

Abstract
Stat3 is a latent transcription factor that promotes cell survival and proliferation and is often constitutively active in multiple cancers. Inhibition of Stat3 signaling pathways suppresses cell survival signals and leads to apoptosis in cancer cells, suggesting direct inhibition of Stat3 function is a viable therapeutic approach. Herein, we identify a small molecule, C48, as a selective Stat3-family member inhibitor. To determine its mechanism of action, we used site-directed mutagenesis and multiple biochemical techniques to show that C48 alkylates Cys468 in Stat3, a residue at the DNA-binding interface. We further demonstrate that C48 blocks accumulation of activated Stat3 in the nucleus in tumor cell lines that over-express active Stat3 leading to impressive inhibition of tumor growth in mouse models. Collectively, these findings suggest Cys468 in Stat3 represents a novel site for therapeutic intervention and demonstrates the promise of alkylation as a potentially effective chemical approach for Stat3-dependent cancers.
Keywords: Stat3, small molecule inhibitor, alkylation, cysteine, cancer, drug development
Signal Transducers and Activators of Transcription (STATs) comprise a family of transcription factors that transmit signal responses triggered by cytokines, growth factors and hormones (1-3). Upon ligand engagement, STATs are recruited to cell surface receptors and become phosphorylated at a single carboxy-terminal tyrosine by receptor-intrinsic, receptor-associated or non-receptor tyrosine kinases. Tyrosine phosphorylation of STATs then initiates their nuclear translocation, resulting in binding of activated STAT dimers to specific promoter regions in DNA and regulation of target gene expression (4-7). Although STATs exist as non-phosphorylated and phosphorylated dimers, it has been speculated that the conformational change that occurs after tyrosine phosphorylation stabilizes the dimer and improves its ability to bind to specific DNA regions (8-13).
The STAT family includes seven members, namely Stat1–Stat4, Stat5a, Stat5b, and Stat6 (1). In normal cells, STAT signaling is usually transient and tightly regulated by cytokines and growth factors. However, constitutive activation of some STATs, including Stat3 and Stat5, has been detected in a large number of diverse human cancer cells and primary tumor tissues, including blood malignancies and solid tumors (14, 15). Overwhelming evidence indicates that hyperactivation of Stat3 signaling leads to enhanced tumor cell proliferation and survival (16), and that this signaling pathway is a valid target for the development of cancer therapeutics. Moreover, Stat3 itself has also been identified as an oncoprotein (17) and a valid target for cancer drugs. In contrast, aberrant Stat1 signaling has been associated with suppression of cell growth rather than malignant transformation, and thus can act as a potential tumor suppressor (18). Because of the opposing effects of Stat1 and Stat3 on oncogenesis, and their close structural similarity, it is important to develop drugs that can distinguish between Stat1 and Stat3.
Direct inhibition of the activated Stat3 dimer or the Stat3-DNA interaction is an attractive approach for cancer therapeutics, and does not require knowledge of possible upstream activators of Stat3 in any given tumor type. However, disrupting protein-protein or protein-DNA interactions with small molecules is challenging and also an emerging therapeutic approach. We used molecular dynamics (MD) and modeling studies to identify potential leads that were experimentally tested using electrophoretic mobility shift assay (EMSA) studies. NCI compound NSC-368262 (C48) was found to block DNA-binding of Stat3, but not of Stat1. Additional analogs were tested, but these showed little or no improvement in efficacy of Stat3-DNA inhibition. While no structure-activity relationship could be identified, closer examination of the chemical properties of the parental fragment suggested that C48 and the selected analogs alkylate Stat3. Comparison of the Stat3 and Stat1 structure showed that Stat3 possesses a unique surface cysteine, Cys468, that is located in the DNA-binding region of Stat3. The equivalent residue in Stat1 is a serine. Through mutagenesis, we demonstrate that alkylation of Cys468 inhibits Stat3 DNA-binding. We further show that alkylation of Stat3 by C48 inhibits proliferation and survival of tumor cell lines harboring constitutive Stat3 DNA-binding activity. In addition, C48 shows anti-tumor efficacy in xenograft and syngeneic mouse models using human and mouse breast cancer cells, respectively. Therefore, we have identified C48 as a potential small-molecule inhibitor of aberrant Stat3 signaling. Moreover, we provide evidence that selective alkylation of Stat3 at Cys468 constitutes a viable site for further development of selective Stat3 inhibitors for cancer treatment.
RESULTS
Screening of potential inhibitors of Stat3 DNA-binding activity
To identify potential Stat3 inhibitors, virtual ligand screening (VLS) using a commercial chemical library was performed using a snapshot of Stat3 from a previous MD simulation (12). Regions in Stat3 that showed low sequence similarity to Stat1 were identified as docking sites for VLS to enhance subtype selectivity. Of the 7000 ligands that docked to a distinct site, 437 compounds were selected based on ligand-buried surface area (>75%) and favorable van der Waals and hydrogen bond energies. Side chain rotamers for residues within 5 Å for each docked ligand were minimized. The ligands were sorted by the calculated binding energies (including desolvation energy of the ligand) and clustered by similarity in chemical functionalities, producing 52 compounds representative of the structural diversity.
Each compound was tested by EMSA experimentally and AP-906/42850375 (C36) was identified as a potential Stat3 inhibitor (Fig. 1A). C36 inhibited Stat3 DNA-binding activity with an IC50 of 30–50 μM (not shown). Next, using this compound as a template to search a publicly available 85,563 chemical library, as well as the National Cancer Institute (NCI) chemical library (about 300,000 compounds), we selected 48 new compounds that structurally resembled C36. EMSA identified three additional compounds that inhibited Stat3 DNA-binding activity at concentrations of 100 μM or lower: AP-906/42850385 (C29), AP-906/42850377 (C30) and NSC-368262 (C48) (Fig. 1A). All four compounds inhibited Stat3 DNA-binding activity with IC50 values of 10–50 μM (not shown). Further analysis, however, revealed that compounds C29, C30 and C36 lost their Stat3 inhibitory activity when dissolved and stored in aqueous solutions for 24 h or longer prior to EMSA analysis. In contrast, in EMSA, C48 did not show any loss of activity when stored in aqueous solution for 7 days or longer, even through repeated freeze-thaw cycles.
Figure 1. Structure of Stat3 inhibitor compounds and effect of C48 on STAT DNA-binding activity.
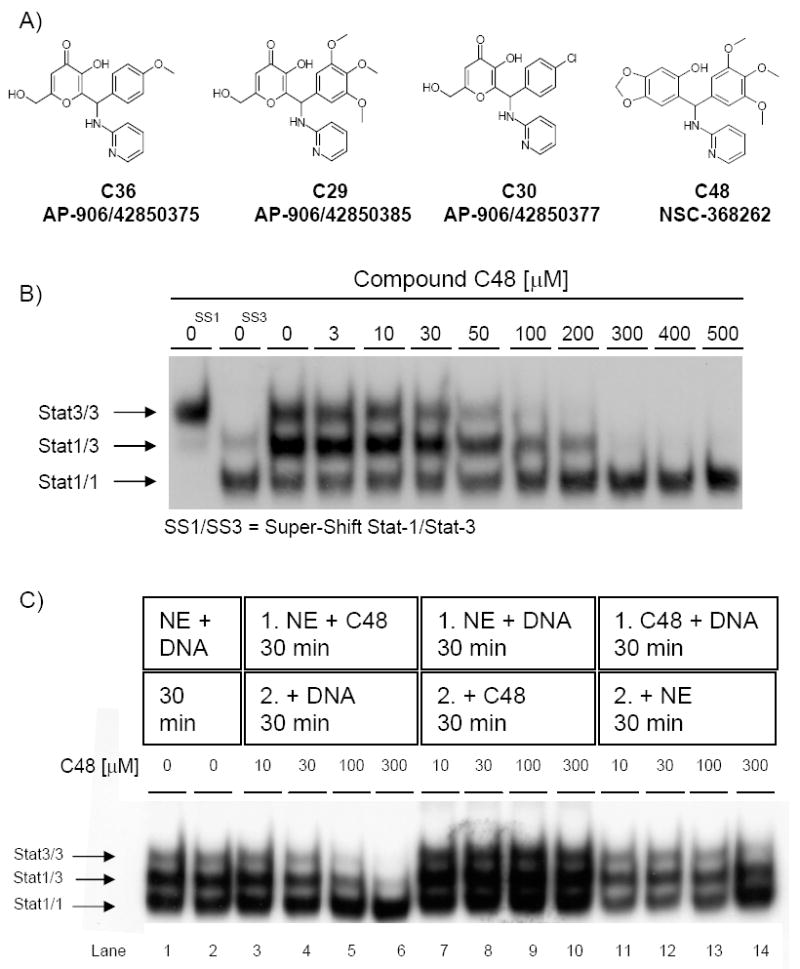
A) Structure of the initial lead Stat3 inhibitor compound (C36) and structures of Stat3 inhibitor compounds identified through a similarity search (C29, C30 and C48). B) Dose-response effect of compound C48 on the DNA-binding activity of Stat1 and Stat3 homodimers, as well as Stat1/3 heterodimers in vitro, as assessed by EMSA. C48 and nuclear extract (NE) that contained activated STAT proteins were pre-incubated for 30 min prior to addition of DNA for 30 min. SS, Super-shifted STAT proteins. C) Mode of action of C48 on STAT DNA-binding. As a control, NE was incubated with DNA for 30 minutes (lanes 1-2), C48 and NE that contained activated STAT proteins were pre-incubated prior to addition of DNA (lanes 3-6), NE and DNA were pre-incubated prior to addition of C48 (lanes 7-10), or C48 and DNA were pre-incubated prior to addition of NE (lanes 11-14).
