

Abstract
Abstract
Chronic inflammatory diseases such as heart failure, cancer and arthritis have secondary effects on skeletal muscle that cause weakness and exercise intolerance. These symptoms exacerbate illness and make death more likely. Weakness is not simply a matter of muscle atrophy. Functional studies show that contractile dysfunction, i.e. a reduction in specific force, makes an equally important contribution to overall weakness. The most clearly defined mediator of contractile dysfunction is tumour necrosis factor (TNF). TNF serum levels are elevated in chronic disease, correlate with muscle weakness, and are a predictor of morbidity and mortality. Research is beginning to unravel the mechanism by which TNF depresses specific force. TNF acts via the TNFR1 receptor subtype to depress force by increasing cytosolic oxidant activity. Oxidants depress myofibrillar function, decreasing specific force without altering calcium regulation or other aspects of myofibrillar mechanics. Beyond these concepts, the intracellular mechanisms that depress specific force remain undefined. We do not know the pathway by which receptor–ligand interaction stimulates oxidant production. Nor do we know the type(s) of oxidants stimulated by TNF, their intracellular source(s), or their molecular targets. Investigators in the field are pursuing these issues with the long-term goal of preserving muscle function in individuals afflicted by chronic disease.
Michael Reid is the Shih-Chun Wang Professor and Chair of the Department of Physiology at the University of Kentucky. Research interests include redox homeostasis in skeletal muscle, chronic inflammation-induced weakness, and interventions to preserve muscle function. His research team was the first to demonstrate that skeletal muscle produces reactive oxygen species, that NO is an endogenous modulator of muscle contraction, and that oxidative stress plays a causal role in human muscle fatigue. Jennifer Moylan is an Assistant Professor in the Department of Physiology at the University of Kentucky. She has a diverse research background, work ranging from mechanisms of chloroplast mRNA processing to innate immunity to skeletal muscle function. Current efforts include investigation of unique aspects of skeletal muscle biology that involve a convergence of lipid and redox signalling, focusing on identifying novel targets for interventions that preserve muscle function.
 |
This article focuses on muscle-derived oxidants as potential mediators of weakness in chronic inflammatory disease. The concept is supported by a large and disparate literature on human pathophysiology. For example, inflammation and oxidative stress are closely linked to muscle atrophy and weakness in conditions that include ageing, myotonic and Duchenne dystrophies, chronic obstructive pulmonary disease, kidney disease, rheumatoid arthritis, sepsis, cancer, type 2 diabetes, liver disease and chronic heart failure (CHF). These clinical associations argue for basic and translational research to define cellular mechanisms and identify effective therapies.
With many chronic inflammatory diseases, muscle weakness runs parallel with, and in part independently of, muscle atrophy. For example, ageing human populations experience a decline in muscle strength that outpaces the loss of muscle mass (Goodpaster et al. 2006; Hairi et al. 2010). Studies of patients with either severe (Levine et al. 2003) or mild (Ottenheijm et al. 2005) chronic obstructive pulmonary disease define the mechanisms of muscle weakness in more detail. Skinned diaphragm fibres from these patients have 25–35% reduced specific force that is independent of muscle mass. Patients with rheumatoid arthritis have significant muscle weakness. Many factors influence the reduction in strength (−25 to −50%, Stenstrom & Minor, 2003): loss of mass, joint deformity, pain. Intrinsic myofibrillar dysfunction has not been established in patients with rheumatoid arthritis (Helliwell & Jackson, 1994). However, mice with collagen-induced arthritis have impaired muscle function that may be linked to oxidative modification of myofibrillar proteins (Yamada et al. 2009). Studies of myofibrillar function in animal models (van Norren et al. 2009) or patients (Weber et al. 2009) with cancer cachexia are limited but contrast with other inflammatory diseases. In populations studied thus far, although absolute strength is reduced, force normalized for loss of muscle mass remains unchanged.
Our laboratory is pursuing this problem in the context of heart failure. Weakness of limb and respiratory muscles plagues individuals who suffer from chronic heart failure (Hammond et al. 1990; Evans et al. 1995; Carmo et al. 2001). Loss of muscle strength exacerbates an array of debilitating symptoms and affects patient survival. Muscle weakness is an independent predictor of survival (Meyer et al. 2001) that correlates with breathlessness (McParland et al. 1992; Mancini et al. 1994; Weiner et al. 1999) and exercise limitation (Chua et al. 1995; Meyer et al. 2001). Muscle weakness is not a simple function of atrophy. Force loss generally exceeds the loss of muscle mass. Specific force (force per cross-sectional area) is depressed in muscle fibres from patients with heart failure (Szentesi et al. 2005) and in animal models of the disease (Supinski et al. 1994; Howell et al. 1995; Stassijns et al. 1999). This represents loss of function independent of muscle mass, i.e. contractile dysfunction.
How does inflammation in a remote organ such as the heart weaken skeletal muscle? The answer appears to be multifactorial with systemic inflammation being a major contributor (Gan et al. 2004; Yende et al. 2006). Circulating pro-inflammatory mediators – notably interleukin-6, C-reactive protein, sphingomyelinase (SMase), and tumour necrosis factor-α (TNF) – are elevated in patients with chronic cardiopulmonary disease and correlate with muscle weakness (Cicoira et al. 2001; Toth et al. 2006; Yende et al. 2006; Doehner et al. 2007). Among these humoral factors, TNF is most strongly implicated. TNF serum concentration is inversely related to muscle strength in patients with heart failure (Cicoira et al. 2001; Toth et al. 2006). Intravenous infusion of TNF decreases pressure generation by the canine diaphragm in vivo, a response evident within 3 h (Wilcox et al. 1994), and exposure to high concentrations of TNF can depress force of excised muscle in vitro (Wilcox et al. 1996). At the tissue level, muscle dysfunction appears to be mediated, at least in part, by oxidative stress. Clinical data show that CHF depresses the activities of major antioxidant enzymes and causes protein oxidation in human muscle (Linke et al. 2005; Vescovo et al. 2008). Laboratory studies confirm oxidative stress as a potential mediator, demonstrating that free radical production by muscle is elevated in experimental heart failure (Li et al. 2000; Supinski & Callahan, 2005; Coirault et al. 2007).
