

Non-technical summary
The striatum is a part of the basal ganglia that receives input from the cerebral cortex, extracts relevant information from background noise and relays that information to other parts of the basal ganglia. It is largely composed of nerve cells known as medium-sized spiny neurons (MSNs), and neurons from the cerebral cortex make synaptic connections with them. This study investigates the function of astrocytes at this synaptic connection, where their role is to remove the neurotransmitters glutamate and GABA that spills out from the synaptic cleft. It appears that astrocytes, via the uptake of neurotransmitters, increase the strength of filtering operated by MSNs.
Abstract
Abstract
The astrocytes, active elements of the tripartite synapse, remove most of the neurotransmitter that spills over the synaptic cleft. Neurotransmitter uptake operated by astrocytes contributes to the strength and timing of synaptic inputs. The striatum, the main input nucleus of basal ganglia, extracts pertinent cortical signals from the background noise and relays cortical information toward basal ganglia output structures. We investigated the role of striatal astrocytic uptake in the shaping of corticostriatal transmission. We performed dual patch-clamp recordings of striatal output neuron (the medium-sized spiny neurons, MSNs)–astrocyte pairs while stimulating the somatosensory cortex. Cortical activity evoked robust synaptically activated transporter-mediated currents (STCs) in 78% of the recorded astrocytes. STCs originated equally from the activities of glutamate transporters and GABA transporters (GATs). Astrocytic STCs reflected here a presynaptic release of neurotransmitters. STCs displayed a large magnitude associated with fast kinetics, denoting an efficient neurotransmitter clearance at the corticostriatal pathway. Inhibition of glutamate transporters type-1 (GLT-1) and GATs decreased the corticostriatal synaptic transmission, through, respectively, desensitization of AMPA receptors and activation of GABAA receptor. STCs displayed a bidirectional short-term plasticity (facilitation for paired-pulse intervals less than 100 ms and depression up to 1 s). We report a genuine facilitation of STCs for high-frequency cortical activity, which could strengthen the detection properties of cortical activity operated by MSNs. MSN EPSCs showed a triphasic short-term plasticity, which was modified by the blockade of GLT-1 or GATs. We show here that neurotransmitter uptake by astrocytes plays a key role in the corticostriatal information processing.
Introduction
Astrocytes are now viewed as active components of neuronal networks, since they are endowed with a great variety of voltage- and ligand-operated ion channels (Verkhratsky & Steinhauser, 2000). There is an increasing body of evidence demonstrating that astrocytes play an active role in synaptogenesis, synaptic transmission and cerebral blood flow, to name but a few (Volterra & Meldolesi, 2005; Haydon & Carmignoto, 2006; Halassa & Haydon, 2010). The existence of bidirectional communication between astrocytes and neurons has led to the concept of the tripartite synapse. The pre- and postsynaptic neuronal elements and the nearby astrocytic processes enveloping the synapse constitute the tripartite synapse (Araque et al. 1999; Haydon, 2001; Halassa et al. 2007). A major role of astrocytes is the uptake of neurotransmitters. Indeed, astrocytic transporters remove most of the glutamate that spills over the synaptic cleft (Kullmann & Asztely, 1998; Danbolt, 2001). To date, five structurally related glutamate transporters have been cloned and constitute a family of voltage-dependent excitatory amino acid transporters (Danbolt, 2001). In rodents, glutamate transporter type-1 (GLT-1), highly expressed by astrocytes, is responsible for the largest proportion of total glutamate uptake in the forebrain (Rothstein et al. 1996; Tanaka et al. 1997). Astrocytes express also, at a lower level, glutamate–aspartate transporter (GLAST). Excitatory amino acid carrier 1 (EAAC1) and excitatory amino acid transporter 4 (EAAT4) are expressed by neurons, and EAAT5 is found only in the retina. Beside glutamate, astrocytes also uptake GABA with GABA transporters (GATs). GATs constitute a family of four Na+/Cl−-dependent transporter subtypes (GAT-1–3 and BGT-1). The GAT-3 subtype is predominately expressed in glia (Borden, 1996; Minelli et al. 1996). The involvement of astrocytes in the synaptic transmission and plasticity has been investigated at the level of the glial receptors or the release of gliotransmitters but few studies have addressed the contribution of astrocytic uptake in synaptic transmission (Lozovaya et al. 1999; Diamond, 2001; Arnth-Jensen et al. 2002).
The striatum, the main input nucleus of the basal ganglia, receives glutamatergic inputs from the entire cerebral cortex. Striatal output neurons (the medium-sized spiny neurons, MSNs) act as detectors of cortical activity and extract relevant information from the background noise (Calabresi et al. 1987; Nisenbaum et al. 1994). We have previously investigated transmission and activity-dependent long-term plasticity at the corticostriatal synapses (Fino et al. 2005, 2008, 2010). Here, we investigated the role of astrocytes in neurotransmitter uptake and their contribution to corticostriatal transmission. Corticostriatal transmission is glutamatergic but is also efficiently modulated by local GABAergic circuits involving striatal GABAergic interneurons (Tepper et al. 2008) or MSN collaterals (Venance et al. 2004). Accordingly, in the present study, we investigated the contribution of both glutamate and GABA transport operated by astrocytes, on corticostriatal transmission and paired-pulse plasticity.
Methods
Ethical approval
Animals, OFA rats (Charles River, L'Arbresle, France) (postnatal days 15–21), were killed by decapitation and brains were immediately removed. All experiments were performed in accordance with local animal welfare committee (Institute of Biology, Center for Interdisciplinary Research in Biology and College de France) and EU guidelines (directive 86/609/EEC). The experiments comply with the policies and regulations of The Journal of Physiology (see Drummond, 2009). Every precaution was taken to minimize stress and the number of animals used in each series of experiments.
Electrophysiological recordings
Connections between the somatosensory cerebral cortex (layer 5) and the dorsal striatum are preserved in a horizontal plane (Fino et al. 2005, 2008). Whole-cell patch-clamp recordings of MSNs and glial cells were performed on horizontal brain slices (330 μm). These horizontal slices included the somatosensory cortical area and the corresponding corticostriatal projection field (Fino et al. 2005) and were prepared with a vibrating blade microtome (VT1000S and VT1200S, Leica Microsystems, Nussloch, Germany). Dual patch-clamp recordings were made as previously described (Venance et al. 2004; Vandecasteele et al. 2008; Meme et al. 2009). Briefly, borosilicate glass pipettes contained (mm): 105 potassium gluconate, 30 KCl, 10 Hepes, 10 phosphocreatine, 4 ATP-Mg, 0.3 GTP-Tris, 0.3 EGTA (adjusted to pH 7.35 with KOH). In a subset of experiments (for the analysis of the phasic and tonic GABAergic inhibition, Fig. 6), the chloride concentration was increased to obtain a reversal potential for Cl− (ECl,rev) of 0 mV, and K+ was replaced by Cs+; the composition of the internal solution was the following (in mm): 135 CsCl, 10 Hepes, 10 phosphocreatine, 4 ATP-Mg, 0.3 GTP-Tris, 0.3 EGTA (adjusted to pH 7.35 with CsOH). The composition of the extracellular solution was (mm): 125 NaCl, 2.5 KCl, 25 glucose, 25 NaHCO3, 1.25 NaH2PO4, 2 CaCl2, 1 MgCl2, 10 μm pyruvic acid bubbled with 95% O2–5% CO2. In a subset of experiments (for the analysis of the NMDA currents), Mg2+ was removed and the composition of the external solution was the following (in mm): 127 NaCl, 2.5 KCl, 25 glucose, 25 NaHCO3, 1.25 NaH2PO4, 2 CaCl2, and 10 μm pyruvic acid. All recordings (single and dual patch-clamp) were performed near physiological temperature at 34°C using a temperature control system (Bath-controller V, Luigs & Neumann, Ratingen, Germany) and slices were continuously superfused at 2–3 ml min−1 with the extracellular solution. Indeed, it has been reported that temperature was critical for the activity of glutamate transporters (Bergles & Jahr, 1998). Individual neurons and glial cells were identified using infrared-differential interference contrast microscopy (BX51, Olympus, Rungis, France) with CCD camera (Hamamatsu C2400-07; Hamamatsu, Japan). Signals were amplified using an EPC10-2 amplifier (HEKA Elektronik, Lambrecht, Germany). Current-clamp recordings were filtered at 2.5 kHz and sampled at 5 kHz and voltage-clamp recordings were filtered at 5 kHz and sampled at 10 kHz using the program Patchmaster v2x32 (HEKA Elektronik). The series resistance was compensated at 75–80%. The distance ranges between neuron–glial cell pairs were between 20 and 50 μm and between astroctyte–astrocyte pairs were between 10 and 20 μm. Cells were typically located at a similar depth within the slice, about 40–50 μm. In control condition, recordings were performed without any pharmacological treatments or ionic modifications to preserve the local striatal microcircuits involved in corticostriatal transmission.
Figure 6. Increased tonic GABA inhibition is not responsible for the effect of nipecotic acid on corticostriatal transmission.
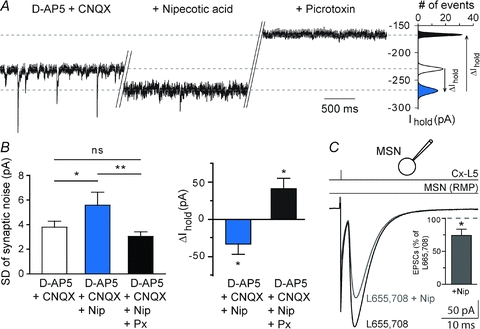
A, the phasic and tonic GABAergic components were estimated, respectively, after inhibition of the ionotropic glutamatergic (d-AP5, 50 μm, CNQX, 10 μm) and GABAergic (picrotoxin, 50 μm) conductances. Nipecotic acid increased the tonic component as illustrated by a larger current injected to maintain the Em and by a reduction of the synaptic noise. B, bar graphs show the significant increase of the average values of the injected current necessary to maintain the MSN at −80 mV, when nipecotic acid (500 μm) was applied. Application of nipecotic acid induced a significant increase of the SD of the synaptic noise (denoting an increase of the tonic inhibition), which was significantly reduced by picrotoxin (denoting the abolition of the tonic inhibition). C, when tonic inhibition was blocked by L655,708 (10 μm), the corticostriatal transmission could still be significantly reduced by nipecotic acid (n = 6). This indicates that phasic and not tonic inhibition was involved in the decrease of the corticostriatal transmission induced by nipecotic acid. Note that inhibition of α5-GABAA receptors with L655,708 did not induce significant effect on corticostriatal transmission (n = 11). ns: not significant, *P < 0.05, **P < 0.01.
