

Abstract
Cell penetrating peptides (CPPs) have tremendous potential for use in gene and drug delivery applications. The selection of new CPPs with desired capabilities from randomized peptide libraries is challenging, since the CPP phenotype is a complex selection target. Here we report the discovery of an unusual new CPP from a randomized peptide library using a functional selection system based on plasmid display (PD). After four rounds of screening of a 14-mer peptide library over PC12 cells, several peptides were identified and tested for their ability to deliver the green fluorescent protein (GFP). One peptide (SG3) exhibited a cell penetrating phenotype, however unlike other well-known CPPs such as TAT or Penetratin, the newly identified peptide was not highly cationic. The PD protocol necessitated the addition of a cationic lipid (Lipofectamine2000), and in the presence of this compound, the SG3 peptide significantly outperformed the well-known TAT CPP in the delivery of GFP to PC12 cells and primary astrocytes. When the SG3 peptide was fused to the pro-apoptotic BH3 peptide from the Bak protein, significant cell death was induced in cultured primary astrocytes, indicating relevant, intracellular delivery of a functional cargo. The PD platform is a useful method for identifying functional new CPPs from randomized libraries with unique delivery capabilities.
Keywords: cell penetrating peptides, non-viral delivery, plasmid display, protein delivery, random peptide library screening
Introduction
Peptides that can gain access to the cytosol are potentially valuable for non-viral mediated gene and drug delivery applications since these peptides can readily be conjugated to a variety of cargoes including fluorochromes, proteins, enzymes, antibodies, DNA, phage particles, liposomes, and nanoparticles (1–6). Several peptides with this ability, termed cell penetrating peptides (CPPs), have been identified from various natural protein domains such as the homeodomain of the transcription factor Antennapeida from Drosophila (called Penetratin) and the TAT transcription factor from the HIV virus (called TAT). In general, CPPs tend to be short and contain mostly basic amino acids. While these CPPs have generated a great deal of enthusiasm, their therapeutic application has been hampered by several limitations including stability, specificity, and an incomplete understanding of their mechanism of action (7–9). While the method by which CPPs, such as TAT, cross the plasma membrane is debated, it is likely that the process is initiated by an electrostatic interaction between the positively charged CPP and negatively charged cell surface proteoglycans, such as heparan sulfate (10–18). However, since the amount of glycosaminoglycan (GAG) on the surface of a cell can vary (18–20), CPP transduction may vary depending on cell type, cell environment, and function (18). Therefore, the development of new CPPs that can be better tailored to a desired target cell or tissue type would be beneficial for drug delivery applications.
Several research groups have worked to develop methods to identify new targeting and delivery peptides with desirable traits, such as cell type specificity, from randomized libraries. Phage display techniques have been used by several groups to identify peptides that associate with different cell and tissue types, which has led to the identification of “homing peptides” that can target different cell types in vivo (21–24). In some cases, these experiments identified peptides that also fortuitously have cell penetrating abilities (25–27). But, a significant limitation of this approach is that the selection protocols employed cannot differentiate between peptides physically associated with the outside of targeted cells and those that have entered the cytosol. Better functional selection systems are needed to identify CPPs with desirable traits for therapeutic applications which require intracellular delivery.
High-throughput selection methods rely on linking the peptides to their cognate DNA sequences for subsequent identification following separation from the randomized library. A number of methods are available for this requirement (28, 29). Plasmid display (PD) is a conceptually simple and underutilized method for this biomolecular association. In this method, the members of the peptide library are expressed as fusions to a DNA binding protein domain which enables the peptides to be non-covalently associated to its encoding expression plasmid (30–32). The PD system used in this work relies on peptide fusions to the p50 DNA-binding domain of the NF-κB protein (30). To modify the PD system into a functional selection platform for selecting new CPPs, the sequence for a fluorescent protein (EYFP) under the control of a mammalian promoter was inserted into the plasmid DNA (33). The fluorescent protein gene acts as a mock cargo that can be delivered to cells by the bound peptides. Therefore, the expression of the fluorescent transgene within a cell indicates that the associated CPP successfully delivered the plasmid DNA cargo into the cell in a functional form which could undergo transcription.
In this study we report the identification of a new CPP from a randomized peptide library. A randomized 14-mer library was displayed on the PD vector, and after four rounds of selection on PC12 cells, a new CPP, named SG3, was identified. The functionality of the peptide was verified using fusions to GFP and to a pro-apoptotic death peptide, the BH3 domain from the Bak protein. Functional delivery was demonstrated in both PC12 cells and primary astrocytes. The sequence of the SG3 peptide is substantially different from other known CPPs, which demonstrates the potential of using PD and combinatorial protein engineering methods for the identification of novel delivery peptides.
Results and Discussion
The main advantage of using the PD platform for identifying CPPs from a random library is the delivery of the transgene incorporated in the plasmid, which indicates that the plasmid cargo has reached the cytosol, traveled to the nucleus intact, and has not degraded (17, 34–36). However, we have been unable to use a known CPP, the TAT peptide, to deliver the large plasmid cargo of the PD complexes to cells unassisted, presumably due to the large negative charge of the plasmid DNA. We have identified two methods for assisting TAT-mediated delivery of the PD complex. First we have demonstrated that a large amount of a p50-GFP-TAT fusion protein (1.89 protein molecules per base pair of DNA) is capable of delivering plasmid DNA to PC12 cells (36). And second, we have recently developed a method whereby two copies of the TAT peptide can be used to deliver the PD system to PC12 cells with the addition of the cationic lipid Lipofectamine2000 to shield the charge of the DNA (33). The expression of the EYFP transgene was observed in the absence of a known nuclear localization sequence. The requirement of a charge-shielding reagent for CPP-mediated plasmid delivery has previously been reported by others. For example, Kilk et al. (37) used PEI to deliver a GFP expression plasmid that was conjugated to the TP10 CPP through a peptide nucleic acid (PNA) linkage. In the development of the PD protocol, we experimented with PEI as a charge shielding reagent, but it was very sensitive to the N/P ratio which was difficult to determine in the PD complexes (33). Therefore in the present work, Lipofectamine2000 was used in the PD selection protocol (Figure 1).
Figure 1.

Schematic of the selection protocol developed for identifying new CPPs from a randomized library using the PD platform.
