

Non-technical summary
Our focus was on whether amyotrophic lateral sclerosis (ALS) might be precipitated by early developmental changes in large spinal motoneurons, which are vulnerable to early die-off in ALS. It has been shown that some electrical properties in motoneurons are profoundly altered soon after birth in mutant superoxide dismutase-1 (SOD1) mice, a standard animal model of ALS. These same properties undergo rapid developmental changes in normal mice during this time period. Our goal was to compare the development of motoneuron electrical properties in normal and SOD1 mice. Properties were measured from birth to 12 days of age, when the mouse is considered juvenile, but long before symptom onset. Most electrical properties in the SOD1 motoneurons showed an accelerated pace of maturation during this early developmental period compared with the normal motoneurons. If this trend persists, it could, along with other disease factors, hasten the onset of normal motoneuron degeneration due to ageing and result in the development of ALS.
Abstract
Abstract
Spinal motoneurons are highly vulnerable in amyotrophic lateral sclerosis (ALS). Previous research using a standard animal model, the mutant superoxide dismutase-1 (SOD1) mouse, has revealed deficits in many cellular properties throughout its lifespan. The electrical properties underlying motoneuron excitability are some of the earliest to change; starting at 1 week postnatal, persistent inward currents (PICs) mediated by Na+ are upregulated and electrical conductance, a measure of cell size, increases. However, during this period these properties and many others undergo large developmental changes which have not been fully analysed. Therefore, we undertook a systematic analysis of electrical properties in more than 100 normal and mutant SOD1 motoneurons from 0 to 12 days postnatal, the neonatal to juvenile period. We compared normal mice with the most severe SOD1 model, the G93A high-expressor line. We found that the Na+ PIC and the conductance increased during development. However, mutant SOD1 motoneurons showed much greater increases than normal motoneurons; the mean Na+ PIC in SOD1 motoneurons was double that of wild-type motoneurons. Additionally, in mutant SOD1 motoneurons the PIC mediated by Ca2+ increased, spike width decreased and the time course of the after-spike after-hyperpolarization shortened. These changes were advances of the normal effects of maturation. Thus, our results show that the development of normal and mutant SOD1 motoneurons follows generally similar patterns, but that the rate of development is accelerated in the mutant SOD1 motoneurons. Statistical analysis of all measured properties indicates that approximately 55% of changes attributed to the G93A SOD1 mutation can be attributed to an increased rate of maturation.
Introduction
Multiple factors have been implicated in the progressive degeneration of motoneurons in amyotrophic lateral sclerosis (ALS), suggesting that motoneurons are particularly vulnerable to homeostatic challenges, both genetic and environmental (Bacman et al. 2006; Van Den Bosch et al. 2006; Rothstein, 2009). A factor that has been given little attention thus far is the role that postnatal development and maturation processes have in the development of the disease state. Though ALS is clearly an adult-onset disease, very early changes in cellular, electrical and anatomical properties have been reported (Pieri et al. 2003a; Kuo et al. 2004, 2005; Amendola & Durand, 2008; van Zundert et al. 2008; Pambo-Pambo et al. 2009) in the standard mouse model of ALS, in which a mutated form of the human gene for superoxide dismutase-1 (SOD1) is over-expressed (Gurney et al. 1994; Dal Canto & Gurney, 1995). In these mice, overt symptoms first appear around postnatal day 90 (P90) as the spinal motoneurons degenerate (Gurney et al. 1994; Chiu et al. 1995). Large α-motoneurons, which have low Ca2+-buffering capacity, are among the most vulnerable (Tandan & Bradley, 1985; Mohajeri et al. 1998; von Lewinski & Keller, 2005; Pun et al. 2006; Hegedus et al. 2008), and their intrinsic electrical excitability is one of the first parameters to become abnormal.
The persistent inward current (PIC) generated by Na+ channels is a major factor influencing net neuronal excitability and is elevated months before symptom onset. This upregulation in the Na+ PIC has been demonstrated in SOD1 lumbar motoneurons, both embryonic (cultured) and at a week old in the slice preparation (Kuo et al. 2004, 2005), in 1-week-old SOD1 brainstem motoneurons in the slice (van Zundert et al. 2008), in cultured embryonic cortical motoneurons (Pieri et al. 2009) and in adult cortical pyramidal neurons (Carunchio et al. 2010). In the brainstem motoneurons, Na+ PIC enhancement is also accompanied by increased background synaptic input, which suggests an increased rate of maturation (van Zundert et al. 2008). In addition, there is a marked increase in branching of proximal dendrites in 1-week-old SOD1 motoneurons, linked to an increase in electrical conductance (Amendola & Durand, 2008; Elbasiouny et al. 2010).
Many electrical properties of motoneurons are known to exhibit characteristic changes in motoneurons during early developmental stages, including an increasing contribution of Ca2+ (Ca2+ PIC) to the total PIC (Jiang et al. 1999; Carrascal et al. 2005). As previous studies on early changes in SOD1 motoneurons have looked only at a few of these properties at limited time points and in a variety of preparations, it has been difficult to assess whether the early changes in SOD1 motoneuron excitability have any relationship to normal development. Therefore, in this study a large number of normal and SOD1 motoneurons were examined during maturation from neonatal (postnatal day 0; P0) to juvenile (postnatal day 12; P12) stages to compare the development of multiple parameters of their electrical properties.
Methods
All mice were used according to Northwestern University's Animal Care and Use Committee guidelines. Transgenic mice overexpressing the human SOD1G93A gene or the wild-type (WT) human SOD1WT gene were provided by the laboratory of T.S. (Davee Department of Neurology and Clinical Neurosciences). The G93A and wild-type SOD1 genes were identified using standard PCR techniques (Rosen et al. 1993). Briefly, 20–25 mg of tissue was used for the DNA extraction. The primers for amplification are SOD1P7: CAT CAG CCC TAA TCC ATC TGA and SOD1P8: CGC GAC TAA CAA TCA AAG TGA. All PCR work was done by the laboratory of T.S. after the experiments and analyses had already performed. A total of 58 mice were used for this study.
Surgery
Mice, postnatal day 0–12, were deeply anaesthetized with isofluorane, decapitated and eviscerated. The lumbar and sacral spinal cord was quickly removed and embedded in 2.5% w/v agar. The agar block was then superglued to a stainless-steel slicing bath, and 350-μm-thick transverse slices were made using a Leica 1000 vibratome as described previously (Theiss & Heckman, 2005). During both spinal cord isolation and slicing, the spinal cord was immersed in 1–4°C high-osmolarity dissecting solution containing (mm): sucrose, 234.0; KCl, 2.5; CaCl2.2H2O, 0.1; MgSO4.7H2O, 4.0; Hepes, 15.0; glucose, 11.0; and Na2PO4, 1.0; pH 7.35 when bubbled with 95% O2–5% CO2. After cutting, the slices were incubated for >1 h at 28–34°C in incubating solution containing (mm): NaCl, 126.0; KCl, 2.5; CaCl2.2H2O, 2.0; MgCl2.6H2O, 2.0; NaHCO3, 26.0; and glucose, 10.0; pH 7.4 when bubbled with 95% O2–5% CO2.
Electrophysiology
Whole-cell patch clamp was performed on neurons in slices from the ventral lumbar and sacral spinal cord segments using 1–4 MΩ glass electrodes. Electrodes were positioned using a Sutter Instrument (Novato, CA, USA) MP-285 motorized micromanipulator. Whole-cell patch clamp measurements were performed at room temperature using the Multiclamp 700A amplifier (Molecular Devices, Burlingame, CA, USA). Recordings were performed in current- and voltage-clamp modes (more details available in Theiss et al. 2007). Briefly, slices were perfused with a modified Ringer's solution containing (mm): NaCl, 111; KCl, 3.09; NaHCO3, 25.0; KH2PO4, 1.10; MgSO4, 1.26; CaCl2, 2.52; and glucose, 11.1. The solution was oxygenated with 95% O2–5% CO2 and the perfusion rate was 2.5–3.0 ml min−1. Cellular currents were sometimes blocked using 10 mm TEA-Cl (K+), 1 μm TTX (Na+) and 10 μm isradipine (ISR; Ca2+) when indicated. Patch electrodes contained (mm): potassium gluconate, 138; Hepes, 10; ATP-Mg, 5; and GTP-Li, 0.3 (all from Sigma, St Louis, MO, USA). In voltage-clamp mode, fast and slow capacitance transients as well as whole-cell capacitance was compensated using the automatic capacitance compensation on the Multiclamp. Holding potential was set at either −75 or −80 mV, and neurons were subjected to slow, depolarizing voltage ramps of 20 mV s−1, bringing the cell to 5 or 0 mV in 4 s, and then back to the holding potential in the following 4 s. In current clamp, neurons were subjected to depolarizing steps to test for the after-hyperpolarization (AHP), activity-dependent hyperpolarization (ADH) and spike frequency adaptation. Depolarizing current ramps were also used for testing Ion (the current level at firing onset), Ioff (the current level at cessation of firing), deltaI (Ion-Ioff) and the frequency–current (F–I) relationship. Negative current was occasionally necessary to prevent continued action potential (AP) firing, but generally this was temporary and neurons needed no constant holding current.
Neuron selection
The ventrolateral motoneuron pools could be easily visualized in the slice preparation using differential interference contrast optics, and electrodes were positioned above this area. Individual neurons were targeted based on large soma diameter (>20 μm long axis), and only neurons with an input resistance <115 MΩ were included in this study, in order to target the large spinal motoneurons, which are among the most vulnerable to this disease (Tandan & Bradley, 1985; Mohajeri et al. 1998; Kuo et al. 2005; von Lewinski & Keller, 2005). Additionally, only neurons maintaining a resting potential <−50 mV, an action potential height crossing 0 mV and a series resistance of <25 MΩ were used. Neurons were eliminated from analysis if series resistance or resting potential varied more than 10 MΩ or 10 mV, respectively, throughout the recording period.
