. Histochem. Cell Biol. 2001;115:205–214. doi: 10.1007/s004180100250. [DOI] [PubMed] [Google Scholar]
- Nguyen HX, Tidball JG. Null mutation of gp91phox reduces muscle membrane lysis during muscle inflammation in mice. J Physiol. 2003;553:833–841. doi: 10.1113/jphysiol.2003.051912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Percival JM, Anderson KN, Gregorevic P, Chamberlain JS, Froehner SC. Functional deficits in nNOSmu-deficient skeletal muscle: myopathy in nNOS knockout mice. PLoS One. 2008;3:e3387. doi: 10.1371/journal.pone.0003387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pescatori M, Broccolini A, Minetti C, Bertini E, Bruno C, D'amico A, et al. Gene expression profiling in the early phases of DMD: a constant molecular signature characterizes DMD muscle from early postnatal life throughout disease progression. FASEB J. 2007;21:1210–1226. doi: 10.1096/fj.06-7285com. [DOI] [PubMed] [Google Scholar]
- Peterson JM, Kline W, Canan BD, Ricca DJ, Kaspar B, Delfín DA, et al. Peptide-based inhibition of NF-κB rescues diaphragm muscle contractile dysfunction in a murine model of Duchenne muscular dystrophy. Mol Med. 2011 doi: 10.2119/molmed.2010.00263. DOI 10.2119/molmed.2010.00263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers SK, Kavazis AN, McClung JM. Oxidative stress and disuse muscle atrophy. J Appl Physiol. 2007;102:2389–2397. doi: 10.1152/japplphysiol.01202.2006. [DOI] [PubMed] [Google Scholar]
- Puttini S, Lekka M, Dorchies OM, Saugy D, Incitti T, Ruegg UT, et al. Gene-mediated restoration of normal myofiber elasticity in dystrophic muscles. Mol Ther. 2009;17:19–25. doi: 10.1038/mt.2008.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaswamy KS, Palmer ML, van der Meulen JH, Renoux A, Kostrominova TY, Michele DE, Faulkner JA. Lateral transmission of force is impaired in skeletal muscles of dystrophic mice and very old rats. J Physiol. 2011 doi: 10.1113/jphysiol.2010.201921. DOI 10.1113/jphysiol.2010.201921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rando TA. The dystrophin-glycoprotein complex, cellular signaling, and the regulation of cell survival in muscular dystrophies. Muscle Nerve. 2001;24:1575–1594. doi: 10.1002/mus.1192. [DOI] [PubMed] [Google Scholar]
- Rando TA, Disatnik MH, Yu Y, Franco A. Muscle cells from mdx mice have an increased susceptibility to oxidative stress. Neuromuscul Disord. 1998;8:14–21. doi: 10.1016/s0960-8966(97)00124-7. [DOI] [PubMed] [Google Scholar]
- Rice KM, Preston DL, Neff D, Norton M, Blough ER. Age-related dystrophin-glycoprotein complex structure and function in the rat extensor digitorum longus and soleus muscle. J Gerontol A Biol Sci Med Sci. 2006;11:1119–1129. doi: 10.1093/gerona/61.11.1119. [DOI] [PubMed] [Google Scholar]
- Roelofs RI, de Arango GS, Law PK, Kinsman D, Buchanan DC, Park JH. Treatment of Duchenne's muscular dystrophy with penicillamine. Results of a double-blind trial. Arch Neurol. 1979;36:266–268. doi: 10.1001/archneur.1979.00500410044005. [DOI] [PubMed] [Google Scholar]
- Rousseau J, Dumont N, Lebel C, Quenneville SP, Côté CH, Frenette J, Tremblay JP. Dystrophin expression following the transplantation of normal muscle precursor cells protects mdx muscle from contraction-induced damage. Cell Transplant. 2010;19:589–596. doi: 10.3727/096368910X4863235. [DOI] [PubMed] [Google Scholar]
- Sandri M, El Meslemani AH, Sandri C, Schierling P, Vissing K, Andersen JL, et al. Caspase-3 expression correlates with skeletal muscle apoptosis in Duchenne and vacioscapulo human muscular dystrophy. A potential target for pharmacological treatment? J Neuropath Exp Neurol. 2001;60:302–312. doi: 10.1093/jnen/60.3.302. [DOI] [PubMed] [Google Scholar]
- Sandri M, Carraro U. Apoptosis of skeletal muscle during development and disease. Int J Biochem Cell Biol. 1999;21:1373–1390. doi: 10.1016/s1357-2725(99)00063-1. [DOI] [PubMed] [Google Scholar]