The peptide library was created by randomizing 14 consecutive amino acids followed by an invariant glycine. An oligonucleotide with this randomized sequence was used to replace the TAT sequence in the pTAT PD vector (33) so that the randomized sequence was added to the C-terminus of the p50 gene. This peptide length was chosen since it is intermediate to the TAT CPP (11 amino acids) (6) and the Penetratin CPP (16 amino acids) (8). Theoretically, there would be 1.18×1021 unique DNA sequences from the complete randomized library made with 14 (NNS, where N=A/T/C/G and S=G/C) codons, which would require at least 1.96 mmols of randomized DNA oligonucleotides. In this study, only a fraction of the potential randomized library was created and used in the screening experiments (~106 sequences). DNA sequencing of 60 randomly selected members of the library showed the expected diversity in both DNA and amino acid sequences (SI). In some of the randomly selected clones from the library, part of the TAT peptide sequence was found after the inserted library peptide due to a frame shift coupled with the incomplete digestion of the pTAT vector. Therefore the initial library contained a fraction of positively charged TAT-like CPP sequences that could potentially be selected in addition to the randomized peptides.
Protein/DNA complexes from the randomized library were expressed in E. coli and partially purified (33). The complexes were mixed with Lipofecamine2000 and applied to PC12 cells. Cells expressing the fluorescent transgene (EYFP) included in the PD plasmid were selected using fluorescent activated cell sorting (FACS), and PCR was used to amplify a chosen region of the delivered plasmids (~600bp) which contained the CPP sequences (52bp). The doubly digested PCR product containing the CPP sequences (64bp) was ligated back into the PD vector which was used to prepare more protein/DNA complexes (Figure 1). In each round of selection, the concentration of the protein/DNA complexes was increased while the Lipofectamine2000 concentration was held constant to increase the stringency of the selections.
This PD-based selection process is substantially different from phage display biopanning in which phage are isolated from the target cells and are used for re-infection of E. coli (24). Phage display favors isolation of intact phage and selects against phage particles that are degraded following cellular entry. The PCR amplification step of the PD process is time consuming, but it is required to amplify candidate CPP sequences that delivered functional genes to the cytosol.
After each round of screening, 20 colonies were randomly selected for DNA sequencing. Interestingly, the TAT-containing peptides that were an artifact of the library production process were selected out of the library by the second round of selection. The initial library also contained a high level of truncations due to stop codon insertions, but by the second and third round of selection, most of the sequences were found to be full-length. Surprisingly, basic amino acids were not found to be enriched in the peptides during the selection procedure (SI).
After four rounds of selection, there was a significant reduction in clones yielding readable and full-length sequences, and so the selection protocol was halted. The library did not appear to converge to a single optimal sequence, but three peptide sequences were observed several times in the colonies that were sequenced after each round. SG1 (N′-VKRLMRWGQELGRC-C′) was found in a colony after the second round and in a colony after the third round of screening. SG2 (N′-YNKHEGTTGGRTEI-C′) was found in two colonies after the third round of screening. And, the peptide SG3 (N′-RLSGMNEVLSFRWL-C′) was found in a colony after the third round and in a colony after the fourth round of screening. These three peptides were chosen for further evaluation.
It is not unexpected that the library did not converge to a single optimal sequence. In many peptide selection schemes, peptides with affinity to some target are sought, and the selection is repeated until the optimal sequence for this purpose is identified. Since the CPP mechanism is not thought to be a receptor-mediated process, there is no a priori reason why a single sequence should necessarily be optimal. It is possible that many different peptides may be good CPPs as evidenced by the wide variety in sequence of the known functional CPPs (1, 3, 5). The optimal amino acids for the CPP phenotype may not need to be adjacent to each other in the sequence to be functional, and important features like positive charge may be fulfilled by multiple different side chain configurations.
The PD system is not optimal for quantifying the transduction abilities of the selected peptides since the yields of the complexes are low, the samples are difficult to fully purify, the transduction efficiencies are low due to the large DNA cargo, and experiments require time for expression of the fluorescent transgene reporter (33). Therefore, we chose to use the simpler method of direct fluorescent protein delivery (GFP) by the three selected peptides to more thoroughly assess their performance. The DNA sequences encoding each of the three peptides were genetically fused to the C-terminus of the GFP protein gene, and these fusion proteins were expressed and purified as previously described (38). We were unable to express GFP-SG1 in E. coli in a functional form even after various expression conditions were explored. Therefore, this peptide was not investigated further. The purity of the remaining fusion proteins as well as GFP and GFP-TAT was assessed by SDS-PAGE (SI). The GFP-SG2 peptide was found to undergo some proteolytic degradation, but the majority of the protein in the GFP-SG2 samples was full-length. No degradation was observed with either GFP-TAT or GFP-SG3.
The purified fusion proteins and controls (GFP, GFP-TAT, GFP-SG2 and GFP-SG3) were incubated with PC12 cells for 4 hours followed by trypsinization for 10 min to remove any fluorescent fusion protein associated with the external cell membrane before analysis via confocal microscopy and flow cytometry. Forward-scatter and side-scatter plots were used to identify and gate out dead cells and debris. For all four proteins and the untreated controls, approximately 80% of the recorded events were suitable for analysis, with no significant difference between the five groups. This indicated that none of the fusion protein constructs had a negative effect on cell viability.
The transduction of the fusion proteins into PC12 cells as assessed by flow cytometry is shown in Figure 2A. GFP-TAT and GFP-SG3 exhibited significant transduction to PC12 cells compared to GFP and GFP-SG2, which did not transduce the PC12 cells. Both GFP-TAT and GFP-SG3 transduced PC12 cells in a dose dependent manner (Figure 2B) indicating that SG3 is a bona fide new CPP sequence. In the absence of Lipofectamine2000, GFP-TAT exhibited significantly higher transduction ability than that of GFP-SG3 at each loading dose. Confocal microscopy studies support these results as GFP-TAT (Figure 2C) and GFP-SG3 (Figure 2D) were found within the PC12 cells with a similar punctate appearance, while GFP (Figure 2E) was not.
Figure 2.