Data analysis
Data were collected using Winfluor software (University of Strathclyde, Glasgow, UK) and analysed using Spike2 software (Cambridge Electronic Design, Cambridge, UK). Graphs and statistics were performed using IGOR Pro software (Wavemetrics, Oswego, OR, USA), SPSS (Chicago, IL, USA) and Microsoft Excel (Redmond, WA, USA). Successive slow voltage ramp files that were collected in control and drug-treated conditions were subtracted from each other using Spike2.
Statistics
Factorial ANOVAs with a two (age) by two (transgene) design were performed on all data. Levene's test of equality of error variance was used to determine normality of the data. The natural logarithm of non-normal data was taken, followed by a factorial ANOVA. Variables that were found to be non-normally distributed were total PIC amplitude, Ion, Ioff, capacitance, conductance and AP duration. Bonferroni adjustments were made to account for multiple comparisons. Significant P values were set at 0.05 for ANOVA comparisons. Student's Unpaired t tests, with equal variance not assumed, were performed on comparisons between data categorized as high or low PIC amplitude for SOD1G93A neurons with significance at P = 0.05. For the overall analysis for Figs 5 and 7, we first normalized all parameters to their overall mean (of all the measured values for that parameter), so that an increase in a given parameter with age (or with SOD1 mutation) would yield a positive value, such as 0.5 for a 50% increase in that parameter, or a negative value for a decrease. The simplified formulae for this calculation are as follows, with Mean ALL referring to the mathematical mean of all the values for a particular parameter measured in this study:
Figure 5. Overall patterns of mutation and ageing.
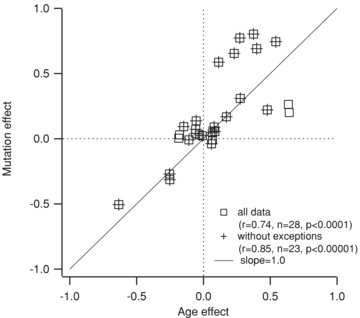
The y-axis is the average difference in an electrical property induced by the SOD1 mutation, normalized by the average of values from all cells (all ages, both WT and SOD1). The x-axis is the averaged normalized difference for age, such that if a property were showing a 40% increase with both age and mutation it would fall precisely on the slope = 1 line at (0.4, 0.4). For all data, r2= 0.548, n = 28, P < 0.0001; and without exceptions, r2= 0.723, n = 23, P < 0.00001.
Figure 7. Overall patterns of high-PIC SOD1 characteristics and ageing.

A, the y-axis is the average difference for high-PIC SOD1 cells compared with low-PIC SOD1 cells (normalized by the average for all SOD1 cells). The x-axis is the averaged normalized difference for age, such that if a property were showing a 40% increase in high-PIC SOD1 motoneurons versus low-PIC SOD1 and a 40% increase with ageing, it would fall precisely on the slope = 1 line at (0.4, 0.4). For all data, r2= 0.608, n = 28, P < 0.0001; and without exceptions, r2= 0.723, n = 223, P < 0.00001.
Results
We analysed a diverse set of electrical properties across the age range 0–12 days postnatal (P0–12), a period when many developmental changes are taking place. We focused on large motoneurons (soma diameter >20 μm). Standard patch-clamp methods were applied in slices of the lumbosacral region of the cord. We studied over 100 cells, divided into two samples, a mutant sample obtained from mice overexpressing the G93A mutation of human SOD1 (SOD1 motoneurons; Gurney et al. 1994) and a WT sample obtained from either non-transgenic littermates or mice overexpressing the normal, wild-type human SOD1 (no significant differences were observed between these two control groups). To detect effects of maturation, these two groups were subdivided into younger (P0–5) and older (P6–12) subgroups, giving four basic groups (WT younger, WT older, SOD1 younger and SOD1 older). All data samples were checked for normality using Levene's test, and statistical analyses were performed using ANOVAs, with age and mutation as factors (see Methods) unless otherwise noted. Tables 1 and 2 provide means, standard deviations and sample sizes for all measured parameters, with arrows indicating significance.
Table 1.
Changes in persistent inward current (PIC) parameters with maturation and superoxide dismutase-1 (SOD1) mutation
| WT P0–5 | SOD1 P0–5 | WT P6+ | SOD1 P6+ | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Mean | SD n | Mean | SD n | Mean | SD n | Mean | SD n | Significance of age | Significance of SOD1 | |
| Total PIC onset voltage (mV) | −46 | ±5 | −46 | ±6 | −42 | ±10 | −45 | ±8 | — | — |
| 13 | 17 | 20 | 20 | |||||||
| Total PIC peak (mV) | −22 | ±7 | −22 | ±5 | −18 | ±9 | −19 | ±5 | ↑ | — |
| 13 | 17 | 20 | 20 | |||||||
| Total PIC amplitude (pA) | −149 | ±96 | −294 | ±153 | −214 | ±127 | −450 | ±349 | ↑ | ↑↑↑ |
| 13 | 17 | 20 | 20 | |||||||
| Na+ PIC onset voltage (mV) | −44 | ±5 | −45 | ±6 | −43 | ±6 | −44 | ±6 | — | — |
| 14 | 17 | 19 | 16 | |||||||
| Na+ PIC peak (mV) | −17 | ±5 | −20 | ±4 | −18 | ±7 | −15 | ±5 | — | — |
| 14 | 17 | 19 | 16 | |||||||
| Na+ PIC amplitude (pA) | −132 | ±75 | −229 | ±126 | −179 | ±99 | −450 | ±343 | ↑ | ↑↑↑ |
| 14 | 17 | 19 | 16 | |||||||
| Ca2+ PIC onset voltage (mV) | −43 | ±8 | −43 | ±7 | −40 | ±12 | −43 | ±9 | — | — |
| 15 | 19 | 21 | 24 | |||||||
| Ca2+ PIC peak (mV) | −25 | ±9 | −26 | ±9 | −22 | ±11 | −21 | ±7 | ↑ | — |
| 15 | 19 | 21 | 24 | |||||||
| Ca2+ PIC amplitude (pA) | −47 | ±38 | −79 | ±56 | −49 | ±49 | −109 | ±69 | — | ↑↑↑ |
| 15 | 19 | 21 | 24 | |||||||
| Total PIC/C (pA pF−1) | −0.52 | ±0.27 | −1.20 | ±1.04 | −0.70 | ±0.40 | −1.56 | ±1.39 | — | ↑↑↑ |
| 13 | 17 | 20 | 20 | |||||||
| Na+ PIC/C (pA pF−1) | −0.48 | ±0.25 | −1.00 | ±1.11 | −0.61 | ±0.34 | −1.56 | ±1.36 | — | ↑↑ |
| 14 | 17 | 19 | 16 | |||||||
| Ca2+ PIC/C (pA pF−1) | −0.18 | ±0.18 | −0.28 | ±0.21 | −0.16 | ±0.14 | −0.35 | ±0.23 | — | ↑↑ |
| 15 | 19 | 20 | 24 | |||||||
| Capacitance (pF) | 273 | ±81 | 324 | ±156 | 321 | ±83 | 326 | ±93 | — | — |
| 24 | 25 | 31 | 31 | |||||||
The rows contain the values for the total, Na+ and Ca2+ PIC, and PIC normalized to capacitance (C). The PIC properties include the voltage of onset, the voltage of its peak and the amplitude of the current at peak. Also shown are the values for whole-cell capacitance. The mean value ± SD for each group is listed with the number of samples (n). The number of arrows in the rightmost two columns indicates significance, as follows: ↑ indicates 0.05 > P≥ 0.01; ↑↑ indicates 0.01 > P≥ 0.001; and ↑↑↑ indicates P < 0.001.
Table 2.