TNF and contractile dysfunction
TNF plays dual roles in skeletal muscle. Autocrine release of TNF is crucial for inducing myogenesis during both injury-induced regeneration (Chen et al. 2005; Liu et al. 2010) and mechanical stimulation of myogenesis (Chen et al. 2005; Zhan et al. 2007; Liu et al. 2010). The level of TNF increases in injured muscle through synthesis by myofibres and by release from infiltrating inflammatory cells (Collins & Grounds, 2001; Warren et al. 2002). Mechanical stimulation induces myofibre release of TNF by activation of TNF-converting enzyme and processing of membrane-bound pro-TNF (Zhan et al. 2007). TNF exerts its action through stimulation of p38 MAP kinase, phosphorylation of MEF2 (myocyte enhancer factor 2) transcription factors (de Angelis et al. 2005) and E47 (Lluis et al. 2005). Phosphorylation of E47 in turn stimulates MyoD transactivation (Puri et al. 2000). Thus TNF is an important mediator of muscle myogenesis in response to injury, disease, or training. In contrast, chronic exposure to TNF promotes muscle weakness (Li et al. 2000; Tisdale, 2008).
TNF-induced weakness has long been attributed to loss of muscle mass or cachexia and was originally designated ‘cachectin’ in recognition of its catabolic action (Reid & Li, 2001). However, the weakness is not entirely attributable to loss of muscle mass. When force measurements are normalized to muscle mass, TNF depresses specific force of muscle in intact animals. We first observed this phenomenon in transgenic mice engineered for stable, cardiac-specific overexpression of TNF (Li et al. 2000) which modestly elevates circulating TNF levels (250–350 pg ml−1). These animals are a genetic model of heart failure and we intended to study the changes associated with cardiac cachexia. Instead, adult TNF transgenic mice were phenotypically normal; animal growth, excised muscle weights, fibre bundle cross-sectional areas, and muscle fibre ultrastructure were indistinguishable from wild-type animals. However, functional studies identified a 40% decrement in the specific force of diaphragm fibre bundles. Thus, specific force was depressed by TNF serum levels that were too low to cause atrophy. This identified contractile dysfunction as a separate process from TNF-induced catabolism. Extrapolating to humans, these data predict that contractile dysfunction in patients with chronic heart failure may be more prevalent than appreciated and may precede cachexia.
TNF causes a similar loss of function in muscles of wild-type mice (Hardin et al. 2008). We determined this by injecting adult animals with recombinant TNF and harvesting muscle fibre bundles for functional assessment at various time points. As in transgenic animals, TNF depressed specific force of wild-type muscle across a broad range of stimulus frequencies. The force loss was comparable to the decrement observed in CHF patients (Fig. 1). It occurred within one hour and persisted for at least 48 h. This experimental model further enabled us to test receptor specificity of TNF effects. Respiratory and limb skeletal muscles express both the 55 kDa TNF receptor subtype 1 (TNFR1) and the 75 kDa subtype TNFR2 (De Bleecker et al. 1999). We tested TNF effects in mice that were genetically deficient in either subtype and found that TNFR1 deficiency abolished the response to TNF, preserving specific force, whereas TNFR2 deficiency did not (Fig. 1). Thus, TNF appears to act via TNFR1 to cause contractile dysfunction in intact animals.
Figure 1. Heart failure and TNF depress specific force.
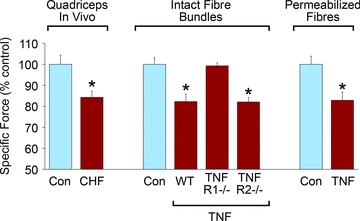
Comparison of CHF-induced weakness in human quadriceps muscle during knee extension efforts (left) versus effects of in vivo TNF treatment on murine diaphragm fibre bundles from wild-type, TNFR1-deficient, and TNFR2-deficient animals (centre) and on permeabilized single fibres from murine diaphragm (right); data adapted from original reports (Harrington et al. 1997; Hardin et al. 2008).
Loss of specific force is not simply an indirect systemic effect of TNF, e.g. on hormonal status, vascular function, or nutrition. TNF can act directly on skeletal muscle to depress function. Incubation of wild-type muscle with recombinant TNF in vitro decreases specific force within hours, a response seen in both limb muscle (Reid et al. 2002) and respiratory muscle (Li et al. 2000; Reid et al. 2002). These observations confirm a direct effect of the cytokine on muscle (Wilcox et al. 1996; Alloatti et al. 2000; Li et al. 2000; Reid et al. 2002) and justify studies of the underlying cell biology.
Research using single muscle fibres show that TNF depresses specific force by altering myofibrillar function. In intact single fibres, we determined that TNF decreases specific force of tetanic contractions without altering calcium regulation (Reid et al. 2002). TNF had no effect on resting calcium, peak tetanic calcium, or the shape of tetanic calcium waveforms. These findings suggested that the site of TNF-induced dysfunction is downstream of the calcium transient, i.e. at the myofibrillar level. Studies of permeabilized muscle fibres confirmed this thesis (Hardin et al. 2008). Animals were treated with TNF or buffer for one hour. Diaphragms were harvested and single fibres were isolated and chemically permeabilized, enabling direct activation of myofilaments using exogenous calcium. Permeabilized fibres from TNF-treated animals showed depressed specific force (Fig. 1) over a wide range of calcium activation. Thus TNF induces modifications in vivo that are maintained in a membrane-free environment. The effect on specific force was highly selective. TNF did not alter other aspects of myofibrillar mechanics; the calcium concentration required for half-maximal activation, Hill coefficient, rate of tension recovery, and maximal shortening velocity were unaltered. These findings are consistent with troponin, myosin heavy chain, or tropomyosin as potential molecular targets of TNF/TNFR1 signalling.