Delivery of GFP to PC12 cells using peptides identified from the library selection. A. Fusion proteins (2.8 nmol) were added to PC12 cells in a 24-well plate and analyzed by flow cytometry. B. Dose-dependent uptake of fluorescent fusions by PC12 cells: GFP (■), GFP-TAT (▲) and GFP-SG3 (○). In both panels, each experiment was performed in triplicate (n=3) and error bars represent standard errors. ‘*’ indicates the value is significantly different from the GFP treated control cells. C–E. Confocal microscopy analysis of fusion protein uptake by PC12 cells (Green: GFP signal; Red: CellTracker Red CMTPX; Yellow arrows: a typical spot on the image of Z plane, orthogonal X and Y plane). C: incubation for 4 hours with GFP-TAT, resulting in an overlap coefficient of 0.983; D: incubation for 4 hours with GFP-SG3, resulting in an overlap coefficient of 0.994; E: incubation for 4 hours with GFP, resulting in an overlap coefficient of 0.978.
The mechanism of action of CPPs has been a subject of much debate in the literature. We have recently demonstrated that TAT-mediated delivery of GFP into PC12 cells was dependent on the presence of GAG, in particular heparin sulfate, on the surface of the cell, and that TAT-mediated delivery can be competitively inhibited by pre-treatment of PC12 cells with heparin (18). Interestingly, the SG3 peptide was not inhibited by heparin pre-treatment (100μg/mL for 1 hour) (Figure 3), suggesting that the mechanism of cell penetration of SG3 is different from that of the TAT peptide and likely does not involve an electrostatic interaction with cell surface GAGs. It has been proposed in the literature that the TAT CPP is internalized through an energy dependent macropinocytosis mechanism or clathrin-mediated endocytosis (17). These mechanisms may also be used by SG3, but the lack of effect of an interaction with heparin suggests that at least the initial interactions between the peptides and membrane before internalization may be different from the TAT peptide.
Figure 3.
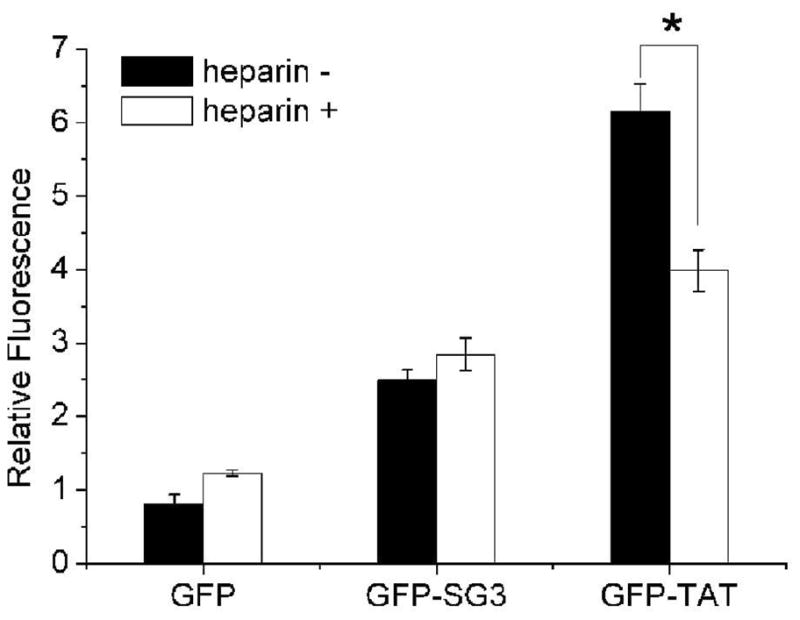
Effect of heparin treatment on CPP-mediated delivery of the fluorescent fusion proteins to PC12 cells. Black bars: untreated cells incubated with 2.8 nmol of protein for 4 hours at 37°C; White bars: cells pre-treated with 100ug/mL of heparin before incubation with each protein. Each experiment was performed in triplicate (n=3) and error bars represent standard error. ‘*’ indicates the value is significantly different from the heparin treated cells.
The addition of a cationic lipid was required for the PD-based selection procedure, and thus the experiments were repeated in the presence of Lipofactamine2000 to determine if the selection procedure resulted in a peptide with a dependency on this compound. The addition of the cationic lipid dramatically increased the uptake of GFP-SG3 by the PC12 cells, and under these conditions, the SG3 peptide significantly outperformed the TAT peptide in a dose dependent manner (Figure 4A).
Figure 4.
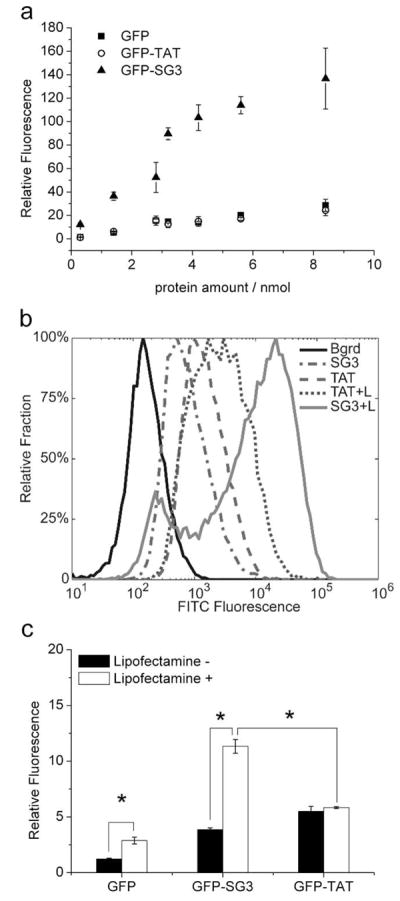
Effect of the cationic lipid, Lipofectamine2000 on CPP-mediated fusion protein delivery. A. Dose-dependent delivery of proteins to PC12 cells in the presence of 10μl Lipofectamine2000. After 4 hours of incubation with the protein/Lipofectamine2000 complex, cells were analyzed by flow cytometry. GFP (■), GFP-TAT (○) and GFP-SG3 (▲). B. Sample histograms from flow cytometry analysis of GFP and GFP-TAT with and without Lipofectamine2000. Protein (2.8nmol) with or without Lipofectamine was incubated with primary astrocytes under the same transduction conditions as panel A. C. Comparison of replicate flow cytometry results as described in panel B. For panels A and C, experiments were performed in triplicate (n=3) and error bars represent standard error.
