

Abstract
Although protein kinases have recently emerged as important drug targets, the anti-infective potential of protein kinase inhibitors has not been developed extensively. We identified the mammalian PDK1 inhibitor KP-372-1 as a potent antifungal molecule with activity against yeast and fungal biofilms using a screening strategy for protein kinase inhibitors that block the cell wall stress response in yeast. Genetic and biochemical studies indicate that KP-372-1 inhibits fungal PDK1 orthologs (Pkh kinases) as part of its mode of action and support a role for Pkh kinases in eisosome assembly. Two other structurally distinct molecules that inhibit PDK1, OSU-03012 and UCN-01, also have antifungal activity. Taken together, these data indicate that fungal PDK1 orthologs are promising targets for new antifungal drug development.
INTRODUCTION
Protein kinases have emerged as one of the most important classes of drug targets with applications to a wide variety of therapeutic areas including oncology, diabetes, and rheumatology (1). Following G-protein coupled receptors, protein kinases are thought to be the second largest class of drug targets and it is estimated to that 20–30% of drugs candidates currently in clinical development are protein kinase inhibitors (PKIs). The emergence of PKIs as useful drugs was catalyzed by the approval of Imatinab, an inhibitor of the oncogene bcr-abl fusion protein, for the treatment of chronic myleogenous leukemia. Since the approval of imatinab in 2001, a number of additional PKIs have received FDA-approval or have reached late-stage clinical development. As a result, protein kinases are now regarded as a highly “druggable” set of cellular targets with applications to a wide range of disease processes (1).
One therapeutic area where PKIs have not yet been extensively studied is anti-infectives. Recently, however, a large library of PKIs was screened for compounds with anti-bacterial activity (2). This campaign yielded a new mechanistic class of antibiotics that target biotin carboxylase, a bacterial enzyme with an ATP binding site very similar to eukaryotic protein kinases. Importantly, sufficient structural differences exist within the ATP binding site of biotin carboxylase to allow the identification of molecules with considerable specificity for the bacterial enzymes relative to host protein kinases (2). Indeed, the realization that small structural differences within the ATP binding sites of protein kinases can be effectively exploited to generate highly selective protein kinase inhibitors has been an important impetus for the re-emergence of protein kinases as viable drug targets (1).
Fungi are eukaryotic pathogens and, as such, have many protein kinase-based signaling pathways that are well conserved with mammalian systems. Indeed, the study of eukaryotic signaling pathway in the model yeast S. cerevisiae has been instrumental in establishing many of the mechanistic paradigms of eukaryotic signal transduction (3). It follows, then, that PKIs active toward human protein kinases may also have activity toward fungal protein kinases. Consistent with this notion, the canonical non-specific protein kinase C inhibitor staurosporine is highly toxic to both human and fungal cells.
For PKIs to be useful anti-fungal drugs, such molecules must be selective for fungal kinases or target fungal kinases structurally divergent from human orthologs. Fortunately, many yeast kinases display significant sequence and structural differences as compared to their human orthologs. For example, human PDK1 is 556 aa and has a pleckstrin homology domain while the C. albicans PDK1 homolog Pkh1 is 944 aa and has no pleckstrin homology domain (4). In addition, the two PDK1 proteins have only 50% identity at the active site and much less in other regions. Therefore, it may be possible to exploit the structural differences between human and fungal kinases in the development of antifungal PKIs.
Invasive fungal infections are life-threatening opportunistic infections that are an increasingly important cause of morbidity and mortality in patients with compromised immune function (5). One of the reasons for the high mortality rate of invasive fungal infections is that the number of clinically useful antifungal drugs is extremely limited, particularly when compared to the number of agents available for the treatment of bacterial infections (6). In the last thirty years, the echinocandins (1,3-β-glucan synthase inhibitors) have been the only new mechanistic class of antifungal drugs introduced into clinical practice. Although the echinocandins are an important addition to the antifungal armamentarium, these drugs have a number of limitations including ineffectiveness against C. neoformans and a variety of other medically important fungal pathogens and poor oral bioavailability (6). Furthermore, as the number of patients with invasive fungal infections increase, resistance to currently used agents inevitably develops. Indeed, isolates with resistance to each class of antifungal drugs have been described. Therefore, the identification of new antifungal drug targets and antifungal small molecules is an important goal of current anti-infective research.
Although the number of studies designed to identify fungal specific PKIs pale in comparison to other areas, PKIs with specificity for fungal protein kinases have been reported (7,8). For example, researchers at Lilly used a high throughput screening (HTS) to identify cercosporamide and subsequently showed that it is selective for C. albicans protein kinase C relative to human PKC isozymes (7). Fungal PKCs function within the cell wall integrity (CWI) signaling pathway and, thereby, regulate cell wall biosynthesis (9). The fungal cell wall has recently emerged as a very attractive antifungal drug target because it is unique to fungi and since molecules that target the cell wall directly kill the fungi by causing cell lysis. Although the potential of PKIs to interfere with CWI signaling is well-recognized (10), few drug discovery efforts directed toward identifying PKIs that target this pathway have been reported.