6-Cyano-7-nitroquinoxaline-2,3-dione (CNQX, 10 μm) (Tocris, Ellisville, MO, USA), dl-2-amino-5-phosphono-pentanoic acid (d-AP5, 50 μm) (Tocris), dihydrokainic acid (DHK, 300 μm) (Tocris), nipecotic acid (Nip, 500 μm) (Sigma, Saint Quentin, France), bicuculline methiodide (BMI, 10 μm) (Sigma) and l-trans-pyrrolidine-2,4-dicarboxylic acid (PDC, 300 μm) (Sigma) were dissolved directly in the extracellular solution and bath applied. Cyclothiazide (CTZ, 100 μm) (Tocris) and picrotoxin (Px, 50 μm) (Sigma) were dissolved in DMSO and ethanol, respectively, and then added in the external solution at a final dilution of DMSO and ethanol of 1/1000. BAPTA (10 mm) (Sigma) was dissolved directly into the intracellular solution and applied via the patch-clamp pipette.
We tested the effects of DHK and nipecotic acid on MSN and astrocyte resting membrane potential (RMP). In the whole-cell configuration, DHK (300 μm) induced a significant variation of injected current needed to maintain constant the RMP in MSNs but not in astrocytes (ΔIinjected in MSNs: −93.6 ± 11.5 pA, P < 0.0001, n = 22, and ΔIinjected in astrocytes: −20.9 ± 21.4 pA, P > 0.05, n = 11). Recordings were made in voltage-clamp mode, and therefore MSN RMP was held at a constant value (i.e. their RMP in control condition) during DHK treatment. Nipecotic acid (500 μm) did not have any significant effect on MSN or astrocyte RMP (ΔIinjected in MSNs: −7.6 ± 3.9 pA, P > 0.05, n = 22, and ΔIinjected in astrocytes: −18.5 ± 16.3 pA, P > 0.05, n = 17). Lastly, we recorded fast-spiking GABAergic interneurons in the presence of DHK (300 μm); only one out of five displayed a firing activity during DHK application.
Biocytin filling and histochemistry
Biocytin (Sigma) 5 mg ml−1 was dissolved into the patch-clamp pipette solution and cells were filled during 20 min of recording (performed at 34°C). Subsequently, slices were fixed overnight in 2% paraformaldehyde at 4°C. Biocytin-filled cells were visualized using the avidin–biotin–horseradish peroxidase reaction (ABC Elite peroxidase kit; Vector Laboratories, Burlingame, CA, USA) according to the instructions of the manufacturer, or in a 1/1600 diluted streptavidin–Alexa 555 (Invitrogen, Carlsbad, CA, USA), incubated 2 h at room temperature. NG2 immunostaining was performed by incubation of the slices in a 1/500 diluted rabbit anti-NG2 monoclonal antibody (Chemicon) overnight at 4°C. Goat anti-rabbit secondary antibody, coupled to Alexa 488 (IgG Invitrogen) was incubated at dilution 1/1000, 2 h at room temperature.
Cortical stimulations and paired-pulse protocols
Electrical stimulations were performed with a bipolar electrode (Phymep, Paris, France) placed in the superficial part of the layer 5 of the somatosensory cerebral cortex by applying a monophasic and constant current (duration: 100–150 μs) (ISO-Flex stimulator controlled by a Master-8, A.M.P.I., Jerusalem, Israel) (Fino et al. 2005, 2008). Repetitive control stimuli were applied at 0.1 Hz, a frequency for which neither short- nor long-term synaptic efficacy changes in EPSC amplitudes were induced (Fino et al. 2005). Inter-stimulus intervals (ISIs) for paired-pulse experiments ranged from 4 ms to 10 s. For ISIs of ≤10 ms for MSNs and ISIs of ≤25 ms for astrocytes, a single stimulation was performed 10 s before each paired pulse to serve as the control. Indeed, for these short duration ISIs, the second EPSC or STC affected the proper measurement of the first EPSC or STC.
Drugs were applied in the bath, after recording at least 5 min of baseline in control. In these conditions, drugs were allowed to diffuse into the bath for at least 5 min before starting the recording for the 10 min duration in which their effects were estimated.
Series resistance and input resistance (Ri) were monitored throughout the experiments and a variation of more than 20% led to the rejection of the experiment. Ri was calculated from the response to a hyperpolarizing potential step (−5 mV in neurons and NG2+ cells, and −2 mV in astrocytes) during each sweep.
Data analysis
Off-line analysis was performed using Igor-Pro 6.0.3 (Wavemetrics, Lake Oswego, OR, USA) and Mini Analysis 6.0.7 software (Synaptosoft, Fort Lee, NJ, USA). All results were expressed as means ± SEM and statistical significance was assessed using Student's t test or Wilcoxon's non-parametric signed-rank test where appropriate at the significance level (P) indicated. EPSC or STC mean amplitudes were the average of 30 evoked EPSCs or STCs. Paired-pulse ratio was calculated by the mean (EPSC2 amplitude/EPSC1 amplitude) for MSNs and the mean (STC2 amplitude/STC1 amplitude) for astrocytes; averages were of 12 EPSC2/EPSC1 or 12 STC2/STC1. Intercellular variability of paired-pulse plasticity was estimated by classifying cells depending on their paired-pulse ratio (PPR) values into three groups: PPR <95%, 95–105% and >105%.
Spontaneous phasic and tonic GABAA currents were measured in a subset of experiments using a Cs+–high-chloride based intracellular solution (see composition above). Phasic and tonic GABAergic components were estimated after inhibition of ionotropic glutamatergic receptors by adding D-AP5 (50 μm) and CNQX (10 μm) at the beginning of the experiment. Phasic and tonic components were analysed during 10 s recording segments before and after pharmacological treatments. Concerning phasic current, spontaneous IPSCs (sIPSCs) were identified using semi-automated amplitude threshold based detection software (Mini Analysis 6.0.7 program) and were visually confirmed. Concerning tonic current, we sampled the holding current every 100 ms for a 10 s period preceding drug application and discarded points landing on IPSCs. The corresponding distribution, not skewed by synaptic events, was fitted by a Gaussian and the peak indicated the mean holding current (Ihold) required to maintain the membrane potential at −80 mV. After pharmacological treatment, we determined a new Ihold, and ΔIhold corresponded to the tonic component affected by the drug. Picrotoxin was systematically added at the end of the experiment to estimate the magnitude of the tonic GABAergic inhibition.
Results
Characterization of striatal output neurons, astrocytes and NG2+ cells
MSNs were identified based on electrophysiological properties (Kawaguchi, 1993; Venance et al. 2004; Fino et al. 2005). Briefly, MSNs displayed a hyperpolarized RMP (−74.2 ± 0.3 mV, n = 250), a high Ri (275 ± 7.8 MΩ), an inward rectifying I–V curve, a long delay to first spike (387 ± 5 ms, for 500 ms depolarizing pulses) evoked at rheobase and a medium firing rate (14.7 ± 0.3 Hz at +30 pA above rheobase) (Fig. 1A). As previously described (Adermark & Lovinger, 2006; Même et al. 2009), astrocytes were distinguished from neurons by their small (5–10 μm) oval shaped somata and by electrophysiological features: a hyperpolarized RMP (−75.3 ± 0.3 mV, n = 201), a low Ri (51.2 ± 3.0 MΩ), a linear I–V relationship and an absence of action potentials in response to depolarizing current injections (Fig. 1Ba). Moreover, in voltage-clamp mode, the lack of typical large off-set currents allows the distinguishing of astrocytes from oligodendrocytes and glial precursors (Chvatal et al. 1995). To assess the occurrence of gap junctional communication, astrocytes (n = 17) were filled with biocytin, a low molecular weight tracer that permeates gap junction channels. After 20 min of whole-cell recording, biocytin was systematically detected (n = 17) in a large number of surrounding cells (222 ± 41 cells) (Fig. 1Bb) indicating an extensive network of connected astrocytes in the striatum (Adermark & Lovinger, 2008).
Figure 1. Characterization of striatal output neurons, astrocytes and NG2+ cells.
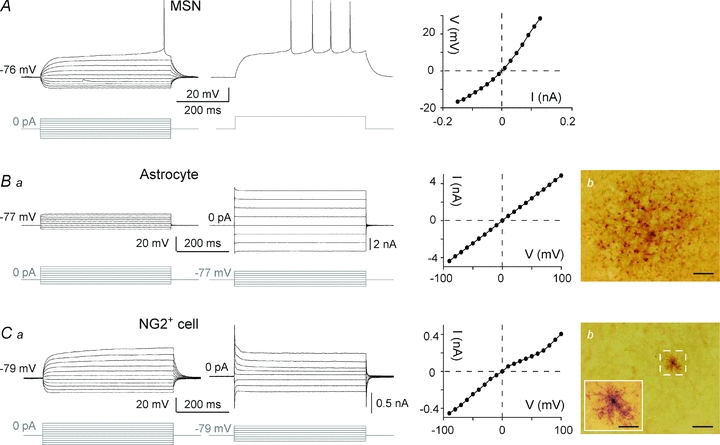
A, electrophysiological characteristics of MSNs: a hyperpolarized RMP, an inward rectification (illustrated by the steady-state I–V relationship, right panel) and a long depolarizing ramp to the action potential threshold leading to a delayed spike discharge. Raw traces show voltage responses to 500 ms current pulses from −130 pA to +140 pA with 30 pA steps (left panel) and to +30 pA above rheobase (middle panel). Ba, characteristic membrane properties of an astrocyte in voltage and current clamp modes. Hyperpolarizing and depolarizing steps (raw traces show individual responses to series of 500 ms voltage or current pulses from −200 pA or −90 mV with 100 pA or 30 mV increasing steps) activated large time- and current/voltage-independent membrane voltages/currents that resulted in a linear steady-state I–V relationship typical of astrocytes (right panel). Bb, example of biocytin intercellular coupling between astrocytes. An electrophysiologically identified astrocyte located in the dorsal striatum was filled with an intercellular tracer (biocytin) that diffused into a large number of neighbouring cells (326 cells in this example) through gap junctions (scale bar: 50 μm). Ca, membrane properties of a NG2+ cell: a hyperpolarized RMP, a high input resistance, an outward rectification (illustrated in the steady-state I–V relationship, middle panel) and a lack of spiking activity. Raw traces show individual voltage responses to series of 500 ms current pulses from −130 pA to +170 pA with 30 pA increasing current steps (left panel) or current responses to series of 500 ms pulses from −80 mV to +130 mV with 30 mV increasing voltage steps (right side). Cb, high-magnification view of an electrophysiologically identified complex cell filled with biocytin (scale bar: 50 μm); note the characteristic absence of dye-coupling. Complex cell showing a small cell body with restricted ramifications (inset, scale bar: 20 μm).
Besides astrocytes, other glial cells, the NG2+ cells, identified by the specific expression of NG2, a chondroitin sulfate proteoglycan, were observed in various areas of the brain (Nishiyama et al. 2002). Electrophysiologically, these cells are referred to as complex glial cells because of their non-linear I–V relationship (Zhou & Kimelberg 2000). Among all the recorded glial cells, we observed 18% (n = 43) of complex glial cells and immunohistochemistry demonstrated their NG2+ phenotype (see online Supplemental Material, Suppl. Fig. 1). Briefly, the NG2+ cells were characterized by an outward rectifying I–V relationship, a high Ri (157.4 ± 12.1 MΩ, significantly different from the astrocyte Ri, P < 0.0001), a very hyperpolarized RMP (−79.3 ± 0.9 mV; Fig. 1Ca) and a lack of junctional communication (n = 14 NG2+ cells injected with biocytin) (Fig. 1Cb).