To assay for Stat3 subtype selectivity of compound C48, we performed EMSA using nuclear extract derived from Sk-Mel-5 human melanoma cells stimulated with IFN-γ. This extract contains activated Stat3/Stat3 and Stat1/Stat1 homodimers, as well as activated Stat1/Stat3 heterodimers, all of which bind to the same radiolabeled DNA probe. C48 blocked the DNA-binding activity of phosphorylated Stat3 homodimers (IC50 10–50 μM), without inhibiting the DNA-binding of phosphorylated Stat1 homodimers, even at concentrations as high as 500 μM (Fig. 1B). Moreover, C48 also disrupted the DNA-binding activity of Stat1/Stat3 heterodimers, although at a slightly higher concentration (IC50 50–100 μM). These data show that C48 specifically blocks DNA-binding activity of phosphorylated Stat3 homodimers and Stat1/Stat3 heterodimers, and suggest that C48 possesses subtype selectivity for Stat3 over Stat1.
Order-of-addition is critical for inhibition of Stat3-DNA complex formation by compound C48
To further examine the mode of action of C48 on Stat3 DNA-binding, we performed EMSA analysis as described above, while varying the order-of-addition of the interaction components. Pre-incubation of C48 with nuclear extract that contained activated Stat3 and Stat1, followed by addition of the oligonucleotide probe, prevented de novo binding of Stat3 homodimers and Stat1/Stat3 heterodimers to DNA (Fig. 1C, lanes 3-6). In contrast, pre-incubation of the nuclear extract with DNA, followed by addition of C48, did not disrupt pre-existing Stat3 DNA-binding (Fig. 1C, lanes 7-10). Similarly, pre-incubation of C48 with DNA, followed by addition of the nuclear extract, did not markedly affect STAT DNA-binding activity (Fig. 1C, lanes 11-14). These results demonstrate that compound C48 inhibits Stat3 DNA-binding activity only when allowed to interact with Stat3 protein before Stat3 protein encounters its DNA-binding site. Although these data suggest that compound C48 directly interacts with activated Stat3 protein, the order-of-addition requirement to inhibit the Stat3-DNA interaction suggests that compound C48 binds to a Stat3 site that is only accessible before Stat3 binds to DNA.
Compound C48 alkylates cysteine 468 of Stat3
Examination of compound C48, as well as C36 and the other compounds, indicated that the benzylic carbon is potentially reactive, suggesting that these compounds might alkylate cysteine residues. Furthermore, examination of the Stat3 crystal structure indicated several surface exposed cysteines that could be modified (Fig. 2A) (19). Significantly, Cys468 of Stat3 is at the Stat3-DNA interface and its modification would sterically block DNA binding (Fig. 2B). Moreover, serine occupies the equivalent position in Stat1 and Stat5 suggesting a potential mechanism for the isoform specificity of C48 (Fig. 2C). To test whether Cys468 is susceptible to alkylation by C48, this residue was mutated to serine resulting in the generation of phosphorylated C468S Stat3 (e.g., phosphorylated Tyr705) mutant. As a control, phosphorylated wildtype Stat3 was utilized. We first demonstrated that both phosphorylated recombinant proteins, wildtype and C468S Stat3 mutant, bound to DNA (Fig. 3). Next, the wildtype and C468S phospho-Stat3 proteins were treated with increasing concentrations of compound C48. EMSA studies revealed no significant effect of C48 on DNA binding of C468S Stat3 (Fig. 3A, second row).
Figure 2. Surface representation of Stat3 bound to DNA.
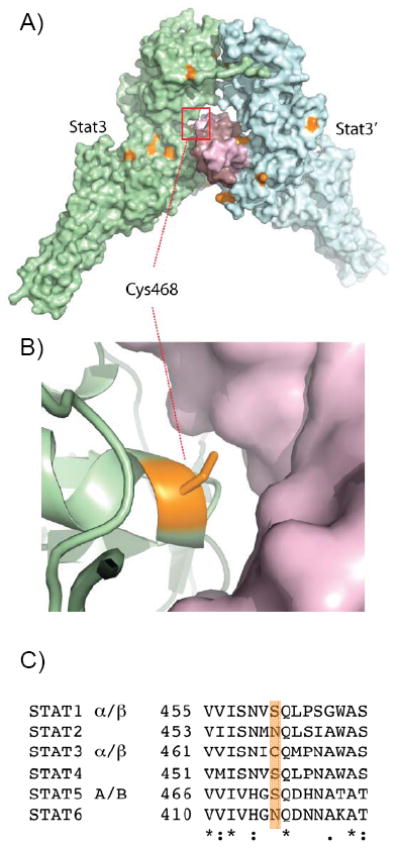
A) The crystal structure of phosphorylated Stat3-β showing the Stat3 homodimer (cyan and lime) bound to DNA (pink) (PDB:2BG1). All 11 cysteine residues in Stat3-β are colored orange. B) Cys468 at the DNA interface is within 4.2 Å of direct contact with DNA. C) Sequence alignment of all human STAT family members showing that the cysteine at position 468 is unique to Stat3 (orange highlight).
Figure 3. Effect of C48 on wildtype and mutant STAT homologs.
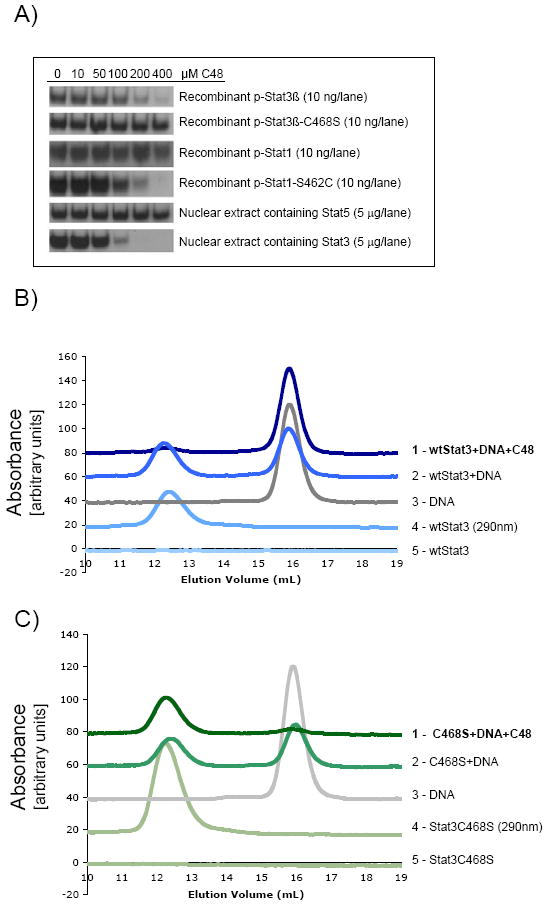
A) Dose-response effect of compound C48 on DNA-binding activity of wildtype and mutant STAT homologs in vitro, as assessed by EMSA. Recombinant STATs or nuclear extract that contained activated STAT proteins was pre-incubated with C48 for 30 min prior to addition of DNA for 30 min. DNA-binding properties of STATs as assessed by size exclusion chromatography: B) Wildtype Stat3 and C) C468S mutant Stat3 in the presence and absence of 100 μM C48. Unlike the C468S Stat3 mutant (trace 1 in panel C), formation of wildtype Stat3-DNA complex was inhibited upon incubation with C48 (trace 1 in B). Each chromatogram was monitored at 495 nm except trace 4 in both panels.
