

Abstract
The expanded CAG repeat that causes striatal cell vulnerability in Huntington's disease (HD) encodes a polyglutamine tract in full-length huntingtin that is correlated with cellular [ATP] and [ATP/ADP]. Since striatal neurons are vulnerable to energy deficit, we have investigated, in Hdh CAG knock-in mice and striatal cells, the hypothesis that decreased energetics may affect neuronal (N)-cadherin, a candidate energy-sensitive adhesion protein that may contribute to HD striatal cell sensitivity. In vivo, N-cadherin was sensitive to ischemia and to the effects of full-length mutant huntingtin, progressively decreasing in HdhQ111 striatum with age. In cultured striatal cells, N-cadherin was decreased by ATP depletion and STHdhQ111 striatal cells exhibited dramatically decreased N-cadherin, due to decreased Cdh2 mRNA and enhanced N-cadherin turnover, which was partially normalized by adenine supplementation to increase [ATP] and [ATP/ADP]. Consistent with decreased N-cadherin function, STHdhQ111 striatal cells displayed profound deficits in calcium-dependent N-cadherin-mediated cell clustering and cell–substratum adhesion, and primary HdhQ111 striatal neuronal cells exhibited decreased N-cadherin and an abundance of immature neurites, featuring diffuse, rather than clustered, staining for N-cadherin and synaptic vesicle markers, which was partially rescued by adenine treatment. Thus, mutant full-length huntingtin, via energetic deficit, contributes to decreased N-cadherin levels in striatal neurons, with detrimental effects on neurite maturation, strongly suggesting that N-cadherin-mediated signaling merits investigation early in the HD pathogenic disease process.
INTRODUCTION
The CAG expansion mutation that causes Huntington's disease (HD) elongates a polymorphic polyglutamine segment in the huntingtin protein. Full-length huntingtin with a polyglutamine region of more than ∼37 residues initiates a disease process that culminates in the loss of neurons, especially in the striatum, and the onset of the motor, psychiatric and cognitive symptoms (1,2). Understanding the rate-limiting events that contribute to the early vulnerability of striatal neurons would guide efforts to track the natural history of the disease and may provide new avenues for therapeutic development.
Studies investigating the earliest consequences of full-length mutant huntingtin, in HD patient cells and tissues and in genetically accurate Hdh CAG knock-in mouse cells and tissues, have revealed perturbations in membrane vesicle trafficking, gene transcription, intracellular signaling pathways (3–6), as well as altered energetics, characterized by decreased [ATP] and [ATP/ADP], which is correlated with the size of the polyglutamine repeat (7–9). We have been studying the effects of altered energetics because the correlation of energetic measures with the polyglutamine repeat in full-length huntingtin implies a dominant effect that conforms to the genetic features of the HD trigger mechanism, and energetic defects, thought to be important to striatal cells, may be evident throughout the lifetime of the cell (7–9). Certainly, early weight loss in HD and a systemic metabolic defect in branched chain amino acids are consistent with a systemic attempt to compensate for an early energy deficit (10).
Neuronal (N)-cadherin, which is intimately involved in neuronal cell adhesion, signaling, differentiation and synapse function (11,12), is a prime candidate for being affected by energy deficit. Members of the cadherin family exhibit selective degradation in response to renal ischemia and ATP depletion (13–15), and in normal rat kidney cells via cleavage by membrane-type 1 matrix metalloprotease (MT-MMP) (16). However, N-cadherin has not been studied either in acute neuronal ischemia and ATP depletion or in response to the HD mutation, which elicits a chronic energy deficit in a process that culminates in neurodegeneration.
N-cadherin is a transmembrane cell adhesion glycoprotein composed of an extracellular domain, a single-pass transmembrane region and a cytoplasmic tail (17). N-cadherin molecules make calcium-dependent homophilic bonds between their extracellular domains (18). The cytoplasmic domain contains two main binding regions, the C-terminal domain (CTD) and the juxtamembrane domain (JMD). The CTD binds β- and γ-catenin, which in turn associate with the actin cytoskeleton to modulate cell adhesion and mobility via α-catenin (19,20). The JMD interacts with p120-catenin and with presenilin 1, which has emerged as a potential regulator of cell adhesion and neuronal physiology (21,22).
Here, we have assessed the candidacy of N-cadherin as an energy-sensitive contributor to the striatal cell vulnerability that ensues from the HD mutation. Specifically, we have investigated N-cadherin in HdhQ111 CAG knock-in mouse striatum and cultured HdhQ111 striatal neuronal cells, which express endogenous full-length 111-glutamine mutant huntingtin. We first tested whether N-cadherin was sensitive to acute ATP depletion/ischemia and to the chronic effects of full-length mutant huntingtin protein and then we evaluated immortalized STHdhQ111 and primary HdhQ111 striatal neurons to explore N-cadherin ATP sensitivity and the phenotypic consequences of decreased N-cadherin function. Our findings reveal that N-cadherin is an ATP-sensitive protein that is associated with altered HD CAG striatal cell adhesion and neuritogenesis.
RESULTS
Striatal N-cadherin was sensitive to acute ischemia and to the HD CAG mutation
To assess whether N-cadherin might be affected in a mild ischemic brain injury paradigm where the striatum displays early vulnerability, we performed transient middle cerebral artery (MCA) occlusion with 12-month-old wild-type HdhQ7/Q7 and mutant HdhQ111/Q111 knock-in mice. For both genotypes, immunoblot of protein extracts at 24 h after reperfusion (Fig. 1A and B) revealed robustly decreased N-cadherin in the ischemic, compared with the contralateral, striatal hemispheres. Both the reduction in N-cadherin and the infarct size (at 30 and 60 min) were similar in wild-type and mutant brain, consistent with a previous report demonstrating that CAG expansion did not sensitize to acute ischemic injury (23). In contrast, full-length wild-type 7-glutamine huntingtin and 111-glutamine mutant huntingtin cleaved by calpains in response to severe ischemia (24), α-catenin and β-catenin were not decreased, though p120 was slightly reduced by ischemia, attesting to the mildness of the lesion and demonstrating the exquisite sensitivity of N-cadherin in the striatum to ischemia.
Figure 1.

ATP-sensitive N-cadherin stability. (A) A representative coronal section of wild-type HdhQ7/Q7 mouse brain, following MCA occlusion/reperfusion, stained with 2,3,5-triphenyltetrazolium chloride (TTC), to demonstrate the infarct region (white area) in the left hemisphere (L), as well as the contralateral right hemisphere (R). The infarct sizes (mm3 and mean ± standard errors) were measured from mice brains of both genotypes at 30 and 60 min MCA occlusion as 33.9 ± 3.64 and 35.2 ± 3.55 for wild-type brain at 30 and 60 min and 41.5 ± 5.97 and 43.1 ± 6.81 for mutant brain and showed no significant differences by genotypes and times (n= 3), consistent with the previous report (23). (B) Immunoblot showing bands of N-cadherin, β-catenin, α-catenin and p120 detected in protein extracts of striata from the MCA-occluded hemisphere (L) and contralateral hemisphere (R) for wild-type HdhQ7/Q7 (Hdh7/7) and mutant HdhQ111/Q111 (Hdh111/111) mice, with 30 min MCA occlusion (30′) or 60 min MCA occlusion (60′), demonstrating decreased N-cadherin on the lesion side, relative to the α-tubulin loading control band. However, the reduction in N-cadherin was not statistically different between two genotypes or two occlusion times (n= 3). (C) Immunoblot showing bands of N-cadherin, β-catenin, α-catenin and p120 detected in protein extracts of wild-type STHdhQ7/Q7 (ST7/7) and mutant STHdhQ111/Q111 (ST111/111) cells, at time 0 (cont), and 0.5, 1 and 2 h in ATP-depletion medium. The adjacent graph plots the relative band intensity of N-cadherin normalized to α-tubulin in the same lane (y-axis), with the time in ATP-depleting medium (x-axis), illustrating decreased N-cadherin in mutant cells at baseline and ATP-sensitive degradation of N-cadherin in cells of both genotypes. Bars denote the standard deviation from three independent experiments. (D) Immunoblot showing N-cadherin in extracts of STHdhQ111/Q111 cells, after incubation in medium supplemented with 10 μm adenine for 0, 2, 4 and 6 days (D), relative to the α-tubulin band. The adjacent bar graph plots the relative N-cadherin intensities normalized to the α-tubulin band intensities (y-axis) for the time points analyzed (x-axis). Relative to the untreated control, N-cadherin levels were significantly elevated by day 4 (*P < 0.05, n= 3).