Here, we describe a screening strategy designed to identify molecules that cause yeast cell lysis and block CWI-pathway signaling. Application of this strategy to a focused library of mechanistically characterized PKIs led to the identification of PDK1-inhibitors as antifungal molecules with excellent activity against pathogenic yeasts in both planktonic and biofilm growth stages. Chemical genetics-guided mechanistic studies indicate that mammalian PDK1 inhibitors also target fungal PDK1 orthologs as part of their mode of action and, thus, validate fungal PDK1s as promising targets for antifungal drug discovery. In addition, our studies have shown that PDK1 inhibitors are valuable mechanistic probes for the study of PDK1 orthologs in yeast. Since two of the PDK1 inhibitors (OSU-03012 and UCN-01) identified in this study have been, or are currently being, studied in human clinical trials, PDK1 inhibitors appear to be a promising class of molecules for future antifungal drug development.
RESULTS AND DISCUSSION
PDK1 inhibitor KP-372 blocks cell wall integrity signaling in yeast
To identify PKIs that disrupt yeast cell wall integrity, we designed a four-part screening strategy as outlined in Fig. 1A. The primary screen in our approach detects molecules that cause yeast cells to lyse, a characteristic phenotype of yeast cell wall damage, by use of an assay that detects the release of adenylate kinase (AK) to the growth medium as a reporter of yeast cell lysis. In previous work, we have validated the AK assay in HTS format and shown that it can detect as few as 500 lysed yeast cells in a sample of 105 cells (11). PKIs that cause yeast cells lysis (hits) were then re-confirmed by AK dose-response assays and tested for in vitro antifungal activity against the human fungal pathogen C. albicans using standard microdilution susceptibility testing (12). Lastly, to select PKIs specific for the CWI signaling pathway, we tested the set of hits for their ability to block the activation of a transcriptional reporter of the CWI-signaling pathway (9).
Figure 1.

Screen of protein kinase library identifies fungilytic molecules. a. Schematic of screening strategy: AK = adenylate kinase; CWI = cell wall integrity. b. Scatter plot of primary AK assay screening data. Data points above line represent >3-fold increase in relative light units (RLU) of AK activity and were scored as positive. c. List of positive scoring molecules following re-test: MIC = minimum inhibitory concentration against C. albicans as determined by microdilution methods.
To rapidly survey a sample of well-characterized PKIs, we utilized the commercially available InhibitorSelect collection, a library of 80 PKIs with diverse mechanistic and structural characteristics. C. albicans clinical reference strain SC5314 was screened against the InhibitorSelect library for molecules that caused a release of AK into the growth medium using our recently reported protocol (11). Compounds were screened at 5 μM and 50 μM concentrations and a compound was scored as positive if it induced a three-fold increase in extracellular AK activity relative to DMSO-treated cells (11). A scatter plot of the raw screening data is shown in Fig. 1B. As summarized in Fig. 1C, eight protein kinase inhibitors caused C. albicans cell lysis (hit rate 10%) and possessed good in vitro activity against C. albicans by growth assays (Minimum Inhibitory Concentration (MIC), 3–20 μg/mL). Four hits were PKIs previously shown to have antifungal activity (staurosporine, chelerythine, rapamycin, and heribmycin) and these served to confirm the validity of our primary screening approach. The set of four novel hits included three molecules (Akt IV, Akt V, & KP-372-1) that target the AGC family-derived PIK3/PDK1/Akt signaling network in mammalian cells (13) and one molecule that targets tyrosine kinases (Syk II, 14). Although the library contained a number of MAPK inhibitors, none were identified in our screen. This is likely due to the fact that MAPKs are not essential genes in either S. cerevisiae or C. albicans (9).
The novel, antifungal PKIs identified in the primary screen were evaluated for their ability to block CWI pathway signaling using a reporter construct that contains two copies of the consensus binding site for the CWI-pathway-regulated transcription factor Rlm1 fused to the β-galactosidase gene (15). A plasmid containing RLM1-lacZ was transformed into the model yeast S. cerevisiae. The chitin binding agent Calcofluor white (CFW), a well-characterized inducer of cell wall stress, was used to activate reporter activity. Sub-inhibitory concentrations of both KP-372-1 and SykII completely abolished reporter activity induced by CFW (Fig. 2A). The Akt inhibitors had no effect on reporter activity (data not shown), a finding consistent with the fact that the yeast Akt homolog, Sch9, has not been previously linked to CWI-pathway signaling (16). Since our goal was to identify molecules that interfere with CWI-pathway signaling and since the Akt homolog Sch9 is not essential in yeast, we did not study the Akt inhibitors further. These data validate the utility of our screening strategy and provide two new structural classes of potential cell wall-targeted antifungal small molecules.