- Senf SM, Dodd SL, McClung JM, Judge AR. Hsp70 overexpression inhibits NF-κB and Foxo3a transcriptional activities and prevents skeletal muscle atrophy. FASEB J. 2008;22:3836–3845. doi: 10.1096/fj.08-110163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiao T, Fond A, Deng B, Wehling-Henricks M, Adams ME, Froehner SC, Tidball JG. Defects in neuromuscular junction structure in dystrophic muscle are corrected by expression of a NOS transgene in dystrophin-deficient muscles, but not in muscles lacking α- and β1-syntrophins. Hum Mol Genet. 2004;13:1873–1884. doi: 10.1093/hmg/ddh204. [DOI] [PubMed] [Google Scholar]
- Skrabek RQ, Anderson JE. Metabolic shifts and myocyte hypertrophy in deflazacort treatment of mdx mouse cardiomyopathy. Muscle Nerve. 2001;24:192–202. doi: 10.1002/1097-4598(200102)24:2<192::aid-mus40>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- Smith LW, Smith JD, Criswell DS. Involvement of nitric oxide synthase in skeletal muscle adaptation to chronic overload. J Appl Physiol. 2002;92:2005–2011. doi: 10.1152/japplphysiol.00950.2001. [DOI] [PubMed] [Google Scholar]
- Song W, Kwak HB, Lawler JM. Exercise training modulates the NOS profile in skeletal muscle from old rats. J Gerontol Ser A Biol Sci Med Sci. 2009;64:540–549. doi: 10.1093/gerona/glp021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer MJ, Tidball JG. Do immune cells promote the pathology of dystrophin-deficient myopathies? Neuromuscul Disord. 2001;11:556–564. doi: 10.1016/s0960-8966(01)00198-5. [DOI] [PubMed] [Google Scholar]
- Spurney CF, Knoblach S, Pistilli EE, Nagaraju K, Martin GR, Hoffman EP. Dystrophin-deficient cardiomyopathy in mouse: expression of Nox4 and Lox are associated with fibrosis and altered functional parameters in the heart. Neuromuscul Disord. 2008;18:371–381. doi: 10.1016/j.nmd.2008.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern LZ, Ringel SP, Ziter FA, Menander-Huber KB, Ionasescu V, Pellegrino RJ, Snyder RD. Drug trial of superoxide dismutase in Duchenne's muscular dystrophy. Arch Neurol. 1982;39:342–346. doi: 10.1001/archneur.1982.00510180020004. [DOI] [PubMed] [Google Scholar]
- Stern LZ, Ringel SP, Ziter FA, Menander-Huber KB, Ionasescu V, Pellegrino RJ, Snyder RD. Drug trial of superoxide dismutase in Duchenne's muscular dystrophy. Arch Neurol. 1982;39:342–346. doi: 10.1001/archneur.1982.00510180020004. [DOI] [PubMed] [Google Scholar]
- Stevens ED, Faulkner JA. The capacity of the mdx mouse diaphragm muscle to do oscillatory work. J Physiol. 2000;522:457–466. doi: 10.1111/j.1469-7793.2000.t01-3-00457.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunada Y, Ohi H, Hase A, Ohi H, Hosono T, Arata S, et al. Transgenic mice expressing mutant caveolin-3 show severe myopathy associated with increased nNOS activity. Hum Mol Gen. 2001;10:173–178. doi: 10.1093/hmg/10.3.173. [DOI] [PubMed] [Google Scholar]
- Sussman M. Duchenne muscular dystrophy. J Amer Acad Orthoped Surg. 2002;10:138–151. doi: 10.5435/00124635-200203000-00009. [DOI] [PubMed] [Google Scholar]
- Suzuki N, Motohashi N, Uezumi A, Fukada S, Yoshimura T, Itoyama Y, et al. NO production results in suspension-induced muscle atrophy through dislocation of neuronal NOS. J Clin Invest. 2007;117:2468–2476. doi: 10.1172/JCI30654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki N, Motohashi N, Uezumi A, Fukada S, Yoshimura T, Itoyama Y, Aoki M, Miyagoe Suzuki Y, Takeda S. NO production results in suspension-induced muscle atrophy through dislocation of neuronal NOS. J Clin Invest. 2007;117:2468–2476. doi: 10.1172/JCI30654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarnopolsky MA, Mahoney DJ, Vaisar J, Rodriguez C, Doherty TJ, Roy BD, Biggar D. Creatine monohydrate enhances strength and body composition in Duchenne muscular dystrophy. Neurology. 2004;62:1771–1777. doi: 10.1212/01.wnl.0000125178.18862.9d. [DOI] [PubMed] [Google Scholar]