The immunoblot results also revealed reduced N-cadherin in contralateral HdhQ111/Q111 striata compared with wild-type striata, suggesting an effect of the CAG mutation. This was confirmed by immunoblot analysis of wild-type and HdhQ111/Q111 striatal tissues at different ages, which revealed a progressive decrease in N-cadherin (normalized to α-tubulin) from 3 to 5 months of age that reached statistical significance by 12 months of age (Supplementary Material, Fig. S1).
Notably, N-cadherin (Cdh2) mRNA levels were not reduced in total striatal tissue, following ischemic reperfusion or in response to the CAG mutation (data not shown), implying that cell-specific and/or multiple mechanisms may contribute to the energy-dependent N-cadherin decrease in vivo.
Striatal cell N-cadherin was sensitive to ATP depletion and the HD CAG mutation
We then assessed whether N-cadherin might be sensitive to ATP depletion and to the CAG mutation in cultured STHdhQ7/Q7 and STHdhQ111/Q111 striatal neuronal cells expressing wild-type (7-glutamine) and mutant (111-glutamine) full-length huntingtin, respectively. Cells were treated with 2-deoxyglucose and antimycin A, to inhibit both glycolysis and mitochondrial respiration, and nucleotides in cellular extracts were measured by HPLC analysis. As reported previously (7,8), the baseline [ATP/ADP] was significantly lower in mutant, compared with wild-type, striatal cells, and, for both genotypes, [ATP/ADP] was dramatically decreased within 2 h of energy depletion (Supplementary Material, Fig. S2A), although cell viability was not significantly changed (Supplementary Material, Fig. S2B). Immunoblot analysis revealed that N-cadherin was decreased at baseline in mutant, compared with wild-type, striatal cell extracts, with a progressive reduction over the time course of ATP depletion for both genotypes (Fig. 1C). In contrast, α-catenin, β-catenin and p120 levels were similar for cells of either genotype and were not changed by ATP depletion (Fig. 1C). Thus, the N-cadherin level was reduced concomitant with decreased [ATP/ADP], both due to purposeful energy depletion and in response to ATP deficit due to full-length mutant huntingtin.
Further analyses demonstrated that decreased N-cadherin in mutant, compared with wild-type, striatal cells reflected decreased Cdh2 mRNA, as demonstrated by the results of RT–PCR analysis (Supplementary Material, Fig. S3A), as well as the increased turnover rate of the protein, which had a half-life of ∼2 h in STHdhQ111/Q111 cells compared with a half-life of >4 h in wild-type cells (Supplementary Material, Fig. S3B). To explore the energy sensitivity of these measures, mutant striatal cell culture medium was supplemented with adenine, a precursor of high-energy nucleotides. By 2 days, [ATP/ADP] was mildly but significantly elevated (Supplementary Material, Fig. S2C), and by 4 days of treatment, N-cadherin was modestly but consistently increased (Fig. 1D), though Cdh2 mRNA levels were not altered over the entire 6 day time course (data not shown). These results suggested that enhanced N-cadherin protein turnover in the mutant striatal cells may involve an energy-sensitive process, while altered Cdh2 mRNA may reflect a different underlying process. However, assays to explore the involvement of metalloproteases, activated in ischemia, failed to disclose elevated MT-MMP, and MT-MMP inhibition with GM-6001 did not alter the half-life of N-cadherin in mutant striatal cells (data not shown).
STHdhQ111/Q111 cells exhibited deficits in N-cadherin-mediated cell–cell adhesion
The potential functional consequences of decreased N-cadherin in mutant striatal cells were then evaluated, beginning with confocal microscopy to investigate N-cadherin subcellular localization. Wild-type STHdhQ7/Q7 cells exhibited prominent N-cadherin immunostain at cell–cell contacts, whereas mutant striatal cells displayed a decreased N-cadherin signal that was not prominent at cell–cell contacts (Fig. 2A), strongly implying a defect in calcium-dependent N-cadherin-mediated cell–cell adhesion (25,26). To assess this possibility, wild-type STHdhQ7/Q7 and mutant STHdhQ111/Q111 cells were evaluated in a cell-clustering assay, which measured the time taken for detached single cells to form clusters (Fig. 2B). Within 30 min of dispersal to single cells, 70% of the wild-type cells, but only 30% of the mutant striatal cells, formed clusters in Ca2+-containing media, though cells of both genotypes exhibited similar Ca2+-independent (in EGTA) cell clustering. This phenotype was N-cadherin dependent, as exogenous expression of N-cadherin, after transfection, raised N-cadherin levels to ∼50% of wild-type levels (Supplementary Material, Fig. S3C) and partially rescued the mutant cell deficit in Ca2+-dependent cluster formation. At 30 min after being dispersed, ∼20% more N-cadherin-transfected mutant striatal cells formed clusters in Ca2+ media, compared with mutant cells transfected with the control vector (Fig. 2C). In the absence of Ca2+ (in EGTA), the transfection of N-cadherin had no effect on cell–cell adhesion.
Figure 2.

STHdhQ111/Q111 striatal cells exhibited decreased N-cadherin function. (A) Confocal images of the N-cadherin immunostain signal (red) in wild-type STHdhQ7/Q7 (ST7/7) and STHdhQ111/Q111 (ST111/111) striatal cells, indicating decreased N-cadherin signal at STHdhQ111/Q111cell–cell contacts. (B) Plot summarizing the results of three independent cell-clustering assays, showing the proportion of single cells compared with time zero (Nt/N0) (y-axis) relative to time of clustering (x-axis) for STHdhQ7/Q7 (square) and STHdhQ111/Q111 (circle) striatal cells, in the absence (open symbols) and presence (closed symbols) of Ca2+, illustrating that mutant striatal cells exhibit impaired calcium-dependent clustering. Error bars denote the standard deviation. (C) Plot summarizing the results of three independent STHdhQ111/Q111 cell-clustering assays for cells transfected with the phN-cad human N-cadherin expression vector (squares) or with the control vector (circles), performed in the absence (open symbols) and presence (closed symbols) of Ca2+. Error bars denote the standard deviation. (D) Fluorescence images of STHdhQ7/Q7 (ST7/7) and STHdhQ111/Q111 (ST111/111) striatal cells, showing the pattern of DAPI (blue) nuclei, TRITC–phalloidin (red) F-actin signal and anti-cofilin (green) immunostain, illustrating decreased nuclear size, paucity of actin stress fibers and bright perinuclear cofilin stain of the mutant cells, consistent with an elongated rounded-up (less flat) morphology. The adjacent bar graph shows the average phalloidin signal and the total or nuclear area cofilin intensities. The average intensity of the phalloidin signal was significantly reduced in mutant striatal cells (*P < 0.001) and the inset was taken at different exposure conditions to examine the pattern of the phalloidin signal in mutant cells. The average intensity of the total cofilin signal was not altered but its localization was shifted to the nuclear and perinuclear region (**P < 0.001). Error bars represent standard errors.