Figure 2.
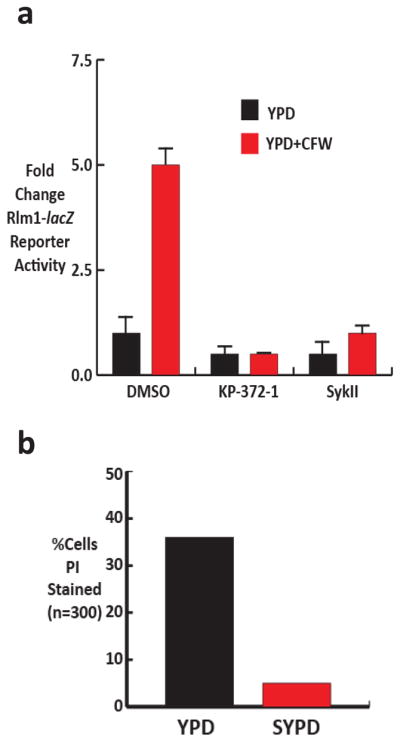
KP-372-1 blocks CWI pathway signaling. a. CWI pathway reporter Rlm1-lacZ activity in S. cerevisiae induced by Calcofluor white +/− sub-lethal concentrations (1/2MIC) KP-372-1 and SykII. Bars indicate mean fold change in Miller Units of β-galactosidase activity relative to logarithmic phase cells without calcfluor or PKI. Error bars indicate standard deviation of three independent trials performed in triplicate. b. C. albicans cells were treated with KP-372-1 at MIC for 2 hr at 37°C in YPD or YPD+1M sorbitol (SYPD) and stained with propidium iodide.
The percentage of cells with PI uptake was determined by fluorescence
KP-372-1 is a dual PDK1/Akt inhibitor with anti-cancer properties (17) while SykII is a tyrosine kinase inhibitor that has been developed as an approach to the treatment of auto-immune diseases (14). Since the main potential of Syk inhibitors is as immunosuppressive agents and the underlying cause of invasive fungal infections is almost invariably immunosuppression, we chose not to pursue SykII further. KP-372-1 has been shown to have potent activity against both leukemic and glioblastoma cells but is tolerated by normal cells at micromolar concentrations without significant cytotoxicity (17). Consistent with its low cytotoxicity toward normal cells, KP-372-1 has been screened against a large panel of human protein kinases and found to have at least 10-fold selectivity for a limited number of human kinases (17). Therefore, we focused on investigating the antifungal activity of KP-372-1 further.
A hallmark phenotype of mutations and drugs that interfere with CWI signaling is that their effects can be suppressed by the addition of osmotic support such as 1M soribitol to the culture medium (9). By equalizing the osmotic gradient across the plasma membrane, cell integrity is less dependent upon an intact cell wall. To further confirm that the fungilytic activity of KP-372-1 was due to disruption of cell wall integrity, we examined the effect of 1M sorbitol on the ability of KP-372-1 to kill C. albicans using propidium iodide uptake as a marker of disrupted cellular integrity. As shown in Fig. 2B, 36% of C. albicans cells were stained by propidium iodide after 2h of KP-372-1 treatment in standard yeast peptone dextrose (YPD) while five-fold fewer cells were stained by propidium iodide in 1M sorbitol-supplemented YPD. This result strongly supports the idea that KP-372-1 targets processes required for yeast cell wall integrity as part of its mode of action.
KP-372-1 is active against non-albicans Candida spp. and C. albicans biofilms
To further characterize the in vitro antifungal activity of KP-372-1, we determined its MIC for a set of pathogenic Candida spp. and C. neoformans (12). As shown in Fig. 3A, it is highly active against the two most common Candida spp., C. albicans and C. glabrata, while less so toward other Candida spp (Fig. 3A). Importantly, KP-372-1 is highly active against C. neoformans. Since the echinocandin class of cell wall-targeted 1,3-β-glucan synthase inhibitors is not active against C. neoformans, this represents one of the few cell wall-targeted molecules with activity against this important pathogen.
Figure 3.

KP-372-1 has activity against pathogenic yeast and C. albicans biofilms. a. Schematic representation of the minimum inhibitory concentration (MIC) of KP-372-1 against pathogenic yeast. Filled circles indicate growth and clear circles indicate no detectable turbidity. The lowest concentration of drug resulting in no growth is defined as the MIC. b. Activity of KP-372-1 against 48-h C. albicans biofilms. Biofilms were generated in 96-well plates by incubation at 37°C for 48h and treated with KP-372-1 for an additional 24 h. Metabolic activity was determined using the XTT assay as described in materials and methods. The sMIC50 (6.3 μg/mL) is defined as the concentration that reduces metabolic activity by 50% relative to untreated control.