Oxidative stress as a mediator
Free radicals and their redox derivatives modulate contractile function of skeletal muscle. Skeletal muscle fibres continually synthesize parent radicals in the two major redox cascades, nitric oxide (NO) derivatives and reactive oxygen species (ROS). Oxidant activity in skeletal muscle is increased by a wide variety of conditions that promote weakness, fatigue, or both. It is likely that overproduction of muscle-derived oxidants mediate TNF effects on muscle function. Myofibrillar proteins are relatively sensitive to oxidative stress and are likely to be the main site of diaphragm dysfunction in chronic disease (Tikunov et al. 1996). Consistent with this view, specific force of intact muscle fibres is depressed by ROS or NO concentrations that are too low to alter tetanic calcium levels (Andrade et al. 1998a,b, 2001). Thus, ROS mimic the biological action of TNF, preferentially acting on targets downstream of the calcium transient, i.e. at the myofibrillar level.
Experimental evidence shows that muscle-derived oxidants are essential mediators of TNF/TNFR1-induced dysfunction. TNF increases cytosolic oxidant activity in skeletal muscle fibres. This is a robust companion to contractile dysfunction which parallels the loss of specific force caused by transgenic overexpression (Li et al. 2000), injection of the recombinant cytokine in vivo (Hardin et al. 2008), and direct exposure by in vitro incubation (Li et al. 2000). The latter example is illustrated in Fig. 2. Like contractile dysfunction, the rise in oxidant activity is also TNFR1-dependent; it is abolished by genetic deficiency in TNFR1 but not TNFR2 (Hardin et al. 2008). In skeletal muscle, large or persistent increases in cytosolic oxidant activity diminish specific force. In the case of TNF, oxidants have been identified as downstream effectors by the use of pharmacological antioxidants that interrupt oxidant signalling. In vivo experiments tested the effect of injecting animals with 6-hydroxy-2,5,7,8-tetramethylchroman-2-carboxylic acid (Trolox; a water-soluble vitamin E derivative) prior to TNF. Trolox pretreatment markedly depressed oxidant activity and prevented contractile dysfunction in muscles of TNF-treated animals. Experiments in vitro have yielded similar results. Muscle fibre bundles preincubated with N-acetylcysteine (NAC; thiol antioxidant, supports glutathione resynthesis) maintained normal specific forces despite direct TNF exposure. Interestingly, incubation with NAC for 30 min increased the specific force of muscles from TNF transgenic animals by almost one-half. Thus, despite life-long elevation of circulating TNF levels, a substantial portion of the contractile dysfunction was acutely reversible. This further supports the thesis that TNF depresses specific force via a post-translational mechanism.
Figure 2. Muscle-derived oxidants mediate TNF action.
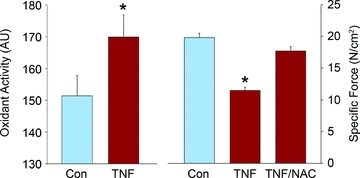
The decrement in rise in oxidant activity of muscle fibres exposed to TNF in vitro (left) is paralleled by a fall in specific force that is abolished by pretreatment with the antioxidant NAC (right); data adapted from original report (Li et al. 2000).
Targets for mechanistic research
The cellular mechanism by which TNF depresses contractile function is a promising area for future research. It is clear that TNF initiates the process by activating TNFR1 (Hardin et al. 2008). But how does this increase oxidant activity in muscle fibres? The existing literature suggests a cascade of potential signalling events that are illustrated in Fig. 3. Early postreceptor signalling is probably mediated by one or more sphingomyelinase isoforms. Neutral (nSMase), acid (aSMase) and secretory (sSMase) sphingomyelinases have distinct biochemical properties, are regulated via unique mechanisms, and can have opposing effects on cell function (Wiegmann et al. 1994). TNFR1 links to nSMase activation via the adaptor proteins FAN (factor associated with neutral sphingomyelinase activation) (Adam-Klages et al. 1997) and RACK1 (receptor for activated C kinase-1) (Tcherkasowa et al. 2002) that bind a cytoplasmic signalling domain upstream of the death domain. nSMase hydrolyses sphingomyelin in the cell membrane to generate ceramide. Clinical data show that sphingomyelinase activity correlates with elevated TNF levels and diminished muscle strength in patients with chronic heart failure (Doehner et al. 2007), suggesting a role for ceramide signalling in inflammation-induced weakness. Consistent with this model, we recently discovered that ceramide depresses the contractile function of skeletal muscle (Ferreira et al. 2010).
Figure 3. Model of signalling events that mediate heart failure-induced weakness.
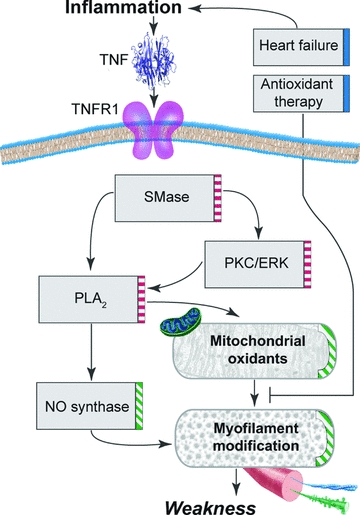
Model shows hypothetical mechanism by which chronic heart failure could depress specific force of skeletal muscle. Boxes below the sarcolemma (horizontal band) depict intracellular events that regulate TNFR1/oxidant signalling (right edge, red dashed bar) and oxidative inhibition of myofilament function (green hatched bar). Boxes above sarcolemma represent extracellular events that modulate the process (blue bar). TNF, tumour necrosis factor; TNFR1, TNF receptor subtype 1; SMase, sphingomyelinase; PKC, phosphokinase C; ERK, extracellular regulatory kinase; PLA2, phospholipase A2; NO, nitric oxide.