To further establish the importance of Cys468 alkylation in Stat3 as a modality for disrupting STAT/DNA interactions, a similar approach was studied using Stat1, which possesses a serine instead of a cysteine in the equivalent position (Fig. 2C). We reasoned that mutating Ser462 in Stat1 to Cys, would cause the mutant Stat1 to become sensitive to C48. In the absence of C48, both recombinant, phosphorylated Stat1 and recombinant, phosphorylated S462C Stat1 bound DNA with similar affinity (Fig. 3A, rows 3 and 4, respectively). However, as concentrations of C48 increased, the S462C mutant lost its ability to interact with DNA (Fig. 3A, row 4). As expected, DNA-binding of wildtype Stat1 was not affected by C48 (Fig. 3A, row 3). Finally, the DNA-binding property of phosphorylated Stat5 (nuclear extract isolated from K562 chronic myelogenous leukemia cells) was also insensitive to C48 exposure (Fig. 3A, row 5), whereas DNA-binding of phosphorylated Stat3 (nuclear extract isolated from Sk-Mel-5 melanoma cells) was inhibited by C48 (Fig. 3A, row 6). Just as for wildtype Stat1, in wildtype Stat5 serine is the equivalent residue to Cys468 in Stat3 (Fig. 2C).
In addition to EMSA, size exclusion chromatography (SEC) was used to further confirm that C48 blocks DNA binding. In these experiments, DNA was fluorescently labeled, which enabled direct monitoring of its hydrodynamic properties at 495 nm (e.g., eliminate spectral overlap of the DNA and protein signal at either 260 nm or 280 nm). SEC analysis of only the fluorescently-labeled DNA produced a peak that eluted at 16 mL (Fig. 3B, trace 3), whereas the admixture of wildtype Stat3 and DNA eluted at 12.5 mL (Fig. 3B, trace 2). SEC analysis of only wildtype phosphorylated Stat3 produced a peak at 12.5 mL when monitored at 280 nm (Fig. 3B, trace 4). Also no signal from Stat3 was observed when monitored at 495 nm (Fig. 3B, trace 5). Finally, treatment of wildtype Stat3 with C48 for 30 min followed by addition of the fluorescent DNA eliminated the peak that eluted at 12.5 mL (Fig. 3B, trace 1). Likewise, in similar SEC experiments, the DNA binding properties of the C468S phospho-Stat3 mutant treated with compound C48 were unaffected (Fig. 3C), whereas wildtype phospho-Stat3 treated with compound C48 failed to bind DNA (Fig. 3A and B). In fact, it appears that the C48 treated C468S phospho-Stat3 mutant binds slightly better than the non-treated mutant. While the precise mechanism for this enhancement in not immediately clear, it further underscores modification of Cys468 in Stat3 abrogates DNA binding. Collectively, this analysis indicates that C48 blocks Stat3 DNA binding.
In addition, mass spectrometry was used to monitor the alkylation of Stat3. The exposure of phosphorylated Stat3 (10 μM) with C48 (600 μM) for 30 minutes produced an ‘envelope’ of peaks, each differing by 317 Dal (Fig. 4A). The most intense peak of the envelope corresponds to six C48 adducts whereas the first peak corresponds to the addition of four C48 adducts. Attempts to identify which cysteine residues in Stat3 are modified by C48 by MS were not successful, possibly due to the nature C48’s benzylic reactivity which may reverse during digestion and work-up. Thus, to confirm that one of these modification of Stat3 is Cys468 and understanding that prolonged exposure to C48 produces an envelope of multiple modifications, we exposed wildtype Stat3 and the C468S Stat3 mutant to C48 for one minute under the same conditions. In the mass spectra, three peaks were observed for wildtype Stat3 that corresponds to the alkylation of three, four, and five C48 residues (Fig. 4B, blue trace). Likewise, three peaks were also observed for mutant Stat3, but these correspond to the formation of two, three and four C48 adducts(Fig. 4B, pink trace). This shift of the envelope to one less adduct suggests that Cys468 is one of the first residues to be modified by C48. As further evidence of the reactive nature of C48 towards sulfhydryl groups, glutathione was used as a model compound. Mixing glutathione with C48 produced a fragment ion spectrum (MS/MS), which confirms alkylation of the glutathione thiol group by C48 (Supplemental Fig. 1).
Figure 4. Modification of Stat3 by C48.
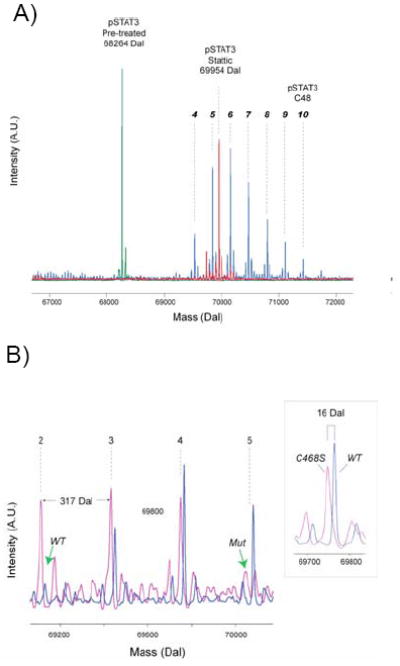
A) Mass spectra of phosphorylated Stat3 treated with C48 and Stattic. Quadrupole time of flight mass spectra of phosphorylated Stat3 before treatment (green trace) and after treatment with C48 (blue trace) and Stattic (red trace). Multiple modifications are observed for Stat3. Based on the expected mass of the C48 fragment, more than 6 modifications were observed corresponding to 4 to 10 C48 adducts. Phosphorylated Stat3 treated with Stattic under the same conditions produce a predominant peak at 69954, which corresponds to the modification of precisely 8 residues.
B) Mutation of Cys468 reduces the number of C48 modifications. Wildtype Stat3 (blue trace) and C468S Stat3 mutant (pink trace), both unphosphorylated, were treated with C48 for one minute at 37 °C and analyzed by mass spectrometry. For wildtype Stat3, the mass spectrum indicates 3 adducts comprising 3, 4 and 5 modifications. For the C468S mutant, the mass spectrum also indicates 3 adducts, but consists of 2, 3 and 4 modifications. The green arrows indicate absence of modifications on wildtype Stat3 (wt) and the mutant Stat3 (mut). The inset shows the mass difference between the wildtype and mutant is precisely 16 daltons, as expected.
Finally, C48 does not affect the dimerization state of phospho-Stat3, as evidenced by SEC and sedimentation equilibrium experiments using analytical ultracentrifugation (Supplemental Fig. 2). These data also point to modification and steric occlusion of the Stat3 DNA-binding site. Collectively, these biochemical studies strongly suggest that the specificity of C48 is due to its ability to alkylate Cys468, which is unique to Stat3.
Stattic alkylates Stat3
Stattic has been reported as a selective inhibitor of Stat3 that is commercially available. This inhibitor possesses a potentially reactive vinyl sulfone moiety. To investigate whether Stattic alkylates Stat3, similar experiments were performed using SEC and mass spectrometry. With SEC, we observe that phosphorylated Stat3 treated with Stattic remains dimeric, but does not bind to DNA (data not shown), which is consistent with C48. Upon mass spectral analysis, we observe a mass increase of 1690 Dal for phosphorylated Stat3 (10 μM) treated with of Stattic (800 μM) after 30 minutes at 37 °C (Fig. 4A). This mass difference corresponds to the addition of eight Stattic molecules which suggests that the mechanism of action by Stattic is through alkylation.
Compound C48 affects Stat3 nuclear localization and Stat3-mediated gene expression
Since C48 is selective for Stat3, we asked whether it would selectively block Stat3-mediated transcription in cell-based assays. An established, stably transfected murine embryonic fibroblast cell line that expressed a Stat3-YFP fusion protein (MEF-Stat3-YFP) (20) was used. This cell line allows for monitoring Stat3 nuclear translocation after stimulation with Oncostatin M (OSM), a known activator of the JAK/STAT pathway (21). Confocal laser scanning microscopy showed that in unstimulated, serum-starved MEF-Stat3-YFP cells, Stat3 is present in nucleus and cytosol and that the majority of Stat3 is located in the cytosol (Fig. 5, left panel). Our observed distribution pattern of Stat3 in unstimulated cells is in agreement with previous findings (22). Upon stimulation with OSM, receptor-associated Janus kinases phosphorylate Stat3 protein at Tyr705, which caused the activated Stat3 dimers to translocate into and accumulate in the nuclei (Fig. 5, second panel from left). Pre-incubation of MEF-Stat3-YFP cells with compound C48 (40 μM) completely prevented OSM-induced nuclear accumulation of Stat3-YFP, as compared to control-treated cells (Fig. 5, fourth panel from left). These data suggest that C48 inhibits Stat3 nuclear accumulation primarily by abrogating its interaction with DNA, permitting its relocation to the cytoplasm. In other words, the productive interaction of phospho-Stat3 with DNA is essential in sequestering Stat3 in the nucleus. In support of this notion, we note that others have observed that Stat3 in non-stimulated cells or Stat3 Y705F mutant continuously shuttles between the cytoplasm and nucleus (22).