Changes in active and passive properties with maturation and SOD1 mutation
| WT P0–5 | SOD1 P0–5 | WT P6+ | SOD1 P6+ | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Mean | SD n | Mean | SD n | Mean | SD n | Mean | SD n | Significance of age | Significance of SOD1 | |
| Vthresh (mV) | −29 | ±5 | −27 | ±5 | −30 | ±7 | −30 | ±5 | — | — |
| 14 | 16 | 18 | 26 | |||||||
| AP overshoot (mV) | 28 | ±7 | 29 | ±10 | 30 | ±11 | 32 | ±7 | — | — |
| 19 | 19 | 20 | 28 | |||||||
| AP duration (ms) | 1.5 | ±0.6 | 1.1 | ±0.3 | 1.1 | ±0.3 | 0.9 | ±0.2 | ↓↓↓ | ↓↓↓ |
| 19 | 19 | 20 | 28 | |||||||
| AP rise (mV ms−1) | 75 | ±26 | 95 | ±33 | 95 | ±31 | 106 | ±20 | ↑↑ | ↑ |
| 19 | 19 | 20 | 28 | |||||||
| AP fall (mV ms−1) | 39 | ±13 | 56 | ±19 | 54 | ±16 | 72 | ±17 | ↑↑↑ | ↑↑↑ |
| 19 | 19 | 20 | 28 | |||||||
| ISI 1–3/SS ISI (%) | 70 | ±34 | 66 | ±22 | 57 | ±32 | 71 | ±25 | — | — |
| 16 | 15 | 17 | 15 | |||||||
| AHP amplitude (mV) | 9.4 | ±2.3 | 8.9 | ±2.3 | 9.2 | ±3.0 | 10.8 | ±2.9 | — | — |
| 15 | 15 | 11 | 17 | |||||||
| AHP τ (ms) | 40 | ±18 | 28 | ±15 | 28 | ±10 | 24 | ±10 | ↓ | ↓ |
| 15 | 14 | 11 | 17 | |||||||
| ADH amplitude (mV) | 5.5 | ±2.7 | 2.9 | ±2.5 | 2.5 | ±1.5 | 1.9 | ±1.6 | ↓↓ | ↓ |
| 7 | 13 | 13 | 17 | |||||||
| Conductance (nS) | 16 | ±6 | 20 | ±10 | 26 | ±10 | 32 | ±14 | ↑↑↑ | ↑ |
| 24 | 25 | 30 | 32 | |||||||
| Ion (pA) | 368 | ±175 | 443 | ±281 | 664 | ±374 | 903 | ±519 | ↑↑↑ | — |
| 16 | 18 | 18 | 27 | |||||||
| Ioff (pA) | 420 | ±201 | 486 | ±293 | 781 | ±425 | 983 | ±583 | ↑↑↑ | — |
| 16 | 18 | 18 | 27 | |||||||
| ΔI (pA) (ms) | 52 | ±134 | 44 | ±86 | 117 | ±195 | 80 | ±243 | — | — |
| 16 | 18 | 18 | 27 | |||||||
| F–Iasc (Hz nA−1) | 35 | ±15 | 40 | ±23 | 31 | ±15 | 33 | ±17 | — | — |
| 15 | 18 | 17 | 27 | |||||||
| F–Idesc (Hz nA−1) | 32 | ±14 | 40 | ±28 | 33 | ±21 | 35 | ±24 | — | — |
| 13 | 18 | 16 | 27 | |||||||
| RMP (mV) | −57 | ±4 | −56 | ±4 | −60 | ±6 | −60 | ±5 | ↓↓↓ | — |
| 16 | 19 | 20 | 27 | |||||||
Shown here are the properties of the action potential (AP), including the voltage at AP threshold (Vthresh), AP overshoot and the interspike intervals (ISIs) of first three APs of a square current pulse normalized to steady state (ISI 1–3/SS ISI). The following rows include conductance, the ascending and descending slope of the F–I curves (F–Iasc, F–Idesc) and the resting membrane potential (RMP). The mean value ± SD for each group is listed with the number of samples (n). The number of arrows in the rightmost two columns indicates significance, as follows: ↑ indicates 0.05 > P≥ 0.01; ↑↑ indicates 0.01 > P≥ 0.001; and ↑↑↑ indicates P < 0.001.
Effects of mutation compared with maturation on motoneuron electrical properties
Previous studies have consistently found an elevation in the Na+ PIC in SOD1 motoneurons, suggesting increased excitability. However, the postnatal development of the Na+ PIC has not been studied in either SOD1 or WT motoneurons. Hence, our first aim was to compare the development of the Na+ PIC in WT and SOD1 motoneurons. In addition to the Na+ PIC, which is probably mediated by Nav1.1 and 1.2 in neonates, and Nav1.1, 1.2 and 1.6 channels in juvenile mice (Boiko et al. 2001, 2003; Rush et al. 2005), motoneurons also have a Ca2+ PIC, probably mediated by Cav1.3 channels (Perrier & Hounsgaard, 2003; Li et al. 2007), which remains small until around P10 in WT motoneurons (Jiang et al. 1999). The Ca2+ PIC is also very important in determining motoneuron excitability (Alaburda et al. 2002; Heckman et al. 2008). Therefore, we measured both PICs from slow voltage ramps (Schwindt & Crill, 1980) and identified each component by pharmacological blockade (TTX for the Na+ PIC and isradipine for the Ca2+ PIC; Fig. 1A and C for a P1 WT motoneuron, Fig. 1B and D for a P9 SOD1 motoneuron).
Figure 1. Measurement of persistent inward currents (PICs).
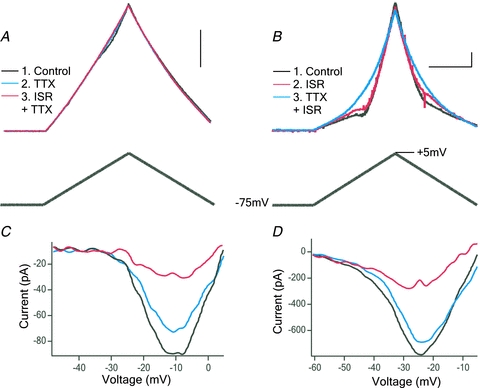
Whole-cell currents were recorded from motoneurons during a slow, depolarizing voltage ramp. A, current trace (top) from a P1 wild-type motoneuron in response to a voltage ramp (bottom trace). First trace is in control conditions (black); next, tetrodotoxin (TTX; 1 μm) was applied to block Na+ (blue), followed by TTX with isradipine (ISR;10 μm) to block Ca2+ (red). B, a similar recording from a P9 SOD1 motoneuron, which was recorded first in control conditions (black), then in ISR (red), followed by ISR and TTX (blue). Scale bars: horizontal 2 s, vertical 0.5 nA. C and D, traces from A and B were subtracted to show the total PIC (black), Na+ PIC (blue) and Ca2+ PIC (red).
Our data revealed that total PIC and Na+ PIC amplitude significantly increase with maturation from P0–5 to P6–12, as shown in Fig. 2 and Table 1. The amplitude of the total PIC and Na+ PIC was also significantly enhanced by mutation, as was expected from previous work (Kuo et al. 2005; van Zundert et al. 2008; Pieri et al. 2009; Fig. 2B). Owing to its delayed appearance, the Ca2+ PIC did not show significant changes with age from P0–5 to P6–12 (see Table 1 for means and significance). However, SOD1 motoneurons had significantly larger Ca2+ PICs than WT motoneurons, an important and novel finding (Fig. 2C and Table 1). The total PIC was dominated by the Na+ PIC during this time window in both WT and SOD1 motoneurons (Fig. 2A). The amplitude of each component was measured at its peak, and because the Ca2+ PIC and Na+ PIC peaked at slightly different voltages (see Table 1), the total PIC is not simply peak Na+ PIC + peak Ca2+ PIC. Although there was considerable variation within each group, the increase in total, Na+ and Ca2+ PIC amplitudes in SOD1 motoneurons was substantial, with the total PIC of young SOD1 motoneurons being double that of the young WT motoneurons, and the older SOD1 motoneurons having an average total PIC of three times that of the young WTs. Maturation had small but significant effects on the voltages for the peak of the Ca2+ and total PICs, which were not matched by the SOD1 mutation. In 18 motoneurons, TEA was applied before measuring the PIC in order to block the K+ currents, which could contaminate the PIC measurement. These 18 motoneurons did not show significantly different PIC amplitudes from the other 66 neurons recorded (data not shown).
Figure 2. Changes in PIC with age and mutation.
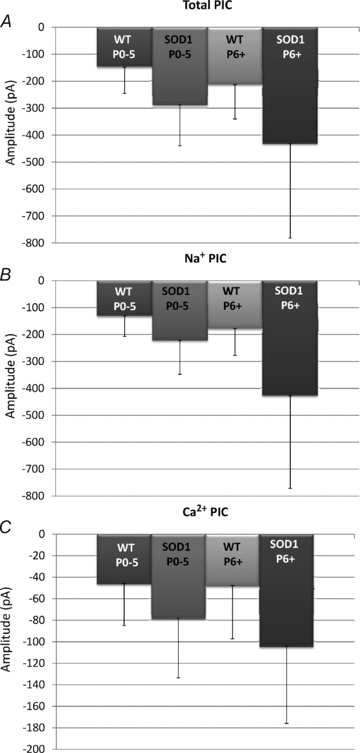
The peak amplitude of the total PIC (A), Na+ PIC (B) and Ca2+ PIC (C) was plotted for groups based on age [postnatal day 0–5 (P0–5) and postnatal day 6 and older (P6+)] and mutation: wild type (including both nontransgenic and SOD1WT transgenic mice, WT) and SOD1G93A (SOD1). Error bars represent the standard deviation.
In the next analysis, we examined a feature characteristic of postnatal development in spinal motoneurons, the change in duration of the AP and the AHP after a single spike. The AHP is a major determinant of AP firing frequency. In addition, we also measured ADH, the hyperpolarizing potential that occurs after a burst of action potentials (Disterhoft & Oh, 2007). With maturation, the time constant (τ) of the AHP decay decreases, shortening the time course of the AHP without any change in its amplitude. In addition, the rates of the AP rise and fall both increase, resulting in a shorter duration AP (Fig. 3 and Table 2). These changes are all consistent with previous studies of maturation (Carrascal et al. 2005). The ADH was measured after 1 s depolarizing pulses that elicited firing rates of 20–30 Hz. The amplitude of the ADH was found to be significantly smaller in both older and SOD1 motoneurons (Table 2). These results show that mutation significantly enhanced the effects of maturation on the ADH, AHP and AP, resulting in even shorter duration APs with faster decaying AHPs followed by smaller amplitude ADHs. As was the case for PICs, these changes were substantial. For example, AP width (measured at half-peak) narrowed from ∼1.5 to ∼1.1 ms with age (P0–5 compared with P6–12 in WT) and was further decreased to ∼ 0.9 ms in the older SOD1 motoneurons (P6–12; Fig. 3).
Figure 3. Maturation of the action potential (AP) and after-hyperpolarization (AHP).
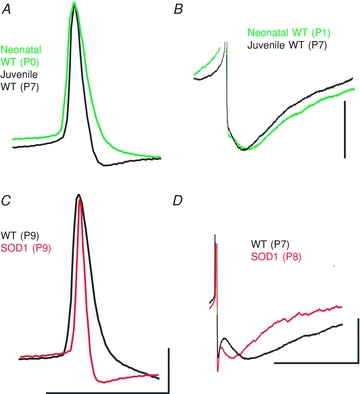
A, during normal postnatal development in WT neurons, the AP narrows. B, the time constant of the medium AHP decreases, shortening the duration of the AHP. With SOD1 mutation, the AP shows advanced narrowing (C) and the AHP shows an increased rate of decay (D). Scale bars for A and C: horizontal, 0.1 s; vertical, 20 mV. Scale bars for B and D: horizontal, 0.1 s; B vertical, 5 mV; D vertical, 20 mV.