In previous studies, we have shown that the CPP transduction efficiency can significantly differ between cell lines and primary cells (18), depending on the GAG content on the membrane of specific type of cells. In this study, however, similar delivery efficiencies were observed in primary astrocytes: in the absence of the cationic lipid, the GFP-SG3 fusion was delivered to cells better than GFP alone, but it was not as effective as the TAT fusion protein (Figure 4B & 4C). And, upon the addition of Lipofectamine2000, a significant increase in the delivery ability of the SG3 peptide was again observed. The addition of Lipofectamine2000 did improve the delivery of GFP but it did not have a significant effect on the delivery of GFP-TAT (Figure 4C) which may be due to the more neutral charge of GFP-TAT as compared to the negatively charged GFP (SI). These results are consistent with the selection experiment results where, in the presence of Lipofectamine2000, the TAT-containing peptides found in the PD library were selected out of the library by the second round presumably because the TAT peptide is a less efficient CPP in the presence of the cationic lipid.
In another recent study it has been shown that a tryptophan side chain found in the full-length TAT protein is involved in membrane insertion and protein translocation from the endosome to the cytosol following endocytosis while the basic domain of the TAT protein, which contains the TAT CPP, is more important for TAT binding to the phosphate groups of the bilayer phospholipids for the induction of endocytosis (39). Although molecular dynamics simulations suggest a potential mechanism for translocation of TAT peptide across lipid membranes (40), no experimental evidence has demonstrated TAT translocation through model membranes (39). Interestingly, attachment of a tryptophan residue to the TAT CPP was able to induce membrane insertion into model lipid membranes (41). Taken together, this work implies that a tryptophan amino acid may be important for CPP-mediated delivery, and the tryptophan residue in SG3 may play a similar role. The addition of Lipofectamine2000 likely promotes endocytosis, and these synergistic effects may explain the selection of the SG3 peptide in the presence of the cationic lipid.
To demonstrate the ability of the SG3 peptide to deliver a protein cargo intracellularly and in a functional (undegraded) state, we chose to deliver a pro-apoptotic peptide, BH3, to primary astrocytes to induce cell death, an application which may have implications for cancer treatment. The Bcl-2 homology 3 (BH3) domain is crucial for the death-inducing and dimerization properties of pro-apoptotic members of the Bcl-2 protein family, including Bak, Bax, and Bad (42). Previously, the TAT CPP has been used to deliver the BH3 domain from PUMA (p53 upregulated modulator of apoptosis) (43) and the amino-terminus of Bax (p3Bax) into cancer cells (44). Furthermore, the Penetratin CPP has been used to deliver the BH3 domain of Bak to HeLa cells (42). We purchased synthetic peptides with SG3, SG2 and the TAT sequence fused to the C-terminus of the BH3 domain of Bak (N′-MGQVGRQLAIIGDDINRRY-C′). The SG2 peptide was chosen as a size-matched negative control since it was unable to deliver GFP to the cells (and is therefore not a CPP). The solubility of all peptide constructs was increased with DMSO and a nonionic surfactant, Pluronic F-127. In the absence of Lipofectamine2000, the fusion peptide BH3-TAT rapidly induced cell death in primary astrocytes whereas BH3-SG2 and BH3-SG3 did not (Figure 5). The results with the SG2 and SG3 fusion peptides were similar to what has been reported with the BH3 peptide alone, where no effect was observed in HeLa cells, likely due to the inability of the BH3 peptide to transduce the plasma membrane on its own (42). When Lipofectamine2000 was added, the delivery of BH3-TAT improved which was different from what was seen with GFP-TAT, and this effect was likely due to the dramatic differences in charge and size between GFP and BH3 (SI). In addition, as was observed with GFP fusions, the addition of Lipofectamine2000 significantly enhanced the delivery of the BH3-SG3 construct, which enabled it to outperform the TAT fusion protein as the BH3-SG3 construct induced the greatest amount of cell death (Figure 5).
Figure 5.

CPP-mediated delivery of the BH3 death peptide into primary astrocytes. The number of dead cells was compared to the number of dead cells obtained upon treatment with solvent or solvent plus Lipofectamine2000 alone. Each experiment was performed in triplicate (n=3) and error bars represent standard error. ‘*’ indicates the value is significantly different from the cells transduced by fusion peptides without the presence of cationic lipid.
In order to rule out a toxic effect of SG3 alone, free SG3 peptide was incubated with the cells with and without Lipofectamine2000. The cell viability was no different from the control cells indicating that the SG3 peptide was non-toxic under these conditions (Figure 5).
Intracellular therapeutic delivery is a multistep process which first requires the construct to be invaginated by the plasma membrane forming an intracellular vacuole. These often form endosomes which degrade the internalized contents. For a cargo to be therapeutically active, it must then escape the endosome prior to degradation. The punctate appearance of the confocal images in Figure 2 suggests that a significant amount of the GFP-TAT and GFP-SG3 fusion proteins were located in vesicles such as endosomes. However, when combined with Lipofectamine2000, the SG3 peptide must necessarily possess both functions (induction of internalization followed by endosome escape) since the peptide cargo, the BH3 domain, was capable of inducing cell death. Retention of its biological effect was evidence that this construct possesses both functions suggesting the possibility that SG3 could be fused to other peptide cargoes with other specific intracellular functions. Future work will explore whether SG3 is a universal delivery peptide and whether it has innate nuclear localization ability.
The SG3 peptide is notably different from most other known CPPs. A BLAST search of the SG3 sequence did not result in significant homology to any known CPPs or trans-membrane proteins. Conventional and recently identified CPPs, including TAT, polyarginine (45), Penetratin (46), Transportan 10 (46), M918 (46), PTD-DRBD (47) and CADY (48), generally contain a large number of positively charged amino acids (the TAT sequence N′-YGRKKRRQRRR-C′ contains 8 such amino acids) but the SG3 peptide (N′-RLSGMNEVLSFRWL-C′) has just two arginine residues and a single glutamic acid residue resulting in a net charge of +1 only. Since SG3 is able to deliver GFP to cells (even in the absence of Lipofectamine2000), this result demonstrates that a significant positive charge is not an absolute requirement for an effective CPP.