Figure 5. Effect of C48 on Stat3 nuclear translocation.
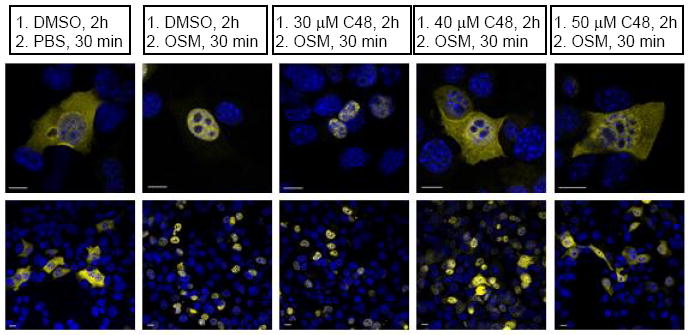
Effect of C48 on Oncostatin M (OSM)-induced activation and nuclear localization of Stat3-YFP protein (yellow) in serum-starved, MEF-Stat3-YFP expressing cells. The nuclei (blue) are stained with DAPI. Not all cells are transfected with the Stat3-YFP construct. Upper panel: 100x, lower panel 10x magnification. One representative result from 2 independent experiments is shown.
To confirm these results, HeLa cells genetically engineered to carry a chromosomal integration of a luciferase reporter gene for which expression is regulated by either activated Stat3 or activated Stat1 was used. These cell lines allow for monitoring Stat1 or Stat3 transcriptional activity through measurement of luciferase activity (i.e., luminescence emission). When compared to OSM-induced Stat3-dependent luciferase activity, pre-treatment of cells with compound C48 prior to stimulation with OSM resulted in dose-dependent inhibition of luciferase activity with an IC50 of 3–10 μM (Fig. 6A). At 20 μM C48, the luminescence emission was significantly lower than that of cells that were not treated with OSM, suggesting there is basal Stat3 activity in unstimulated cells. These results indicate that the effect of C48 on Stat3 DNA-binding activity and Stat3-induced gene-expression in cells can be detected at C48 concentrations as low as 3–10 μM; however, a higher concentration (up to 20 μM) is required to completely abolish Stat3-dependent transcriptional activity. In contrast, pre-treatment of cells with C48 did not dramatically reduce IFN-γ-induced Stat1-mediated luciferase activity as compared to the luciferase activity of untreated IFN-γ-stimulated cells (Fig. 6B). Taken together, the results obtained from these cell-based assays, combined with the in vitro EMSA results (Fig. 1B), suggest that compound C48 disrupts DNA-binding of activated Stat3, but does not significantly affect Stat1-induced signaling.
Figure 6. Effect of C48 on Stat3- and Stat1-mediated transcriptional activity.
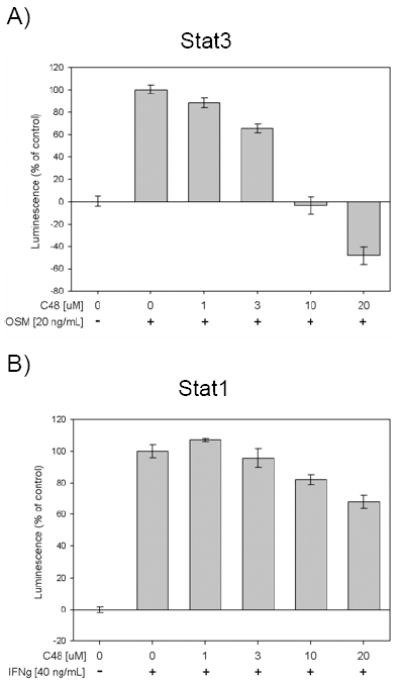
A) Effect of C48 on Oncostatin M (OSM)-induced, Stat3-mediated expression of luciferase as a measure of transcription activity. Serum-starved HeLa-Stat3-Luc cells were pre-incubated with C48 1 h prior to stimulation with OSM. Luminescence was measured 8 h post stimulation. B) Effect of C48 on IFNγ-induced, Stat1-mediated expression of luciferase as a measure of transcription activity. Serum-starved HeLa-Stat1-Luc cells were pre-incubated with C48 1 h prior to stimulation with IFNγ. Luminescence was measured 8 h post stimulation. One representative result is shown (n=3, in triplicate).
C48 inhibits Stat3 signaling and induces apoptosis in human cancer cells
The human breast cancer cell lines MDA-MB-468 and MDA-MB-231 harbor constitutive phosphorylation of Stat3 Tyr705, and thus, constitutive Stat3 DNA-binding activity. These cell lines have been shown to undergo apoptosis upon inhibition of Stat3 signaling using small-molecule inhibitors (23). In contrast, the human prostate cancer cell line LNCaP does not exhibit constitutive Stat3 DNA-binding activity, and therefore, does not rely on Stat3 signaling for survival. We treated these cell lines with C48 and measured cell viability by AnnexinV/PI staining as assessed by flow cytometry. MDA-MB-468 and MDA-MB-231, but not LNCaP cells, underwent apoptosis following a 48 h treatment with 1–20 μM C48 (Fig. 7A, B and C), and the IC50 for induction of apoptosis was 10–20 μM C48, a concentration range similar to that needed to block Stat3 DNA-binding activity in vitro and to inhibit Stat3 transcriptional activity in vivo. Western blot analysis of lysates from MDA-MB-468 cells treated for 48 h with C48 revealed much less Stat3 Tyr705 phosphorylation in cells treated with 10 μM C48 compared to untreated cells, and complete inhibition of phosphorylation with 20 μM C48 (Fig. 7D). In contrast, the activation state of p44/p42 MAP kinases was not significantly inhibited by C48, even when used at 20 μM. In addition, expression of Mcl-1, a pro-survival gene and known downstream target of Stat3, was fully blocked in cells treated with 20 μM C48. In addition, when treated with 20 μM C48, the majority of MDA-MB-468 cells are undergoing apoptosis, as indicated by the appearance of cleaved PARP fragment, which is comparable to the Annexin/PI results (Fig. 7A). These data suggest that compound C48 exerts its ability to kill human tumor cells at least in part through inhibition of Stat3 signaling.
Figure 7. Effect of C48 on cell viability and Stat3 signaling pathway proteins.
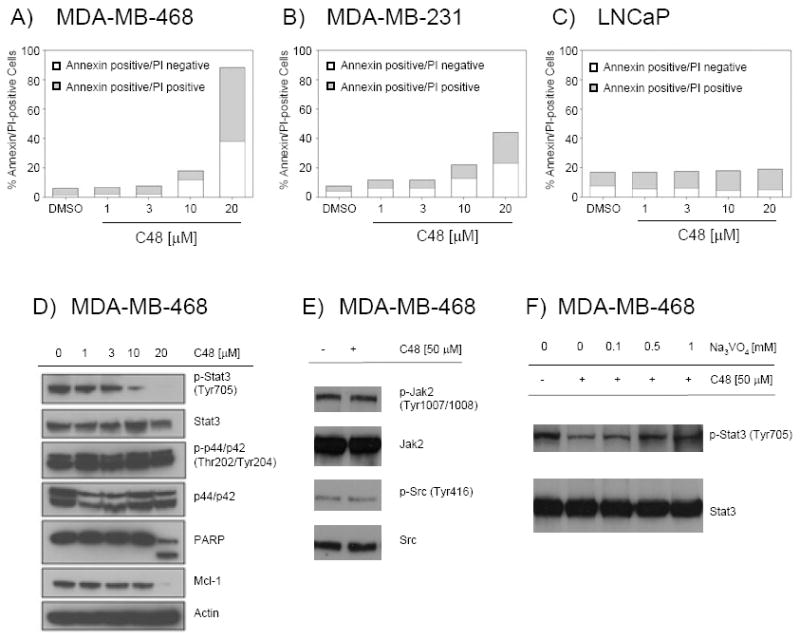
A-C) Effect of C48 on viability of the Stat3-pTyr705-positive cell lines MDA-MB-468 (A) and MDA-MB-231 (B) and the Stat3-pTyr705-negative cell line LNCaP (C). Cells were treated for 48 h with C48 or DMSO control and analyzed for Annexin V/PI staining. One representative result per cell line is shown (n=2, in triplicate). D-F) Western blot of total protein lysate from MDA-MB-468 cells treated with C48. D) Cells were treated for 48 h with C48 in the concentrations indicated prior to analysis for signaling proteins. E) Cells were treated for 4 h with 50 μM C48 prior to analysis of Jak2 and Src family phosphorylation. F) Cells were pre-treated for 30 min with Na3VO4 in the concentrations indicated, followed by addition of 50 μM C48 for 2 h. Cells were then analyzed for Stat3-pTyr705 levels.