Normally, motoneuron size increases during maturation, resulting in increases in total electrical conductance and capacitance (Carrascal et al. 2005). In our data, total conductance increased significantly from younger to older motoneuron groups and in SOD1 motoneurons, but there was no increase in total capacitance, perhaps because of the restricted age range or loss of processes in the slice preparation (see the Discussion for further comments). Maturation also induced a small but significant hyperpolarization of resting potential, which was not matched by effects of mutation (see Tables 1 and 2).
Net excitability of motoneurons is usually measured from the threshold and slope of the relationship between firing frequency and injected current (the F–I function; Powers & Binder, 2001). We used a triangular current waveform to evaluate the amount of current at firing onset (Ion), ascending and descending slopes of the F–I relationship, and current at firing offset (Ioff; see examples in Fig. 4). In adult motoneurons, the F–I slope is decreased and Ion and Ioff are increased compared with young motoneurons (Manuel et al. 2009; Meehan et al. 2010b). In the time window of the present study, neither maturation nor mutation had a significant impact on F–I slope, as can be seen in Fig. 4. However, Ion and Ioff were significantly increased with maturation but not mutation, an exception to the pattern of similar effects of maturation and mutation. The difference between Ion and Ioff (ΔI) is a useful index, which can indirectly confirm the presence of the Ca2+ PIC; however, it is not as sensitive as direct measurements performed here, and by itself it does not prove absence of a PIC (Bennett et al. 2001). Overall, neither maturation nor mutation increased net excitability during this time window.
Figure 4. Firing frequency–current relationships.
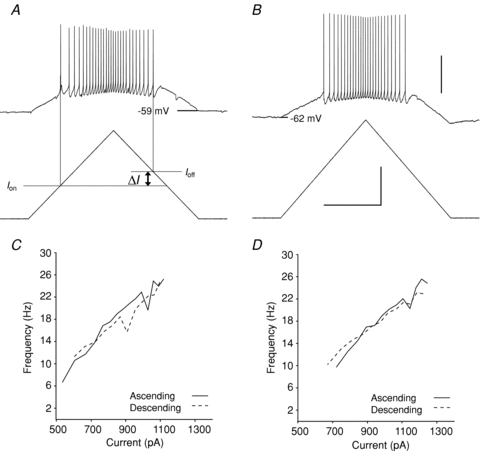
Depolarizing ramps of current were injected into motoneurons. Shown are examples from a young (P3) WT (A) and older (P10) SOD1 motoneuron (B). The current level was measured at the onset (Ion) and cessation of firing (Ioff); ΔI=Ion−Ioff. The relationship between firing and current is plotted in C for the motoneuron in A and in D for the motoneuron in B. Scale bars: horizontal, 1.0 s; vertical: 40 mV for top traces in A and B and 0.5 nA for bottom traces.
Overall pattern
To obtain an overview of how well the profile of electrical properties induced by mutation matched that of maturation, we plotted the change in each electrical parameter due to the mutation versus the change due to maturation (changes were normalized by the average values for each parameter across both age and mutation). In the plot shown in Fig. 5, exactly equal effects of maturation and mutation would generate a line with a slope of 1.0 (continuous line). Despite the few exceptions where age had a significant effect but mutation did not (i.e. Ion, Ioff, resting potential, voltages at peak total and Ca2+ PICs; see Tables 1 and 2), Fig. 5 shows that an overall positive correlation between maturation and SOD1 mutation emerged across electrical parameters (r = 0.74, P < 0.0001, n = 28; and when the exceptions were eliminated, r = 0.85, P < 0.00001, n = 23). Thus, the overall similarity between the patterns of mutation and maturation is highly significant even with the noted exceptions. Our results show that overall the changes in SOD1 motoneuron electrical properties are very similar to the changes taking place during postnatal development. Based on the value r2= 0.55 across all electrical properties, roughly half of the changes observed in SOD1 motoneurons could be attributed to this pattern of accelerated development.
A more pronounced phenotype in a subset of SOD1 motoneurons
The substantial variation in PICs meant that some of the SOD1 motoneurons had much larger PICs than WT motoneurons, while PICs of other SOD1 motoneurons were within the WT range. If these large PICs are indicative of an especially mature state, then the other electrical properties in these large PIC cells should likewise be more advanced in development than the SOD1 cells with smaller PICs. To evaluate this possibility, we selected the motoneurons in which the total PIC amplitude was greater than 1 SD above the mean of total PIC for all motoneurons recorded (total PIC >–523 pA). A subset of nine SOD1 motoneurons met this criterion (no WT cells had PICs this large). The high-PIC SOD1 motoneurons were then compared with the remaining 36 low-PIC SOD1 motoneurons. As expected from this selection criterion based on total PIC, the high-PIC SOD1 motoneurons also had significantly elevated Na+ and Ca2+ PIC component current amplitudes compared with the low-PIC SOD1 motoneurons (Table 3). This subset of neurons did not only display larger PICs, they also exhibited more mature APs and AHPs than the other SOD1 motoneurons. They had the fastest AP parameters of all motoneurons, having significantly shorter duration APs and faster decaying AHPs than low-PIC SOD1 and WT motoneurons (Table 3 and Fig. 6). Figure 7 is analogous to Fig. 5, showing a significant correlation between the change in high-PIC versus low-PIC SOD1 cells versus change due to ageing (r = 0.78, P < 0.00001, n = 28). In summary, the cells with the largest PICs were all from the SOD1 sample and overall had the most mature profile of electrical properties.
Table 3.
Changes in active and passive properties between the high-PIC and low-PIC SOD1 motoneurons
| Low-PIC SOD1 | High-PIC SOD1 | ||||
|---|---|---|---|---|---|
| Mean | SD n | Mean | SD n | Significance | |
| Total onset voltage (mV) | −46 | ±7 | −44 | ±6 | — |
| 28 | 9 | ||||
| Total peak (mV) | −22 | ±4 | −16 | ±6 | ↑ |
| 28 | 9 | ||||
| Total amplitude (pA) | −246 | ±109 | −791 | ±265 | ↑↑↑ |
| 28 | 9 | ||||
| Na+ PIC onset voltage (mV) | −44 | ±6 | −46 | ±6 | — |
| 24 | 9 | ||||
| Na+ PIC peak (mV) | −18 | ±5 | −17 | ±4 | — |
| 24 | 9 | ||||
| Na+ PIC amplitude (pA) | −200 | ±95 | −696 | ±275 | ↑↑↑ |
| 24 | 9 | ||||
| Ca2+ PIC onset voltage (mV) | −44 | ±8 | −39 | ±9 | — |
| 34 | 9 | ||||
| Ca2+ PIC peak (mV) | −24 | ±8 | −18 | ±7 | ↑ |
| 34 | 9 | ||||
| Ca2+ PIC amplitude (pA) | −87 | ±67 | −129 | ±39 | ↑ |
| 34 | 9 | ||||
| Total PIC/C (pA pF−1) | −0.91 | ±0.77 | −2.90 | ±1.22 | ↑↑↑ |
| 28 | 9 | ||||
| Na+ PIC/C (pA pF−1) | −0.79 | ±0.91 | −2.55 | ±1.18 | ↑↑ |
| 24 | 9 | ||||
| Ca2+ PIC/C (pA pF−1) | −0.28 | ±0.21 | −0.48 | ±0.20 | ↑ |
| 34 | 9 | ||||
| Capacitance (pF) | 331 | ±130 | 294 | ±86 | — |
| 47 | 9 | ||||
| Vthresh (mV) | −29 | ±5 | −29 | ±4 | — |
| 34 | 8 | ||||
| RMP (mV) | −58 | ±5 | −61 | ±5 | — |
| 37 | 9 | ||||
| AP overshoot (mV) | 30 | ±9 | 33 | ±4 | — |
| 38 | 9 | ||||
| AP duration (ms) | 1.0 | ±0.3 | 0.7 | ±0.1 | ↓↓↓ |
| 38 | 9 | ||||
| AP rise (mV ms−1) | 100 | ±28 | 111 | ±13 | — |
| 38 | 9 | ||||
| AP fall (mV ms−1) | 62 | ±18 | 81 | ±15 | ↑↑ |
| 38 | 9 | ||||
| ISI 1–3/SS ISI (%) | 66 | ±20 | 81 | ±32 | — |
| 24 | 6 | ||||
| AHP amplitude (mV) | 10.0 | ±2.6 | 9.3 | ±3.8 | — |
| 27 | 5 | ||||
| AHP τ (ms) | 28 | ±13 | 17 | ±5 | ↓ |
| 26 | 5 | ||||
| ADH amplitude (mV) | 2.6 | ±2.2 | 1.1 | ±0.7 | ↓ |
| 25 | 5 | ||||
| Conductance (nS) | 25 | ±13 | 33 | ±17 | — |
| 48 | 9 | ||||
| Ion (pA) | 659 | ±475 | 958 | ±512 | — |
| 36 | 9 | ||||
| Ioff (pA) | 737 | ±531 | 974 | ±580 | — |
| 36 | 9 | ||||
| ΔI (pA) (ms) | 78 | ±203 | 15 | ±159 | — |
| 36 | 9 | ||||
| F–Iasc (Hz nA−1) | 36 | ±16 | 37 | ±30 | — |
| 36 | 9 | ||||
| F–Idesc (Hz nA−1) | 37 | ±25 | 36 | ±28 | — |
| 36 | 9 | ||||
Shown here are the same properties as those in Tables 1 and 2, but comparing those motoneurons that had the largest total PIC amplitude (more than 1 SD above the mean) with the rest of the SOD1 motoneurons. The mean value ± SD for each group is listed with number of samples (n). The number of arrows in the rightmost column indicates significance, as follows: ↑ indicates 0.05 > P≥ 0.01; ↑↑ indicates 0.01 > P≥ 0.001; and ↑↑↑ indicates P < 0.001.