Ceramide mediates the rise in oxidant production stimulated by TNF in other cell types (Suematsu et al. 2003), a response attributed to mitochondria in studies of cardiac myocytes (Suematsu et al. 2003). We postulate a homologous role for ceramide in skeletal muscle fibres. Ceramide-induced weakness is mediated by a rise in muscle-derived oxidant activity (Ferreira et al. 2010). The rise in oxidant activity may be triggered by direct interaction of ceramide with protein kinase Cς (PKCς). Ceramide binding activates PKCς and stimulates translocation (Fox et al. 2007). In turn, PKCς phosphorylates extracellular regulatory kinases 1 and 2 (ERK1/2), increasing ERK1/2 activity and stimulating ERK1/2-dependent signal transduction (Schonwasser et al. 1998).
Phospholipase A2 (PLA2) is the putative link between ERK1/2 and oxidant production. Our studies (Gong et al. 2006) and those of other investigators (Zuo et al. 2004) indicate that PLA2 regulates diaphragm oxidant production in an isoform-specific manner. Murine muscle expresses three isoforms of PLA2: the 85 kDa cytosolic cPLA2 (Group IV) isoform, 85 kDa calcium-independent iPLA2 (Group VI), and 14 kDa secretory sPLA2 (Group VII) (Schaloske & Dennis, 2006). TNFR1 signalling activates cPLA2 which synthesizes arachidonic acid from membrane phospholipids and appears to be essential for TNF-stimulated oxidant production (Woo et al. 2000). ERK1/2 phosphorylates cPLA2 (Lin et al. 1993), increasing cPLA2 activity and stimulating arachidonic acid release. These findings identify cPLA2 as a potential target of TNFR1/ERK1/2 signalling and a potential source of arachidonic acid to stimulate oxidant production.
These data do not identify the source or composition of TNF-stimulated oxidants. Skeletal muscle continually generates NO derivatives. These may derive from at least two constitutively-expressed NO synthase (NOS) isoforms. A muscle-specific neuronal NOS isoform (nNOSμ) localizes to the dystrophin complex in fast-type fibres whereas the endothelial isoform (eNOS) may be associated with mitochondria. Muscle also generates ROS which are detectable in the cytosol and extracellular space. Putative sources of ROS within muscle fibres include the mitochondrial electron transport chain, a sarcolemmal NAD(P)H oxidase complex, and xanthine oxidase.
TNF/TNFR1 signalling can increase production of either NO or ROS, responses that appear to be cell type specific. Our data do not yet identify the dominant mechanism in skeletal muscle. Live-cell measurements of oxidant activity were made using 2,3-dichlorofluorescin diacetate (DCFH-DA), a fluorescence probe that detects both NO derivatives and ROS, and antioxidant interventions (Trolox, NAC) were selected to buffer both cascades. Nor does the functional response to TNF help discriminate between NO and ROS. Like TNF, direct exposure to either NO or ROS decreases specific force of intact muscle fibres via effects on myofibrillar proteins. Thus, the redox cascade that mediates TNF-induced weakness remains an open question.
The mechanism by which oxidant activity depresses myofibrillar function is particularly interesting. Acute TNF administration to mice depresses specific force by 30–40% within 60 min (Hardin et al. 2008) while the weakness caused by long-term TNF overexpression is partially reversed by brief incubation in an antioxidant solution (Li et al. 2000). The rapid nature of these responses suggests that TNF depresses specific force via a post-translational mechanism. The mechanism may involve direct reaction of TNF-stimulated oxidants with myofilament proteins; for example, the myosin head (Burke et al. 1976), tropomyosin (Williams & Swenson, 1982), troponin C (Putkey et al. 1993) and actin (Liu et al. 1990) have regulatory sulfhydryls that are sensitive to oxidation and could alter myofilament interactions. Alternatively, we and our colleagues have proposed that redox control may occur upstream of the myofibrils, e.g. redox-sensitive kinases or phosphatases that alter the phosphorylation state of myofibrillar proteins and thereby influence force (Andrade et al. 2001). Other post-translational mechanisms may include carbonylation (Coirault et al. 2007), ubiquitination (Dalla Libera et al. 2005; van Hees et al. 2007), and myosin degradation (Tikunov et al. 1996; van Hees et al. 2007).
Summary and conclusion
Chronic inflammatory diseases decrease specific force of skeletal muscle. This contractile dysfunction appears to be mediated via oxidant effects on myofilament proteins. TNF is strongly implicated as an important molecule in the process. Beyond these basic concepts, the intracellular mechanisms that depress specific force remain largely undefined. We do not know the pathway by which receptor–ligand interaction stimulates oxidant production. Nor do we know the type(s) of oxidants stimulated by TNF, their intracellular sources, or the myofilament proteins that are affected. Research is needed to define the cellular mechanism and identify novel therapies. Skeletal muscle weakness is a major clinical problem. It promotes exercise intolerance and breathlessness that plague individuals with heart failure, cancer, arthritis and other chronic diseases. It complicates chronic disease, diminishes the quality of life, and makes death more likely. Effective strategies to preserve skeletal muscle function would have broad clinical significance. Discovery and implementation of such therapies is a long-term goal for scientists in this field of muscle biology.
Acknowledgments
This manuscript is dedicated to the late Brian Hardin for his invaluable contributions to our research on this topic. We thank Jeff Smith for technical assistance and Tom Dolan for artistic support. This research is supported by National Institutes of Health grant no. R01 AR055974.