STHdhQ111/Q111 cells exhibited deficits in cell–substratum adhesion
N-cadherin is known to regulate cell–substratum adhesion (27) and actin cytoskeleton dynamics (28,29). Consequently, co-stained striatal cells were monitored by epifluorescence microscopy to detect DAPI-stained nuclei and filamentous actin (F-actin) cytoskeletal-binding proteins (Fig. 2D). Compared with wild-type STHdhQ7Q/7 cells, the mutant STHdhQ111/Q111 striatal cells appeared smaller with elongated shapes, and decreased stress fiber-like rhodamine–phalloidin signal, though in the inset the pattern of the phalloidin signal resembled that of wild-type cells (Fig. 2D). Consistent with the elongated (less flat) morphology, mutant striatal cells exhibited smaller DAPI-stained nuclei and the significant intense cofilin stain of the mutant cells was localized to the nuclear and perinuclear region, although quantification demonstrated that the total cofilin signal in mutant and wild-type cells was similar (Fig. 2D). The mutant striatal cells also exhibited a similar nuclear/perinuclear immunostaining pattern for profilin, another F-actin-binding protein, which, like cofilin, is involved in actin filament structure and dynamics (30) (data not shown). Notably, immunoblot revealed similar levels of F-actin, and actin monomer (G-actin), in extracts of mutant and wild-type striatal cells (data not shown). Thus, rather than lacking an actin cytoskeleton, the altered subcellular patterns of actin-associated proteins, along with decreased N-cadherin, were consistent with the elongated, less flat morphology of the striatal cells expressing full-length mutant huntingtin and strongly implied deficits in cell–substrate adhesion, as well as cell–cell adhesion.
HdhQ111/Q111 primary striatal neuronal cells exhibited decreased N-cadherin and immature neuritis
N-cadherin-mediated cell–substrate adhesion promotes neurite outgrowth (27) and N-cadherin is required for proper vesicle clustering essential for neurite formation and maturation (31–34). To evaluate the potential impact of full-length mutant huntingtin on N-cadherin function in developing striatal neurons, we examined primary neuronal cell cultures from the striatum of E14 control and HdhQ111/Q111 knock-in mouse embryos. At day 10 of differentiation (days in vitro, div10), F-actin rhodamine–phalloidin binding was assessed by confocal microscopy, as a surrogate for proper actin cytoskeleton/cell–substratum adhesion, and cultures were immunostained to detect MAP2, a neuronal cell microtubule-associated protein, to evaluate the developing neuronal cell projections. Compared with wild-type primary striatal neuronal cells, the primary HdhQ111/Q111 cells exhibited decreased rhodamine–phalloidin stain and an abnormally robust network of fine MAP2-stained projections, which confirmed neuronal cell differentiation, while implying altered adhesion and development (Fig. 3).
Figure 3.

Primary HdhQ111/Q111 striatal neurons exhibit altered cytoskeletal elements. Confocal images of primary HdhQ7/Q7 (Hdh7/7) and HdhQ111/Q111 (Hdh111/111) striatal cell cultures at 10 days (div10), showing the pattern of rhodamine–phalloidin F-actin signal (left) and anti-MAP2 microtubule immunostain (right), illustrating decreased phalloidin signal and an abundance of fine MAP2-positive projections of the mutant neuronal cells, consistent with impaired adhesion and altered neuronal cell development. The bar graph below plots the fold intensity of the signal in mutant cells relative to wild-type cells (*P < 0.001). Error bars represent standard errors.
Consistent with decreased N-cadherin function, co-staining revealed dramatically decreased N-cadherin signal, detected in β-III-tubulin-positive HdhQ111/Q111 striatal neuronal cells, compared with the intense signal of wild-type striatal cells (Fig. 4A), with only weak diffuse stain along the projections instead of the bright punctate pattern of wild-type cells (Fig. 4A inset, quantified in Supplementary Material, Fig. S4A and B). This observation implied that the synaptic vesicles that transport N-cadherin were not clustered, as expected of mature neurites, but instead were distributed along the processes in the diffuse pattern characteristic of immature neurons (35).
Figure 4.

Primary HdhQ111/Q111 striatal neurons displayed decreased N-cadherin signal and abnormally extensive neurite networks. (A) Confocal images of primary HdhQ7/Q7 (Hdh7/7) and HdhQ111/Q111 (Hdh111/111) striatal cell cultures, at 10 days (div10), showing the pattern of N-cadherin immunostain (green), β-III-tubulin microtuble immunostain (red) and DAPI-signal marking nuclei (blue), with an overlay of all layers (merge), showing in mutant cells decreased N-cadherin signal intensity and paucity of bright puncta (inset), concomitant with an extensive network of β-III-tubulin-positive extensions (neurites). (B) Bar graph plotting the total β-III-tubulin-positive neurite length per cell, which was significantly increased in the div10 primary HdhQ111/Q111 (Hdh111/111) striatal neuronal cells compared with the wild-type HdhQ7/Q7 (Hdh7/7) cells (*P < 0.005). Error bars represent standard errors. (C) As a consequence of HD mutation, several markers of synaptic development showed decreased signal intensity, such as N-cadherin (green) and presynaptic markers, synapsin 1 (red in the left column) and synaptophysin (red in the right column). Along with a decrease in the markers for synaptic development in the HdhQ111/Q111 neurons, there was also a diffuse staining pattern in contrast to a punctuated pattern in the wild-type processes.
To explore the potential developmental deficits, β-III-tubulin-positive neuronal cell processes (here called neurites) of div10 primary neuronal cells were quantified. Automated image analysis revealed that mutant cells exhibited a 3-fold increase in total length of β-III-tubulin-positive neurites, compared with wild-type striatal neuronal cells (Fig. 4B, quantification in Supplementary Material, Fig. S4C), implying delayed maturation of the neurite network. Moreover, co-staining to detect N-cadherin and markers of synaptic vesicles, synapsin and synaptophysin, confirmed this interpretation. The decreased N-cadherin signal, as well as the synapsin and synaptophysin vesicle signals, was distributed diffusely within the processes of the mutant HdhQ111/Q111 striatal neuronal cells, rather than assuming the bright punctuate matured appearance of these markers in wild-type HdhQ7/Q7 striatal cells (Fig. 4C). This immature pattern, which prominently featured decreased N-cadherin (35), was consistent with decreased N-cadherin function and delayed maturation of the network of developing neurites and synapses elaborated by the mutant primary neuronal cells.
HdhQ111/Q111 primary striatal developmental deficit was partially rescued with adenine
As adenine treatment of mutant STHdhQ111/Q111-immortalized striatal cells partially elevated [ATP/ADP] (Supplementary Material, Fig. S2C) and partially normalized the N-cadherin level (Fig. 1D), we determined whether adenine treatment might modulate the propensity of primary div10 E14 HdhQ111/Q111 striatal neurons to form abnormally extensive neurite networks. As shown in Figure 5, after treatment with adenine for 10 days, the div10 HdhQ111/Q111 striatal cultures exhibited MAP2-positive neuronal cells with fewer neurites per cell body compared with DMSO vehicle-treated cultures, though the number of cell bodies was not appreciably altered, as shown by DAPI nuclear staining. Consistent with the ameliorative effect of adenine on energy measures and N-cadherin levels demonstrated in STHdhQ111/Q111 cells, these findings suggest that the delayed development of mutant primary neuronal cells may, at least in part, reflect decreased function of the ATP-sensitive N-cadherin adhesion molecule.
Figure 5.

Adenine ameliorated altered HdhQ111/Q111 primary striatal neuron neuritogenesis. Confocal images of primary HdhQ111/Q111 striatal cell cultures at 10 days (div10), after culture in media supplemented with vehicle (DMSO) or with adenine (Adenine), showing MAP2-immunostained microtubules (red) and DAPI-stained nuclei (blue) and overlapped images (merge), illustrating that adenine-treated neuronal cells displayed a less branched neurite network than vehicle-treated cells. The bar graph below shows that adenine supplementation significantly decreased the total length of neurites/cell by 25% (*P = 0.001), while a similar number of DAPI-stained nuclei confirmed that adenine did not result in toxicity or loss of cells. Error bars represent standard errors.
DISCUSSION
HD is a progressive neurodegenerative disorder, with marked loss of the major population of neurons in the striatum (medium-sized spiny neurons), which are vulnerable to acute energetic challenge. The neuronal specificity of HD stems from the effects of the HD CAG repeat, encoding a polyglutamine repeat in the full-length huntingtin protein of more than ∼37 residues. We and others have discovered that the polyglutamine repeat modulates the, as yet unknown, role of full-length huntingtin in negatively regulating measures of energy metabolism in human cells and in genetically accurate CAG knock-in mice and striatal cells (7,8,36). In the current study, in order to guide in vivo investigations of striatal cell vulnerability, we have explored the hypothesis that chronically decreased energetics, due to endogenous full-length mutant huntingtin, may affect critical aspects of the biology of cultured striatal neuronal cells.