Cortical activity triggers robust inward currents in striatal glial cells
Using paired recordings of MSN–astrocyte, we recorded the responses evoked in the dorsal striatum by the electrical stimulation in layer 5 of the somatosensory cortex (Fig. 2A). Cortical stimulations evoked glutamatergic excitatory postsynaptic currents (EPSCs) (inhibited by CNQX 10 μm and d-AP5 50 μm, n = 5) in MSNs with a success rate of 93% (n = 250) (Fig. 2B). Transmission was monosynaptic since latency SD was less than 1 ms (0.33 ± 0.03 ms, n = 43). A single cortical stimulation evoked simultaneously an EPSC in MSN and a robust inward current in a neighbouring astrocyte with an occurrence of 78% (n = 201). Once the corticostriatal transmission occurred, no failure was observed in MSNs or astrocytes indicating very reliable and efficient inward current responses. MSN–astrocyte paired recordings also allowed us to control the strength of the cortical stimulation and kept MSN at a subthreshold state in order to record evoked responses in astrocytes without overstimulating. The astrocytic inward current had a rapid onset (latency: 4.2 ± 0.2 ms, n = 69), rose to a peak (60 ± 5 pA, n = 69) in 11.7 ± 0.4 ms (n = 69) and decayed with a fast phase (298 ± 46 ms, n = 17) followed by a slow component (7.8 ± 0.6 s, n = 17) representing 32.1 ± 4.0% of the peak amplitude (Fig. 2B and D). The cortically evoked inward current (fast and slow phases) in astrocytes was strictly action potential dependent since it was totally blocked by tetrodotoxin (TTX, 0.5 μm, n = 4) (Fig. 2B). In addition, bath application of kynurenic acid (0.5–1 mm, n = 5) had no significant effect on either the latency (2.9 ± 0.4 ms in control vs. 2.7 ± 0.3 ms with kynurenic acid) or the amplitude (49.0 ± 9.5 pA in control vs. 44.5 ± 11.4 pA with kynurenic acid) of cortically evoked inward current at astrocytes (Suppl. Fig. 2), excluding a putative neuronal field component that would contaminate the recording of astrocytes.
Figure 2. Cortically evoked responses at MSNs, astrocytes and NG2+ cells.
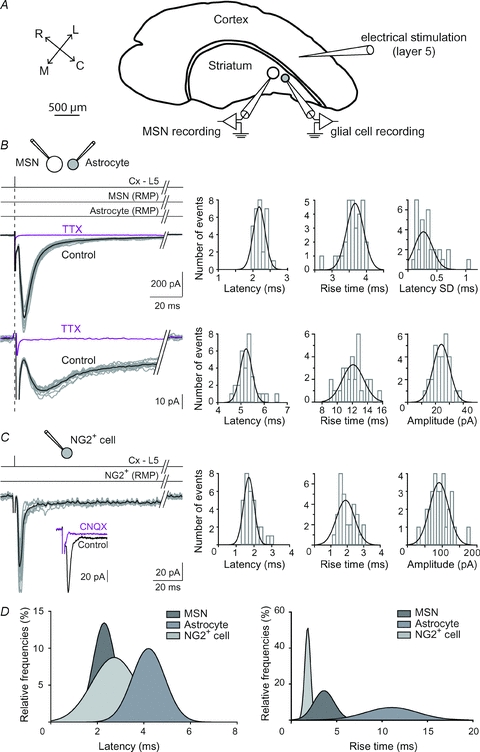
A, shematic view of the horizontal corticostriatal slice preparation. The stimulation electrode was placed in the layer 5 of the somatosensory cerebral cortex and double patch-clamp recordings were performed in the functionally related dorsal striatum. R, C, L and M: rostral, caudal, lateral and median. B, dual patch-clamp recordings of MSN EPSCs and astrocyte inward currents evoked by cortical layer 5 (Cx-L5) stimulations. Black trace represents the average of 10 consecutive raw traces. Latency and rise time distributions (well fitted by Gaussian curves centred on 2.2 and 3.6 ms, respectively) of 30 EPSCs recorded in the same MSN shown in the left panel. Latency SD distribution was centred on 0.33 ms (n = 31 MSNs). Latency, rise time and amplitude distributions (well fitted by Gaussian curves centred on 5.3 and 12.0 ms and 23.8 pA, respectively) of astrocyte evoked responses. Note the short latency centred on 5.3 ms and the very low variability of response amplitudes. TTX treatment inhibited EPSCs as well as astrocyte evoked responses. C, cortically evoked responses in NG2+ cells. Inset: NG2+ cells EPSCs were entirely AMPA receptor mediated because they were totally inhibited by CNQX (10 μm). Latency, rise time and amplitude distributions (centred around 1.8 ms, 2.0 ms and 93 pA, respectively). D, surimposition of latency and rise time of MSNs (n = 43), astrocytes (n = 68) and NG2+ (n = 12) cells. Horizontal lines illustrate the experimental procedures: a single stimulation (vertical line) was imposed in the cortex layer 5 (Cx-L5) while recording MSNs and astrocytes hold at their resting membrane potential (RMP).
Cortical stimulations evoked an inward current in NG2+ cells with a success rate of 48% (n = 40). Latency, rise time and amplitudes displayed narrow distributions centred on 2.7 ± 0.2 ms, 2.0 ± 0.2 ms and 41.5 ± 11.0 pA (n = 12), respectively (Fig. 2C). Cortical stimulation evoked an AMPA receptor-activation-dependent EPSC in NG2+ cells since it was abolished by CNQX (10 μm, n = 5) (Fig. 2C) (Steinhäuser & Gallo, 1996). The fast kinetic characteristics of NG2+-cell-evoked responses differed considerably from those recorded in astrocytes but were similar to those evoked in MSNs. Namely, latency and rise time were significantly shorter from those recorded in cortically evoked responses in astrocytes (P < 0.0001 in both cases) but not significantly different from MSN EPSC latency and rise time (Fig. 2D). The aim of this study was the analysis of the contribution of neurotransmitters transport by glial cells on neurotransmission. Because NG2+ cells lacked detectable transport current, we focused our investigation on involvement of astrocytes in neurotransmitter uptake.
Electrogenic activities of GLT-1 and GATs underlie cortically evoked currents in astrocytes
Synaptically activated transporter currents (STCs) generated by the activation of electrogenic carriers responsible for glutamate and GABA transport present a rectifying inward current (at peak), large at negative potentials, reduced on polarizing to positive potentials, but with no reversal of current (Brew & Attwell, 1987; Cammack & Schwartz, 1993; Bergles & Jahr, 1997; Barakat & Bordey, 2002). Therefore, we investigated the voltage dependence of the cortically evoked inward current in astrocytes by gradually changing the holding potential of the recorded astrocytes from −100 to +40 mV (n = 6) while applying single electrical stimulation in the cortex repeated at low frequency (0.1 Hz) (Fig. 3A). The rectifying inward current (at peak) was large at negative potentials and reduced on polarizing to positive potentials, but no reversal of current was observed. Such a voltage relationship of the cortically evoked inward current in astrocytes suggests that the inward currents are STCs.
Figure 3. Synaptically activated transporter currents (STCs) were generated by the activation of glutamate and GABA transporters.
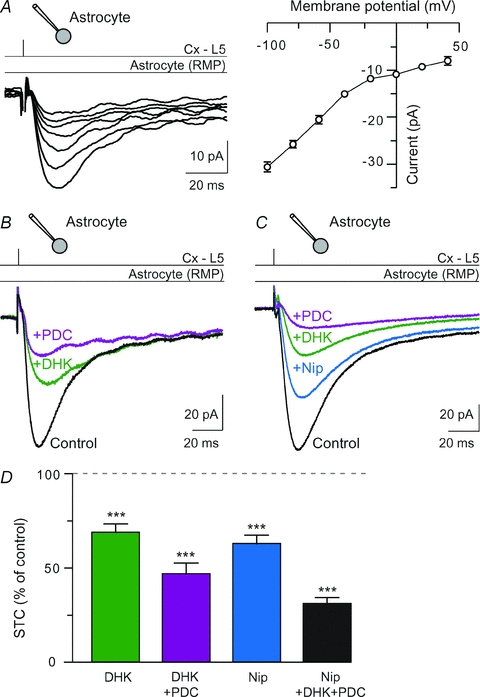
A, cortically induced currents in astrocytes recorded at different holding potentials from −100 to +40 mV. A representative I–V curve illustrates the voltage dependence of the cortically evoked inward current in astrocytes. Between −100 and +40 mV, the cortically evoked current at astrocytes was rectifying, strictly inward and no reversal of current was observed. This suggests the activities of electrogenic carriers. B, representative recordings of the inhibition of STCs after DHK (300 μm) treatments and DHK–PDC (300 μm) co-applications. The contribution of glutamate uptake currents to the fast and slow components of STCs is, respectively, 39 and 38% (DHK), and 60 and 71% (DHK and PDC). C, GATs are involved in STCs. Nipecotic acid (500 μm) reduced significantly STCs. The contribution of GABA and glutamate uptake currents to the fast and slow components of STCs is, respectively, 37 and 17% (nipecotic acid), 62 and 58% (nipecotic acid and DHK) and 83 and 84% (nipecotic acid, DHK and PDC). D, glutamate transporters underlie 53.3 ± 5.8% (DHK+PDC, n = 10) of STCs. Nipecotic acid (500 μm, n = 17) reduced significantly STCs in control conditions (−37.2 ± 4.6%, n = 17). ***P < 0.001. Thus, glutamate and GABA transport represents most of the cortically evoked STCs.
Because of the glutamatergic nature of the corticostriatal pathway, we first tested if the inward current in astrocytes was due to the electrogenic activity of GLT-1, a glutamate transporter highly expressed by astrocytes and responsible for the largest proportion of total glutamate uptake in the forebrain (Rothstein et al. 1996; Tanaka et al. 1997; Bergles & Jahr, 1997; Bergles et al. 1999; Schousboe et al. 2004). GLT-1 is essential for maintaining low extracellular glutamate. For this purpose, we applied dihydrokainate (DHK, 300 μm), a glutamate analogue that binds selectively to GLT-1 but is not transported across membrane (Pines et al. 1992; Arriza et al. 1994; Rothstein et al. 1994). The inward currents were significantly reduced by DHK (−31.2 ± 4.6%, n = 13, P < 0.0001) (Fig. 3B and D). Beside GLT-1, astrocytes also expressed GLAST but at a lower level (Rothstein et al. 1994). Indeed, l-trans-pyrrolidine-2,4-dicarboxylic acid (PDC, 300 μm), a non-specific competitive inhibitor of glutamate transporter, decreased significantly the amplitude of the STCs, when applied after DHK treatment, by 24.8 ± 2.3% (n = 4, P < 0.005). Co-application of DHK and PDC inhibited the STCs by 54.0 ± 6.5% (n = 9, P < 0.0001) (Fig. 3B and D). DHK did not affect significantly (−14.5 ± 11.3%, n = 13, P > 0.05) the slow component of STC amplitude, while PDC alone or co-applied with DHK exerted a significant decrease (−24.3 ± 3.8%, n = 4, P < 0.0001 and −58.0 ± 5.9%, n = 8, P < 0.0001, respectively).