Glossary
Abbreviations
- CHF
chronic heart failure
- cPLA2
cytosolic phospholipase A2
- ERK1/2
extracellular regulatory kinases 1 and 2
- NAC
N-acetylcysteine
- NO
nitric oxide
- nSMase
neutral sphingomyelinase
- PLA2
phospholipase A2
- PKCς
protein kinase Cς
- ROS
reactive oxygen species
- SMase
sphingomyelinase
- TNF
tumour necrosis factor-α
- TNFR1 and TNFR2
TNF receptor subtype 1 and 2
References
- Adam-Klages S, Schwandner R, Adam D, Kreder D, Bernardo K, Kronke M. Distinct adapter proteins mediate acid versus neutral sphingomyelinase activation through the p55 receptor for tumor necrosis factor. J Leukoc Biol. 1997;63:678–682. doi: 10.1002/jlb.63.6.678. [DOI] [PubMed] [Google Scholar]
- Alloatti G, Penna C, Mariano F, Camussi G. Role of NO and PAF in the impairment of skeletal muscle contractility induced by TNF-α. Am J Physiol Regul Integr Comp Physiol. 2000;279:R2156–R2163. doi: 10.1152/ajpregu.2000.279.6.R2156. [DOI] [PubMed] [Google Scholar]
- Andrade FH, Reid MB, Allen DG, Westerblad H. Effect of hydrogen peroxide and dithiothreitol on contractile function of single skeletal muscle fibres from the mouse. J Physiol. 1998a;509:565–575. doi: 10.1111/j.1469-7793.1998.565bn.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrade FH, Reid MB, Allen DG, Westerblad H. Effect of nitric oxide on single skeletal muscle fibres from the mouse. JPhysiol. 1998b;509:577–586. doi: 10.1111/j.1469-7793.1998.577bn.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrade FH, Reid MB, Westerblad H. Contractile response to low peroxide concentrations: myofibrillar calcium sensitivity as a likely target for redox modulation of skeletal muscle function. FASEB J. 2001;15:309–311. doi: 10.1096/fj.00-0507fje. [DOI] [PubMed] [Google Scholar]
- Burke M, Reisler F, Harrington WF. Effect of bridging the two essential thiols of myosin on its spectral and actin-binding properties. Biochemistry. 1976;15:1923–1927. doi: 10.1021/bi00654a020. [DOI] [PubMed] [Google Scholar]
- Carmo MM, Barbara C, Ferreira T, Branco J, Ferreira S, Rendas AB. Diaphragmatic function in patients with chronic left ventricular failure. Pathophysiology. 2001;8:55–60. doi: 10.1016/s0928-4680(01)00065-7. [DOI] [PubMed] [Google Scholar]
- Chen SE, Gerken E, Zhang Y, Zhan M, Mohan RK, Li AS, Reid MB, Li YP. Role of TNF-α signaling in regeneration of cardiotoxin-injured muscle. Am J Physiol Cell Physiol. 2005;289:C1179–C1187. doi: 10.1152/ajpcell.00062.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua TP, Anker SD, Harrington D, Coats AJ. Inspiratory muscle strength is a determinant of maximum oxygen consumption in chronic heart failure. Br Heart J. 1995;74:381–385. doi: 10.1136/hrt.74.4.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cicoira M, Bolger AP, Doehner W, Rauchhaus M, Davos C, Sharma R, Al-Nasser FO, Coats AJ, Anker SD. High tumour necrosis factor-α levels are associated with exercise intolerance and neurohormonal activation in chronic heart failure patients. Cytokine. 2001;15:80–86. doi: 10.1006/cyto.2001.0918. [DOI] [PubMed] [Google Scholar]
- Coirault C, Guellich A, Bargry T, Samuel JL, Riou B, Lecarpentier Y. Oxidative stress of myosin contributes to skeletal muscle dysfunction in rats with chronic heart failure. Am J Physiol Heart Circ Physiol. 2007;292:H1009–H1017. doi: 10.1152/ajpheart.00438.2006. [DOI] [PubMed] [Google Scholar]
- Collins RA, Grounds MD. The role of tumor necrosis factor-alpha (TNF-α) in skeletal muscle regeneration. Studies in TNF-α(-/-) and TNF-α(-/-)/LT-α(-/-) mice. J Histochem Cytochem. 2001;49:989–1001. doi: 10.1177/002215540104900807. [DOI] [PubMed] [Google Scholar]
- Dalla Libera L, Ravara B, Gobbo V, Danieli Betto D, Germinario E, Angelini A, Vescovo G. Skeletal muscle myofibrillar protein oxidation in heart failure and the protective effect of Carvedilol. J Mol Cell Cardiol. 2005;38:803–807. doi: 10.1016/j.yjmcc.2005.02.023. [DOI] [PubMed] [Google Scholar]
- de Angelis L, Zhao J, Andreucci JJ, Olson EN, Cossu G, McDermott JC. Regulation of vertebrate myotome development by the p38 MAP kinase-MEF2 signaling pathway. Dev Biol. 2005;283:171–179. doi: 10.1016/j.ydbio.2005.04.009. [DOI] [PubMed] [Google Scholar]
- De Bleecker JL, Meire VI, Declercq W, Van Aken EH. Immunolocalization of tumor necrosis factor-alpha and its receptors in inflammatory myopathies. Neuromuscul Disord. 1999;9:239–246. doi: 10.1016/s0960-8966(98)00126-6. [DOI] [PubMed] [Google Scholar]
- Doehner W, Bunck AC, Rauchhaus M, von Haehling S, Brunkhorst FM, Cicoira M, Tschope C, Ponikowski P, Claus RA, Anker SD. Secretory sphingomyelinase is upregulated in chronic heart failure: a second messenger system of immune activation relates to body composition, muscular functional capacity, and peripheral blood flow. Eur Heart J. 2007;28:821–828. doi: 10.1093/eurheartj/ehl541. [DOI] [PubMed] [Google Scholar]