In a candidate approach, we have evaluated N-cadherin, an integrator of adhesion and cytoskeletal signaling required for proper neuronal cell development and synaptic function. N-cadherin was sensitive to acute ATP depletion, in striatal tissue and cultured striatal neuronal cells, and was strikingly sensitive to the effects of full-length mutant huntingtin. N-cadherin was progressively reduced with age in HdhQ111 knock-in mouse striatum and, consistent with a defect from birth, was dramatically decreased both in cultured STHdhQ111 immortalized neuronal cells, generated from embryonic HdhQ111 striatal primordia (37), and in primary cultures of embryonic HdhQ111 striatal neurons.
The molecular basis of decreased N-cadherin in response to full-length mutant huntingtin is not yet clear, but in STHdhQ111 striatal cells appears to involve regulation of Cdh2 mRNA, as well as N-cadherin protein stability. Chronic energy deficit appears to contribute to the latter, but not the former, as adenine nucleotide was able to elevate both [ATP/ADP] and N-cadherin levels, and the rate of N-cadherin turnover was increased under ATP-depletion conditions, while these manipulations did not affect Cdh2 mRNA levels, which instead may reflect a role for full-length huntingtin in mediating some aspect of Cdh2 gene expression or mRNA stability. Enhanced N-cadherin turnover in STHdhQ111 striatal cells did not appear to involve MMP-1, which mediates ischemia-induced turnover of cadherin (15,16). MMP-1 levels were not increased in mutant STHdhQ111 striatal cells and N-cadherin turnover was unaffected by GM-6001, a specific MMP inhibitor (data not shown). Rather, elevated N-cadherin instability may entail altered GTPase-regulated N-cadherin phosphorylation (38,39), consistent with decreased STHdhQ111 striatal cell GTP and [GTP/GDP], as well as ATP and [ATP/ADP] (8). However, other possibilities include: altered calcium-dependent ligand or catenin binding and internalization, consistent with full-length huntingtin/syntaxin 1A regulation of N-type calcium channels (40), impaired N-cadherin membrane association due to altered cholesterol metabolism (41,42) or abnormal N-cadherin precursor processing, trafficking and/or glycosylation, as implied by the findings that STHdhQ111 and primary HdhQ111 neuronal cells demonstrated a paucity of clustered N-cadherin and synaptic marker-positive vesicles. Certainly, STHdhQ111 striatal cells display diverse membrane trafficking defects, involving endocytic vesicles (43,44), autophagic vacuole cargo engulfment (45) and the ER/Golgi network (37), attesting to the impact of the polyglutamine repeat on full-length huntingtin function in regulating membrane trafficking (37,43,46).
N-cadherin-mediated calcium-dependent adhesion/cytoskeletal organization and signaling are especially needed for normal neuronal cell development and functionality, for example to achieve proper neurite outgrowth, synaptic vesicle clustering in maturing neurons, and synapse formation, maturation and dynamics (27,29,47–49). Mutant striatal cells exhibited impaired cell–cell, as well as cell–substrate adhesion, deficits in N-cadherin, synaptophysin and synapsin vesicle clustering and immature neurite networks, strongly suggesting delayed developmental maturation, though this interpretation remains to be tested. In part, the chronic energetic deficit contributed to impaired N-cadherin function, perhaps via decreased N-cadherin half-life. Adenine, which partially normalized STHdhQ111 striatal cell ATP and N-cadherin levels, significantly rescued primary HdhQ111 striatal cell neurite development, consistent with the hypothesis that the energetic state may be a prominent factor in determining N-cadherin levels and function in cells expressing full-length mutant huntingtin.
HD is typically assumed to be due to a disease process that begins later in life. However, our observation of decreased N-cadherin and delayed development of embryonic HdhQ111 striatal neurons in vitro offers a possible explanation for impaired development of the striatum in E13.5–15.5 HdhQ111 embryos (50), and for decreased measures of brain neurodevelopment in humans with expanded CAG repeats (51), which demonstrate effects of the HD CAG repeat that become manifest even before birth. Later in life, N-cadherin progressively decreased with age in HdhQ111 striatum, implying chronic synaptic dysfunction, as N-cadherin orchestrates activity-modulated CNS synapses (35). Indeed, decreased N-cadherin may contribute to impaired actin polymerization and long-term potentiation detected in HdhQ92 mice (52,53).
Accumulated evidence now supports CAG-dependent energetic deficits in human cells and tissues, before onset of overt clinical symptoms, as well as in the brains of symptomatic individuals (7,10,54–56). Therefore, it will be important to determine whether decreased levels of N-cadherin may contribute to the HD pathogenic process, in a manner that is specific to striatal neurons, thereby contributing to the early vulnerability of the striatum, compared with other brain regions. Furthermore, it will be of interest to determine whether subtly altered development may sensitize striatal cells, and perhaps other neuronal cells, to the disease process or may represent a rate-limiting step in the disease process that is initiated by the impact of the polyglutamine repeat on full-length huntingtin.
MATERIALS AND METHODS
Mice and striatal neuronal cell cultures
HdhQ111/Q111 knock-in mice have been described previously (57). Striata were dissected from genotyped homozygous mutant HdhQ111/Q111 and wild-type HdhQ7/Q7 littermates from heterozygous HdhQ111/Q7 matings, at various ages. Conditionally immortalized wild-type STHdhQ7/Q7 striatal cells and homozygous mutant STHdhQ111/Q111 striatal cells, generated from HdhQ111/Q111 and HdhQ7/Q7 littermate embryos, were described previously (37). The striatal cells were grown at 33°C in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum, 1% non-essential amino acids, 2 mm l-glutamine and 400 μg/ml G418 (Geneticin; Invitrogen). Primary neuronal cell cultures were set up from striata dissected from E14 wild-type HdhQ7/Q7 and mutant HdhQ111/Q111 embryos, from heterozygous HdhQ111/Q7 matings. The striata were dissociated with trypsin (0.5%), trypsin inhibitor (0.1%) and DNase (228 U/ml) treatments for 1 min each in a 37°C warm water bath. Dissociated cells were plated onto poly-l-lysine (0.1 mg/ml) and laminin (50 µg/ml) (Sigma) double-coated coverslips in neurobasal medium (GIBCO) supplemented with 2% B27 and 1% penicillin/streptomycin/gentamycin. Cells were cultured for 10 days (div10) at 37°C and 5% CO2.
Focal ischemia
All focal ischemia experiments were conducted in accordance with the National Institutes of Health and Massachusetts General Hospital institutional guidelines on animal experimentation. Three wild-type HdhQ7/Q7 and three HdhQ111/Q111 mice (12 months of age) were anesthetized with 2% halothane in 70% N2O and 30% O2, and then maintained on 1% halothane in a similar gaseous mixture. Transient focal cerebral ischemia was performed using an 8-0 nylon monofilament coated with silicone, which was introduced into the internal carotid artery via the external carotid artery and then advanced 10 mm distal to the carotid bifurcation to occlude the MCA as described (58). Laser Doppler flowmetry (PF2B; Perimed Stockholm) of relative cerebral blood fluid (CBF) was used to verify successful occlusion (<20% baseline value). The MCA was occluded for 0.5 and 1 h followed by withdrawal of filament and reperfusion for 24 h. Relative CBF returned to >95% of baseline values, indicating almost complete reperfusion without residual occlusion. After reperfusion, right (control hemisphere) and left (infarct hemisphere) striata from each mouse brain were dissected for tissue extraction and immunoblot analysis.