Striatum is mainly composed of GABAergic cells, MSNs and GABAergic interneurons (Kawaguchi, 1993), which are contacted monosynaptically by cortical pyramidal cells (Fino et al. 2008, 2009). Cortical stimulations efficiently recruit striatal GABAergic microcircuits. Therefore, we investigated the involvement of GATs in the STCs. Astrocytes express GAT-1/2/3 subtypes (Borden, 1996; Conti et al. 2004). Nipecotic acid (500 μm), a competitive inhibitor of GAT-1/2/3 subtypes (Schousboe et al. 1979; Liu et al. 1993), significantly inhibited STCs by 37.2 ± 4.6%(n = 16, P < 0.0001) (Fig. 3C and D). This indicates an efficient transport of GABA in astrocytes following a single cortical activation. Co-application of nipecotic acid and DHK, or nipecotic acid, DHK and PDC inhibited STCs by 50.5 ± 5.3% (n = 6, P < 0.0001) and 69.2 ± 3.4% (n = 12, P < 0.0001), respectively (Fig. 3C and D). Nipecotic acid alone, or co-applied with DHK and PDC, reduced significantly the slow component of STC amplitude (−15.1 ± 5.5%, n = 15, P < 0.05 and −61.2 ± 3.4%, n = 11, P < 0.0001, respectively).
We investigated the involvement of other channels or receptors in cortically evoked STCs. No significant inhibition of residual STCs was observed with a fast Ca2+ buffer, BAPTA (10 mm), applied intracellularly through the patch-clamp pipette (residual STC: 30.8 ± 3.4%, n = 12, in control vs 38.6 ± 13.7%, n = 5, with BAPTA) or with bath applied CNQX (10 μm) (+3.6 ± 10.9%, n = 6), or with bicuculine (20 μm) (+8.0 ± 13.3%, n = 7). Therefore, glutamate and GABA transport represents most of the cortically evoked STCs.
STCs report changes in presynaptic evoked glutamate release
We tested if STCs were reliably sensitive to changes in glutamate release as previously reported in hippocampus (Diamond et al. 1998; Luscher et al. 1998). For this purpose, we performed three standard manipulations that alter the number of releasing site or the probability of release. First, electrical stimulation strength was gradually increased (in a subthreshold range for MSNs) to recruit more synapses and evoked responses were recorded in MSN–astrocyte pairs. Both MSN EPSCs and astrocytic STCs increased linearly (r = 0.90, n = 7 pairs) (Fig. 4A). Second, decreasing presynaptic release by lowering the Ca2+:Mg2+ ratio from 2:1 to 0.5:2.5 diminished both EPSCs and STCs (EPSCs: −59.6 ± 7.6% and STCs: −68.0 ± 3.4%, n = 6) (Fig. 4B). Third, paired-pulse facilitation, which occurred for paired-pulse intervals of 25 and 50 ms, was observed in both MSN EPSCs and astrocytic STCs. Indeed, EPSCs increased 1.15 ± 0.04- and 1.10 ± 0.04-fold (n = 21) and STCs 1.23 ± 0.02- and 1.18 ± 0.03-fold (n = 6) (Fig. 4C). Altogether, these results indicate that the STC amplitude is a reliable reporter of changes in the amount of glutamate release due to changes in either the number of releasing sites or the probability of release.
Figure 4. STCs report changes in presynaptic evoked glutamate release.

A, MSN EPSCs and astrocytic STCs increased linearly upon increasing cortical stimulation strength (n = 7 MSN–astrocyte pairs; each symbol represents one MSN–astrocyte pair). B, lowering the external Ca2+ (ratio Ca2+:Mg2+ from 2:1 to 0.5:2.5) significantly decreased the amplitude of MSNs EPSCs and astrocytic STCs (n = 6 MSNs and 4 astrocytes). C, paired-pulse intervals of 25 and 50 ms induced EPSC facilitation (n = 21 MSNs), as well as a significant increase of STCs (n = 6 astrocytes). *P < 0.05, ***P < 0.001.
Glutamate and GABA transports by astrocytes control corticostriatal transmission
We investigated the relative impact of astrocytic glutamate and GABA uptake on corticostriatal transmission by blocking either GLT-1 or GATs. When astrocytic glutamate transport was blocked with DHK (300 μm), we observed a significant decrease of the mean value of the EPSC amplitude (−26.4 ± 8.0%, n = 12, P < 0.01) (Fig. 5A). Corticostriatal activity produces predominantly AMPA receptor mediated EPSCs in MSNs. Indeed, the AMPA:NMDA current ratios were 89.3 ± 5.0%, (n = 15) at MSN resting membrane potential. AMPA receptors desensitize within a few milliseconds in the sustained presence of glutamate (Trussell et al. 1988). The excess of glutamate in the synaptic cleft, induced by the blockade of GLT-1, could greatly increase the AMPA receptor desensitization process. To test this hypothesis, we applied cyclothiazide (CTZ), a blocker of AMPA receptor desensitization (Partin et al. 1996). CTZ (100 μm) treatment increased corticostriatal EPSC duration (151 ± 16%, n = 8, P < 0.05), consistent with the ability of CTZ to prevent desensitization of AMPA receptors, but did not affect significantly EPSC amplitude (122.7 ± 12.8%, n = 9, P > 0.05) (Fig. 5A) (Akopian & Walsh, 2007). DHK, when co-applied with CTZ, did not decrease significantly EPSC amplitudes (98.7±12.7%, n = 9, P > 0.05). In conclusion, the glutamate accumulation due to GLT-1 blockade increased the AMPA receptor desensitization and therefore led to a significant decrease of the corticostriatal transmission.
Figure 5. Blockade of GLT-1 or GATs decreased the magnitude of corticostriatal glutamatergic transmission.
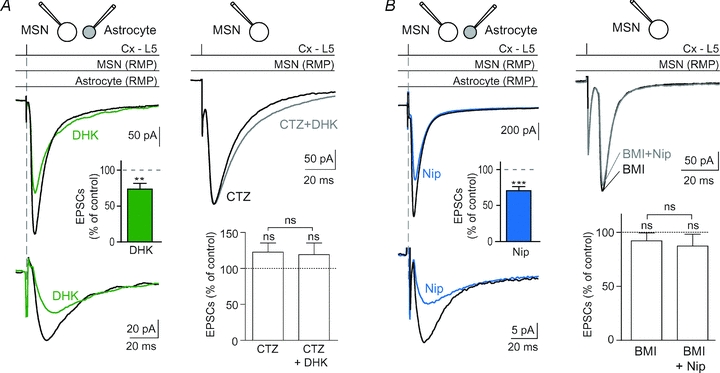
A, MSN–astrocyte double patch-clamp recording illustrates the decrease of corticostriatal EPSCs as well as STCs when GLT-1 was blocked with DHK (300 μm) treatment. Indeed, DHK induced a significant decrease of the EPSC amplitude (−26.4 ± 8.0%, n = 12, P < 0.01 when compared to control). CTZ (100 μm), which prevents the desensitization of AMPA receptors, totally abolished the effect of DHK on corticostriatal transmission (n = 9). Indeed, no significant effect of DHK was observed anymore in the presence of CTZ. Note that CTZ alone increased the duration of corticostriatal EPSCs but did not alter significantly corticostriatal EPSC amplitude (n = 9). B, dual patch-clamp recording of a MSN–astrocyte pair illustrating the decrease of corticostriatal EPSCs as well as STCs when GATs were blocked with nipecotic acid (500 μm). Nipecotic acid induced a significant decrease of corticostriatal EPSC amplitude (−29.2 ± 5.6%, n = 16, P < 0.0001 when compared to control). Pretreatment with bicuculline (BMI) (10 μm), which did not displayed significant effects by itself on EPSCs (n = 7), totally prevented the effects induced by nipecotic acid (n = 7). Indeed, in the presence of bicuculline and nicecotic acid, EPSCs were not significantly different from those recorded in control condition. ns: not significant, **P < 0.01, ***P < 0.001.
The blockade of GATs with nipecotic acid (500 μm) induced a significant decrease of corticostriatal EPSC amplitude (−29.2 ± 5.6%, n = 16, P < 0.0001) (Fig. 5B). When nipecotic acid was applied after bicuculline (10 μm) pretreatment, no significant modification of corticostriatal transmission could be observed (EPSC amplitude: 93.4 ± 5.2%, n = 7, P > 0.05) (Fig. 5B). Bicuculline alone had no significant effect on the EPSC average amplitude (92.1 ± 7.5%, n = 7). Therefore, the blockade of GATs induces a GABA accumulation responsible for the activation of GABAA receptors and a decrease of corticostriatal transmission. In the corticostriatal pathway, GABAA receptors are located postsynaptically, in MSNs (Galvan et al. 2006). Co-application of nicopetic acid and DHK induced a significant decrease of corticostriatal EPSC amplitude (−60.7 ± 1.6%, n = 4, P < 0.0001).
GABAA receptors can be activated either phasically or tonically depending on the concentration of ambient GABA (Farrant & Nusser, 2005; Glykys & Mody, 2007). GATs have been shown to modulate both tonic and phasic GABAergic signalling (Overstreet et al. 2000; Nusser & Mody, 2002; Rossi et al. 2003; Semyanov et al. 2003). Accordingly, we aimed at deciphering the impact of phasic and tonic GABAergic signalling, modified by nipecotic acid, at corticostriatal transmission. Spontaneous GABAergic events, isolated after application of antagonists of ionotropic glutamatergic receptors (d-AP5 50 μm and CNQX 10 μm), were blocked by picrotoxin (50 μm, n = 12) (Fig. 6A). Nipecotic acid induced a significant decrease of the spontaneous IPSC mean frequency (−64.8 ± 12.9%, P < 0.005, n = 12) but did not affect significantly the mean amplitude of the remaining spontaneous IPSCs. As previously reported (Kirmse et al. 2008), nipecotic acid induced a significant increase of the SD of the synaptic noise (before nipecotic acid: 3.8 ± 0.5 pA, and after: 5.6 ± 1.1 pA, P < 0.05, n = 11) with a significant increase of the tonic GABAA receptor-mediated conductances (ΔIhold: +33 ± 14 pA, n = 11, P < 0.05) (Fig. 6B). Ambient GABA can generate tonic inhibition at high-affinity extrasynaptic GABAA receptors composed by the α5-subunit in striatum (Ade et al. 2008). Inhibition of α5-GABAA receptors with L655,708 (10 μm) did not have a significant effect on corticostriatal transmission (EPSC mean amplitude in control: 127 ± 9 pA vs. in L655,708: 132 ± 13 pA, n = 11, P > 0.05). In the presence of L655,708, nipecotic acid could still decrease significantly the EPSC amplitude (−26.7 ± 9.7%, n = 6, P < 0.05) (Fig. 6C). This implies that the increase of the tonic GABAergic signalling did not cause the decrease of the corticostriatal transmission.