The fact that 6 of the 14 amino acids in the SG3 peptide are hydrophobic, including a tryptophan, is notable. CPPs must interact with cellular membranes, and some CPPs also have some hydrophobic content in addition to extensive positive charge. It is tempting to propose that the improvement observed upon addition of Lipofectamine2000 to SG3 is caused by hydrophobic interactions between the peptide and the cationic lipid, thereby producing a cationic, cell penetrating, macromolecular complex.
There has been increasing interest in structural studies of CPPs as mounting evidence suggests that these peptides can be intrinsically disordered, but that structural transitions may also be important in the CPP delivery mechanisms (49). Therefore, the Hierarchical Neural Network (HNN) method (50) was used to predict the presence of secondary structure in the new peptides (SI). The TAT peptide was predicted to posses some helical content, while the Penetratin peptide was predicted to have beta sheet content; these predictions are consistent with the experimental results of other researchers (49). When the method was applied to the new peptides, SG2 was predicted to be disordered, while SG3 was predicted to have both alpha helical and beta sheet structure. When the peptides were fused to the BH3 domain, the TAT peptide was predicted to gain helical content, the SG2 peptide was predicted to remain disordered, and the SG3 peptide was predicted to retain both alpha helical and beta sheet character. These results suggest that at least structurally, SG3 could have some resemblance to Penetratin. We performed circular dichroism CD experiments to try to verify these secondary structure predictions with the free SG3 peptide but the results indicated that the SG3 peptide is predominantly in a random coil configuration in solution (data not shown).
We have successfully used the PD platform to identify a new CPP from a randomized peptide library. There are several advantages and disadvantages in using the PD platform for this purpose as compared to other genotype/phenotype linkage techniques. As discussed above, the biggest advantage is that the expression of the fluorescent transgene in the target cells unequivocally indicates that the plasmid DNA has been delivered to the cytosol intact and transported to the nucleus for transcription. In addition, the fluorescent transgene could easily be replaced with another functional gene so that the PD system with a selected peptide can easily be used for non-viral gene delivery to targeted cells. Another advantage is that the DNA sequences encoding the library peptides are recovered by PCR after each round of selection and subcloned back into the PD vector for amplification and re-expression. In many display systems, including phage display, there is selective pressure for the truncation or deletion of the displayed proteins. Although the PCR recovery of the CPP sequences form the cells was time consuming, it helped to ensure that the PD plasmid was unaltered during the selection process, and it eliminates the clonal selection problem that can plague other amplification procedures where members of the library with truncations and deletions can be amplified asymmetrically.
Two significant limitations of the PD platform are the limited yields and the small library sizes. The yield of plasmid-protein complexes is quite low by design in order to reduce the production of proteins not complexed with the expression plasmids. In addition, since the association between the proteins and plasmids is non-covalent, care must be taken during the purification process to ensure that the genotype/phenotype linkage is not disturbed. The affinity of the p50 domain for the target DNA sequence is quite high (Kd ~10 pM) (51), and it does not appear that the purification procedure significantly interferes with this non-covalent interaction as the complexes must have remained intact for the identification of the SG3 CPP. By the same logic it does not appear that the addition of the cationic lipid affects the stability of the protein/DNA complexes; further work will be required to determine if the presence of the cationic lipid has any effect on the genotype/phenotype linkage of the PD platform. The second limitation was the library size as the libraries in the PD platform are made by transforming bacteria with plasmids containing a randomized DNA sequence. The size of these libraries is limited by the electroporation efficiency of the cells which is much lower than what can be obtained with other methods such as ribosome display. However, in this report we were able to identify a potentially valuable new sequence from a small PD library, which suggests that this process could be used to find other peptides with interesting CPP phenotypes.
Conclusions
We have used a new PD-based selection platform to identify an unusual new CPP, SG3, by screening a randomized peptide library in PC12 cells. By itself, the SG3 peptide was not as effective as the TAT CPP, but the addition of Lipofectamine2000 to the SG3 peptide rendered it more effective than the TAT peptide under the same conditions. The successful selection of SG3 demonstrates the PD system as a potent selection platform for identifying novel peptides or proteins with complex functions. The SG3 peptide may be a useful new vector for cellular gene and drug delivery.
Methods
Library construction
The plasmid vector expressing a randomized library of peptides, pLIB, was constructed by replacing the sequence of the TAT peptide in the pTAT vector (33) with a random DNA sequence (NNS)14.
Preparation and purification of plasmid display complexes
The CPP DNA library, pLIB, was electroporated into electrocompetent M1061 cells and was expressed and purified as previously described (33). The plasmid (pLIB) concentration in the pdLIB protein/DNA complex samples was evaluated by q-PCR using the neat pTAT plasmid as a standard.
Cell culture
PC12 cells were cultured as previously described (18, 38). Approximately 6 × 106 cells were plated onto a 24-well culture plate coated with polylysine a day before transfection or transduction experiments were performed. The cells in each well (~250,000) were transfected or incubated with a sample (based on the plasmid concentration) with or without 10μl of Lipofectamine2000 following the manufacturer’s protocol.
Primary astrocytes were isolated from the cortices of 8–10 day old Sprague-Dawley rat pups and cultured as previously described (18). All procedures involving animals were approved by the Columbia University IACUC. After culturing for two weeks, the astrocytes were trypsinized and seeded onto uncoated tissue culture 6-well plates at a density of 1 × 105 cells per well. The cells were grown for 3–5 days until ~50% confluent for transduction experiments.
Screening of the library
The screening procedure is summarized in Figure 1. Each well containing ~250,000 PC12 cells was exposed to a partially purified sample of the pdLIB library along with 10μl Lipofectamine2000. Multiple replicate transfections were performed to accommodate the entire pdLIB sample. Control transfections were also performed using neat plasmid pCON and Lipofectamine2000. After 4 hours of incubation, the transfection medium was changed with fresh growth medium, and the cells cultured for an additional 20–24 hours to express the EYFP transgene. The cells were trypsinized and replicates were combined for FACS using a Becton Dickinson FACSAria. Gates were set using untreated PC12 cells, and positive control cells transfected with neat plasmid. Positive cells with yellow fluorescence from the pLIB transfected cells were collected in PBS. The cell resuspension was distributed and pelleted into PCR tubes with each tube containing ~500 cells. PCR was performed using Phusion polymerase on a Thermocycler (Eppendorf, Westbury, NY). The PCR products were agarose gel purified and sequentially digested with ScaI and BsaI before being cloned back into the doubly digested parent plasmid vector pTAT to make a new pLIB. To increase the stringency of the selections, the ratio of the Lipofectamine2000 to the pLIB concentration was varied. In the first round, each well received a pdLIB sample with 5ng of plasmid DNA, pLIB. This was increased to 10ng in the second round, 15ng in the third, and 20ng in the fourth round.