Immunoblot analysis
Whole-cell protein extracts were prepared from harvested striata and striatal cells by lysis on ice for 30 min in a buffer containing 20 mm HEPES (pH 7.6), 1 mm EDTA, 0.5% Triton X-100, protease inhibitor mixture (Roche, Indianapolis, IN, USA) and 1 mm phenylmethyl sulfonyl fluoride (PMSF), mixed by tapping every 10 min. The total lysates were cleared by centrifugation at 14 000g for 30 min and the supernatants were collected. Protein concentration was determined by the Bio-Rad (detergent compatible) protein assay. An amount of 25 μg of protein extract was mixed with 4× SDS sample buffer, boiled for 2 min and subjected to 6 or 10% SDS–PAGE. After electrophoresis, the proteins were transferred to nitrocellulose membranes (Schleicher & Schuell) and incubated for 30 min in blocking solution containing 5% non-fat powdered milk in TBS-T (50 mm Tris–HCl, 150 mm NaCl, pH 7.4, 0.1% Tween 20). The blots were probed overnight at 4°C with primary antibodies: MAB2166 for huntingtin (Chemicon), N-cadherin (BD-bioscience), α-catenin (Chemicon), β-catenin (Sigma), p120 (Sigma) and α-tubulin (Sigma). After four washes of 10 min each in TBS-T, the blots were incubated for 1 h at room temperature with horseradish peroxidase-conjugated anti-mouse or anti-rabbit antisera. After a 30 min wash, the membranes were processed using an ECL chemiluminescence substrate kit (New England Biolabs, Beverly, MA, USA) and exposed to autoradiographic film (Hyperfilm ECL; Amersham Bioscience). Quantification of the immunoreactive bands was performed by scanning and analysis using the GS-800 Calibrated Densitometer and the Quantity One software (BioRad, Hercules, CA, USA).
Immunocytochemistry
Wild-type STHdhQ7/Q7 and homozygous mutant STHdhQ111/Q111 cells were grown on 4-chamber glass slides at a density of 2 × 105 cells/well. The cells were fixed in 4% paraformaldehyde for 20 min, permeabilized for 5 min in 0.1% Triton X-100 in phosphate-buffered saline (PBS), treated for 30 min in blocking buffer [2% bovine serum albumin (BSA) in PBS] and incubated for 2 h in blocking solution containing anti-N-cadherin. After several washes in PBS (3 × 5 min), cells were incubated for 1 h in blocking solution containing the anti-mouse fluorescent secondary antibody. Cells were imaged with a laser confocal microscope (Leica) using a 60× oil objective. Primary cells were fixed with 4% formaldehyde for 10 min, followed by 20 min of 100% MeOH for permeabilization. Single or double incubation of neurons with various primary antibodies was performed overnight at 4°C. The primary antibodies in this study were N-cadherin antibody (Abcam; 1:200), β-III-tubulin (Chemicon; 1:500), Cofilin (Sigma: 1:1000), Synapsin-1 (Synaptic Systems: 1:1000), Synaptophysin-1 (Synaptic Systems 1:200) and MAP2 (EnCor Biotechnology Inc: 1:10 000). For visualization, 1 h treatment with Alexa-488 and Alexa-568 secondary antibodies (both Molecular Probes, 1:500) was performed. The Phalloidin–TRITC (Sigma: 1:1000) signal was visualized without the secondary antibody. Coverslips were fixed onto object glasses using ProLong Gold antifade reagent containing DAPI (Invitrogen) for subsequent laser confocal microscopy (Leica) or epifluorescence microscopy (Zeiss).
Cell-clustering assay
Sub confluent striatal cell cultures were incubated for 10 min in cold PBS and the cells were then collected with a scraper. After washing twice with PBS containing 10 mm HEPES–NaOH (pH 7.4), cells were resuspended in PBS containing 10 mm HEPES–NaOH (pH 7.4), 1 mg/ml BSA, 1 mm EGTA and maintained at 4°C. Before the start of clustering assays, cells were carefully resuspended to ensure single-cell suspensions, and viability of cells (>90%) was confirmed by trypan blue exclusion. Clustering assays, which were performed at 33°C with rotation in non-tissue culture 24-well Falcon dishes to prevent cell–dish attachment, were started by addition of 50 μl of cell suspension (5 × 106 cells/ml) to 500 μl of pre-warmed (33°C) PBS containing 1 mg/ml BSA, 10 mm HEPES–NaOH (pH 7.4) and either 2 mm CaCl2 or 2 mm EGTA. Incubations were terminated by addition of 500 μl of 5% glutaraldehyde in PBS and particle numbers were determined on a Coulter Counter Model Z2 (Beckman Coulter, Fullerton, CA, USA). Clustering is expressed as the fractional loss of particle number, Nt/N0, where N0 is the particle number at time 0 and Nt is the particle number after any given time point.
RNA extraction and quantitative RT–PCR
Harvested striatal cells or dissected striata were extracted with TRIzol reagent (Invitrogen) to isolate total RNA according to the manufacturer's instructions. cDNA was synthesized using Oligo(dT)15 primer and Reverse Transcription System (Promega) according to the manufacturer's instructions. Amplification by PCR was performed with 10 μl aliquots of cDNA in a total volume of 50 μl using iQTM SYBR Green Supermix (Bio Rad) with a Bio-Rad iCycler (Hercules, CA, USA). Expression of Cdh2 was specifically detected by using two primers: Cdh2 forward, 5′-AGAGGCCTATCCATGCTGAG-3′ and Cdh2 reverse, 5′-AGCAGCTTTAAGGCCCTCAT-3′. The thermocycling program used began with incubation at 95°C for 1 min, followed by 30 cycles of 95°C for 15 s, 56°C for 20 s and 72°C for 15 s. A primer set of β-actin, forward, 5′-GACGGCCAGGTCATCACTAT-3′, reverse, 5′-ATGCCACAGGATTCCATACC-3′, was used as a positive control to ensure the integrity and quantity of RNA. PCR amplification was performed for β-actin under the following conditions: 95°C for 1 min; 30 cycles of 95°C for 15 s, 54°C for 20 s and 72°C for 20 s. The ΔΔCT method was used to calculate gene expression levels from quantitative RT–PCR (59).
N-cadherin half-life
STHdhQ7/Q7 and STHdhQ111/Q111 cells plated on 6-well dishes were incubated in growth medium containing cycloheximide at a final concentration of 30 μg/ml for 0, 1, 2 and 4 h. At each time point, the cells were washed once with ice-cold PBS and lysed by incubation for 30 min in a buffer containing 20 mm HEPES (pH 7.6), 1 mm EDTA, 0.5% Triton X-100, protease inhibitor mixture (Roche, Indianapolis, IN, USA) and 1 mm PMSF, followed by tapping every 10 min. The total lysates were then cleared by centrifugation at 14 000g for 30 min and the supernatants were collected. The protein concentration was determined by the Bio-Rad (detergent compatible) protein assay and equal amounts of protein from each lysate were resolved by 10% SDS–PAGE. The proteins were transferred to nitrocellulose membranes, blocked in 5% non-fat milk TBS-T and incubated overnight at 4°C with a monoclonal anti N-cadherin antibody. The immunoblot was then probed with horseradish peroxidase-conjugated secondary antibody and visualized by ECL reagents.
ATP depletion
STHdhQ7/Q7 and STHdhQ111/Q111 cells plated on 6-well dishes were washed twice with PBS and then were incubated at 33°C in either normal growth medium (control cells) or glucose-free DMEM containing 10 mm 2-deoxyglucose and 10 μm antimycin A to yield ATP-depleted cells. After incubation for 0.5, 1 and 2 h, cells were collected with a scraper and extracts were generated for immunoblot analysis.
After ATP depletion, viability was assessed by the MTS assay kit (Promega, Madison, WI, USA). Prior to the assay, cells were washed three times with PBS. MTS [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt] solution was added to each well, followed by incubation at 33°C for 2 h. Absorbance was measured at 490 nm on a multi-well spectrophotometer (Molecular Devices). For measuring altered ATP nucleotide in cells after ATP depletion, HPLC analysis was performed as described previously (7).