EPSCs and STCs displayed, respectively, a triphasic and biphasic short-term plasticity
In vivo, MSNs receive distributed discharges with a wide range of frequencies from cortical areas that will not be integrated similarly. Indeed, a strong feature of MSNs is that they act as detectors and integrators of cortical activity (Graybiel et al. 1994). We explored evoked responses for very short inter-stimulus intervals (ISIs), from 4 to 10 ms, to test specifically synchronized cortical inputs. To estimate the frequency dependence of corticostriatal synaptic transmission and STCs, we recorded EPSCs and STCs evoked by paired-pulse stimulations at various frequencies with MSN–astrocyte patch-clamp recordings (n = 21) (Fig. 7). There exist some medial-to-lateral differences in expression of paired-pulse plasticity in the striatum (Partridge et al. 2000; Smith et al. 2001). To avoid such a source of variation, we recorded MSNs and astrocytes in a restricted area located in the caudal and dorsolateral part of the striatum. MSN EPSCs were depressed at most paired-pulse frequencies tested except for two ISIs (25 and 50 ms) for which a significant facilitation could be observed (Fig. 7). No significant paired-pulse plasticity was observed for ISIs of >1 s. Therefore, MSNs displayed triphasic paired-pulse plasticity: first, a depression (for 4–10 ms ISI), and then a brief facilitation (25 and 50 ms) followed by a depression for longer ISIs (250 ms to 1 s). Astrocytic STCs displayed similar short-term plasticity to MSN EPSCs, with the notable exception of an extended facilitation (for 4–10 ms ISI), while MSN EPSCs showed depression. Similar to MSN EPSCs, for ISIs of >1 s no more significant paired-pulse plasticity was observed. STCs displayed, therefore, biphasic paired-pulse plasticity. Interestingly, although STC magnitude depends on presynaptic release, paired-pulse plasticity of STCs displayed facilitation for ISIs of <25 ms, while EPSCs displayed a marked depression for ISIs of <25 ms. Then, we investigated the origin of STC facilitation observed for ISIs of 4–10 ms. Such an increase could arise from a greater presynaptic release of glutamate leading to AMPA receptor desensitization or from a genuine paired-pulse facilitation of STCs allowing a greater uptake of glutamate that may account for the observed paired-pulse EPSC depression. For 4 and 6 ms ISIs, a significant paired-pulse depression of EPSCs was still observed with CTZ (100 μm) (Fig. 7C). This indicates that the paired-pulse depression of EPSCs did not originate from a desensitization of postsynaptic AMPA receptor but from a genuine paired-pulse facilitation of astrocytic glutamate uptake. Accordingly, STC plasticity could participate in the coincidence detection strength operated by MSNs. For ISI = 50 ms, CTZ increased significantly the facilitation observed in control (P < 0.001, n = 5) (Fig. 7C). Therefore, the magnitude of the facilitation is diminished by a desensitization of AMPA receptors in control conditions. Lastly, the depression observed for ISIs of 250 and 500 ms was not due to a desensitization of AMPA receptors because CTZ did not affect it significantly (n = 5 for ISI of 250 ms and n = 5 for ISI of 500 ms). In conclusion, depression observed for ISIs of <25 ms and ISIs of >100 ms was not due to a desensitization of postsynaptic AMPA receptor, whereas facilitation was significantly dismished by such desensitization.
Figure 7. EPSCs and STCs exhibit, respectively, a triphasic and biphasic short-term plasticity.
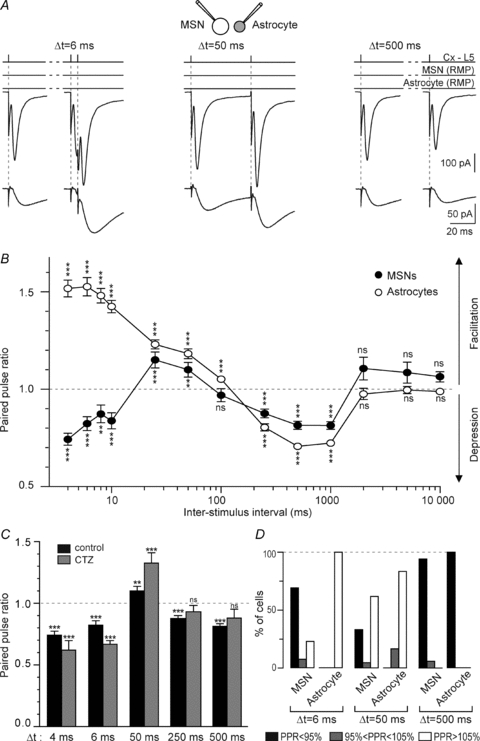
A, representative experiments of paired-pulse stimulation in the cortex at 6, 50 and 500 ms ISIs illustrate the triphasic (MSN EPSCs) and biphasic (astrocyte STCs) short-term plasticity in a representative MSN–astrocyte pair. Current traces represent the average of 15 consecutive EPSCs and STCs raw traces. B, MSN and astrocytes displayed marked paired-pulse plasticity. Patch-clamp recordings of striatal MSN–astrocyte pairs (n = 17) showed the occurrence of the triphasic short-term plasticity of MSN EPSCs and a biphasic paired-pulse plasticity of the astrocyte STCs. C, CTZ pretreatment (100 μm) did not impair significantly the paired-pulse depression of MSN EPSCs observed for 4 and 6 ms ISIs (n = 4 and 7, respectively) or for depression for ISIs of 250 ms (n = 5) and 500 ms (n = 5). Note that facilitation observed for ISI = 50 ms was significantly increased by CTZ (n = 5). D, occurrence of short-term plasticity (PPR < 95% indicated a depression, PPR 95–105% an absence of short-term plasticity and PPR > 105% a facilitation) in MSNs and astrocytes for 6, 50 and 500 ms ISIs. ns: not significant, **P < 0.01, ***P < 0.001.
To estimate the intercellular variability of the paired-pulse plasticity, cells were categorized as displaying EPSC or STC facilitation (paired-pulse ratio, PPR >105%), no change (PPR of 95–105%) or depression (PPR <95%). When compared to depression, EPSC facilitation occurrence varied considerably between MSNs studied (Fig. 7D). Indeed, even though a significant facilitation was observed for ISIs of 50 ms, 62% of the MSNs (n = 21) showed a facilitation while 33% exhibited a depression. Depression appeared more reliable since 94% of the MSNs displayed a depression for ISIs of 500 ms. Concerning astrocyte short-term plasticity, STCs seemed to be much more reliable (Fig. 7D). Indeed, there is almost no intercellular variability, since the plasticity of STCs was always oriented in one direction (facilitation or depression) with very few failures (PPR of 95–105%). Therefore, short-term plasticity of astrocytic STCs appears to be more reliable than EPSC paired-pulse plasticity.
Glutamate and GABA uptakes control EPSC and STC paired-pulse plasticity
We demonstrated that glutamate and GABA transporters significantly controlled the corticostriatal transmission. Therefore, we explored their contribution to the short-term plasticity, by blocking GLT1 or GATs. Blockade of either GLT-1 (DHK 300 μm, n = 8) or GATs (nicopetic acid 500 μm, n = 7) increased significantly EPSC paired-pulse depression (for 4 and 6 ms ISIs) and facilitation (for 50 ms ISIs) (Fig. 8A). Namely, synaptic depression was increased for ISIs of 4 ms (2.1- and 1.8-fold, for DHK and nipecotic acid, respectively) and 6 ms (1.7- and 2.2-fold, for DHK and nipecotic acid, respectively). Synaptic facilitation at ISI = 50 ms was increased 2.6- and 3.8-fold, for DHK and nipecotic acid, respectively. In addition, DHK and nipecotic acid treatment induced EPSC facilitation for ISI = 100 ms (+20.8 ± 6.6% and +17.3 ± 6.1%, for DHK and nipecotic acid, respectively); note that in control, a paired-stimulation at ISI = 100 ms did not induce any significant short-term plasticity (−3.1 ± 3.4%). In conclusion, blockade of GLT-1 or GATs increased the magnitude of the short-term plasticity for ISIs subject to the most prominent depression (ISIs of 4 and 6 ms) or facilitation, while the other ISIs remained unaffected.
Figure 8. Effects of GLT-1 or GATs blockade on paired-pulse plasticity.
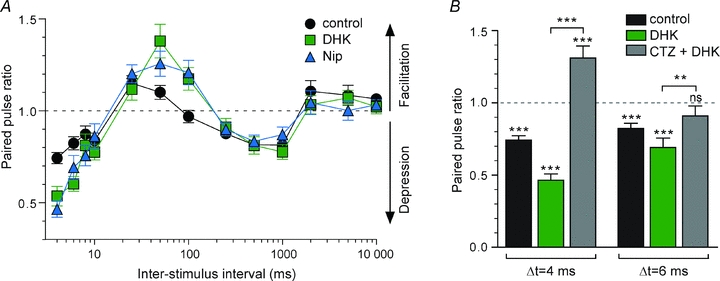
A, paired-pulse plasticity of MSN EPSCs recorded in control (n = 17), with DHK (300 μm, n = 8) and nipecotic acid (500 μm, n = 7). Both treatment DHK and nipecotic acid increased significantly EPSC depression for 4 and 6 ms ISIs and facilitation for ISI = 50 ms. DHK and nipecotic acid induced facilitation (which was not induced in control) for ISI = 100 ms. EPSC depression, for ISIs from 250 ms to 1 s, was not significantly affected by DHK or nipecotic acid treatments. B, effect of desensitization of AMPA receptors induced by DHK was investigated for 4 and 6 ms ISIs, by reducing the AMPA receptor desensitization with CTZ (100 μm). Paired-pulse depression was significantly increased when glutamate clearance was blocked with DHK (n = 8). CTZ (n = 7 and 9 for ISI of 4 and 6 ms, respectively) totally abolished the paired-pulse depression. ns: not significant, **P < 0.01, ***P < 0.001.
Incomplete clearance of glutamate before the second release of neurotransmitter could accentuate the desentization of AMPA receptors and contribute to paired-pulse depression. We tested this hypothesis for short ISIs, of 4 and 6 ms, by reducing the AMPA receptor desensitization with CTZ (100 μm). Such short ISIs would correspond to synchronized cortical activities leading to MSN activation. Paired-pulse depression was significantly increased when we blocked glutamate clearance with DHK (Fig. 8B). Coapplication of CTZ with DHK totally abolished the paired-pulse depression previously observed for 4 and 6 ms ISIs since the paired-pulse ratio was +31.2 ± 8.3% (n = 7, P < 0.001) in control and −9.1 ± 7.0% (n = 9, P > 0.05) after CTZ and DHK co-application.
In conclusion, it appears that astrocytes, via the uptake of neurotransmitters, increased the strength of filtering operated by MSNs to extract relevant information from the cortical background noise.