- Evans SA, Watson L, Hawkins M, Cowley AJ, Johnston ID, Kinnear WJ. Respiratory muscle strength in chronic heart failure. Thorax. 1995;50:625–628. doi: 10.1136/thx.50.6.625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira LF, Smith JD, Moylan JS, Reid MB. Sphingomyelinase and ceramide increase reactive species and depress maximal skeletal muscle force in vitro. FASEB J. 2010;24:801.16. [Google Scholar]
- Fox TE, Houck KL, O'Neill SM, Nagarajan M, Stover TC, Pomianowski PT, Unal O, Yun JK, Naides SJ, Kester M. Ceramide recruits and activates protein kinase C ζ (PKCζ) within structured membrane microdomains. J Biol Chem. 2007;282:12450–12457. doi: 10.1074/jbc.M700082200. [DOI] [PubMed] [Google Scholar]
- Gan WQ, Man SFP, Senthilselvan A, Sin DD. Association between chronic obstructive pulmonary disease and systemic inflammation: a systematic review and a meta-analysis. Thorax. 2004;59:574–580. doi: 10.1136/thx.2003.019588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong MC, Arbogast S, Guo Z, Mathenia J, Su W, Reid MB. Calcium-independent phospholipase A2 modulates cytosolic oxidant activity and contractile function in murine skeletal muscle cells. J Appl Physiol. 2006;100:399–405. doi: 10.1152/japplphysiol.00873.2005. [DOI] [PubMed] [Google Scholar]
- Goodpaster BH, Park SW, Harris TB, Kritchevsky SB, Nevitt M, Schwartz AV, Simonsick EM, Tylavsky FA, Visser M, Newman AB. The loss of skeletal muscle strength, mass, and quality in older adults: the health, aging and body composition study. J Gerontol A Biol Sci Med Sci. 2006;61:1059–1064. doi: 10.1093/gerona/61.10.1059. [DOI] [PubMed] [Google Scholar]
- Hairi NN, Cumming RG, Naganathan V, Handelsman DJ, Le Couteur DG, Creasey H, Waite LM, Seibel MJ, Sambrook PN. Loss of muscle strength, mass (sarcopenia), and quality (specific force) and its relationship with functional limitation and physical disability: the Concord Health and Ageing in Men Project. J Am Geriatr Soc. 2010;58:2055–2062. doi: 10.1111/j.1532-5415.2010.03145.x. [DOI] [PubMed] [Google Scholar]
- Hammond MD, Bauer KA, Sharp JT, Rocha RD. Respiratory muscle strength in congestive heart failure. Chest. 1990;98:1091–1094. doi: 10.1378/chest.98.5.1091. [DOI] [PubMed] [Google Scholar]
- Hardin BJ, Campbell KS, Smith JD, Arbogast S, Smith J, Moylan JS, Reid MB. TNF-α acts via TNFR1 and muscle-derived oxidants to depress myofibrillar force in murine skeletal muscle. J Appl Physiol. 2008;104:694–699. doi: 10.1152/japplphysiol.00898.2007. [DOI] [PubMed] [Google Scholar]
- Harrington D, Anker SD, Chua TP, Webb-Peploe KM, Ponikowski PP, Poole-Wilson PA, Coats AJ. Skeletal muscle function and its relation to exercise tolerance in chronic heart failure. J Am Coll Cardiol. 1997;30:1758–1764. doi: 10.1016/s0735-1097(97)00381-1. [DOI] [PubMed] [Google Scholar]
- Helliwell PS, Jackson S. Relationship between weakness and muscle wasting in rheumatoid arthritis. Ann Rheum Dis. 1994;53:726–728. doi: 10.1136/ard.53.11.726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell S, Maarek JM, Fournier M, Sullivan K, Zhan WZ, Sieck GC. Congestive heart failure: differential adaptation of the diaphragm and latissimus dorsi. J Appl Physiol. 1995;79:389–397. doi: 10.1152/jappl.1995.79.2.389. [DOI] [PubMed] [Google Scholar]
- Levine S, Nguyen T, Kaiser LR, Rubinstein NA, Maislin G, Gregory C, Rome LC, Dudley GA, Sieck GC, Shrager JB. Human diaphragm remodeling associated with chronic obstructive pulmonary disease: clinical implications. Am J Respir Crit Care Med. 2003;168:706–713. doi: 10.1164/rccm.200209-1070OC. [DOI] [PubMed] [Google Scholar]
- Li X, Moody MR, Engel D, Walker S, Clubb FJ, Jr, Sivasubramanian N, Mann DL, Reid MB. Cardiac-specific overexpression of tumor necrosis factor-α causes oxidative stress and contractile dysfunction in mouse diaphragm. Circulation. 2000;102:1690–1696. doi: 10.1161/01.cir.102.14.1690. [DOI] [PubMed] [Google Scholar]
- Lin LL, Wartmann M, Lin AY, Knopf JL, Seth A, Davis RJ. cPLA2 is phosphorylated and activated by MAP kinase. Cell. 1993;72:269–278. doi: 10.1016/0092-8674(93)90666-e. [DOI] [PubMed] [Google Scholar]
- Linke A, Adams V, Schulze PC, Erbs S, Gielen S, Fiehn E, Mobius-Winkler S, Schubert A, Schuler G, Hambrecht R. Antioxidative effects of exercise training in patients with chronic heart failure: increase in radical scavenger enzyme activity in skeletal muscle. Circulation. 2005;111:1763–1770. doi: 10.1161/01.CIR.0000165503.08661.E5. [DOI] [PubMed] [Google Scholar]