Transduction of recombinant fluorescent proteins into PC12 cells and astrocytes
The fusion proteins (GFP-TAT, GFP-SG2 and GFP-SG3) and GFP were incubated with cells as previously described (18). Briefly, the proteins and cells were incubated for 4 hours at 37°C before evaluation of the tranduced cells using a flow cytometer (Becton Dickinson FACSCantoII). For Lipofectamine2000 assisted transduction, 10μl of Lipofectamine2000 was used. The results were presented as the fold increase in geometric mean fluorescence of the live population compared to untreated, live, control cells.
Confocal Microscopy
To analyze the sub-cellular localization of the fusion proteins in live PC12 cells, the cells were stained with CellTracker Red CMTPX during the last 30min of the incubation with fluorescent fusion proteins before being supplemented with growth medium and immediately visualized under a Leica TCS SP5 confocal microscope (Wetzlar, Germany).
Transduction of a pro-apoptotic peptide to primary astrocytes
The cell penetrating phenotype of the selected CPPs, SG2 and SG3, was further validated by delivering the BH3 domain peptide (N′-MGQVGRQLAIIGDDINRRY-C′) of the Bak protein to primary astrocytes in a fusion peptide construct. The fusion peptides (with the CPPs fused to the C-terminus of BH3 peptide) were purchased from Neopeptide (Cambridge, MA). Primary astroctytes were incubated with 50μM of the peptides in 1mL DMEM containing 0.1% DMSO and 4% Pluronic F-127 for 4 hours before the cells were trypsinized, washed twice with ice cold PBS and stained with 7-Aminoactinomycin D (7-AAD) following the manufacturer’s protocol. The stained cells were kept on ice and the viability of the cells was immediately evaluated by flow cytometry. The live and dead cell populations were gated based on untreated cells and cells treated with 0.4mM H2O2 overnight. The percent of dead cells in each sample was normalized to the percent of dead cells in the sample treated by the solvents alone (DMSO and Pluronic F-127). For Lipofectamine2000-assisted transduction, the peptide stock solution was added to 50μl DMEM containing the solvents and incubated with 50μl DMEM containing 1ul Lipofectamine2000 at room temperature for 20 minutes before bringing the total volume up to 1mL and incubating with the cells for 4 hours. As a control, the cells were also incubated with the mixture of solvent in DMEM and Lipofectamine2000 in DMEM. After incubation, the cells were harvested and stained with 7-AAD before being analyzed by flow cytometry as described above. The percent of dead cells in each sample was normalized to the percent of dead cells in the sample treated by the mixture of solvent containing DMEM and Lipofectamine2000.
Statistical analysis
All statistical analysis was calculated using Student’s t-test or 1-way ANOVA with p<0.05 being considered significant.
Supplementary Material
Acknowledgments
Financial support provided by the Brain Trust (SB and BM), NIH R21 MH080024 (SB and BM), and an NSF Graduate Research Fellowship (MJS). We thank J. Blackburn for the kind gift of the plasmid display vector pRES115 which was used to create the pTAT plasmid. We also thank Dr. Ben Elkin for assisting us with the confocal microscopy imaging and Dr. Matthew Lluis for assisting us with the circular dichroism spectroscopy studies.
Footnotes
Supporting Information Available: This material is free via the Internet.
References
- 1.Dietz GP, Bahr M. Delivery of bioactive molecules into the cell: the Trojan horse approach. Mol Cell Neurosci. 2004;27:85–131. doi: 10.1016/j.mcn.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 2.Torchilin VP. Tat peptide-mediated intracellular delivery of pharmaceutical nanocarriers. Adv Drug Deliv Rev. 2008;60:548–558. doi: 10.1016/j.addr.2007.10.008. [DOI] [PubMed] [Google Scholar]
- 3.Joliot A, Prochiantz A. Transduction peptides: from technology to physiology. Nat Cell Biol. 2004;6:189–196. doi: 10.1038/ncb0304-189. [DOI] [PubMed] [Google Scholar]
- 4.Zorko M, Langel U. Cell-penetrating peptides: mechanism and kinetics of cargo delivery. Adv Drug Deliv Rev. 2005;57:529–545. doi: 10.1016/j.addr.2004.10.010. [DOI] [PubMed] [Google Scholar]
- 5.Deshayes S, Morris MC, Divita G, Heitz F. Cell-penetrating peptides: tools for intracellular delivery of therapeutics. Cell Mol Life Sci. 2005;62:1839–1849. doi: 10.1007/s00018-005-5109-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schwarze SR, Ho A, Vocero-Akbani A, Dowdy SF. In vivo protein transduction: delivery of a biologically active protein into the mouse. Science. 1999;285:1569–1572. doi: 10.1126/science.285.5433.1569. [DOI] [PubMed] [Google Scholar]
- 7.Vives E. Present and future of cell-penetrating peptide mediated delivery systems: “Is the Trojan horse too wild to go only to Troy? J Control Release. 2005;109:77–85. doi: 10.1016/j.jconrel.2005.09.032. [DOI] [PubMed] [Google Scholar]
- 8.Trehin R, Merkle HP. Chances and pitfalls of cell penetrating peptides for cellular drug delivery. Eur J Pharm Biopharm. 2004;58:209–223. doi: 10.1016/j.ejpb.2004.02.018. [DOI] [PubMed] [Google Scholar]