Discussion
Basal ganglia are involved in procedural learning and motor habit formation (Graybiel, 2005; Yin & Knowlton, 2006). MSNs act as detectors of cortical activity and have the ability to extract relevant information from the background noise. MSNs, very hyperpolarized at rest, display a low level of spontaneous activity explained by non-linear electrical membrane properties. These intrinsic MSN properties allow an efficient filtering of the small and uncorrelated synaptic events (Calabresi et al. 1987; Nisenbaum et al. 1994). The strength of the detection operated by MSNs is strongly modulated by local inhibitory networks (Tepper et al. 2008) and the clearance of neurotransmitters by astrocytes as demonstrated in this study.
We reported here that corticostriatal STCs evoked in astrocytes by single cortical stimulation (even at low frequencies of stimulation, 0.1 Hz) displayed fast kinetics and large amplitude, denoting an important expression of transporters in the striatum (Furuta et al. 1997). Here we showed that corticostriatal STCs mainly originated from glutamate and GABA transporter activities. The residual current, insensitive to glutamatergic or GABAergic transporter inhibitors, could arise from cortically evoked release of neuromodulators such as dopamine, serotonine or choline (Inazu et al., 1999, 2001, 2005). Here, we report for the first time in the striatum a large STC underlain by astrocytic GABA transport. This observation is in accordance with the GABAergic nature of the striatum since all striatal neurons (except the 2% that are cholinergic interneurons) are GABAergic and directly contacted by cortical pyramidal cells (Kawaguchi, 1993; Tepper et al. 2008). Astrocytes potentially express all GAT subtypes (Schousboe et al. 2004). Nipecotic acid (500 μm) blocks all GABA transporters, which allowed us to characterize the effect of GABA transport in general, but it did not permit determination of the GAT subtype that could be preferentially involved in this astrocytic GABA uptake. Concerning glutamate uptake, we demonstrated pharmacologically that GLT-1 and GLAST were responsible for the glutamate uptake operated by astrocytes. GLT-1 is highly expressed in astrocytes and is in charge of most of the clearance of glutamate from the synaptic cleft (Rothstein et al. 1996; Tanaka et al. 1997). We observed very large glutamatergic STCs in astrocytes denoting a high expression of glutamate transporters. Indeed, GLT-1 is particularly expressed in striatum when compared to other brain structures (Rothstein et al. 1994; Lehre et al. 1995; Furuta et al. 1997).
The inward current generated by the uptake of one molecule of glutamate or GABA relies on the entry of two and one positive charges, respectively (Kanner & Schuldiner, 1987; Kavanaugh et al. 1992; Zerangue & Kavanaugh, 1996; Danbolt, 2001; but see Guastella et al. 1990 and Loo et al. 2000 for different GAT stoechiometry). The clearance of GABA and glutamate appears to be extremely efficient since inhibition of either decreases significantly the corticostriatal transmission. We now consider the number and density of transporters involved in corticostriatal STCs and therefore the potentiality of this system. Astrocyte STCs exhibited a mean value of 60 pA while MSN EPSC amplitude was ∼400 pA. Based on our results, on average GLT-1 electrogenic activity represents 31% of STCs and thus 19 pA current. Similarly, 32 pA out of 60 pA can be attributed to the astrocytic glutamate clearance operated by both GLT-1 and GLAST. Note that the GLT-1 ratio is probably underestimated because PDC, although unspecific, has a greater affinity for GLT-1 than DHK (Arriza et al. 1994; Wang et al. 1998). For two charges entering per carrier cycle (Zerangue & Kavanaugh, 1996; Danbolt, 2001), and a cycle time of 15 s−1 at −80 mV (close to astrocyte RMP) (Wadiche et al. 1995), our data imply that there are 4.0 × 106 GLT-1 and 6.8 × 106 GLT-1/GLAST transporters involved in cortically evoked STCs. Concerning GABA transport, if we assumed that GABA uptake is accompanied by one charge entering per carrier cycle (Kanner & Schuldiner, 1987; Kavanaugh et al. 1992) and display a cycle time of 5.8 s−1 at −80 mV (Mager et al. 1993), our data imply that there are 24.0 × 106 GATs involved in astrocytic STCs. Note that these values were obtained for non-saturating glutamate and/or GABA concentration. When single cortical stimulation strength was increased (from ∼2 to 6 μA), STCs could reach amplitudes of ∼650 pA. In these conditions, 43 × 106 GLT-1, 70 × 106 GLT-1/GLAST and 260 × 106 GATs transporters were involved in cortically evoked STCs denoting a high capacity of the astrocytes for glutamate and GABA clearance at corticostriatal synapses. Lastly, we evaluated the density of glutamatergic transporters involved in cortically evoked STCs. Considering the maximal glutamatergic STCs recorded during a single cortical stimulation (∼350 pA; the present study), the neuropilar volume occupied by an astrocyte (66×10−3μm3; Bushong et al. 2002) and the astroglial surface density (1.4 μm2/μm3; Lehre & Danbolt, 1998), we estimated a density of 760 active glutamatergic transporters per μm2. However, a saturating concentration of glutamate revealed that density of glutamate transporters in hippocampus is 3.3-fold higher (2500 glutamate transporters per μm2; Bergles & Jahr, 1997). This suggests that glutamatergic clearance of striatal astrocytes is potentially 3.3-fold higher than that observed in the present study, meaning a glutamatergic STC of 1.2 nA. Such large glutamate clearance is consistent with in vivo data showing that a very high concentration of glutamate (30 mm) is required to activate MSNs (Chevalier et al. 1985).
Interestingly, both glutamate and GABA astrocytic transports have a permissive effect on corticostriatal transmission, since their inhibition lead to a significant decrease of this transmission. Indeed, excess of glutamate in the synaptic cleft induced a desensitization of AMPA receptors while GABA accumulation caused an activation of postsynaptic GABAA receptors. Because bicuculline restored the whole corticostriatal transmission, we can exclude the alternative hypothesis that nipecotic acid could reduce the quantal size of glutamatergic events because there is less GABA that can be used to support glutamate synthesis. A recent study reported that dl-threo-β-benzyloxyaspartate (TBOA), a broad spectrum inhibitor of glutamate transporters, induced an increase of corticostriatal transmission and the authors concluded that neuronal glutamate transporters, rather than glial, shape EPSCs (Beurrier et al. 2009). The discrepancy with our study may arise from the use of TBOA, since with DHK they observed a decrease of corticostriatal EPSCs. DHK induces significant changes in MSN holding current. To counteract such an effect, MSNs were recorded in voltage-clamp mode to hold their resting membrane potential at a constant value. Depending on the brain structures, blockade of astrocytic glutamate or GABA transporters affects synaptic transmission differently (for example see Hestrin et al. 1990 in hippocampus and Gonzalez-Burgos et al. 2009 in cortex). Such effects could rely on various structures of the tripartite synapse. Namely, the contribution of neurotransmitter uptake to synaptic transmission depends on the extent of astrocyte ensheatment of synapses, and the nature and location of neuronal receptors and astrocytic transporters vary a lot among CNS structures.
The paired-pulse plasticity determines the neuronal responses to patterned presynaptic activities, and thus is crucial in understanding the basic principles of information processing. Depending on the frequency of cortical activity, astrocytes behave as followers of presynaptic release or play an active role in neurotransmitter clearance. Indeed, EPSCs and STCs showed similar paired-pulse plasticity, with the notable exception of ISIs of 4–10 ms where EPSCs displayed a depression while STCs exhibited a facilitation. Such opposite paired-pulse plasticity of EPSCs and STCs could be explained by a genuine facilitation of glutamate uptake leading to less glutamate available for postsynaptic activation. Indeed, when desensitization of AMPA receptors was prevented with CTZ, the depression of MSN EPSCs was not impaired. MSNs select synchronized cortical events, corresponding to high frequency signals. It is precisely for high frequencies (100–250 Hz) that astrocytes display a marked facilitation of neurotransmitter uptake. Accordingly, astrocytes might reinforce the strength of the detection of cortical activity operated by MSNs. Endogenous molecules affecting neurotransmitter transport, such as arachidonic acid or adenosine (Volterra et al. 1992; Cristovao-Ferreira et al. 2009), or diseases modifying the transporter expression in striatum (Huntington and Parkinson's diseases) (Arzberger et al. 1997; Dervan et al. 2004) should therefore alter the detection properties of cortical events by MSNs.
Acknowledgments
We would like to thank Anne-Marie Godeheu for technical assistance. This work was supported by the INSERM, ANR Mobil and College de France.
Glossary
Abbreviations
- CNQX
6-cyano-7-nitroquinoxaline-2,3-dione
- CTZ
cyclothiazide
- d-AP5
dl-2-amino-5-phosphono-pentanoic acid
- DHK
dihydrokainic acid
- EAAC
excitatory amino acid carrier
- EAAT
excitatory amino acid transporter
- EPSC
excitatory postsynaptic current
- GAT
GABA transporter
- GLAST
glutamate-aspartate transporter
- GLT-1
glutamate transporter type-1
- ISI
inter-stimulus interval
- MSN
medium-sized spiny neuron
- PDC
l-trans-pyrrolidine-2,4-dicarboxylic acid
- RMP
resting membrane potential
- STC
synaptically activated transporter-mediated current
Author contributions
Conception and design of the experiments: L.V. Collection, analysis and interpretation of data: V.G., and L.V. Drafting the article and revising it critically for important intellectual content: V.G., E.F. and L.V.