- Liu DF, Wang D, Stracher A. The accessibility of the thiol groups on G- and F-actin of rabbit muscle. Biochem J. 1990;266:453–459. doi: 10.1042/bj2660453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Chen SE, Jin B, Carson JA, Niu A, Durham W, Lai JY, Li YP. TIMP3: a physiological regulator of adult myogenesis. J Cell Sci. 2010;123:2914–2921. doi: 10.1242/jcs.057620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lluis F, Ballestar E, Suelves M, Esteller M, Munoz-Canoves P. E47 phosphorylation by p38 MAPK promotes MyoD/E47 association and muscle-specific gene transcription. EMBO J. 2005;24:974–984. doi: 10.1038/sj.emboj.7600528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McParland C, Krishnan B, Wang Y, Callagher CG. Inspiratory muscle weakness and dyspnea in chronic heart failure. Am Rev Resp Dis. 1992;146:467–472. doi: 10.1164/ajrccm/146.2.467. [DOI] [PubMed] [Google Scholar]
- Mancini DM, Henson D, LaManca J, Levine S. Evidence of reduced respiratory muscle endurance in patients with heart failure. J Am Coll Cardiol. 1994;24:972–981. doi: 10.1016/0735-1097(94)90858-3. [DOI] [PubMed] [Google Scholar]
- Meyer FJ, Borst MM, Zugck C, Kirschke A, Schellberg D, Kübler W, Haass M. Respiratory muscle dysfunction in congestive heart failure: clinical correlation and prognostic significance. Circulation. 2001;103:2153–2158. doi: 10.1161/01.cir.103.17.2153. [DOI] [PubMed] [Google Scholar]
- Ottenheijm CAC, Heunks LMA, Sieck GC, Zhan WZ, Jansen SM, Degens H, de Boo T, Dekhuijzen PNR. Diaphragm dysfunction in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2005;172:200–205. doi: 10.1164/rccm.200502-262OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puri PL, Wu Z, Zhang P, Wood LD, Bhakta KS, Han J, Feramisco JR, Karin M, Wang JY. Induction of terminal differentiation by constitutive activation of p38 MAP kinase in human rhabdomyosarcoma cells. Genes Dev. 2000;14:574–584. [PMC free article] [PubMed] [Google Scholar]
- Putkey JA, Dotson DG, Mouawad P. Formation of inter- and intramolecular bonds can activate cardiac troponin C. J Biol Chem. 1993;268:6827–6830. [PubMed] [Google Scholar]
- Reid MB, Lännergren J, Westerblad H. Respiratory and limb muscle weakness induced by tumor necrosis factor-α: involvement of muscle myofilaments. Am J Respir Crit Care Med. 2002;166:479–484. doi: 10.1164/rccm.2202005. [DOI] [PubMed] [Google Scholar]
- Reid MB, Li YP. Tumor necrosis factor-α and muscle wasting: a cellular perspective. Respir Res. 2001;2:269–272. doi: 10.1186/rr67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaloske RH, Dennis EA. The phospholipase A2 superfamily and its group numbering system. Biochim Biophys Acta. 2006;1761:1246–1259. doi: 10.1016/j.bbalip.2006.07.011. [DOI] [PubMed] [Google Scholar]
- Schonwasser DC, Marais RM, Marshall CJ, Parker PJ. Activation of the mitogen-activated protein kinase/extracellular signal-regulated kinase pathway by conventional, novel, and atypical protein kinase C isotypes. Mol Cell Biol. 1998;18:790–798. doi: 10.1128/mcb.18.2.790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stassijns G, Gayan-Ramirez G, De Leyn P, de Bock V, Dom R, Lysens R, Decramer M. Effects of dilated cardiomyopathy on the diaphragm in the Syrian hamster. Eur Respir J. 1999;13:391–397. doi: 10.1183/09031936.99.13239199. [DOI] [PubMed] [Google Scholar]
- Stenstrom CH, Minor MA. Evidence for the benefit of aerobic and strengthening exercise in rheumatoid arthritis. Arthritis Rheum. 2003;49:428–434. doi: 10.1002/art.11051. [DOI] [PubMed] [Google Scholar]
- Suematsu N, Tsutsui H, Wen J, Kang D, Ikeuchi M, Ide T, Hayashidani S, Shiomi T, Kubota T, Hamasaki N, Takeshita A. Oxidative stress mediates tumor necrosis factor-α-induced mitochondrial DNA damage and dysfunction in cardiac myocytes. Circulation. 2003;107:1418–1423. doi: 10.1161/01.cir.0000055318.09997.1f. [DOI] [PubMed] [Google Scholar]
- Supinski G, DiMarco A, Dibner-Dunlap M. Alterations in diaphragm strength and fatiguability in congestive heart failure. J Appl Physiol. 1994;76:2707–2713. doi: 10.1152/jappl.1994.76.6.2707. [DOI] [PubMed] [Google Scholar]
- Supinski GS, Callahan LA. Diaphragmatic free radical generation increases in an animal model of heart failure. J Appl Physiol. 2005;99:1078–1084. doi: 10.1152/japplphysiol.01145.2004. [DOI] [PubMed] [Google Scholar]
- Szentesi P, Bekedam MA, van Beek-Harmsen BJ, van der Laarse WJ, Zaremba R, Boonstra A, Visser FC, Stienen GJM. Depression of force production and ATPase activity in different types of human skeletal muscle fibers from patients with chronic heart failure. J Appl Physiol. 2005;99:2189–2195. doi: 10.1152/japplphysiol.00542.2005. [DOI] [PubMed] [Google Scholar]
- Tcherkasowa AE, Adam-Klages S, Kruse ML, Wiegmann K, Mathieu S, Kolanus W, Krönke M, Adam D. Interaction with factor associated with neutral sphingomyelinase activation, a WD motif-containing protein, identifies receptor for activated C-kinase 1 as a novel component of the signaling pathways of the p55 TNF receptor. J Immunol. 2002;169:5161–5170. doi: 10.4049/jimmunol.169.9.5161. [DOI] [PubMed] [Google Scholar]