Figure 6. More pronounced SOD1 phenotype of the AP and AHP in a subset of SOD1 neurons.
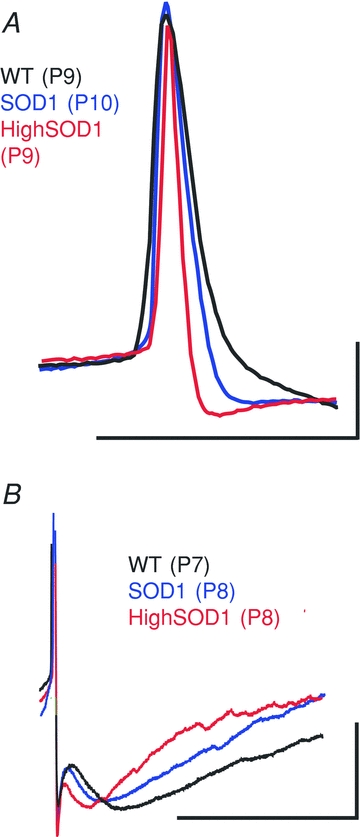
A, the APs in the high-PIC SOD1 group were significantly shorter in duration than the rest of the SOD1 group. B, the time constant of the medium AHP decreased more drastically in the high-PIC SOD1 group, shortening the duration of the AHP. Scale bars: A horizontal, 0.1 s; A vertical, 20 mV; B horizontal, 0.1 s; B vertical, 20 mV.
Discussion
Our results provide an exhaustive analysis of over 25 cell properties in over 100 spinal motoneurons in the most severe mouse model of ALS, the SOD1G93A mouse. Overall, the electrical changes fit a profile of an increased rate of maturation.
Comparison of SOD1 animal models with ALS
The SOD1 mouse takes advantage of the human mutant SOD1 gene that causes ALS. Familial ALS (fALS) caused specifically by SOD1 mutations makes up 2% of all cases, or 20% of all fALS (Rosen et al. 1993), while sporadic ALS (sALS) accounts for 90% of all ALS cases. Transgenic mice overexpressing a mutated human SOD1 gene undergo progressive neurodegeneration similar to ALS in humans (Gurney et al. 1994), marked by muscle weakness and paralysis (Kong & Xu, 1998). Spinal motoneurons acquire pathological features seen in ALS, including axonal spheroids and fragmentation of the Golgi apparatus (Dal Canto & Gurney, 1995; Mourelatos et al. 1996). Furthermore, evidence for cortical hyperexcitability and increased Na+ conductance in motoneurons and axons has been shown in both SOD1 mice and in fALS patients (Mogyoros et al. 1998; Vucic et al. 2008; Carunchio et al. 2010; Vucic & Kiernan, 2010). Increased cortical excitability, similar to that seen in sALS patients, has been observed to precede the symptom onset in fALS, suggesting similar pathophysiology for ALS whether the cause is genetic or environmental (Vucic et al. 2008). Although the neurodegeneration in SOD1 mice and fALS may be triggered by separate mechanisms, the final common pathways to neurodegeneration are probably shared. Hopefully, use of the SOD1 animal model will soon lead to useful interventions applicable in human ALS.
Comparison with the ageing process
Motoneurons are highly vulnerable to the normal ageing process, with a progressive die-off occurring in healthy humans without ALS starting at around 60 years of age (Campbell et al. 1973; Tomlinson & Irving, 1977). Thus, while the ability of a young motoneuron to maintain homeostasis in non-disease states is remarkable (Marder & Goaillard, 2006), the processes leading to the degeneration of motoneurons in ALS could involve genetic or environmental stresses earlier in life that trigger a pathological acceleration of the normal ageing process. Results from this study would support this possibility, although it remains speculative and further investigation at later time points is needed.
Progression over time
Changes in SOD1 motoneuron electrical and anatomical properties occur remarkably early, within the first 10 days of life (Kuo et al. 2005; Bories et al. 2007; Amendola & Durand, 2008; van Zundert et al. 2008). An important question is, what happens as the motoneuron progresses past P12 into the adult state? Some of these parameters have been measured in WT adult mouse spinal motoneurons and show a continuation of the changes observed in this study. The AP and AHP, found here to shorten significantly in duration, are even faster in adult WT spinal motoneurons, with an average AP duration of 0.3 ± 0.05 ms and time constant for the medium AHP of 10 ± 3 ms in adults (Manuel et al. 2009), compared with 0.9 ± 0.2 ms APs and 24 ± 10 ms AHP time constants found here in the older SOD1 group (cf. Table 2). Input conductance and PIC amplitudes, shown here to increase from P0 to P12, are likewise reported to be even higher in adult rodent spinal motoneurons, although recorded in varying conditions (Li & Bennett, 2003; Manuel et al. 2009; Meehan et al. 2010a,b;). Our finding that the threshold currents for repetitive firing (Ion and Ioff) increase less with mutation than with age may indicate that the SOD1 motoneurons will become hyperexcitable with more time. Consistent with this possibility, SOD1 motoneurons grown in cell culture have markedly enhanced excitability (Pieri et al. 2003a; Kuo et al. 2004, 2005). Taken together, these findings indicate that the trends we found from P0 to P12 are likely to continue into adulthood. The high-PIC SOD1 group in the present study may attain hyperexcitability first, contributing to excitotoxicity, increasing metabolic demands, and possibly becoming the first cells to degenerate. An alternative hypothesis to accelerated maturation is that SOD1 motoneurons are instead expressing an exaggerated amount of the normal complement of ion channels. A detailed analysis of channel subtypes and their order of appearance during maturation would be needed to investigate this hypothesis fully (Boiko et al. 2001, 2003); however, if this were the case one might expect to see an overexpression of the SK channels that produce the AHP (Lape & Nistri, 2000), for example. This would result in an increased AHP amplitude in SOD1 motoneurons, which was not the case (Fig. 3 and Table 2).
Comparison with brainstem SOD1 motoneurons
Brainstem SOD1G93A motoneurons in the same age range (P4–10) as our sample and recorded using similar methods (patch clamp in slices) exhibited increased F–I gain compared with WT (van Zundert et al. 2008), whereas our spinal motoneurons did not. However, normal brainstem motoneurons also differ from spinal motoneurons in this behaviour, with F–I gain increasing with age (Carrascal et al. 2005) instead of decreasing as for spinal motoneurons (Manuel et al. 2009; Meehan et al. 2010b). Thus, an increased gain is consistent with accelerated maturation of SOD1 brainstem motoneurons. Significant decreases in spike duration, AHP time constant or input conductance did not occur in these brainstem motoneurons, but the sample size was small (n = 7; van Zundert et al. 2008) compared with the present study (n = 42 for WT, 51 for SOD1). These brainstem SOD1 motoneurons also exhibited increased levels of spontaneous input and dendritic pruning characteristic of a more mature state (van Zundert et al. 2008).
Comparison with SOD1 models that have a slower disease progression
Studies have also been performed on SOD1 models with a slower rate of disease progression than the high-expressor SOD1G93A model used here. In the work of Pambo-Pambo et al. (2009) on the low-expressor SOD1G93A and SOD1G85R models, motoneurons do not appear to be in a more mature state, with the SOD1G85R motoneurons having normal spike widths and the low-expressor SOD1G93A motoneurons having wider, i.e. more immature, spike widths than WT motoneurons. Just as the asymptomatic period is roughly twice as long in the low-expressor SOD1G93A and in the SOD1G85R animals, it may be that the developmental abnormalities are likewise varied. In the SOD1 animals, as in the ALS patients, the disease onset, time course and early indicators are highly variable. While our SOD1G93A high-expressor motoneurons appear to have an increased rate of maturation, we do not suggest that this is the only early developmental abnormality that could set the motoneurons on a course for premature degeneration.
In all three models, high-expressor SOD1G93A, which we used in our studies, low-expressor SOD1G93A and SOD1G85R, there is a significant increase in input conductance which, in the case of the SOD1G85R, has been shown to be due to increased dendritic branching (Amendola & Durand, 2008) as well as increased specific membrane conductance (Elbasiouny et al. 2010). It is not yet known whether these excess branches occur in the SOD1G93A high-expressor line or whether the branching results in increased synaptic contacts, nor is it clear whether the mechanism causing these excess distal branches in spinal motoneurons is related to the more extensive pruning of distal branches seen in brainstem SOD1G93A motoneurons (van Zundert et al. 2008). As our studies were performed in the slice preparation, it was not ideal to make any anatomical comparisons, owing to a possible loss of processes. If the SOD1G93A high-expressor line also has increased distal dentritic branching similar to the SOD1G85R motoneurons, our measurements of input conductance, though significantly larger in SOD1 and older motoneurons, may have been an underestimate of the true difference due to the lost processes. In addition, the same could be said of the Ca2+ PIC, which is also significantly larger in SOD1 motoneurons and most probably generated by Ca2+ channels on the dendrites. Therefore, the Ca2+ PIC in SOD1 motoneurons may be even larger if measured in motoneurons with fully intact processes.