Supplementary material
supplementary figure 1
supplementary figure 2
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer-reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors
References
- Ade KK, Janssen MJ, Ortinski PI, Vicini S. Differential tonic GABA conductances in striatal medium spiny neurons. J Neurosci. 2008;28:1185–1197. doi: 10.1523/JNEUROSCI.3908-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adermark L, Lovinger DM. Ethanol effects on electrophysiological properties of astrocytes in striatal brain slices. Neuropharmacology. 2006;51:1099–1108. doi: 10.1016/j.neuropharm.2006.05.035. [DOI] [PubMed] [Google Scholar]
- Akopian G, Walsh JP. Reliable long-lasting depression interacts with variable short-term facilitation to determine corticostriatal paired-pulse plasticity in young rats. J Physiol. 2007;580:225–240. doi: 10.1113/jphysiol.2006.115790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araque A, Parpura V, Sanzgiri RP, Haydon PG. Tripartite synapses: glia, the unacknowledged partner. Trends Neurosci. 1999;22:208–215. doi: 10.1016/s0166-2236(98)01349-6. [DOI] [PubMed] [Google Scholar]
- Arnth-Jensen N, Jabaudon D, Scanziani M. Cooperation between independent hippocampal synapses is controlled by glutamate uptake. Nat Neurosci. 2002;5:325–331. doi: 10.1038/nn825. [DOI] [PubMed] [Google Scholar]
- Arriza JL, Fairman WA, Wadiche JI, Murdoch GH, Kavanaugh MP, Amara SG. Functional comparisons of three glutamate transporter subtypes cloned from human motor cortex. J Neurosci. 1994;14:5559–5569. doi: 10.1523/JNEUROSCI.14-09-05559.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arzberger T, Krampfl K, Leimgruber S, Weindl A. Changes of NMDA receptor subunit (NR1, NR2B) and glutamate transporter (GLT1) mRNA expression in Huntington's disease – an in situ hybridization study. J Neuropathol Exp Neurol. 1997;56:440–454. doi: 10.1097/00005072-199704000-00013. [DOI] [PubMed] [Google Scholar]
- Barakat L, Bordey A. GAT-1 and reversible GABA transport in Bergmann glia in slices. J Neurophysiol. 2002;88:1407–1419. doi: 10.1152/jn.2002.88.3.1407. [DOI] [PubMed] [Google Scholar]
- Bergles DE, Jahr CE. Synaptic activation of glutamate transporters in hippocampal astrocytes. Neuron. 1997;19:1297–1308. doi: 10.1016/s0896-6273(00)80420-1. [DOI] [PubMed] [Google Scholar]
- Bergles DE, Jahr CE. Glial contribution to glutamate uptake at Schaffer collateral-commissural synapses in the hippocampus. J Neurosci. 1998;18:7709–7716. doi: 10.1523/JNEUROSCI.18-19-07709.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergles DE, Diamond JS, Jahr CE. Clearance of glutamate inside the synapse and beyond. Curr Opin Neurobiol. 1999;9:293–298. doi: 10.1016/s0959-4388(99)80043-9. [DOI] [PubMed] [Google Scholar]
- Beurrier C, Bonvento G, Kerkerian-Le Goff L, Gubellini P. Role of glutamate transporters in corticostriatal synaptic transmission. Neuroscience. 2009;158:1608–1615. doi: 10.1016/j.neuroscience.2008.11.018. [DOI] [PubMed] [Google Scholar]
- Borden LA. GABA transporter heterogeneity: pharmacology and cellular localization. Neurochem Int. 1996;29:335–356. doi: 10.1016/0197-0186(95)00158-1. [DOI] [PubMed] [Google Scholar]
- Brew H, Attwell D. Electrogenic glutamate uptake is a major current carrier in the membrane of axolotl retinal glial cells. Nature. 1987;327:707–709. doi: 10.1038/327707a0. [DOI] [PubMed] [Google Scholar]
- Bushong EA, Martone ME, Jones YZ, Ellisman MH. Protoplasmic astrocytes in CA1 stratum radiatum occupy separate anatomical domains. J Neurosci. 2002;22:183–192. doi: 10.1523/JNEUROSCI.22-01-00183.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabresi P, Misgeld U, Dodt HU. Intrinsic membrane properties of neostriatal neurons can account for their low level of spontaneous activity. Neuroscience. 1987;20:293–303. doi: 10.1016/0306-4522(87)90021-2. [DOI] [PubMed] [Google Scholar]
- Cammack JN, Schwartz EA. Ions required for the electrogenic transport of GABA by horizontal cells of the catfish retina. J Physiol. 1993;472:81–102. doi: 10.1113/jphysiol.1993.sp019938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevalier G, Vacher S, Deniau JM, Desban M. Disinhibition as a basic process in the expression of striatal functions. I. The striato-nigral influence on tecto-spinal/tecto-diencephalic neurons. Brain Res. 1985;334:215–226. doi: 10.1016/0006-8993(85)90213-6. [DOI] [PubMed] [Google Scholar]
- Chvatal A, Pastor A, Mauch M, Sykova E, Kettenmann H. Distinct populations of identified glial cells in the developing rat spinal cord slice: ion channel properties and cell morphology. Eur J Neurosci. 1995;7:129–142. doi: 10.1111/j.1460-9568.1995.tb01027.x. [DOI] [PubMed] [Google Scholar]
- Conti F, Minelli A, Melone M. GABA transporters in the mammalian cerebral cortex: localization, development and pathological implications. Brain Res Brain Res Rev. 2004;45:196–212. doi: 10.1016/j.brainresrev.2004.03.003. [DOI] [PubMed] [Google Scholar]
- Cristovao-Ferreira S, Vaz SH, Ribeiro JA, Sebastiao AM. Adenosine A2A receptors enhance GABA transport into nerve terminals by restraining PKC inhibition of GAT-1. J Neurochem. 2009;109:336–347. doi: 10.1111/j.1471-4159.2009.05963.x. [DOI] [PubMed] [Google Scholar]
- Danbolt NC. Glutamate uptake. Prog Neurobiol. 2001;65:1–105. doi: 10.1016/s0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- Dervan AG, Meshul CK, Beales M, McBean GJ, Moore C, Totterdell S, Snyder AK, Meredith GE. Astroglial plasticity and glutamate function in a chronic mouse model of Parkinson's disease. Exp Neurol. 2004;190:145–156. doi: 10.1016/j.expneurol.2004.07.004. [DOI] [PubMed] [Google Scholar]
- Diamond JS, Bergles DE, Jahr CE. Glutamate release monitored with astrocyte transporter currents during LTP. Neuron. 1998;21:425–433. doi: 10.1016/s0896-6273(00)80551-6. [DOI] [PubMed] [Google Scholar]
- Diamond JS. Neuronal glutamate transporters limit activation of NMDA receptors by neurotransmitter spillover on CA1 pyramidal cells. J Neurosci. 2001;21:8328–8338. doi: 10.1523/JNEUROSCI.21-21-08328.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond GB. Reporting ethical matters in The Journal of Physiology: standarts and advice. J Physiol. 2009;587:713–719. doi: 10.1113/jphysiol.2008.167387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrant M, Nusser Z. Variations on an inhibitory theme: phasic and tonic activation of GABA(A) receptors. Nat Rev Neurosci. 2005;6:215–229. doi: 10.1038/nrn1625. [DOI] [PubMed] [Google Scholar]
- Fino E, Glowinski J, Venance L. Bidirectional activity-dependent plasticity at corticostriatal synapses. J Neurosci. 2005;25:11279–11287. doi: 10.1523/JNEUROSCI.4476-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fino E, Deniau JM, Venance L. Cell-specific spike-timing-dependent plasticity in GABAergic and cholinergic interneurons in corticostriatal rat brain slices. J Physiol. 2008;586:265–282. doi: 10.1113/jphysiol.2007.144501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fino E, Paille V, Deniau JM, Venance L. Asymmetric spike-timing dependent plasticity of striatal nitric oxide-synthase interneurons. Neuroscience. 2009;160:744–754. doi: 10.1016/j.neuroscience.2009.03.015. [DOI] [PubMed] [Google Scholar]
- Fino E, Paille V, Cui Y, Morera-Herreras T, Deniau JM, Venance L. Distinct coincidence detectors govern the corticostriatal spike timing-dependent plasticity. J Physiol. 2010;588:3045–3062. doi: 10.1113/jphysiol.2010.188466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuta A, Rothstein JD, Martin LJ. Glutamate transporter protein subtypes are expressed differentially during rat CNS development. J Neurosci. 1997;17:8363–8375. doi: 10.1523/JNEUROSCI.17-21-08363.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvan A, Kuwajima M, Smith Y. Glutamate and GABA receptors and transporters in the basal ganglia: what does their subsynaptic localization reveal about their function? Neuroscience. 2006;143:351–375. doi: 10.1016/j.neuroscience.2006.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glykys J, Mody I. Activation of GABAA receptors: views from outside the synaptic cleft. Neuron. 2007;56:763–770. doi: 10.1016/j.neuron.2007.11.002. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Burgos G, Rotaru DC, Zaitsev AV, Povysheva NV, Lewis DA. GABA transporter GAT1 prevents spillover at proximal and distal GABA synapses onto primate prefrontal cortex neurons. J Neurophysiol. 2009;101:533–547. doi: 10.1152/jn.91161.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graybiel AM, Aosaki T, Flaherty AW, Kimura M. The basal ganglia and adaptive motor control. Science. 1994;265:1826–1831. doi: 10.1126/science.8091209. [DOI] [PubMed] [Google Scholar]
- Graybiel AM. The basal ganglia: learning new tricks and loving it. Curr Opin Neurobiol. 2005;15:638–644. doi: 10.1016/j.conb.2005.10.006. [DOI] [PubMed] [Google Scholar]
- Guastella J, Nelson N, Nelson H, Czyzyk L, Keynan S, Miedel MC, Davidson N, Lester HA, Kanner BI. Cloning and expression of a rat brain GABA transporter. Science. 1990;249:1303–1306. doi: 10.1126/science.1975955. [DOI] [PubMed] [Google Scholar]
- Halassa MM, Fellin T, Haydon PG. The tripartite synapse: roles for gliotransmission in health and disease. Trends Mol Med. 2007;13:54–63. doi: 10.1016/j.molmed.2006.12.005. [DOI] [PubMed] [Google Scholar]
- Halassa MM, Haydon PG. Integrated brain circuits: astrocytic networks modulate neuronal activity and behavior. Annu Rev Physiol. 2010;72:335–355. doi: 10.1146/annurev-physiol-021909-135843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haydon PG. GLIA: listening and talking to the synapse. Nat Rev Neurosci. 2001;2:185–193. doi: 10.1038/35058528. [DOI] [PubMed] [Google Scholar]
- Haydon PG, Carmignoto G. Astrocyte control of synaptic transmission and neurovascular coupling. Physiol Rev. 2006;86:1009–1031. doi: 10.1152/physrev.00049.2005. [DOI] [PubMed] [Google Scholar]
- Hestrin S, Sah P, Nicoll RA. Mechanisms generating the time course of dual component excitatory synaptic currents recorded in hippocampal slices. Neuron. 1990;5:247–253. doi: 10.1016/0896-6273(90)90162-9. [DOI] [PubMed] [Google Scholar]
- Inazu M, Kubota N, Takeda H, Zhang J, Kiuchi Y, Oguchi K, Matsumiya T. Pharmacological characterization of dopamine transport in cultured rat astrocytes. Life Sci. 1999;64:2239–2245. doi: 10.1016/s0024-3205(99)00175-7. [DOI] [PubMed] [Google Scholar]