Since we observed a decrease in the phosphorylation level of Stat3 upon treatment with C48, the question whether compound C48 could inhibit upstream activators of STAT proteins was addressed. Western blot analysis of lysates from MDA-MB-468 cells treated for 4 h with 50 μM C48 revealed no inhibition of phospho-Jak2 (Tyr1007/1008) and phospho-Src family (Tyr416) proteins (Fig. 7E). In addition, in vitro kinase activity assays were performed on Jak1 and Jak2 kinases. As shown in Supplemental Figure 3, the IC50 of compound C48 for inhibition of Jak1 and Jak2 kinases > 400 μM, the highest concentration used in this assay. Staurosporine was used as a positive control and inhibited the activity of both Jak1 and Jak2 kinase with an IC50 of < 1.0 nM. These data suggest that compound C48 does not inhibit the STAT upstream activators Jak1 and Jak2.
To test whether blocking the Stat3-DNA interaction would make phosphorylated Stat3 more susceptible to phosphatase activity, MDA-MB-468 cells were treated with compound C48 in the presence and absence of sodium orthovanadate, a phosphatase inhibitor. We observe that phosphorylation levels of Stat3 in MDA-MB-468 cells pre-treated for 30 min with 1 mM sodium orthovanadate prior to the addition of C48 for 2 is similar to that of untreated cells. These data suggest that treatment of cells with compound C48 facilitates the dephosphorylation of Stat3.
C48 suppresses the in vivo growth of MDA-MB-468 human breast cancer cells in a xenograft mouse model
Several studies have shown that use of small molecule inhibitors to interfere with Stat3 signaling results in growth inhibition of human tumor xenografts (23). We extended these studies to evaluate the antitumor efficacy of C48 in a nude mouse model with subcutaneously established MDA-MB-468 human breast tumor xenografts with constitutively active Stat3. Tumor-bearing mice were given i.p. injections of C48 (200 mg kg-1) or vehicle control (ddH20 supplemented with 2% Tween-80) daily for 5 days per week, 8 weeks total, and tumor measurements were taken twice a week. As compared with tumors from control-treated mice, which continued to grow, tumors from C48-treated mice displayed significant growth-inhibition (Fig. 8A).
Figure 8. Effect of C48 on in vivo growth of a human breast tumor xenograft and a syngeneic mouse model of murine breast adenocarcinoma.
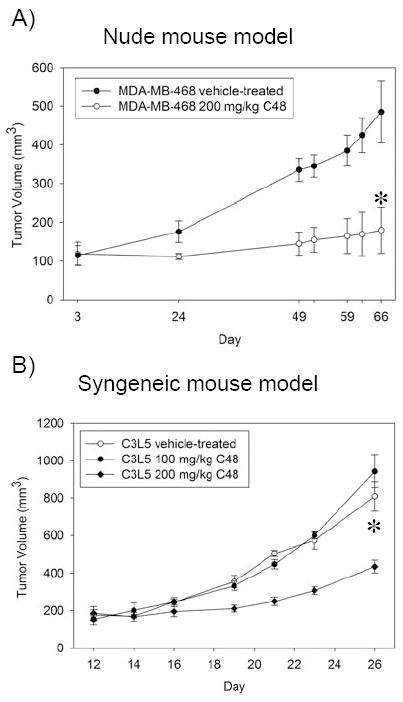
A) Breast tumor xenograft mouse model. Tumor volume of MDA-MB-468 tumor-bearing mice treated once daily for 5 days a week with C48 (white circle) at 200 mg kg-1 (delivered i.p.) or vehicle (black circle) until termination of the experiment (day 66). B) Syngeneic mouse model. Tumor volume of C3L5 tumor-bearing mice treated (i.p.) once daily for 5 days a week with C48 at 100 mg kg-1 (black circle) or 200 mg kg-1 (black diamond), or vehicle control (white circle) until termination of the experiment (day 26). Results are presented as mean tumor volume (n=8). Bars represent standard error. *, P< 0.05 when vehicle-treated control group is compared to 200 mg kg-1 C48-treated group.
C48 suppresses the growth of C3L5 murine breast cancer cells in a syngeneic mouse model
We also tested the effect of C48 in a syngeneic mouse model of breast cancer. C3L5 murine breast adenocarcinoma cells that possess constitutively active Stat3 were injected subcutaneously in C3H/HeJ mice. Tumor-bearing mice were i.p. injected with C48 (100 mg kg-1 or 200 mg kg-1) or vehicle control (ddH20 supplemented with 2% Tween-80) every day for 5 days per week. The tumors were measured every other day for 26 days. Similar to the xenograft model, tumors from control-treated mice continued to grow, while tumors from mice treated with 200 mg kg-1 C48 showed significant growth-inhibition (Fig. 8B).
DISCUSSION
As a central point to integrate and amplify oncogenic signals, transcription factors represent attractive targets to develop therapies broadly applicable to different forms of cancer and other diseases. However, successfully blocking the activity of transcription factors remains challenging. More specifically, protein-protein or protein-DNA interfaces are generally spread over large, relatively flat surfaces, and often do not provide sufficient cavities to develop high affinity, small molecule antagonists. Targeted, chemical modification of a residue located at or in the immediate vicinity of a protein-protein or protein-DNA interface, on the other hand, offers a potential solution to antagonize interactions characterized by extensive interfaces.
C48 was originally identified by VLS to bind to a novel site near the SH2 domain. While this cannot be ruled out, the main mechanism of action is through alkylation of Cys468. Furthermore, C48 abrogates Stat3 DNA-binding activity in vitro and induces apoptosis in two human tumor cell lines with constitutively active Stat3 signaling, but not in a human tumor cell line that lacks Stat3 activation. Finally, C48 was found to possess significant in vivo anti-tumor activity in both a human breast cancer xenograft mouse model and a syngeneic mouse model of breast cancer that harbored persistent Stat3 activation. Collectively, these data provide compelling evidence that C48 is a potential lead compound to antagonize Stat3 activity and that Cys468 is a viable site for further development of irreversible Stat3 inhibitors.
The results of this study may have broader implications to other known Stat3 inhibitors as well. Recently, a number of small molecule inhibitors of Stat3 have appeared in the literature. These include stattic, curcubitacins, withacnistin, and S3I-201, some of which were also identified via molecular modeling and virtual screening. What these molecules have in common is a reactive chemical moiety. Stattic, withacnisitin, and curbubitacins can be classified as Michael acceptors possessing vinyl sulfone and α,β unsaturated ketone functionalites. Withacnistin also contains a potentially reactive epoxide. S3I-201 possesses an α-keto tosylate group which is a potent alkylating agent in general. While we have shown that stattic readily alkylates Stat3 (Fig 4A), all of these reagents are capable of modifying cysteine residues even though they were not reported as such nor has this mode of action been ruled out. Despite being identified as SH2 inhibitors through VLS, we suspect that some of these previously reported Stat3 inhibitors are most likely acting through alkylation of Cys468.
While irreversible inhibitors are necessarily reactive and may potentially react with other targets, the clinical application of irreversible inhibitors and their continued development is promising. For instance, exemestane is a potent, irreversible aromatase inhibitor used for the treatment of estrogen-dependent breast cancer (24, 25). Likewise, the irreversible EGFR/HER2 inhibitor, BIBW 2992, developed by Boehringer-Ingelheim, is in phase III clinical trials for non-small cell lung cancer (26, 27). Recently, the modification of Cys179 in IKKβ by CDDO (2-cyano-3,12-dioxooleana-1,9,-dien-28-oic acid) was shown to inhibit IKKβ kinase activity, which in turn fails to activate NF-κB (28, 29). As such, CDDO sensitizes cells to apoptotic pathways and is currently in clinical trails to treat lymphoma and solid tumors (trial ID: NCT00352040). Our data strongly suggest that Stat3 is very sensitive towards alkylation resulting in potent inhibition of tumor growth in animal models. There are other cysteines in Stat3 for potential targeting as well, including Cys712. This cysteine is located in the SH2 domain of the tyrosine binding site and provides an excellent opportunity for developing selective Cys712 alkylators by way of SH2 binding. Studies along these lines are currently in progress. The successful application and continued development of “irreversible” inhibitors for Stat3 offers a promising approach to a new class of therapeutic agents for the treatment of cancer.