- Tidball JG, Wehling-Henricks M. Expression of a NOS transgene in dystrophin-deficient muscle reduces muscle membrane damage without increasing the expression of membrane-associated cytoskeletal proteins. Mol Genet Metab. 2004;82:312–320. doi: 10.1016/j.ymgme.2004.06.006. [DOI] [PubMed] [Google Scholar]
- Tidball JG, Wehling-Henricks M. The role of free radicals in the pathophysiology of muscular dystrophy. J Appl Physiol. 2007;102:1677–1686. doi: 10.1152/japplphysiol.01145.2006. [DOI] [PubMed] [Google Scholar]
- Tkatchenko AV, Le Cam G, Leger JJ, Dechesne CV. Large-scale analysis of differential gene expression in hindlimb muscle and diaphragm of mdx mouse. Biochim Biophys Acta. 2000;1500:17–30. doi: 10.1016/s0925-4439(99)00084-8. [DOI] [PubMed] [Google Scholar]
- Ushio-Fukai M. Compartmentalization of redox signaling through NADPH oxidase-derived ROS. Antioxid Redox Signal. 2009;11:1289–1299. doi: 10.1089/ars.2008.2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Gammeren D, Betters JL, Shanely RA, Falk DJ, DeRuisseau KC, Powers SK, Criswell DS. Trolox attenuates mechanical ventilation-induced contractile dysfunction and proteasome activity of the diaphragm. FASEB J. 2004;18:A746. doi: 10.1164/rccm.200407-939OC. [DOI] [PubMed] [Google Scholar]
- Vasilaki A, Mansouri A, Van Remmen H, Van der Meulen JH, Larkin L, Richardson AG, et al. Free radical generation by skeletal muscle of adult and old mice: effect of contractile function. Aging Cell. 2006;5:109–117. doi: 10.1111/j.1474-9726.2006.00198.x. [DOI] [PubMed] [Google Scholar]
- Venema VJ, Ju H, Zou R, Venema RC. Interaction of neuronal nitric-oxide synthase with caveolin-3 in skeletal muscle. Identification of a novel caveolin scaffolding/inhibitory domain. J Biol Chem. 1997;272:28187–28190. doi: 10.1074/jbc.272.45.28187. [DOI] [PubMed] [Google Scholar]
- Walton JN, Nattrass FJ. On the classification, natural history and treatment of the myopathies. Brain. 1954;77:169–231. doi: 10.1093/brain/77.2.169. [DOI] [PubMed] [Google Scholar]
- Warren GL, Hayes DA, Lowe DA, Prior BM, Armstrong RB. Materials fatigue initiates eccentric contraction-induced injury in rat soleus muscle. J Physiol. 1993;464:477–488. doi: 10.1113/jphysiol.1993.sp019646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe N. Clinical significance of measurement of circulating tumor necrosis factor. Jap J Clin Pathol. 2001;49:829–833. [PubMed] [Google Scholar]
- Wehling M, Spencer MJ, Tidball JG. A nitric oxide synthase transgene ameliorates muscular dystrophy in mdx mice. J Cell Biol. 2001;155:123–131. doi: 10.1083/jcb.200105110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitehead NP, Pham C, Gervasio OL, Allen DG. N-Acetylcysteine ameliorates skeletal muscle pathophysiology in mdx mice. J Physiol. 2008;586:2003–2014. doi: 10.1113/jphysiol.2007.148338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitehead NP, Streamer M, Lusambili LI, Sachs F, Allen DG. Streptomycin reduces stretch-induced membrane permeability in muscles from mdx mice. Neuromuscul Disord. 2006;16:845–854. doi: 10.1016/j.nmd.2006.07.024. [DOI] [PubMed] [Google Scholar]
- Whitehead NP, Yeung EW, Froehner SC, Allen DG. Skeletal muscle NADPH oxidase is increased and triggers stretch-induced damage in the mdx mouse. PLoS One. 2010;5:e15354. doi: 10.1371/journal.pone.0015354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams IA, Allen DG. The role of reactive oxygen species in the hearts of dystrophin-deficient mdx mice. Am J Physiol Heart Circ Physiol. 2007;293:H1969–H1977. doi: 10.1152/ajpheart.00489.2007. [DOI] [PubMed] [Google Scholar]
- Yang B, Jung D, Motto D, Meyer J, Koretzky G, Campbell KP. SH3 domain-mediated interaction of dystroglycan and Grb2. J Biol Chem. 1995;270L:11711–11714. doi: 10.1074/jbc.270.20.11711. [DOI] [PubMed] [Google Scholar]