Comparison with other potential mechanisms of disease onset
Unlike the channelopathies that lead to disease states through mutations that alter the function of a specific ion channel (Kullmann & Waxman, 2010), the pathways leading to symptom onset and disease progression in SOD1 animals could arise from non-cell autonomous pathways as well as from mechanisms within the motoneurons. Excitotoxicity could be a factor in both of these pathways (Rothstein, 1996). Increased Ca2+-permeable glutamate receptors on the motoneurons (Pieri et al. 2003b) could result in excitation-mediated damage to motoneurons, as could alterations in glutamate handling by SOD1 astrocytes (Nagai et al. 2007; Van Damme et al. 2007; Yang et al. 2009, 2010). In fact, glutamate neurotransmission is probably a significant factor in symptom onset, because reducing excitatory sensory input delayed disease onset in SOD1G93A mice (Ilieva et al. 2008). Likewise, the changes in motoneuron maturation observed in this study could be a reflection of early alterations in activity in the spinal networks, which are a hallmark of normal development (Landmesser & O'Donovan, 1984; Marder & Rehm, 2005; Gonzalez-Islas & Wenner, 2006; Blankenship & Feller, 2010). Initial evidence for altered network activity has been shown in SOD1G93A hypoglossal neurons (van Zundert et al. 2008). In addition to alterations in the local network activity, cortical degeneration and spinal motoneuron degeneration could be also be related. Evidence of cortical hyperexcitability has been shown at the same early time points as in spinal motoneurons (Kuo et al. 2005; Pieri et al. 2009), suggesting that these neurons are developing abnormally concomitantly. It would be interesting to investigate whether more global changes in cortical and spinal networks are present, and whether this would include altered glutamatergic neurotransmission, and a more mature phenotype in the interneurons that synapse onto these neurons, as might be suggested by the increased activity observed in adult spinal interneurons (Jiang et al. 2009). Within the motoneurons, it would be interesting to investigate whether other cellular properties are also shifted to a more mature state, such as those of mitochondria (Bacman et al. 2006) and axon transport systems (Saxena et al. 2009). Loss of motor unit force appears to occur at the neuromuscular junction before the motoneuron degenerates (Balice-Gordon et al. 2000; Rich et al. 2002; Pun et al. 2006), possibly involving deficits in axonal transport (Saxena et al. 2009). Our results and previous studies clearly show that electrical properties of motoneuron cell bodies are altered long before force is lost at about P50 (Hegedus et al. 2007, 2008; Gordon et al. 2009). The questions of whether these very early changes in electrical properties are in response to still earlier transport or neuromuscular junction deficits or instead produce problems at the neuromuscular junction cannot be answered with the present data. A comparative analysis of maturation in these two parts of the motor unit may be very helpful in elucidating mechanisms of disease onset.
Clinical implications
A promising future direction is to use agents that modify the altered electrical properties of SOD1 motoneurons. Riluzole, the current treatment of ALS, slightly prolongs life in both mouse models and in human patients (Bensimon et al. 1994; Gurney et al. 1996). Although riluzole has many actions at higher concentrations, previous studies have shown that, at the low serum concentrations attainable by oral administration (1–2 μm), riluzole is a specific blocker of the Na+ PIC, as well as reducing glutamate release from synaptosomes and increasing glutamate uptake by astrocytes (Hebert et al. 1994; Lacomblez et al. 1996; Huang et al. 1997; Zona et al. 1998; Azbill et al. 2000; Urbani & Belluzzi, 2000; Rao & Weiss, 2004; Kuo et al. 2005; Lamanauskas & Nistri, 2008). The other properties that are shown here to be altered (Ca2+ PIC, AHP and AP) could also be worthwhile targets for intervention with other pharmaceutical agents. Another approach might be to manipulate neuromodulatory input to the spinal cord. Serotonin and noradrenaline have potent effects on motoneurons, including increasing PIC amplitude, hyperpolarizing spike thresholds, decreasing AHP amplitude and depolarizing the resting potential (Powers & Binder, 2001; Alaburda et al. 2002; Hultborn et al. 2004; Heckman et al. 2008). It has been suggested that blocking excessive Na+ PICs at this early stage in life may slow disease onset (van Zundert et al. 2008); if so, it will be interesting to see whether drugs that reduce PICs either by direct channel block or via neuromodulatory actions can slow disease progression by ‘resetting’ the motoneurons to a younger phenotype (cf. Chan et al. 2007).
Conclusions
Motoneurons of SOD1G93A mice show very early alterations in postnatal development of the electrical properties, including Na+ and Ca2+ PIC, input conductance, AP and AHP. These properties appear to be in a more mature state in SOD1 motoneurons than in WT motoneurons. This early advancement in maturation could, along with other disease mechanisms, set the motoneurons on a course towards vulnerability in later life.
Acknowledgments
This work was supported by grants from National Institutes of Health/National Institute of Neurological Disorders and Stroke (NS050162 and NS071951 to C.J.H. and NS050641 to T.S.), and individual NRSAs F32 NS063535 to K.A.Q. and F31 NS060532 to J.E.S. Authors made use of equipment supported by NS054850. Thanks to S. M. Elbasiouny and M. Manuel for comments on the manuscript.
Glossary
Abbreviations
- ADH
activity-dependent hyperpolarization
- AHP
after-hyperpolarization
- ALS
amyotrophic lateral sclerosis
- AP
action potential
- Ca2+ PIC
calcium persistent inward current
- ISR
isradipine
- Na+ PIC
sodium persistent inward current
- P
postnatal day
- PIC
persistent inward current
- SOD1
superoxide dismutase-1
- WT
wild-type
Author contributions
K.A.Q. helped to design the research, conducted the research and performed most of the data analysis. Statistics were performed by J.E.S. All breeding and genotyping of mice was performed by R.F. T.S. contributed to interpretation of results. C.J.H. helped to design the research and interpret the results, and bore primary responsibility for writing the manuscript. All authors discussed results and contributed to writing the manuscript.
References
- Alaburda A, Perrier JF, Hounsgaard J. Mechanisms causing plateau potentials in spinal motoneurones. Adv Exp Med Biol. 2002;508:219–226. doi: 10.1007/978-1-4615-0713-0_27. [DOI] [PubMed] [Google Scholar]
- Amendola J, Durand J. Morphological differences between wild-type and transgenic superoxide dismutase 1 lumbar motoneurons in postnatal mice. J Comp Neurol. 2008;511:329–341. doi: 10.1002/cne.21818. [DOI] [PubMed] [Google Scholar]
- Azbill RD, Mu X, Springer JE. Riluzole increases high-affinity glutamate uptake in rat spinal cord synaptosomes. Brain Res. 2000;871:175–180. doi: 10.1016/s0006-8993(00)02430-6. [DOI] [PubMed] [Google Scholar]
- Bacman SR, Bradley WG, Moraes CT. Mitochondrial involvement in amyotrophic lateral sclerosis: trigger or target? Mol Neurobiol. 2006;33:113–131. doi: 10.1385/MN:33:2:113. [DOI] [PubMed] [Google Scholar]
- Balice-Gordon RJ, Smith DB, Goldman J, Cork LC, Shirley A, Cope TC, Pinter MJ. Functional motor unit failure precedes neuromuscular degeneration in canine motor neuron disease. Ann Neurol. 2000;47:596–605. [PubMed] [Google Scholar]
- Bennett DJ, Li Y, Siu M. Plateau potentials in sacrocaudal motoneurons of chronic spinal rats, recorded in vitro. J Neurophysiol. 2001;86:1955–1971. doi: 10.1152/jn.2001.86.4.1955. [DOI] [PubMed] [Google Scholar]
- Bensimon G, Lacomblez L, Meininger V. A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/Riluzole Study Group. N Engl J Med. 1994;330:585–591. doi: 10.1056/NEJM199403033300901. [DOI] [PubMed] [Google Scholar]
- Blankenship AG, Feller MB. Mechanisms underlying spontaneous patterned activity in developing neural circuits. Nat Rev Neurosci. 2010;11:18–29. doi: 10.1038/nrn2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boiko T, Rasband MN, Levinson SR, Caldwell JH, Mandel G, Trimmer JS, Matthews G. Compact myelin dictates the differential targeting of two sodium channel isoforms in the same axon. Neuron. 2001;30:91–104. doi: 10.1016/s0896-6273(01)00265-3. [DOI] [PubMed] [Google Scholar]
- Boiko T, Van Wart A, Caldwell JH, Levinson SR, Trimmer JS, Matthews G. Functional specialization of the axon initial segment by isoform-specific sodium channel targeting. J Neurosci. 2003;23:2306–2313. doi: 10.1523/JNEUROSCI.23-06-02306.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bories C, Amendola J, Lamotte d’Incamps B, Durand J. Early electrophysiological abnormalities in lumbar motoneurons in a transgenic mouse model of amyotrophic lateral sclerosis. Eur J Neurosci. 2007;25:451–459. doi: 10.1111/j.1460-9568.2007.05306.x. [DOI] [PubMed] [Google Scholar]