METHODS
Virtual ligand screening
Virtual ligand screening (VLS) analysis was performed on the Stat3 dimer by docking 85,563 compounds from a City of Hope database of commercially available chemical libraries using the GLIDE-HTVS procedure from Schrodinger (30). A box size of 34 Å × 34 Å × 34 Å that covered the entire SH2 domain of the Stat3 dimer was used for the VLS procedure. We used a snapshot of the DNA-bound Stat3 dimer extracted from the MD simulations (12). The van der Waals radii were scaled for the ligand atoms by 0.5. Poses with sum of Coulomb and van der Waals energy greater than 100.0 kcal/mol were rejected. The docked poses from the VLS procedure were first sorted and filtered by buried surface area of the ligands calculated using Connolly surface area calculations (31). Ligand poses greater than 75% buried surface were retained and were further filtered by van der Waals and hydrogen bonding energies. Side chain rotamers of residues within 5 Å of the ligand in the binding site were reassigned using Prime module from Schrodinger Inc. (30). The binding energies of the resulting optimized conformations were calculated as Ebinding = Potential energy (ligand in protein) − Potential energy (ligand in water). The potential energy of the ligand was calculated by fixing the protein and using all atom forcefield OPLS using the Macromodel module (30). Finally, the ligand docked conformations were sorted by binding energies and then clustered by structural similarity.
Molecular biology
Stat3β (residues 127-722) and Stat1β (residues 132-712) were amplified from murine and human cDNA, respectively. The corresponding genes were inserted between BamH1 and Xho1 sites of a modified pET28b vector encoding an N-terminal 6-His tagged SMT3 protein. The construct was verified by DNA sequencing (City of Hope DNA Sequencing Core). The Stat3 C468S and Stat1 S462C mutants were generated using the QuikChange Site-Directed Mutagenesis protocol (Stratagene) and verified by DNA sequencing.
To express recombinant phospho-Stat3 and phospho-Stat1 (wildtype and mutants), each plasmid was transformed into Escherichia coli (BL21(DE3) TK [TKB1] strain) (Stratagene), which harbors an inducible tyrosine kinase that can phosphorylate STAT proteins. A two-step induction protocol was followed during the expression procedure. Cells were grown to a density of A600 = 0.5−0.7, at which point IPTG was added to a final concentration of 0.25 mM. After 3 h of induction, cells expressing Stat3 or Stat1 were harvested and resuspended in TK induction medium containing 53 μM indoleacrylic acid. The tyrosine kinase-expressing culture was harvested after 4 h of incubation, resuspended in 1X PBS and stored at -80 °C.
Protein constructs were purified using standard methods. Briefly, cells were lysed by French press, clarified by centrifugation and loaded onto a Ni-NTA column (Qiagen). After an extensive wash (20 column volumes), the protein was eluted with an imidazole gradient. The SMT3 fusion was cleaved by Ulp1 protease, precipitated with 40% ammonium sulfate, dialyzed, and applied to a Superdex G200 preparative column (GE Healthcare). We observed that phospho-Stat3 eluted earlier than purified non-phospho-Stat3. The fractions were analyzed by a 10% SDS-polyacrylamide gel and fractions about the center of the peak were flash frozen at -80 °C. Each construct was purified in a similar manner.
Analytical size exclusion chromatography
6-FAM™– (Fluorescein, Ex: 495 nm, Em: 520 nm) labeled DNA oligonucleotides (Fl-DNA) were ordered from Integrated DNA Technologies with the following sequences: 5’TCATTTCCCGTAAATCCCTA3’ and 5’TAGGGATTTACGGGAAATGA3’. The two strands were annealed and stored at -20 °C. Phospho-Stat3 (wildtype or C468S mutant; 10 mM) was incubated with Fl-DNA (30 min, 37 °C) before being loaded onto an analytical Superdex G200 column (GE Healthcare). The elution profile was monitored at 290 and 495 nm. To determine the effect of C48, C48 (100 μM) was incubated (30 min, 37 °C) with the phospho-Stat3-Fl-DNA complex and analyzed as above.
Mass Spectrometry LC/MS analysis
Samples were analyzed using a Synapt G2 quadrupole time of flight mass spectrometer and a NanoAcquity HPLC (Waters). Samples were separated using a 25mm × 150 μm ID column home-packed with Intrada WP-RP silica (Imtakt, Philadelphia, PA). The samples were loaded in a trapping only mode at 4 μl/min, then eluted into the mass spectrometer at 600 nl/min. Samples were eluted using a gradient from 3% B to 40% B in 9 minutes, followed by a ramp from 40% B to 95% B over 2 minutes. Buffer A was 0.1% Formic acid in water. Buffer B was 0.1% Formic acid in acetonitrile. Protein spectra were averaged over the width of the elution peak and deconvoluted using the Waters provided Maximum Entropy deconvolution software.
Cell lines and cell culture
The human breast cancer cell lines MDA-MB-468 and MDA-MB-231, the human prostate cancer cell line LNCaP, the human melanoma cell line Sk-Mel-5 and the human chronic myelogenous leukemia cell line K562 were purchased from the American Type Culture Collection. The murine breast adenocarcinoma cell line C3L5 is maintained in the laboratory of Dr. John Yim (City of Hope). The Stat1 and Stat3 human cervical cancer reporter cell lines HeLa-Stat1-Luc and HeLa-Stat3-Luc were obtained from Promega. MEF-Stat3-YFP mouse embryonic fibroblast cells that express YFP-labeled, but not wildtype Stat3, were produced by Dr. Andreas Herrmann (City of Hope). All cell lines were maintained in DMEM supplemented with 5% FBS, 50 U mL-1 Penicillin and 50 μg mL-1 Streptomycin. Hygromycin B (500 μg mL-1) was used as selection marker for the reporter cell lines and MEF cells.
Western blot analysis
For Western blotting, cells were washed in 1X PBS containing 1 mM Na-ortho-vanadate and lysed in TGH buffer (1% Triton X-100, 10% glycerol, 50 mM NaCl, 50 mM HEPES, 1 mM EGTA, 1% Na-deoxycholate, 1 mM Na-ortho-vanadate, 2 μg/mL aprotinin, 0.5 μg/mL leupeptin, 50 μg/mL antipain and 1 mM PMSF). Total protein amount was determined using the Bio-Rad Protein Assay reagent, and equal amounts of total protein were loaded in each lane of a 10% SDS-polyacrylamide gel. Following electrophoresis, the proteins were transferred to PVDF membrane, washed with PBS/0.1% Tween-20, blocked with PBS/5% milk and incubated in 1X PBS/5% BSA at 4 °C overnight with the first antibody. The membrane was then washed with PBS/0.1% Tween-20, incubated for 1 h at room temperature with alkaline phosphatase-linked anti-rabbit or anti-mouse secondary antibodies and visualized using SuperSignal West Pico Reagent (Pierce). For detection of total Stat3, total Src, total Jak2 and total p44/p42, the corresponding phospho blots were incubated with stripping buffer (2% SDS, 64 mM Tris, pH 6.7, 0.7% β-Mercaptoethanol) and reblotted. Primary antibodies: total-p44/p42 MAP Kinase (Erk1 and Erk2, Cat#9102), phospho-p44/p42 MAP Kinase (Thr202/Tyr204, Cat#9101), phospho-Stat3 (Tyr705, Cat#9131), Mcl-1 (Cat#4572), total Jak2 (D2E12, Cat#3230), phospho-Jak2 (Tyr1007/1008, Cat#3771S), phospho-Src (Tyr416, Cat# 2101S), and PARP (Cat#9542) were from Cell Signaling. Total Stat3 antibody (C-20, Cat#sc-482) was from Santa Cruz Biotechnology. Anti-Src (GD11, Cat# 05-184) was from Upstate. Anti-β-actin antibody was from Sigma (Cat#A5441).
Nuclear translocation assay and confocal microscopy
MEF-Stat3-YFP cells were plated on poly-L-Lysine-coated coverslips (24 well). After 24 h, the cells were pre-incubated for 2 h with up to 50 μM compound C48 in serum-free medium, followed by stimulation with Oncostatin M (25 ng/mL) for 30 min. Cells were fixed in formaldehyde and analyzed by confocal laser scanning microscopy as described (32). Briefly, slides were mounted with Vectashield hardSet mounting medium containing DAPI (Vector laboratories). Confocal imaging was carried out on a Zeiss LSM 510Meta confocal microscope equipped with a 63X 1.2 NA Zeiss water immersion objective. YFP signals were detected as previously described (22). DAPI was visualized using a two photon laser exciting at 435–485 nm. The images shown represent confocal slices of approximately 1 μm thickness.