- 9.Chauhan A, Tikoo A, Kapur AK, Singh M. The taming of the cell penetrating domain of the HIV Tat: Myths and realities. J Control Release. 2007;117:148–162. doi: 10.1016/j.jconrel.2006.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Duchardt F, Fotin-Mleczek M, Schwarz H, Fischer R, Brock R. A comprehensive model for the cellular uptake of cationic cell-penetrating peptides. Traffic. 2007;8:848–866. doi: 10.1111/j.1600-0854.2007.00572.x. [DOI] [PubMed] [Google Scholar]
- 11.Poon GM, Gariepy J. Cell-surface proteoglycans as molecular portals for cationic peptide and polymer entry into cells. Biochem Soc Trans. 2007;35:788–793. doi: 10.1042/BST0350788. [DOI] [PubMed] [Google Scholar]
- 12.Tyagi M, Rusnati M, Presta M, Giacca M. Internalization of HIV-1 tat requires cell surface heparan sulfate proteoglycans. J Biol Chem. 2001;276:3254–3261. doi: 10.1074/jbc.M006701200. [DOI] [PubMed] [Google Scholar]
- 13.Vives E. Cellular uptake [correction of utake] of the Tat peptide: an endocytosis mechanism following ionic interactions. J Mol Recognit. 2003;16:265–271. doi: 10.1002/jmr.636. [DOI] [PubMed] [Google Scholar]
- 14.Bugatti A, Urbinati C, Ravelli C, De Clercq E, Liekens S, Rusnati M. Heparin-mimicking sulfonic acid polymers as multitarget inhibitors of human immunodeficiency virus type 1 Tat and gp120 proteins. Antimicrob Agents Chemother. 2007;51:2337–2345. doi: 10.1128/AAC.01362-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kumarasuriyar A, Dombrowski C, Rider DA, Nurcombe V, Cool SM. A novel use of TAT-EGFP to validate techniques to alter osteosarcoma cell surface glycosaminoglycan expression. J Mol Histol. 2007;38:435–447. doi: 10.1007/s10735-007-9136-z. [DOI] [PubMed] [Google Scholar]
- 16.Nakase I, Tadokoro A, Kawabata N, Takeuchi T, Katoh H, Hiramoto K, Negishi M, Nomizu M, Sugiura Y, Futaki S. Interaction of arginine-rich peptides with membrane-associated proteoglycans is crucial for induction of actin organization and macropinocytosis. Biochemistry. 2007;46:492–501. doi: 10.1021/bi0612824. [DOI] [PubMed] [Google Scholar]
- 17.Wadia JS, Stan RV, Dowdy SF. Transducible TAT-HA fusogenic peptide enhances escape of TAT-fusion proteins after lipid raft macropinocytosis. Nat Med. 2004;10:310–315. doi: 10.1038/nm996. [DOI] [PubMed] [Google Scholar]
- 18.Simon MJ, Gao S, Kang WH, Banta S, Morrison B., 3rd TAT-mediated intracellular protein delivery to primary brain cells is dependent on glycosaminoglycan expression. Biotechnol Bioeng. 2009;104:10–19. doi: 10.1002/bit.22377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Warda M, Toida T, Zhang F, Sun P, Munoz E, Xie J, Linhardt RJ. Isolation and characterization of heparan sulfate from various murine tissues. Glycoconj J. 2006;23:555–563. doi: 10.1007/s10719-006-7668-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.David G. Integral membrane heparan sulfate proteoglycans. FASEB J. 1993;7:1023–1030. doi: 10.1096/fasebj.7.11.8370471. [DOI] [PubMed] [Google Scholar]
- 21.Ruoslahti E. Drug targeting to specific vascular sites. Drug Discov Today. 2002;7:1138–1143. doi: 10.1016/s1359-6446(02)02501-1. [DOI] [PubMed] [Google Scholar]
- 22.Ruoslahti E, Rajotte D. An address system in the vasculature of normal tissues and tumors. Annu Rev Immunol. 2000;18:813–827. doi: 10.1146/annurev.immunol.18.1.813. [DOI] [PubMed] [Google Scholar]
- 23.Kolonin MG, Sun J, Do KA, Vidal CI, Ji Y, Baggerly KA, Pasqualini R, Arap W. Synchronous selection of homing peptides for multiple tissues by in vivo phage display. Faseb J. 2006;20:979–981. doi: 10.1096/fj.05-5186fje. [DOI] [PubMed] [Google Scholar]
- 24.Pasqualini R, Ruoslahti E. Organ targeting in vivo using phage display peptide libraries. Nature. 1996;380:364–366. doi: 10.1038/380364a0. [DOI] [PubMed] [Google Scholar]
- 25.Sheng J, Oyler G, Zhou B, Janda K, Shoemaker CB. Identification and characterization of a novel cell-penetrating peptide. Biochem Biophys Res Commun. 2009;382:236–240. doi: 10.1016/j.bbrc.2008.11.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kamada H, Okamoto T, Kawamura M, Shibata H, Abe Y, Ohkawa A, Nomura T, Sato M, Mukai Y, Sugita T, Imai S, Nagano K, Tsutsumi Y, Nakagawa S, Mayumi T, Tsunoda S. Creation of novel cell-penetrating peptides for intracellular drug delivery using systematic phage display technology originated from Tat transduction domain. Biol Pharm Bull. 2007;30:218–223. doi: 10.1248/bpb.30.218. [DOI] [PubMed] [Google Scholar]
- 27.Shadidi M, Sioud M. Identification of novel carrier peptides for the specific delivery of therapeutics into cancer cells. Faseb J. 2003;17:256–258. doi: 10.1096/fj.02-0280fje. [DOI] [PubMed] [Google Scholar]
- 28.Lin H, Cornish VW. Screening and selection methods for large-scale analysis of protein function. Angew Chem Int Ed Engl. 2002;41:4402–4425. doi: 10.1002/1521-3773(20021202)41:23<4402::AID-ANIE4402>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 29.Doi N, Yanagawa H. Genotype-phenotype linkage for directed evolution and screening of combinatorial protein libraries. Comb Chem High Throughput Screen. 2001;4:497–509. doi: 10.2174/1386207013330878. [DOI] [PubMed] [Google Scholar]
- 30.Speight RE, Hart DJ, Sutherland JD, Blackburn JM. A new plasmid display technology for the in vitro selection of functional phenotype-genotype linked proteins. Chem Biol. 2001;8:951–965. doi: 10.1016/s1074-5521(01)00066-7. [DOI] [PubMed] [Google Scholar]
- 31.Choi YS, Pack SP, Yoo YJ. Development of a plasmid display system using GAL4 DNA binding domain for the in vitro screening of functional proteins. Biotechnology Letters. 2005;27:1707–1711. doi: 10.1007/s10529-005-2735-4. [DOI] [PubMed] [Google Scholar]