- Campbell MJ, McComas AJ, Petito F. Physiological changes in ageing muscles. J Neurol Neurosurg Psychiatry. 1973;36:174–182. doi: 10.1136/jnnp.36.2.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrascal L, Nieto-Gonzalez JL, Cameron WE, Torres B, Nunez-Abades PA. Changes during the postnatal development in physiological and anatomical characteristics of rat motoneurons studied in vitro. Brain Res Brain Res Rev. 2005;49:377–387. doi: 10.1016/j.brainresrev.2005.02.003. [DOI] [PubMed] [Google Scholar]
- Carunchio I, Curcio L, Pieri M, Pica F, Caioli S, Viscomi MT, Molinari M, Canu N, Bernardi G, Zona C. Increased levels of p70S6 phosphorylation in the G93A mouse model of Amyotrophic Lateral Sclerosis and in valine-exposed cortical neurons in culture. Exp Neurol. 2010;226:218–230. doi: 10.1016/j.expneurol.2010.08.033. [DOI] [PubMed] [Google Scholar]
- Chan CS, Guzman JN, Ilijic E, Mercer JN, Rick C, Tkatch T, Meredith GE, Surmeier DJ. ‘Rejuvenation’ protects neurons in mouse models of Parkinson's disease. Nature. 2007;447:1081–1086. doi: 10.1038/nature05865. [DOI] [PubMed] [Google Scholar]
- Chiu AY, Zhai P, Dal Canto MC, Peters TM, Kwon YW, Prattis SM, Gurney ME. Age-dependent penetrance of disease in a transgenic mouse model of familial amyotrophic lateral sclerosis. Mol Cell Neurosci. 1995;6:349–362. doi: 10.1006/mcne.1995.1027. [DOI] [PubMed] [Google Scholar]
- Dal Canto MC, Gurney ME. Neuropathological changes in two lines of mice carrying a transgene for mutant human Cu,Zn SOD, and in mice overexpressing wild type human SOD: a model of familial amyotrophic lateral sclerosis (FALS) Brain Res. 1995;676:25–40. doi: 10.1016/0006-8993(95)00063-v. [DOI] [PubMed] [Google Scholar]
- Disterhoft JF, Oh MM. Alterations in intrinsic neuronal excitability during normal aging. Aging cell. 2007;6:327–336. doi: 10.1111/j.1474-9726.2007.00297.x. [DOI] [PubMed] [Google Scholar]
- Elbasiouny SM, Amendola J, Durand J, Heckman CJ. Evidence from computer simulations for alterations in the membrane biophysical properties and dendritic processing of synaptic inputs in mutant superoxide dismutase-1 motoneurons. J Neurosci. 2010;30:5544–5558. doi: 10.1523/JNEUROSCI.0434-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Islas C, Wenner P. Spontaneous network activity in the embryonic spinal cord regulates AMPAergic and GABAergic synaptic strength. Neuron. 2006;49:563–575. doi: 10.1016/j.neuron.2006.01.017. [DOI] [PubMed] [Google Scholar]
- Gordon T, Ly V, Hegedus J, Tyreman N. Early detection of denervated muscle fibers in hindlimb muscles after sciatic nerve transection in wild type mice and in the G93A mouse model of amyotrophic lateral sclerosis. Neurol Res. 2009;31:28–42. doi: 10.1179/174313208X332977. [DOI] [PubMed] [Google Scholar]
- Gurney ME, Cutting FB, Zhai P, Doble A, Taylor CP, Andrus PK, Hall ED. Benefit of vitamin E, riluzole, and gabapentin in a transgenic model of familial amyotrophic lateral sclerosis. Ann Neurol. 1996;39:147–157. doi: 10.1002/ana.410390203. [DOI] [PubMed] [Google Scholar]
- Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- Hebert T, Drapeau P, Pradier L, Dunn RJ. Block of the rat brain IIA sodium channel alpha subunit by the neuroprotective drug riluzole. Mol Pharmacol. 1994;45:1055–1060. [PubMed] [Google Scholar]
- Heckman CJ, Johnson M, Mottram C, Schuster J. Persistent inward currents in spinal motoneurons and their influence on human motoneuron firing patterns. Neuroscientist. 2008;14:264–275. doi: 10.1177/1073858408314986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegedus J, Putman CT, Gordon T. Time course of preferential motor unit loss in the SOD1 G93A mouse model of amyotrophic lateral sclerosis. Neurobiol Dis. 2007;28:154–164. doi: 10.1016/j.nbd.2007.07.003. [DOI] [PubMed] [Google Scholar]
- Hegedus J, Putman CT, Tyreman N, Gordon T. Preferential motor unit loss in the SOD1 G93A transgenic mouse model of amyotrophic lateral sclerosis. J Physiol. 2008;586:3337–3351. doi: 10.1113/jphysiol.2007.149286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CS, Song JH, Nagata K, Yeh JZ, Narahashi T. Effects of the neuroprotective agent riluzole on the high voltage-activated calcium channels of rat dorsal root ganglion neurons. J Pharmacol Exp Ther. 1997;282:1280–1290. [PubMed] [Google Scholar]
- Hultborn H, Brownstone RB, Toth TI, Gossard JP. Key mechanisms for setting the input-output gain across the motoneuron pool. Prog Brain Res. 2004;143:77–95. doi: 10.1016/s0079-6123(03)43008-2. [DOI] [PubMed] [Google Scholar]
- Ilieva HS, Yamanaka K, Malkmus S, Kakinohana O, Yaksh T, Marsala M, Cleveland DW. Mutant dynein (Loa) triggers proprioceptive axon loss that extends survival only in the SOD1 ALS model with highest motor neuron death. Proc Natl Acad Sci U S A. 2008;105:12599–12604. doi: 10.1073/pnas.0805422105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang M, Schuster JE, Fu R, Siddique T, Heckman CJ. Progressive changes in synaptic inputs to motoneurons in adult sacral spinal cord of a mouse model of amyotrophic lateral sclerosis. J Neurosci. 2009;29:15031–15038. doi: 10.1523/JNEUROSCI.0574-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Z, Rempel J, Li J, Sawchuk MA, Carlin KP, Brownstone RM. Development of L-type calcium channels and a nifedipine-sensitive motor activity in the postnatal mouse spinal cord. Eur J Neurosci. 1999;11:3481–3487. doi: 10.1046/j.1460-9568.1999.00765.x. [DOI] [PubMed] [Google Scholar]
- Kong J, Xu Z. Massive mitochondrial degeneration in motor neurons triggers the onset of amyotrophic lateral sclerosis in mice expressing a mutant SOD1. J Neurosci. 1998;18:3241–3250. doi: 10.1523/JNEUROSCI.18-09-03241.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kullmann DM, Waxman SG. Neurological channelopathies: new insights into disease mechanisms and ion channel function. J Physiol. 2010;588:1823–1827. doi: 10.1113/jphysiol.2010.190652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo JJ, Schonewille M, Siddique T, Schults ANA, Fu RG, Bär PR, Anelli R, Heckman CJ, Kroese ABA. Hyperexcitability of cultured spinal motoneurons from presymptomatic ALS mice. J Neurophysiol. 2004;91:571–575. doi: 10.1152/jn.00665.2003. [DOI] [PubMed] [Google Scholar]
- Kuo JJ, Siddique T, Fu R, Heckman CJ. Increased persistent Na+ current and its effect on excitability in motoneurones cultured from mutant SOD1 mice. J Physiol. 2005;563:843–854. doi: 10.1113/jphysiol.2004.074138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacomblez L, Bensimon G, Leigh PN, Guillet P, Powe L, Durrleman S, Delumeau JC, Meininger V. A confirmatory dose-ranging study of riluzole in ALS. ALS/Riluzole Study Group-II. Neurology. 1996;47:S242–S250. doi: 10.1212/wnl.47.6_suppl_4.242s. [DOI] [PubMed] [Google Scholar]
- Lamanauskas N, Nistri A. Riluzole blocks persistent Na+ and Ca2+ currents and modulates release of glutamate via presynaptic NMDA receptors on neonatal rat hypoglossal motoneurons in vitro. Eur J Neurosci. 2008;27:2501–2514. doi: 10.1111/j.1460-9568.2008.06211.x. [DOI] [PubMed] [Google Scholar]
- Landmesser LT, O'Donovan MJ. Activation patterns of embryonic chick hind limb muscles recorded in ovo and in an isolated spinal cord preparation. J Physiol. 1984;347:189–204. doi: 10.1113/jphysiol.1984.sp015061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lape R, Nistri A. Current and voltage clamp studies of the spike medium afterhyperpolarization of hypoglossal motoneurons in a rat brain stem slice preparation. J Neurophysiol. 2000;83:2987–2995. doi: 10.1152/jn.2000.83.5.2987. [DOI] [PubMed] [Google Scholar]
- Li X, Murray K, Harvey PJ, Ballou EW, Bennett DJ. Serotonin facilitates a persistent calcium current in motoneurons of rats with and without chronic spinal cord injury. J Neurophysiol. 2007;97:1236–1246. doi: 10.1152/jn.00995.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Bennett DJ. Persistent sodium and calcium currents cause plateau potentials in motoneurons of chronic spinal rats. J Neurophysiol. 2003;90:857–869. doi: 10.1152/jn.00236.2003. [DOI] [PubMed] [Google Scholar]
- Manuel M, Iglesias C, Donnet M, Leroy F, Heckman CJ, Zytnicki D. Fast kinetics, high-frequency oscillations, and subprimary firing range in adult mouse spinal motoneurons. J Neurosci. 2009;29:11246–11256. doi: 10.1523/JNEUROSCI.3260-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marder E, Goaillard JM. Variability, compensation and homeostasis in neuron and network function. Nat Rev Neurosci. 2006;7:563–574. doi: 10.1038/nrn1949. [DOI] [PubMed] [Google Scholar]