Image analysis of primary cultures
Quantification of immunostain was performed using ImageJ, image analysis software available through the NIH website (http://rsb.info.nih.gov/ij/). To determine the area for cells (cell bodies and neurites separately), the number of β-III-positive pixels were counted within a field of view. For cell bodies, β-III-tubulin staining defined the edges of the cell body, so that the total area of the cell body could be determined. Four different fields per genotype were analyzed. For quantification of N-cadherin, intensity histograms of N-cadherin in β-III-tubulin-defined neurites and cell bodies were measured. To obtain the amounts of the N-cadherin signal for a specific genotype and the respective cell part, the products of each intensity over a threshold value multiplied by the percentage of the area covered by that intensity were added up 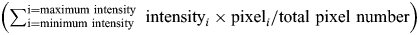 . To compare the four different amounts with one another (wild-type cell body, wild-type neurites, mutant cell body and mutant cell neurites), the signal amount in mutant neurites was defined as one, and the other three signal amounts were expressed as a ratio. The number of N-cadherin signal particles was determined by using the Analyze Particle plug-in of ImageJ. Thirty images of neurites per genotype were analyzed. Images were thresholded six times for different signal intensity intervals (intensity 30–90; 40–90, 50–90, etc., until 80–90). For each thresholded image, the number of particles was calculated (i.e. I30–90, I40–90, I50–90, …,I80–90). To obtain the number of particles with a specific highest intensity (40, 50, …, 90), the number of particles within one intensity interval was subtracted from the one of the next lower interval (i.e. N30–40 = I30–90 – I40–90). The number of particles was also normalized to the length of neurites. In order to determine the total neurite length, in four fields of view per genotype of β-III-tubulin images, the cell bodies were removed. The resulting images were skeletonized and the length of the skeleton was measured. Subsequently, this number was divided by the number of DAPI-positive nuclei in the appropriate field.
. To compare the four different amounts with one another (wild-type cell body, wild-type neurites, mutant cell body and mutant cell neurites), the signal amount in mutant neurites was defined as one, and the other three signal amounts were expressed as a ratio. The number of N-cadherin signal particles was determined by using the Analyze Particle plug-in of ImageJ. Thirty images of neurites per genotype were analyzed. Images were thresholded six times for different signal intensity intervals (intensity 30–90; 40–90, 50–90, etc., until 80–90). For each thresholded image, the number of particles was calculated (i.e. I30–90, I40–90, I50–90, …,I80–90). To obtain the number of particles with a specific highest intensity (40, 50, …, 90), the number of particles within one intensity interval was subtracted from the one of the next lower interval (i.e. N30–40 = I30–90 – I40–90). The number of particles was also normalized to the length of neurites. In order to determine the total neurite length, in four fields of view per genotype of β-III-tubulin images, the cell bodies were removed. The resulting images were skeletonized and the length of the skeleton was measured. Subsequently, this number was divided by the number of DAPI-positive nuclei in the appropriate field.
Statistical analysis
All cell images were quantified in 10 randomly chosen groups comprising at least 100 cells and other experiments, such as immunoblot and [ATP/ADP] measurement, in three independent experiments. The mean, standard deviation (SD) and standard error (SE) were calculated and statistical significance analyzed using an unpaired two-sample t-test.
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported by the National Institute of Neurological Disorders and Stroke (NS32765 to M.E.M., NS070001 to J.K.L.); the Massachusetts HD Center Without Walls [NS16367 (Project 3) to M.E.M.]; the Huntington's Disease Society of America Coalition for the Cure Normal Function Team to M.E.M.; the de Gunzburg Family Foundation at Massachusetts General Hospital to S.Y.S.; National Scientist Develoment Grant of the American Heart Association (0930202N to H.-H.K.); and an anonymous donor.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Dr Anastasios Georgakopoulos from Mount Sinai School of Medicine for a gift of phN-cadherin vector.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Vonsattel J.P., DiFiglia M. Huntington disease. J. Neuropathol. Exp. Neurol. 1998;57:369–384. doi: 10.1097/00005072-199805000-00001. doi:10.1097/00005072-199805000-00001. [DOI] [PubMed] [Google Scholar]
- 2.The Huntington's Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. doi:10.1016/0092-8674(93)90585-E. [DOI] [PubMed] [Google Scholar]
- 3.Gauthier L.R., Charrin B.C., Borrell-Pages M., Dompierre J.P., Rangone H., Cordelieres F.P., De Mey J., MacDonald M.E., Lessmann V., Humbert S., et al. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell. 2004;118:127–138. doi: 10.1016/j.cell.2004.06.018. doi:10.1016/j.cell.2004.06.018. [DOI] [PubMed] [Google Scholar]
- 4.Reddy P.H., Mao P., Manczak M. Mitochondrial structural and functional dynamics in Huntington's disease. Brain Res. Rev. 2009;61:33–48. doi: 10.1016/j.brainresrev.2009.04.001. doi:10.1016/j.brainresrev.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Truant R., Atwal R., Burtnik A. Hypothesis: huntingtin may function in membrane association and vesicular trafficking. Biochem. Cell Biol. 2006;84:912–917. doi: 10.1139/o06-181. doi:10.1139/O06-181. [DOI] [PubMed] [Google Scholar]
- 6.Carnevale F.A., Macdonald M.E., Bluebond-Langner M., McKeever P. Using participant observation in pediatric health care settings: ethical challenges and solutions. J. Child Health Care. 2008;12:18–32. doi: 10.1177/1367493507085616. doi:10.1177/1367493507085616. [DOI] [PubMed] [Google Scholar]
- 7.Seong I.S., Ivanova E., Lee J.M., Choo Y.S., Fossale E., Anderson M., Gusella J.F., Laramie J.M., Myers R.H., Lesort M., et al. HD CAG repeat implicates a dominant property of huntingtin in mitochondrial energy metabolism. Hum. Mol. Genet. 2005;14:2871–2880. doi: 10.1093/hmg/ddi319. doi:10.1093/hmg/ddi319. [DOI] [PubMed] [Google Scholar]
- 8.Gines S., Seong I.S., Fossale E., Ivanova E., Trettel F., Gusella J.F., Wheeler V.C., Persichetti F., MacDonald M.E. Specific progressive cAMP reduction implicates energy deficit in presymptomatic Huntington's disease knock-in mice. Hum. Mol. Genet. 2003;12:497–508. doi: 10.1093/hmg/ddg046. doi:10.1093/hmg/ddg046. [DOI] [PubMed] [Google Scholar]
- 9.Lee J.M., Ivanova E.V., Seong I.S., Cashorali T., Kohane I., Gusella J.F., MacDonald M.E. Unbiased gene expression analysis implicates the huntingtin polyglutamine tract in extra-mitochondrial energy metabolism. PLoS Genet. 2007;3:e135. doi: 10.1371/journal.pgen.0030135. doi:10.1371/journal.pgen.0030135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mochel F., Charles P., Seguin F., Barritault J., Coussieu C., Perin L., Le Bouc Y., Gervais C., Carcelain G., Vassault A., et al. Early energy deficit in Huntington disease: identification of a plasma biomarker traceable during disease progression. PLoS One. 2007;2:e647. doi: 10.1371/journal.pone.0000647. doi:10.1371/journal.pone.0000647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Benson D.L., Colman D.R., Huntley G.W. Molecules, maps and synapse specificity. Nat. Rev. Neurosci. 2001;2:899–909. doi: 10.1038/35104078. doi:10.1038/35104078. [DOI] [PubMed] [Google Scholar]