- 32.Cull MG, Miller JF, Schatz PJ. Screening for Receptor Ligands Using Large Libraries of Peptides Linked to the C-Terminus of the Lac Repressor. Proceedings of the National Academy of Sciences of the United States of America. 1992;89:1865–1869. doi: 10.1073/pnas.89.5.1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gao S, Simon MJ, Morrison B, 3rd, Banta S. A plasmid display platform for the selection of peptides exhibiting a functional cell-penetrating phenotype. Biotechnol Prog. 2010;26:1796–1800. doi: 10.1002/btpr.490. [DOI] [PubMed] [Google Scholar]
- 34.Endoh T, Ohtsuki T. Cellular siRNA delivery using cell-penetrating peptides modified for endosomal escape. Advanced Drug Delivery Reviews. 2009;61:704–709. doi: 10.1016/j.addr.2009.04.005. [DOI] [PubMed] [Google Scholar]
- 35.Magzoub M, Pramanik A, Graslund A. Modeling the endosomal escape of cell-penetrating peptides: Transmembrane pH gradient driven translocation across phospholipid bilayers. Biochemistry. 2005;44:14890–14897. doi: 10.1021/bi051356w. [DOI] [PubMed] [Google Scholar]
- 36.Gao K, Huang L. Nonviral Methods for siRNA Delivery. Molecular Pharmaceutics. 2009;6:651–658. doi: 10.1021/mp800134q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kilk K, El-Andaloussi S, Jarver P, Meikas A, Valkna A, Bartfai T, Kogerman P, Metsis M, Langel U. Evaluation of transportan 10 in PEI mediated plasmid delivery assay. J Control Release. 2005;103:511–523. doi: 10.1016/j.jconrel.2004.12.006. [DOI] [PubMed] [Google Scholar]
- 38.Gao S, Simon MJ, Morrison B, 3rd, Banta S. Bifunctional chimeric fusion proteins engineered for DNA delivery: optimization of the protein to DNA ratio. Biochim Biophys Acta. 2009;1790:198–207. doi: 10.1016/j.bbagen.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yezid H, Konate K, Debaisieux S, Bonhoure A, Beaumelle B. Mechanism for HIV-1 Tat insertion into the endosome membrane. J Biol Chem. 2009;284:22736–22746. doi: 10.1074/jbc.M109.023705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Herce HD, Garcia AE. Molecular dynamics simulations suggest a mechanism for translocation of the HIV-1 TAT peptide across lipid membranes. Proc Natl Acad Sci U S A. 2007;104:20805–20810. doi: 10.1073/pnas.0706574105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thoren PE, Persson D, Esbjorner EK, Goksor M, Lincoln P, Norden B. Membrane binding and translocation of cell-penetrating peptides. Biochemistry. 2004;43:3471–3489. doi: 10.1021/bi0360049. [DOI] [PubMed] [Google Scholar]
- 42.Holinger EP, Chittenden T, Lutz RJ. Bak BH3 peptides antagonize Bcl-xL function and induce apoptosis through cytochrome c-independent activation of caspases. J Biol Chem. 1999;274:13298–13304. doi: 10.1074/jbc.274.19.13298. [DOI] [PubMed] [Google Scholar]
- 43.Liu Y, Li Y, Wang H, Yu J, Lin H, Xu D, Wang Y, Liang A, Liang X, Zhang X, Fu M, Qian H, Lin C. BH3-based fusion artificial peptide induces apoptosis and targets human colon cancer. Mol Ther. 2009;17:1509–1516. doi: 10.1038/mt.2009.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li N, Lin P, Cai C, Pan Z, Weisleder N, Ma J. The amino-terminal peptide of Bax perturbs intracellular Ca2+ homeostasis to enhance apoptosis in prostate cancer cells. Am J Physiol Cell Physiol. 2009;296:C267–272. doi: 10.1152/ajpcell.00390.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kumar P, Wu H, McBride JL, Jung KE, Kim MH, Davidson BL, Lee SK, Shankar P, Manjunath N. Transvascular delivery of small interfering RNA to the central nervous system. Nature. 2007;448:39–43. doi: 10.1038/nature05901. [DOI] [PubMed] [Google Scholar]
- 46.El-Andaloussi S, Johansson HJ, Holm T, Langel U. A novel cell-penetrating peptide, M918, for efficient delivery of proteins and peptide nucleic acids. Mol Ther. 2007;15:1820–1826. doi: 10.1038/sj.mt.6300255. [DOI] [PubMed] [Google Scholar]
- 47.Eguchi A, Meade BR, Chang YC, Fredrickson CT, Willert K, Puri N, Dowdy SF. Efficient siRNA delivery into primary cells by a peptide transduction domain-dsRNA binding domain fusion protein. Nat Biotechnol. 2009;27:567–571. doi: 10.1038/nbt.1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Crombez L, Aldrian-Herrada G, Konate K, Nguyen QN, McMaster GK, Brasseur R, Heitz F, Divita G. A new potent secondary amphipathic cell-penetrating peptide for siRNA delivery into mammalian cells. Mol Ther. 2009;17:95–103. doi: 10.1038/mt.2008.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Eiriksdottir E, Konate K, Langel U, Divita G, Deshayes S. Secondary structure of cell-penetrating peptides controls membrane interaction and insertion. Biochim Biophys Acta. 2010;1798:1119–1128. doi: 10.1016/j.bbamem.2010.03.005. [DOI] [PubMed] [Google Scholar]
- 50.Combet C, Blanchet C, Geourjon C, Deleage G. NPS@: network protein sequence analysis. Trends Biochem Sci. 2000;25:147–150. doi: 10.1016/s0968-0004(99)01540-6. [DOI] [PubMed] [Google Scholar]
- 51.Hart DJ, Speight RE, Cooper MA, Sutherland JD, Blackburn JM. The salt dependence of DNA recognition by NF-kappaB p50: a detailed kinetic analysis of the effects on affinity and specificity. Nucleic Acids Res. 1999;27:1063–1069. doi: 10.1093/nar/27.4.1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.