- Marder E, Rehm KJ. Development of central pattern generating circuits. Curr Opin Neurobiol. 2005;15:86–93. doi: 10.1016/j.conb.2005.01.011. [DOI] [PubMed] [Google Scholar]
- Meehan CF, Moldovan M, Marklund SL, Graffmo KS, Nielsen JB, Hultborn H. Intrinsic properties of lumbar motor neurones in the adult G127insTGGG superoxide dismutase-1 mutant mouse in vivo: evidence for increased persistent inward currents. Acta Physiol (Oxf) 2010a;200:361–376. doi: 10.1111/j.1748-1716.2010.02188.x. [DOI] [PubMed] [Google Scholar]
- Meehan CF, Sukiasyan N, Zhang M, Nielsen JB, Hultborn H. Intrinsic properties of mouse lumbar motoneurons revealed by intracellular recording in vivo. J Neurophysiol. 2010b;103:2599–2610. doi: 10.1152/jn.00668.2009. [DOI] [PubMed] [Google Scholar]
- Mogyoros I, Kiernan MC, Burke D, Bostock H. Strength–duration properties of sensory and motor axons in amyotrophic lateral sclerosis. Brain. 1998;121:851–859. doi: 10.1093/brain/121.5.851. [DOI] [PubMed] [Google Scholar]
- Mohajeri MH, Figlewicz DA, Bohn MC. Selective loss of α motoneurons innervating the medial gastrocnemius muscle in a mouse model of amyotrophic lateral sclerosis. Exp Neurol. 1998;150:329–336. doi: 10.1006/exnr.1998.6758. [DOI] [PubMed] [Google Scholar]
- Mourelatos Z, Gonatas NK, Stieber A, Gurney ME, Dal Canto MC. The Golgi apparatus of spinal cord motor neurons in transgenic mice expressing mutant Cu,Zn superoxide dismutase becomes fragmented in early, preclinical stages of the disease. Proc Natl Acad Sci U S A. 1996;93:5472–5477. doi: 10.1073/pnas.93.11.5472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai M, Re DB, Nagata T, Chalazonitis A, Jessell TM, Wichterle H, Przedborski S. Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat Neurosci. 2007;10:615–622. doi: 10.1038/nn1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pambo-Pambo A, Durand J, Gueritaud JP. Early excitability changes in lumbar motoneurons of transgenic SOD1G85R and SOD1G(93A-Low) mice. J Neurophysiol. 2009;102:3627–3642. doi: 10.1152/jn.00482.2009. [DOI] [PubMed] [Google Scholar]
- Perrier JF, Hounsgaard J. 5-HT2 receptors promote plateau potentials in turtle spinal motoneurons by facilitating an L-type calcium current. J Neurophysiol. 2003;89:954–959. doi: 10.1152/jn.00753.2002. [DOI] [PubMed] [Google Scholar]
- Pieri M, Albo F, Gaetti C, Spalloni A, Bengtson CP, Longone P, Cavalcanti S, Zona C. Altered excitability of motor neurons in a transgenic mouse model of familial amyotrophic lateral sclerosis. Neurosci Lett. 2003a;351:153–156. doi: 10.1016/j.neulet.2003.07.010. [DOI] [PubMed] [Google Scholar]
- Pieri M, Carunchio I, Curcio L, Mercuri NB, Zona C. Increased persistent sodium current determines cortical hyperexcitability in a genetic model of amyotrophic lateral sclerosis. Exp Neurol. 2009;215:368–379. doi: 10.1016/j.expneurol.2008.11.002. [DOI] [PubMed] [Google Scholar]
- Pieri M, Gaetti C, Spalloni A, Cavalcanti S, Mercuri N, Bernardi G, Longone P, Zona C. α-Amino-3-hydroxy-5-methyl-isoxazole-4-propionate receptors in spinal cord motor neurons are altered in transgenic mice overexpressing human Cu,Zn superoxide dismutase (Gly93→Ala) mutation. Neuroscience. 2003b;122:47–58. doi: 10.1016/j.neuroscience.2003.07.003. [DOI] [PubMed] [Google Scholar]
- Powers RK, Binder MD. Input-output functions of mammalian motoneurons. Rev Physiol Biochem Pharmacol. 2001;143:137–263. doi: 10.1007/BFb0115594. [DOI] [PubMed] [Google Scholar]
- Pun S, Santos AF, Saxena S, Xu L, Caroni P. Selective vulnerability and pruning of phasic motoneuron axons in motoneuron disease alleviated by CNTF. Nat Neurosci. 2006;9:408–419. doi: 10.1038/nn1653. [DOI] [PubMed] [Google Scholar]
- Rao SD, Weiss JH. Excitotoxic and oxidative cross-talk between motor neurons and glia in ALS pathogenesis. Trends Neurosci. 2004;27:17–23. doi: 10.1016/j.tins.2003.11.001. [DOI] [PubMed] [Google Scholar]
- Rich MM, Waldeck RF, Cork LC, Balice-Gordon RJ, Fyffe RE, Wang X, Cope TC, Pinter MJ. Reduced endplate currents underlie motor unit dysfunction in canine motor neuron disease. J Neurophysiol. 2002;88:3293–3304. doi: 10.1152/jn.00270.2002. [DOI] [PubMed] [Google Scholar]
- Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- Rothstein JD. Excitotoxicity hypothesis. Neurology. 1996;47:S19–S25. doi: 10.1212/wnl.47.4_suppl_2.19s. discussion S26. [DOI] [PubMed] [Google Scholar]
- Rothstein JD. Current hypotheses for the underlying biology of amyotrophic lateral sclerosis. Ann Neurol. 2009;65(Suppl 1):S3–S9. doi: 10.1002/ana.21543. [DOI] [PubMed] [Google Scholar]
- Rush AM, Dib-Hajj SD, Waxman SG. Electrophysiological properties of two axonal sodium channels, Nav1.2 and Nav1.6, expressed in mouse spinal sensory neurones. J Physiol. 2005;564:803–815. doi: 10.1113/jphysiol.2005.083089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena S, Cabuy E, Caroni P. A role for motoneuron subtype-selective ER stress in disease manifestations of FALS mice. Nat Neurosci. 2009;12:627–636. doi: 10.1038/nn.2297. [DOI] [PubMed] [Google Scholar]
- Schwindt PC, Crill WE. Properties of a persistent inward current in normal and TEA-injected motoneurons. J Neurophysiol. 1980;43:1700–1724. doi: 10.1152/jn.1980.43.6.1700. [DOI] [PubMed] [Google Scholar]
- Tandan R, Bradley WG. Amyotrophic lateral sclerosis: Part 1. Clinical features, pathology, and ethical issues in management. Ann Neurol. 1985;18:271–280. doi: 10.1002/ana.410180302. [DOI] [PubMed] [Google Scholar]
- Theiss RD, Heckman CJ. Systematic variation in effects of serotonin and norepinephrine on repetitive firing properties of ventral horn neurons. Neuroscience. 2005;134:803–815. doi: 10.1016/j.neuroscience.2005.04.041. [DOI] [PubMed] [Google Scholar]
- Theiss RD, Kuo JJ, Heckman CJ. Persistent inward currents in rat ventral horn neurones. J Physiol. 2007;580:507–522. doi: 10.1113/jphysiol.2006.124123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomlinson BE, Irving D. The numbers of limb motor neurons in the human lumbosacral cord throughout life. J Neurol Sci. 1977;34:213–219. doi: 10.1016/0022-510x(77)90069-7. [DOI] [PubMed] [Google Scholar]
- Urbani A, Belluzzi O. Riluzole inhibits the persistent sodium current in mammalian CNS neurons. Eur J Neurosci. 2000;12:3567–3574. doi: 10.1046/j.1460-9568.2000.00242.x. [DOI] [PubMed] [Google Scholar]
- Van Damme P, Bogaert E, Dewil M, Hersmus N, Kiraly D, Scheveneels W, Bockx I, Braeken D, Verpoorten N, Verhoeven K, Timmerman V, Herijgers P, Callewaert G, Carmeliet P, Van Den Bosch L, Robberecht W. Astrocytes regulate GluR2 expression in motor neurons and their vulnerability to excitotoxicity. Proc Natl Acad Sci U S A. 2007;104:14825–14830. doi: 10.1073/pnas.0705046104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Den Bosch L, Van Damme P, Bogaert E, Robberecht W. The role of excitotoxicity in the pathogenesis of amyotrophic lateral sclerosis. Biochim Biophys Acta. 2006;1762:1068–1082. doi: 10.1016/j.bbadis.2006.05.002. [DOI] [PubMed] [Google Scholar]
- van Zundert B, Peuscher MH, Hynynen M, Chen A, Neve RL, Brown RH, Jr, Constantine-Paton M, Bellingham MC. Neonatal neuronal circuitry shows hyperexcitable disturbance in a mouse model of the adult-onset neurodegenerative disease amyotrophic lateral sclerosis. J Neurosci. 2008;28:10864–10874. doi: 10.1523/JNEUROSCI.1340-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Lewinski F, Keller BU. Ca2+, mitochondria and selective motoneuron vulnerability: implications for ALS. Trends Neurosci. 2005;28:494–500. doi: 10.1016/j.tins.2005.07.001. [DOI] [PubMed] [Google Scholar]
- Vucic S, Kiernan MC. Upregulation of persistent sodium conductances in familial ALS. J Neurol Neurosurg Psychiatry. 2010;81:222–227. doi: 10.1136/jnnp.2009.183079. [DOI] [PubMed] [Google Scholar]
- Vucic S, Nicholson GA, Kiernan MC. Cortical hyperexcitability may precede the onset of familial amyotrophic lateral sclerosis. Brain. 2008;131:1540–1550. doi: 10.1093/brain/awn071. [DOI] [PubMed] [Google Scholar]
- Yang Y, Gozen O, Vidensky S, Robinson MB, Rothstein JD. Epigenetic regulation of neuron-dependent induction of astroglial synaptic protein GLT1. Glia. 2010;58:277–286. doi: 10.1002/glia.20922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Gozen O, Watkins A, Lorenzini I, Lepore A, Gao Y, Vidensky S, Brennan J, Poulsen D, Won Park J, Li Jeon N, Robinson MB, Rothstein JD. Presynaptic regulation of astroglial excitatory neurotransmitter transporter GLT1. Neuron. 2009;61:880–894. doi: 10.1016/j.neuron.2009.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zona C, Ferri A, Gabbianelli R, Mercuri NB, Bernardi G, Rotilio G, Carri MT. Voltage-activated sodium currents in a cell line expressing a Cu,Zn superoxide dismutase typical of familial ALS. Neuroreport. 1998;9:3515–3518. doi: 10.1097/00001756-199810260-00033. [DOI] [PubMed] [Google Scholar]