- 12.Takeichi M. The cadherin superfamily in neuronal connections and interactions. Nat. Rev. Neurosci. 2007;8:11–20. doi: 10.1038/nrn2043. doi:10.1038/nrn2043. [DOI] [PubMed] [Google Scholar]
- 13.Mandel L.J., Doctor R.B., Bacallao R. ATP depletion: a novel method to study junctional properties in epithelial tissues. II. Internalization of Na+,K(+)-ATPase and E-cadherin. J. Cell Sci. 1994;107:3315–3324. doi: 10.1242/jcs.107.12.3315. [DOI] [PubMed] [Google Scholar]
- 14.Bush K.T., Tsukamoto T., Nigam S.K. Selective degradation of E-cadherin and dissolution of E-cadherin-catenin complexes in epithelial ischemia. Am. J. Physiol. Renal Physiol. 2000;278:F847–852. doi: 10.1152/ajprenal.2000.278.5.F847. : [DOI] [PubMed] [Google Scholar]
- 15.Covington M.D., Bayless K.J., Burghardt R.C., Davis G.E., Parrish A.R. Ischemia-induced cleavage of cadherins in NRK cells: evidence for a role of metalloproteinases. Am. J. Physiol. Renal Physiol. 2005;289:F280–288. doi: 10.1152/ajprenal.00351.2004. doi:10.1152/ajprenal.00351.2004. [DOI] [PubMed] [Google Scholar]
- 16.Covington M.D., Burghardt R.C., Parrish A.R. Ischemia-induced cleavage of cadherins in NRK cells requires MT1-MMP (MMP-14) Am. J. Physiol. Renal Physiol. 2006;290:F43–51. doi: 10.1152/ajprenal.00179.2005. doi:10.1152/ajprenal.00179.2005. [DOI] [PubMed] [Google Scholar]
- 17.Hatta K., Nose A., Nagafuchi A., Takeichi M. Cloning and expression of cDNA encoding a neural calcium-dependent cell adhesion molecule: its identity in the cadherin gene family. J. Cell Biol. 1988;106:873–881. doi: 10.1083/jcb.106.3.873. doi:10.1083/jcb.106.3.873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Takeichi M. Cadherins: a molecular family important in selective cell–cell adhesion. Annu. Rev. Biochem. 1990;59:237–252. doi: 10.1146/annurev.bi.59.070190.001321. doi:10.1146/annurev.bi.59.070190.001321. [DOI] [PubMed] [Google Scholar]
- 19.Aberle H., Butz S., Stappert J., Weissig H., Kemler R., Hoschuetzky H. Assembly of the cadherin–catenin complex in vitro with recombinant proteins. J. Cell Sci. 1994;107:3655–3663. doi: 10.1242/jcs.107.12.3655. [DOI] [PubMed] [Google Scholar]
- 20.Knudsen K.A., Soler A.P., Johnson K.R., Wheelock M.J. Interaction of alpha-actinin with the cadherin/catenin cell–cell adhesion complex via alpha-catenin. J. Cell Biol. 1995;130:67–77. doi: 10.1083/jcb.130.1.67. doi:10.1083/jcb.130.1.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baki L., Marambaud P., Efthimiopoulos S., Georgakopoulos A., Wen P., Cui W., Shioi J., Koo E., Ozawa M., Friedrich V.L., Jr, et al. Presenilin-1 binds cytoplasmic epithelial cadherin, inhibits cadherin/p120 association, and regulates stability and function of the cadherin/catenin adhesion complex. Proc. Natl Acad. Sci. USA. 2001;98:2381–2386. doi: 10.1073/pnas.041603398. doi:10.1073/pnas.041603398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gumbiner B.M. Regulation of cadherin adhesive activity. J. Cell Biol. 2000;148:399–404. doi: 10.1083/jcb.148.3.399. doi:10.1083/jcb.148.3.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Namura S., Hirt L., Wheeler V.C., McGinnis K.M., Hilditch-Maguire P., Moskowitz M.A., MacDonald M.E., Persichetti F. The HD mutation does not alter neuronal death in the striatum of Hdh(Q92) knock-in mice after mild focal ischemia. Neurobiol. Dis. 2002;11:147–154. doi: 10.1006/nbdi.2002.0532. doi:10.1006/nbdi.2002.0532. [DOI] [PubMed] [Google Scholar]
- 24.Kim M., Roh J.K., Yoon B.W., Kang L., Kim Y.J., Aronin N., DiFiglia M. Huntingtin is degraded to small fragments by calpain after ischemic injury. Exp. Neurol. 2003;183:109–115. doi: 10.1016/s0014-4886(03)00132-8. doi:10.1016/S0014-4886(03)00132-8. [DOI] [PubMed] [Google Scholar]
- 25.Peyrieras N., Hyafil F., Louvard D., Ploegh H.L., Jacob F. Uvomorulin: a nonintegral membrane protein of early mouse embryo. Proc. Natl Acad. Sci. USA. 1983;80:6274–6277. doi: 10.1073/pnas.80.20.6274. doi:10.1073/pnas.80.20.6274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yoshida C., Takeichi M. Teratocarcinoma cell adhesion: identification of a cell-surface protein involved in calcium-dependent cell aggregation. Cell. 1982;28:217–224. doi: 10.1016/0092-8674(82)90339-7. doi:10.1016/0092-8674(82)90339-7. [DOI] [PubMed] [Google Scholar]
- 27.Bixby J.L., Zhang R. Purified N-cadherin is a potent substrate for the rapid induction of neurite outgrowth. J. Cell Biol. 1990;110:1253–1260. doi: 10.1083/jcb.110.4.1253. doi:10.1083/jcb.110.4.1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Goodwin M., Yap A.S. Classical cadherin adhesion molecules: coordinating cell adhesion, signaling and the cytoskeleton. J. Mol. Histol. 2004;35:839–844. doi: 10.1007/s10735-004-1833-2. doi:10.1007/s10735-004-1833-2. [DOI] [PubMed] [Google Scholar]
- 29.Bamji S.X. Cadherins: actin with the cytoskeleton to form synapses. Neuron. 2005;47:175–178. doi: 10.1016/j.neuron.2005.06.024. doi:10.1016/j.neuron.2005.06.024. [DOI] [PubMed] [Google Scholar]
- 30.Muhlrad A., Ringel I., Pavlov D., Peyser Y.M., Reisler E. Antagonistic effects of cofilin, beryllium fluoride complex, and phalloidin on subdomain 2 and nucleotide-binding cleft in F-actin. Biophys. J. 2006;91:4490–4499. doi: 10.1529/biophysj.106.087767. doi:10.1529/biophysj.106.087767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Regalado M.P., Terry-Lorenzo R.T., Waites C.L., Garner C.C., Malenka R.C. Transsynaptic signaling by postsynaptic synapse-associated protein 97. J. Neurosci. 2006;26:2343–2357. doi: 10.1523/JNEUROSCI.5247-05.2006. doi:10.1523/JNEUROSCI.5247-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Togashi H., Abe K., Mizoguchi A., Takaoka K., Chisaka O., Takeichi M. Cadherin regulates dendritic spine morphogenesis. Neuron. 2002;35:77–89. doi: 10.1016/s0896-6273(02)00748-1. doi:10.1016/S0896-6273(02)00748-1. [DOI] [PubMed] [Google Scholar]
- 33.Bamji S.X., Shimazu K., Kimes N., Huelsken J., Birchmeier W., Lu B., Reichardt L.F. Role of beta-catenin in synaptic vesicle localization and presynaptic assembly. Neuron. 2003;40:719–731. doi: 10.1016/s0896-6273(03)00718-9. doi:10.1016/S0896-6273(03)00718-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bozdagi O., Valcin M., Poskanzer K., Tanaka H., Benson D.L. Temporally distinct demands for classic cadherins in synapse formation and maturation. Mol. Cell Neurosci. 2004;27:509–521. doi: 10.1016/j.mcn.2004.08.008. doi:10.1016/j.mcn.2004.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Garner C.C., Waites C.L., Ziv N.E. Synapse development: still looking for the forest, still lost in the trees. Cell Tissue Res. 2006;326:249–262. doi: 10.1007/s00441-006-0278-1. doi:10.1007/s00441-006-0278-1. [DOI] [PubMed] [Google Scholar]
- 36.Clabough E.B., Zeitlin S.O. Deletion of the triplet repeat encoding polyglutamine within the mouse Huntington's disease gene results in subtle behavioral/motor phenotypes in vivo and elevated levels of ATP with cellular senescence in vitro. Hum. Mol. Genet. 2006;15:607–623. doi: 10.1093/hmg/ddi477. doi:10.1093/hmg/ddi477. [DOI] [PubMed] [Google Scholar]