Luciferase assay system for the measurement of Stat3- and Stat1-mediated transcriptional activity
Hela-Stat3-Luc and Hela-Stat1-Luc cells express the luciferase protein under the control of activated Stat3 and Stat1 protein, respectively. To measure the effect of C48 on STAT-mediated transcriptional activity, 10,000 Hela-STAT-Luc cells in DMEM containing 5% FBS were seeded in each well of a 96-well plate. The next day, cells were incubated for 2 h with 1 - 20 μM C48. Oncostatin M (OSM; 25 ng mL-1; Sigma) or Interferon-γ (IFN-γ; 25 ng mL-1; Sigma) was then added to each well to induce Stat3 (OSM) and Stat1 (IFN-γ) activation and luciferase expression. Eight hours later, cells were lysed and luciferase activity was measured as per the manufacturer’s instructions (Promega).
Nuclear extract preparation and EMSA
To detect the DNA-binding activity of Stat1, Stat3, and Stat5 by EMSA, nuclear protein extracts were prepared using high-salt extraction as previously described (33). To test potential inhibitors of Stat3 DNA-binding, nuclear protein (5 μg) from Sk-Mel-5 human melanoma cells (source of activated Stat3/Stat3 homodimers) or Sk-Mel-5 cells stimulated with interferon-γ for 15 min (source of activated Stat1/Stat1 and Stat3/Stat3 homodimers, as well as Stat1/Stat3 heterodimers) was incubated (30 min, 37 °C) with 3-500 μM C48 at the concentrations indicated. Then a 32P-radiolabeled, double-stranded DNA oligonucleotide that binds Stat3/Stat3 and Stat1/Stat1 homo- and Stat1/Stat3 heterodimeric proteins was added to the nuclear protein and incubated for another 30 min. This oligonucleotide is a high-affinity variant of the sis-inducible element (hSIE; sense strand, 5’-AgC-TTC-ATT-TCC-CTG-AAA-TCC-CTA-3’) derived from the c-fos gene promoter, which binds activated Stat3 and Stat1 proteins (34, 35). For Stat5 EMSA, 5 μg of nuclear protein from K562 CML cells was incubated with C48 as indicated above and a 32P-radiolabeled, double-stranded DNA oligonucleotide that binds Stat5/Stat5 homodimers (MGFE probe, mammary gland factor element, derived from the bovine β-casein gene promoter, 5’-AgA-TTT-CTA-ggA-ATT-CAA-3’) was used to detect DNA-binding of Stat5. Anti-Stat3, anti-Stat1, and anti-Stat5 polyclonal antibodies (C20X, Santa Cruz Biotechnology) were used to identify Stat3, Stat1, and Stat5 in “super-shift” assays. For super-shift assays, concentrated antibody (1 μL) was pre-incubated with nuclear protein 20 min prior to the addition of radiolabeled probe (30 min, 37 °C) and separation by non-denaturing polyacrylamide gel-electrophoresis and autoradiographic detection.
Tumor models
Xenograft tumor studies were performed as previously described (36). Briefly, six-week-old athymic mice were purchased from Taconic Laboratories and acclimated for 3+ days prior to tumor implantation. All mice were maintained under specific pathogen-free conditions and were used in compliance with protocols approved by the City of Hope Institutional Animal Care and Use Committee. MDA-MB-468 human breast cancer cells (5 × 107) in a 1:1 mixture of Matrigel (BD Biosciences) and culture medium were subcutaneously implanted in the left flanks of nude mice. Tumor-bearing mice were randomized (8 mice per group) based on tumor volume prior to the initiation of treatment, and treatment was initiated when average tumor volume was at least 65 mm3. C48 (200 mg kg-1) was given intraperitoneally (i.p.) once daily for 5 days per week until termination of the experiment. C48 was dissolved in ddH20 supplemented with 2% Tween-80. At this dose, no lethal toxicity, or weight loss (greater than 10% body weight) was observed among treated animals. Tumors were measured twice per week with vernier calipers, and tumor volumes were calculated by the formula 0.5 * (larger diameter) * (smaller diameter) 2.
In the syngeneic mouse model, 7–9-week-old female C3H/HeJ syngeneic mice were subcutaneously injected with 5 × 105 C3L5 murine breast adenocarcinoma cells as previously described (37). Animals were monitored for tumor growth. Once the tumors were palpable (day 12 after tumor cell injection), animals were grouped (8 mice per group) to receive either i.p. injection of C48 (100 mg kg-1 or 200 mg kg-1) or vehicle in a 5 days on/2 days off cycle for 2 cycles. Tumors were measured every other day, by serial measurements of perpendicular diameters using digital calipers. Two-tailed, paired t test was used to calculate p values.
Flow cytometric analysis of Annexin V/PI
MDA-MB-468, MDA-MB-231 and LNCaP cells (1 × 107 each) were seeded into 10 cm dishes and allowed to attach overnight. The next day, medium was replaced with fresh medium (DMEM/5% FBS) containing either DMSO or 1-20 μM C48. Forty-eight hours later, cells were washed twice with cold PBS and harvested in PBS supplemented with Trypsin/EDTA. Cells were washed twice in PBS and stained using the Annexin-V-FITC apoptosis detection kit according to the manufacturer’s instructions (BD Biosciences). Data acquisition and analysis was performed by the Flow Cytometry Core Facility at City of Hope using FACScalibur.
Note Added in Proof
While this manuscript was under revision, a study reported identified six cysteine residues in Stat3 including Cys468 that are sensitive to hydrogen peroxide (38). The authors showed that 3 mM hydrogen peroxide blocked Stat3 from binding DNA, but not Stat1. These studies further confirm the importance of Cys468 and its modification on the Stat3-DNA interaction.
Supplementary Material
Acknowledgments
Financial support from The Markel-Friedman Ovarian Cancer Research Fund, the National Cancer Institute (R01CA115674 to R. J.) and City of Hope Comprehensive Cancer Center (P30CA33572-27) is gratefully acknowledged. The authors thank the personnel of the Flow Cytometry, Synthetic and Biopolymer Chemistry, Light Microscopy Digital Imaging, High Throughput Screening, Mass Spectrometry, and X-ray Crystallography Core Facilities for their contributions to this work.
Footnotes
Supporting Information Available: This material is available free of charge via the Internet.
References
- 1.Darnell JE., Jr STATs and gene regulation. Science. 1997;277:1630–1635. doi: 10.1126/science.277.5332.1630. [DOI] [PubMed] [Google Scholar]
- 2.Ihle JN. STATs: signal transducers and activators of transcription. Cell. 1996;84:331–334. doi: 10.1016/s0092-8674(00)81277-5. [DOI] [PubMed] [Google Scholar]
- 3.Pellegrini S, Dusanter-Fourt I. The structure, regulation and function of the Janus kinases (JAKs) and the signal transducers and activators of transcription (STATs) Eur J Biochem. 1997;248:615–633. doi: 10.1111/j.1432-1033.1997.00615.x. [DOI] [PubMed] [Google Scholar]
- 4.Bromberg J, Darnell JE., Jr The role of STATs in transcriptional control and their impact on cellular function. Oncogene. 2000;19:2468–2473. doi: 10.1038/sj.onc.1203476. [DOI] [PubMed] [Google Scholar]
- 5.Decker T, Kovarik P. Transcription factor activity of STAT proteins: structural requirements and regulation by phosphorylation and interacting proteins. Cell Mol Life Sci. 1999;55:1535–1546. doi: 10.1007/s000180050393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Darnell JE, Jr, Kerr IM, Stark GR. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science. 1994;264:1415–1421. doi: 10.1126/science.8197455. [DOI] [PubMed] [Google Scholar]
- 7.Bromberg JF, Fan Z, Brown C, Mendelsohn J, Darnell JE., Jr Epidermal growth factor-induced growth inhibition requires Stat1 activation. Cell Growth Differ. 1998;9:505–512. [PubMed] [Google Scholar]
- 8.Liddle FJ, Alvarez JV, Poli V, Frank DA. Tyrosine phosphorylation is required for functional activation of disulfide-containing constitutively active STAT mutants. Biochemistry. 2006;45:5599–5605. doi: 10.1021/bi0525674. [DOI] [PubMed] [Google Scholar]