- Inazu M, Takeda H, Ikoshi H, Sugisawa M, Uchida Y, Matsumiya T. Pharmacological characterization and visualization of the glial serotonin transporter. Neurochem Int. 2001;39:39–49. doi: 10.1016/s0197-0186(01)00010-9. [DOI] [PubMed] [Google Scholar]
- Inazu M, Takeda H, Matsumiya T. Molecular and functional characterization of an Na+-independent choline transporter in rat astrocytes. J Neurochem. 2005;94:1427–1437. doi: 10.1111/j.1471-4159.2005.03299.x. [DOI] [PubMed] [Google Scholar]
- Kanner BI, Schuldiner S. Mechanism of transport and storage of neurotransmitters. CRC Crit Rev Biochem. 1987;22:1–38. doi: 10.3109/10409238709082546. [DOI] [PubMed] [Google Scholar]
- Kavanaugh MP, Arriza JL, North RA, Amara SG. Electrogenic uptake of γ-aminobutyric acid by a cloned transporter expressed in Xenopus oocytes. J Biol Chem. 1992;267:22007–22009. [PubMed] [Google Scholar]
- Kawaguchi Y. Physiological, morphological, and histochemical characterization of three classes of interneurons in rat neostriatum. J Neurosci. 1993;13:4908–4923. doi: 10.1523/JNEUROSCI.13-11-04908.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirmse K, Dvorzhak A, Kirischuk S, Grantyn R. GABA transporter 1 tunes GABAergic synaptic transmission at output neurons of the mouse neostriatum. J Physiol. 2008;586:5665–5678. doi: 10.1113/jphysiol.2008.161943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kullmann DM, Asztely F. Extrasynaptic glutamate spillover in the hippocampus: evidence and implications. Trends Neurosci. 1998;21:8–14. doi: 10.1016/s0166-2236(97)01150-8. [DOI] [PubMed] [Google Scholar]
- Lehre KP, Danbolt NC. The number of glutamate transporter subtype molecules at glutamatergic synapses: chemical and stereological quantification in young adult rat brain. J Neurosci. 1998;18:8751–8757. doi: 10.1523/JNEUROSCI.18-21-08751.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehre KP, Levy LM, Ottersen OP, Storm-Mathisen J, Danbolt NC. Differential expression of two glial glutamate transporters in the rat brain: quantitative and immunocytochemical observations. J Neurosci. 1995;15:1835–1853. doi: 10.1523/JNEUROSCI.15-03-01835.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu QR, Lopez-Corcuera B, Mandiyan S, Nelson H, Nelson N. Molecular characterization of four pharmacologically distinct γ-aminobutyric acid transporters in mouse brain [corrected] J Biol Chem. 1993;268:2106–2112. [PubMed] [Google Scholar]
- Loo DD, Eskandari S, Boorer KJ, Sarkar HK, Wright EM. Role of Cl− in electrogenic Na+-coupled cotransporters GAT1 and SGLT1. J Biol Chem. 2000;275:37414–37422. doi: 10.1074/jbc.M007241200. [DOI] [PubMed] [Google Scholar]
- Lozovaya NA, Kopanitsa MV, Boychuk YA, Krishtal OA. Enhancement of glutamate release uncovers spillover-mediated transmission by N-methyl-D-aspartate receptors in the rat hippocampus. Neuroscience. 1999;91:1321–1330. doi: 10.1016/s0306-4522(98)00638-1. [DOI] [PubMed] [Google Scholar]
- Luscher C, Malenka RC, Nicoll RA. Monitoring glutamate release during LTP with glial transporter currents. Neuron. 1998;21:435–441. doi: 10.1016/s0896-6273(00)80552-8. [DOI] [PubMed] [Google Scholar]
- Mager S, Naeve J, Quick M, Labarca C, Davidson N, Lester HA. Steady states, charge movements, and rates for a cloned GABA transporter expressed in Xenopus oocytes. Neuron. 1993;10:177–188. doi: 10.1016/0896-6273(93)90309-f. [DOI] [PubMed] [Google Scholar]
- Meme W, Vandecasteele M, Giaume C, Venance L. Electrical coupling between hippocampal astrocytes in rat brain slices. Neurosci Res. 2009;63:236–243. doi: 10.1016/j.neures.2008.12.008. [DOI] [PubMed] [Google Scholar]
- Minelli A, DeBiasi S, Brecha NC, Zuccarello LV, Conti F. GAT-3, a high-affinity GABA plasma membrane transporter, is localized to astrocytic processes, and it is not confined to the vicinity of GABAergic synapses in the cerebral cortex. J Neurosci. 1996;16:6255–6264. doi: 10.1523/JNEUROSCI.16-19-06255.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nisenbaum ES, Xu ZC, Wilson CJ. Contribution of a slowly inactivating potassium current to the transition to firing of neostriatal spiny projection neurons. J Neurophysiol. 1994;71:1174–1189. doi: 10.1152/jn.1994.71.3.1174. [DOI] [PubMed] [Google Scholar]
- Nishiyama A, Watanabe M, Yang Z, Bu J. Identity, distribution, and development of polydendrocytes: NG2-expressing glial cells. J Neurocytol. 2002;31:437–455. doi: 10.1023/a:1025783412651. [DOI] [PubMed] [Google Scholar]
- Nusser Z, Mody I. Selective modulation of tonic and phasic inhibitions in dentate gyrus granule cells. J Neurophysiol. 2002;87:2624–2628. doi: 10.1152/jn.2002.87.5.2624. [DOI] [PubMed] [Google Scholar]
- Overstreet LS, Jones MV, Westbrook GL. Slow desensitization regulates the availability of synaptic GABAA receptors. J Neurosci. 2000;20:7914–7921. doi: 10.1523/JNEUROSCI.20-21-07914.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partin KM, Fleck MW, Mayer ML. AMPA receptor flip/flop mutants affecting deactivation, desensitization, and modulation by cyclothiazide, aniracetam, and thiocyanate. J Neurosci. 1996;16:6634–6647. doi: 10.1523/JNEUROSCI.16-21-06634.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partridge JG, Tang KC, Lovinger DM. Regional and postnatal heterogeneity of activity-dependent long-term changes in synaptic efficacy in the dorsal striatum. J Neurophysiol. 2000;84:1422–1429. doi: 10.1152/jn.2000.84.3.1422. [DOI] [PubMed] [Google Scholar]
- Pines G, Danbolt NC, Bjoras M, Zhang Y, Bendahan A, Eide L, Koepsell H, Storm-Mathisen J, Seeberg E, Kanner BI. Cloning and expression of a rat brain L-glutamate transporter. Nature. 1992;360:464–467. doi: 10.1038/360464a0. [DOI] [PubMed] [Google Scholar]
- Rossi DJ, Hamann M, Attwell D. Multiple modes of GABAergic inhibition of rat cerebellar granule cells. J Physiol. 2003;548:97–110. doi: 10.1113/jphysiol.2002.036459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothstein JD, Dykes-Hoberg M, Pardo CA, Bristol LA, Jin L, Kuncl RW, Kanai Y, Hediger MA, Wang Y, Schielke JP, Welty DF. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron. 1996;16:675–686. doi: 10.1016/s0896-6273(00)80086-0. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Martin L, Levey AI, Dykes-Hoberg M, Jin L, Wu D, Nash N, Kuncl RW. Localization of neuronal and glial glutamate transporters. Neuron. 1994;13:713–725. doi: 10.1016/0896-6273(94)90038-8. [DOI] [PubMed] [Google Scholar]
- Schousboe A, Sarup A, Bak LK, Waagepetersen HS, Larsson OM. Role of astrocytic transport processes in glutamatergic and GABAergic neurotransmission. Neurochem Int. 2004;45:521–527. doi: 10.1016/j.neuint.2003.11.001. [DOI] [PubMed] [Google Scholar]
- Schousboe A, Thorbek P, Hertz L, Krogsgaard-Larsen P. Effects of GABA analogues of restricted conformation on GABA transport in astrocytes and brain cortex slices and on GABA receptor binding. J Neurochem. 1979;33:181–189. doi: 10.1111/j.1471-4159.1979.tb11720.x. [DOI] [PubMed] [Google Scholar]
- Semyanov A, Walker MC, Kullmann DM. GABA uptake regulates cortical excitability via cell type-specific tonic inhibition. Nat Neurosci. 2003;6:484–490. doi: 10.1038/nn1043. [DOI] [PubMed] [Google Scholar]
- Smith R, Musleh W, Akopian G, Buckwalter G, Walsh JP. Regional differences in the expression of corticostriatal synaptic plasticity. Neuroscience. 2001;106:95–101. doi: 10.1016/s0306-4522(01)00260-3. [DOI] [PubMed] [Google Scholar]
- Steinhauser C, Gallo V. News on glutamate receptors in glial cells. Trends Neurosci. 1996;19:339–345. doi: 10.1016/0166-2236(96)10043-6. [DOI] [PubMed] [Google Scholar]
- Tanaka K, Watase K, Manabe T, Yamada K, Watanabe M, Takahashi K, Iwama H, Nishikawa T, Ichihara N, Kikuchi T, Okuyama S, Kawashima N, Hori S, Takimoto M, Wada K. Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science. 1997;276:1699–1702. doi: 10.1126/science.276.5319.1699. [DOI] [PubMed] [Google Scholar]
- Tepper JM, Wilson CJ, Koos T. Feedforward and feedback inhibition in neostriatal GABAergic spiny neurons. Brain Res Rev. 2008;58:272–281. doi: 10.1016/j.brainresrev.2007.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trussell LO, Thio LL, Zorumski CF, Fischbach GD. Rapid desensitization of glutamate receptors in vertebrate central neurons. Proc Natl Acad Sci U S A. 1988;85:4562–4566. doi: 10.1073/pnas.85.12.4562-a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandecasteele M, Glowinski J, Deniau JM, Venance L. Chemical transmission between dopaminergic neuron pairs. Proc Natl Acad Sci U S A. 2008;105:4904–4909. doi: 10.1073/pnas.0703121105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venance L, Glowinski J, Giaume C. Electrical and chemical transmission between striatal GABAergic output neurones in rat brain slices. J Physiol. 2004;559:215–230. doi: 10.1113/jphysiol.2004.065672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkhratsky A, Steinhauser C. Ion channels in glial cells. Brain Res Brain Res Rev. 2000;32:380–412. doi: 10.1016/s0165-0173(99)00093-4. [DOI] [PubMed] [Google Scholar]
- Volterra A, Meldolesi J. Astrocytes, from brain glue to communication elements: the revolution continues. Nat Rev Neurosci. 2005;6:626–640. doi: 10.1038/nrn1722. [DOI] [PubMed] [Google Scholar]
- Volterra A, Trotti D, Cassutti P, Tromba C, Galimberti R, Lecchi P, Racagni G. A role for the arachidonic acid cascade in fast synaptic modulation: ion channels and transmitter uptake systems as target proteins. Adv Exp Med Biol. 1992;318:147–158. doi: 10.1007/978-1-4615-3426-6_13. [DOI] [PubMed] [Google Scholar]
- Wadiche JI, Arriza JL, Amara SG, Kavanaugh MP. Kinetics of a human glutamate transporter. Neuron. 1995;14:1019–1027. doi: 10.1016/0896-6273(95)90340-2. [DOI] [PubMed] [Google Scholar]
- Wang GJ, Chung HJ, Schnuer J, Pratt K, Zable AC, Kavanaugh MP, Rosenberg PA. High affinity glutamate transport in rat cortical neurons in culture. Mol Pharmacol. 1998;53:88–96. doi: 10.1124/mol.53.1.88. [DOI] [PubMed] [Google Scholar]
- Yin HH, Knowlton BJ. The role of the basal ganglia in habit formation. Nat Rev Neurosci. 2006;7:464–476. doi: 10.1038/nrn1919. [DOI] [PubMed] [Google Scholar]
- Zerangue N, Kavanaugh MP. Flux coupling in a neuronal glutamate transporter. Nature. 1996;383:634–637. doi: 10.1038/383634a0. [DOI] [PubMed] [Google Scholar]
- Zhou M, Kimelberg HK. Freshly isolated astrocytes from rat hippocampus show two distinct current patterns and different [K+]o uptake capabilities. J Neurophysiol. 2000;84:2746–2757. doi: 10.1152/jn.2000.84.6.2746. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.