- 37.Trettel F., Rigamonti D., Hilditch-Maguire P., Wheeler V.C., Sharp A.H., Persichetti F., Cattaneo E., MacDonald M.E. Dominant phenotypes produced by the HD mutation in STHdh(Q111) striatal cells. Hum. Mol. Genet. 2000;9:2799–2809. doi: 10.1093/hmg/9.19.2799. doi:10.1093/hmg/9.19.2799. [DOI] [PubMed] [Google Scholar]
- 38.Braga V.M. Small GTPases and regulation of cadherin dependent cell–cell adhesion. Mol. Pathol. 1999;52:197–202. doi: 10.1136/mp.52.4.197. doi:10.1136/mp.52.4.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Watanabe T., Sato K., Kaibuchi K. Cadherin-mediated intercellular adhesion and signaling cascades involving small GTPases. Cold Spring Harb. Perspect. Biol. 2009;1:a003020. doi: 10.1101/cshperspect.a003020. doi:10.1101/cshperspect.a003020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Swayne L.A., Chen L., Hameed S., Barr W., Charlesworth E., Colicos M.A., Zamponi G.W., Braun J.E. Crosstalk between huntingtin and syntaxin 1A regulates N-type calcium channels. Mol. Cell Neurosci. 2005;30:339–351. doi: 10.1016/j.mcn.2005.07.016. doi:10.1016/j.mcn.2005.07.016. [DOI] [PubMed] [Google Scholar]
- 41.Markianos M., Panas M., Kalfakis N., Vassilopoulos D. Low plasma total cholesterol in patients with Huntington's disease and first-degree relatives. Mol. Genet. Metab. 2008;93:341–346. doi: 10.1016/j.ymgme.2007.10.002. doi:10.1016/j.ymgme.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 42.Valenza M., Leoni V., Karasinska J.M., Petricca L., Fan J., Carroll J., Pouladi M.A., Fossale E., Nguyen H.P., Riess O., et al. Cholesterol defect is marked across multiple rodent models of Huntington's disease and is manifest in astrocytes. J. Neurosci. 2010;30:10844–10850. doi: 10.1523/JNEUROSCI.0917-10.2010. doi:10.1523/JNEUROSCI.0917-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pardo R., Molina-Calavita M., Poizat G., Keryer G., Humbert S., Saudou F. pARIS-htt: an optimised expression platform to study huntingtin reveals functional domains required for vesicular trafficking. Mol. Brain. 2010;3:17. doi: 10.1186/1756-6606-3-17. doi:10.1186/1756-6606-3-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pal A., Severin F., Lommer B., Shevchenko A., Zerial M. Huntingtin-HAP40 complex is a novel Rab5 effector that regulates early endosome motility and is up-regulated in Huntington's disease. J. Cell Biol. 2006;172:605–618. doi: 10.1083/jcb.200509091. doi:10.1083/jcb.200509091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Martinez-Vicente M., Talloczy Z., Wong E., Tang G., Koga H., Kaushik S., de Vries R., Arias E., Harris S., Sulzer D., et al. Cargo recognition failure is responsible for inefficient autophagy in Huntington's disease. Nat. Neurosci. 2010;13:567–576. doi: 10.1038/nn.2528. doi:10.1038/nn.2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Strehlow A.N., Li J.Z., Myers R.M. Wild-type huntingtin participates in protein trafficking between the Golgi and the extracellular space. Hum. Mol. Genet. 2007;16:391–409. doi: 10.1093/hmg/ddl467. doi:10.1093/hmg/ddl467. [DOI] [PubMed] [Google Scholar]
- 47.Bruses J.L. N-cadherin signaling in synapse formation and neuronal physiology. Mol. Neurobiol. 2006;33:237–252. doi: 10.1385/MN:33:3:237. doi:10.1385/MN:33:3:237. [DOI] [PubMed] [Google Scholar]
- 48.Tan Z.J., Peng Y., Song H.L., Zheng J.J., Yu X. N-cadherin-dependent neuron-neuron interaction is required for the maintenance of activity-induced dendrite growth. Proc. Natl Acad. Sci. USA. 2010;107:9873–9878. doi: 10.1073/pnas.1003480107. doi:10.1073/pnas.1003480107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stan A., Pielarski K.N., Brigadski T., Wittenmayer N., Fedorchenko O., Gohla A., Lessmann V., Dresbach T., Gottmann K. Essential cooperation of N-cadherin and neuroligin-1 in the transsynaptic control of vesicle accumulation. Proc. Natl Acad. Sci. USA. 2010;107:11116–11121. doi: 10.1073/pnas.0914233107. doi:10.1073/pnas.0914233107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Molero A.E., Gokhan S., Gonzalez S., Feig J.L., Alexandre L.C., Mehler M.F. Impairment of developmental stem cell-mediated striatal neurogenesis and pluripotency genes in a knock-in model of Huntington's disease. Proc. Natl Acad. Sci. USA. 2009;106:21900–21905. doi: 10.1073/pnas.0912171106. doi:10.1073/pnas.0912171106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nopoulos P.C., Aylward E.H., Ross C.A., Mills J.A., Langbehn D.R., Johnson H.J., Magnotta V.A., Pierson R.K., Beglinger L.J., Nance M.A., et al. Smaller intracranial volume in prodromal Huntington's disease: evidence for abnormal neurodevelopment. Brain. 2011;134:137–142. doi: 10.1093/brain/awq280. doi:10.1093/brain/awq280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lynch G., Kramar E.A., Rex C.S., Jia Y., Chappas D., Gall C.M., Simmons D.A. Brain-derived neurotrophic factor restores synaptic plasticity in a knock-in mouse model of Huntington's disease. J. Neurosci. 2007;27:4424–4434. doi: 10.1523/JNEUROSCI.5113-06.2007. doi:10.1523/JNEUROSCI.5113-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Simmons D.A., Rex C.S., Palmer L., Pandyarajan V., Fedulov V., Gall C.M., Lynch G. Up-regulating BDNF with an ampakine rescues synaptic plasticity and memory in Huntington's disease knockin mice. Proc. Natl Acad. Sci. USA. 2009;106:4906–4911. doi: 10.1073/pnas.0811228106. doi:10.1073/pnas.0811228106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jenkins B.G., Rosas H.D., Chen Y.C., Makabe T., Myers R., MacDonald M., Rosen B.R., Beal M.F., Koroshetz W.J. 1H NMR spectroscopy studies of Huntington's disease: correlations with CAG repeat numbers. Neurology. 1998;50:1357–1365. doi: 10.1212/wnl.50.5.1357. [DOI] [PubMed] [Google Scholar]
- 55.Saft C., Zange J., Andrich J., Muller K., Lindenberg K., Landwehrmeyer B., Vorgerd M., Kraus P.H., Przuntek H., Schols L. Mitochondrial impairment in patients and asymptomatic mutation carriers of Huntington's disease. Mov. Disord. 2005;20:674–679. doi: 10.1002/mds.20373. doi:10.1002/mds.20373. [DOI] [PubMed] [Google Scholar]
- 56.Sturrock A., Leavitt B.R. The clinical and genetic features of Huntington disease. J. Geriatr. Psychiatry Neurol. 2010;23:243–259. doi: 10.1177/0891988710383573. doi:10.1177/0891988710383573. [DOI] [PubMed] [Google Scholar]
- 57.Wheeler V.C., Auerbach W., White J.K., Srinidhi J., Auerbach A., Ryan A., Duyao M.P., Vrbanac V., Weaver M., Gusella J.F., et al. Length-dependent gametic CAG repeat instability in the Huntington's disease knock-in mouse. Hum. Mol. Genet. 1999;8:115–122. doi: 10.1093/hmg/8.1.115. doi:10.1093/hmg/8.1.115. [DOI] [PubMed] [Google Scholar]
- 58.Endres M., Laufs U., Huang Z., Nakamura T., Huang P., Moskowitz M.A., Liao J.K. Stroke protection by 3-hydroxy-3-methylglutaryl (HMG)-CoA reductase inhibitors mediated by endothelial nitric oxide synthase. Proc. Natl Acad. Sci. USA. 1998;95:8880–8885. doi: 10.1073/pnas.95.15.8880. doi:10.1073/pnas.95.15.8880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Livak K.J., Schmittgen T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. doi:10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.