- Tikunov BA, Mancini D, Levine S. Changes in myofibrillar protein composition of human diaphragm elicted by congestive heart failure. J Mol Cell Cardiol. 1996;28:2537–2541. doi: 10.1006/jmcc.1996.0245. [DOI] [PubMed] [Google Scholar]
- Tisdale MJ. Catabolic mediators of cancer cachexia. Curr Opin Support Palliat Care. 2008;2:256–261. doi: 10.1097/spc.0b013e328319d7fa. [DOI] [PubMed] [Google Scholar]
- Toth MJ, Ades PA, Tischler MD, Tracy RP, LeWinter MM. Immune activation is associated with reduced skeletal muscle mass and physical function in chronic heart failure. Int J Cardiol. 2006;109:179–187. doi: 10.1016/j.ijcard.2005.06.006. [DOI] [PubMed] [Google Scholar]
- van Hees HWH, van der Heijden HFM, Ottenheijm CAC, Heunks LMA, Pigmans CJC, Verheugt FWA, Brouwer RMHJ, Dekhuijzen PNR. Diaphragm single-fiber weakness and loss of myosin in congestive heart failure rats. Am J Physiol Heart Circ Physiol. 2007;293:H819–H828. doi: 10.1152/ajpheart.00085.2007. [DOI] [PubMed] [Google Scholar]
- van Norren K, Kegler D, Argilés JM, Luiking Y, Gorselink M, Laviano A, Arts K, Faber J, Jansen H, van der Beek EM, van Helvoort A. Dietary supplementation with a specific combination of high protein, leucine, and fish oil improves muscle function and daily activity in tumour-bearing cachectic mice. Br J Cancer. 2009;100:713–722. doi: 10.1038/sj.bjc.6604905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vescovo G, Ravara B, Dalla Libera L. Skeletal muscle myofibrillar protein oxidation and exercise capacity in heart failure. Basic Res Cardiol. 2008;103:285–290. doi: 10.1007/s00395-007-0692-x. [DOI] [PubMed] [Google Scholar]
- Warren GL, Hulderman T, Jensen N, McKinstry M, Mishra M, Luster MI, Simeonova PP. Physiological role of tumor necrosis factor α in traumatic muscle injury. FASEB J. 2002;16:1630–1632. doi: 10.1096/fj.02-0187fje. [DOI] [PubMed] [Google Scholar]
- Weber MA, Krakowski-Roosen H, Schroder L, Kinscherf R, Krix M, Kopp-Schneider A, Essig M, Bachert P, Kauczor HU, Hildebrandt W. Morphology, metabolism, microcirculation, and strength of skeletal muscles in cancer-related cachexia. Acta Oncol. 2009;48:116–124. doi: 10.1080/02841860802130001. [DOI] [PubMed] [Google Scholar]
- Weiner P, Waizman J, Magadle R, Berar-Yanay N, Pelled B. The effect of specific inspiratory muscle training on the sensation of dyspnea and exercise tolerance in patients with congestive heart failure. Clin Cardiol. 1999;22:727–732. doi: 10.1002/clc.4960221110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiegmann K, Schutze S, Machleidt T, Witte D, Kronke M. Functional dichotomy of neutral and acidic sphingomyelinases in tumor necrosis factor signaling. Cell. 1994;78:1005–1015. doi: 10.1016/0092-8674(94)90275-5. [DOI] [PubMed] [Google Scholar]
- Wilcox P, Milliken C, Bressler B. High-dose tumor necrosis factor alpha produces an impairment of hamster diaphragm contractility. Attenuation with a prostaglandin inhibitor. Am J Respir Crit Care Med. 1996;153:1611–1615. doi: 10.1164/ajrccm.153.5.8630610. [DOI] [PubMed] [Google Scholar]
- Wilcox PG, Wakai Y, Walley KR, Cooper DJ, Road J. Tumor necrosis factor alpha decreases in vivo diaphragm contractility in dogs. Am J Respir Crit Care Med. 1994;150:1368–1373. doi: 10.1164/ajrccm.150.5.7952566. [DOI] [PubMed] [Google Scholar]
- Williams DL, Jr, Swenson CA. Disulfide bridges in tropomyosin. Effect on ATPase activity of actomyosin. Eur J Biochem. 1982;127:495–499. [PubMed] [Google Scholar]
- Woo CH, Eom YW, Yoo MH, You HJ, Han HJ, Song WK, Yoo YJ, Chun JS, Kim JH. Tumor necrosis factor-α generates reactive oxygen species via a cytosolic phospholipase A2-linked cascade. J Biol Chem. 2000;275:32357–32362. doi: 10.1074/jbc.M005638200. [DOI] [PubMed] [Google Scholar]
- Yamada T, Place N, Kosterina N, Ostberg T, Zhang SJ, Grundtman C, Erlandsson-Harris H, Lundberg IE, Glenmark B, Bruton JD, Westerblad H. Impaired myofibrillar function in the soleus muscle of mice with collagen-induced arthritis. Arthritis Rheum. 2009;60:3280–3289. doi: 10.1002/art.24907. [DOI] [PubMed] [Google Scholar]
- Yende S, Waterer GW, Tolley EA, Newman AB, Bauer DC, Taaffe DR, Jensen R, Crapo R, Rubin S, Nevitt M, Simonsick EM, Satterfield S, Harris T, Kritchevsky SB. Inflammatory markers are associated with ventilatory limitation and muscle dysfunction in obstructive lung disease in well functioning elderly adults. Thorax. 2006;61:10–16. doi: 10.1136/thx.2004.034181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan M, Jin B, Chen SE, Reecy JM, Li YP. TACE release of TNF-α mediates mechanotransduction-induced activation of p38 MAPK and myogenesis. J Cell Sci. 2007;120:692–701. doi: 10.1242/jcs.03372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo L, Christofi FL, Wright VP, Bao S, Clanton TL. Lipoxygenase-dependent superoxide release in skeletal muscle. J Appl Physiol. 2004;97:661–668. doi: 10.1152/japplphysiol.00096.2004. [DOI] [PubMed] [Google Scholar]