- 9.Wenta N, Strauss H, Meyer S, Vinkemeier U. Tyrosine phosphorylation regulates the partitioning of STAT1 between different dimer conformations. Proc Natl Acad Sci U S A. 2008;105:9238–9243. doi: 10.1073/pnas.0802130105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li L, Shaw PE. Elevated activity of STAT3C due to higher DNA binding affinity of phosphotyrosine dimer rather than covalent dimer formation. J Biol Chem. 2006;281:33172–33181. doi: 10.1074/jbc.M606940200. [DOI] [PubMed] [Google Scholar]
- 11.Stancato LF, David M, Carter-Su C, Larner AC, Pratt WB. Preassociation of STAT1 with STAT2 and STAT3 in separate signalling complexes prior to cytokine stimulation. J Biol Chem. 1996;271:4134–4137. doi: 10.1074/jbc.271.8.4134. [DOI] [PubMed] [Google Scholar]
- 12.Lin J, Buettner R, Yuan YC, Yip R, Horne D, Jove R, Vaidehi N. Molecular dynamics simulations of the conformational changes in signal transducers and activators of transcription, Stat1 and Stat3. J Mol Graph Model. 2009;28:347–356. doi: 10.1016/j.jmgm.2009.08.013. [DOI] [PubMed] [Google Scholar]
- 13.Kretzschmar AK, Dinger MC, Henze C, Brocke-Heidrich K, Horn F. Analysis of Stat3 (signal transducer and activator of transcription 3) dimerization by fluorescence resonance energy transfer in living cells. Biochem J. 2004;377:289–297. doi: 10.1042/BJ20030708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yu H, Jove R. The STATs of cancer--new molecular targets come of age. Nat Rev Cancer. 2004;4:97–105. doi: 10.1038/nrc1275. [DOI] [PubMed] [Google Scholar]
- 15.Buettner R, Mora LB, Jove R. Activated STAT signaling in human tumors provides novel molecular targets for therapeutic intervention. Clin Cancer Res. 2002;8:945–954. [PubMed] [Google Scholar]
- 16.Bowman T, Garcia R, Turkson J, Jove R. STATs in oncogenesis. Oncogene. 2000;19:2474–2488. doi: 10.1038/sj.onc.1203527. [DOI] [PubMed] [Google Scholar]
- 17.Bromberg JF, Wrzeszczynska MH, Devgan G, Zhao Y, Pestell RG, Albanese C, Darnell JE., Jr Stat3 as an oncogene. Cell. 1999;98:295–303. doi: 10.1016/s0092-8674(00)81959-5. [DOI] [PubMed] [Google Scholar]
- 18.Bromberg JF, Horvath CM, Wen Z, Schreiber RD, Darnell JE., Jr Transcriptionally active Stat1 is required for the antiproliferative effects of both interferon alpha and interferon gamma. Proc Natl Acad Sci U S A. 1996;93:7673–7678. doi: 10.1073/pnas.93.15.7673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Becker S, Groner B, Muller CW. Three-dimensional structure of the Stat3beta homodimer bound to DNA. Nature. 1998;394:145–151. doi: 10.1038/28101. [DOI] [PubMed] [Google Scholar]
- 20.Herrmann A, Vogt M, Monnigmann M, Clahsen T, Sommer U, Haan S, Poli V, Heinrich PC, Muller-Newen G. Nucleocytoplasmic shuttling of persistently activated STAT3. J Cell Sci. 2007;120:3249–3261. doi: 10.1242/jcs.03482. [DOI] [PubMed] [Google Scholar]
- 21.Hintzen C, Haan C, Tuckermann JP, Heinrich PC, Hermanns HM. Oncostatin M-induced and constitutive activation of the JAK2/STAT5/CIS pathway suppresses CCL1, but not CCL7 and CCL8, chemokine expression. J Immunol. 2008;181:7341–7349. doi: 10.4049/jimmunol.181.10.7341. [DOI] [PubMed] [Google Scholar]
- 22.Pranada AL, Metz S, Herrmann A, Heinrich PC, Muller-Newen G. Real time analysis of STAT3 nucleocytoplasmic shuttling. J Biol Chem. 2004;279:15114–15123. doi: 10.1074/jbc.M312530200. [DOI] [PubMed] [Google Scholar]
- 23.Siddiquee K, Zhang S, Guida WC, Blaskovich MA, Greedy B, Lawrence HR, Yip ML, Jove R, McLaughlin MM, Lawrence NJ, Sebti SM, Turkson J. Selective chemical probe inhibitor of Stat3, identified through structure-based virtual screening, induces antitumor activity. Proc Natl Acad Sci U S A. 2007;104:7391–7396. doi: 10.1073/pnas.0609757104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dixon JM. Exemestane: a potent irreversible aromatase inactivator and a promising advance in breast cancer treatment. Expert Rev Anticancer Ther. 2002;2:267–275. doi: 10.1586/14737140.2.3.267. [DOI] [PubMed] [Google Scholar]
- 25.Hong Y, Yu B, Sherman M, Yuan YC, Zhou D, Chen S. Molecular basis for the aromatization reaction and exemestane-mediated irreversible inhibition of human aromatase. Mol Endocrinol. 2007;21:401–414. doi: 10.1210/me.2006-0281. [DOI] [PubMed] [Google Scholar]
- 26.Li D, Ambrogio L, Shimamura T, Kubo S, Takahashi M, Chirieac LR, Padera RF, Shapiro GI, Baum A, Himmelsbach F, Rettig WJ, Meyerson M, Solca F, Greulich H, Wong KK. BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene. 2008;27:4702–4711. doi: 10.1038/onc.2008.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Spicer JF, Rudman SM. EGFR inhibitors in non-small cell lung cancer (NSCLC): the emerging role of the dual irreversible EGFR/HER2 inhibitor BIBW 2992. Target Oncol. 2010 doi: 10.1007/s11523-010-0140-y. [DOI] [PubMed] [Google Scholar]
- 28.Ahmad R, Raina D, Meyer C, Kharbanda S, Kufe D. Triterpenoid CDDO-Me blocks the NF-kappaB pathway by direct inhibition of IKKbeta on Cys-179. J Biol Chem. 2006;281:35764–35769. doi: 10.1074/jbc.M607160200. [DOI] [PubMed] [Google Scholar]
- 29.Shishodia S, Sethi G, Konopleva M, Andreeff M, Aggarwal BB. A synthetic triterpenoid, CDDO-Me, inhibits IkappaBalpha kinase and enhances apoptosis induced by TNF and chemotherapeutic agents through down-regulation of expression of nuclear factor kappaB-regulated gene products in human leukemic cells. Clin Cancer Res. 2006;12:1828–1838. doi: 10.1158/1078-0432.CCR-05-2044. [DOI] [PubMed] [Google Scholar]
- 30.Schrödinger . Maestro, 7.5 ed. New York: 2005. [Google Scholar]
- 31.Connolly ML. Solvent-accessible surfaces of proteins and nucleic acids. Science. 1983;221:709–713. doi: 10.1126/science.6879170. [DOI] [PubMed] [Google Scholar]
- 32.Herrmann A, Sommer U, Pranada AL, Giese B, Kuster A, Haan S, Becker W, Heinrich PC, Muller-Newen G. STAT3 is enriched in nuclear bodies. J Cell Sci. 2004;117:339–349. doi: 10.1242/jcs.00833. [DOI] [PubMed] [Google Scholar]
- 33.Garcia R, Bowman TL, Niu G, Yu H, Minton S, Muro-Cacho CA, Cox CE, Falcone R, Fairclough R, Parsons S, Laudano A, Gazit A, Levitzki A, Kraker A, Jove R. Constitutive activation of Stat3 by the Src and JAK tyrosine kinases participates in growth regulation of human breast carcinoma cells. Oncogene. 2001;20:2499–2513. doi: 10.1038/sj.onc.1204349. [DOI] [PubMed] [Google Scholar]
- 34.Wagner BJ, Hayes TE, Hoban CJ, Cochran BH. The SIF binding element confers sis/PDGF inducibility onto the c-fos promoter. Embo J. 1990;9:4477–4484. doi: 10.1002/j.1460-2075.1990.tb07898.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yu CL, Meyer DJ, Campbell GS, Larner AC, Carter-Su C, Schwartz J, Jove R. Enhanced DNA-binding activity of a Stat3-related protein in cells transformed by the Src oncoprotein. Science. 1995;269:81–83. doi: 10.1126/science.7541555. [DOI] [PubMed] [Google Scholar]
- 36.Hedvat M, Jain A, Carson DA, Leoni LM, Huang G, Holden S, Lu D, Corr M, Fox W, Agus DB. Inhibition of HER-kinase activation prevents ERK-mediated degradation of PPARgamma. Cancer Cell. 2004;5:565–574. doi: 10.1016/j.ccr.2004.05.014. [DOI] [PubMed] [Google Scholar]
- 37.Kim PK, Armstrong M, Liu Y, Yan P, Bucher B, Zuckerbraun BS, Gambotto A, Billiar TR, Yim JH. IRF-1 expression induces apoptosis and inhibits tumor growth in mouse mammary cancer cells in vitro and in vivo. Oncogene. 2004;23:1125–1135. doi: 10.1038/sj.onc.1207023. [DOI] [PubMed] [Google Scholar]
- 38.Li L, Cheung SH, Evans EL, Shaw PE. Modulation of gene expression and tumor cell growth by redox modification of STAT3. Cancer Research. 2010;70:8222–8232. doi: 10.1158/0008-5472.CAN-10-0894. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.