We also examined the combination of KP-372-1 with the clinically-used antifungal drugs fluconazole and caspofungin using checkerboard interaction assays to determine if they showed synergistic activity toward C. albicans. This assay allows one to compare the activity of a molecule alone and in combination with another molecule by generating a fractional inhibitory concentration (FIC). FIC for two drugs (A & B) is calculated as follows: FIC = MICA/MIC(A+B) + MICB/MIC(A+B). FIC values less than 0.5 are considered synergistic; between 0.5 and 1 are additive; between1–2 are indifferent; and above 2 are antagonistic. The FIC was 0.5 for the combination of KP-372-1 and caspofungin and 1.0 for the combination of KP-372-1 and fluconazole. Therefore, KP-372-1 is not synergistic with either agent.
Next, we determined the effect of KP-372-1 on the viability of biofilms that had matured in microtiter plates for 48h using the established XTT-reduction based assay of metabolic activity (18). As shown in Fig. 3B, KP-372-1 has excellent in vitro activity against C. albicans biofilms with its sMIC50 (3 μg/mL) identical to its MIC against planktonic C. albicans. Since very few antifungals retain activity against fungal biofilms, these results further suggest that KP-372-1 represents an exciting lead compound with many desirable properties as an antifungal agent.
Yeast strains with mutations in PDK1 orthologs are hypersensitive to KP-372-1
As noted above, KP-372-1 has been shown to inhibit both PDK1 and Akt in mammalian cells (17). Orthologs of PDK1 are present in both the model yeast S. cerevisiae and pathogenic fungi and are referred to as PKH genes based on the name assigned to the S. cerevisiae family (4). The closest ortholog of Akt in yeast is Sch9 which is also present in S. cerevisiae as well pathogenic yeast (16). Since Sch9 has not been implicated in yeast cell wall integrity nor is it an essential gene, it seemed unlikely that the antifungal activity of KP-372-1 would be due to specific inhibition of the Akt orthologs. In contrast, deletion of both PKH1 and PKH2 is lethal (4), indicating that the PKH genes carry out essential functions in yeast and suggesting that the PDK1 inhibitory activity of KP-372-1 would be more likely to be responsible for its fungicidal activity than its Akt activity.
Therefore, we focused our initial mechanistic studies on testing the hypothesis that KP-372-1 targets PDK1 orthologs in yeast. Although little is known about the function of PKH genes in pathogenic yeast, S. cerevisiae Pkh1 and Pkh2 has been studied by a number of groups. ScPKH1&2 are a partially redundant pair of essential kinases that function in cell wall integrity (19), flippase regulation (20), endocytosis (21) and eisosome assembly (22, 23). Like mammalian PDK1, Pkh1/2 phosphorylate and activate downstream kinases (24) including the ACG family kinases Ypk1/2p, Sch9p, and Pkc1p (Fig. 4A). Ypk1/2 and Pkc1p are required for cell wall integrity (19) and activation of the CWI signaling pathway (9, 19).
Figure 4.

Yeast strains with mutations in PDK1 orthologs are hypersensitive to KP-372-1. a. Schematic of kinases and cellular functions regulated by S. cerevisiae PDK1 orthologs Pkh1/2. The indicated strains of S. cerevisiae (S.c., b) and C. neformans (C.n., c) were inoculated in top agar of YPD plates and filter discs containing the indicated concentrations of KP-372-1 were placed on the plates. Plates were incubated at 30°C for 3 days prior to photography.
Heterozygous diploid yeast mutants lacking one allele of the gene encoding a putative drug target are frequently hypersensitive to the effects of that drug. This is called drug-induced haploinsufficiency (25). Similarly, if a drug targets the products of two redundant genes, then deletion of one of those genes in a haploid yeast strain will hypersensitize the strain to that drug. Consistent with this phenomena, S. cerevisiae pkh2Δ mutants showed a larger zone of inhibition by disk diffusion assay than wild type or pkh1Δ (Fig. 4B). Pkh1/2 phosphorylate two other AGC protein kinases involved in yeast cell wall integrity, Ypk1&2 (20). As shown in Fig. 4B, neither ypk1Δ nor ypk2Δ is hypersensitive to KP-372-1. The Pkh1/2 kinases function upstream of Ypk1/2 and, thus, if KP-372-1 primarily targets Pkh1/2, then YPK mutants should not be hypersensitive to the drug by epistasis. The fact that the YPK mutants are as sensitive to KP-372-1 as wild type further supports the hypothesis that the drug targets the PDK1 orthologs Pkh1/2.
Two PKH analogs, PKH2-01 and PHK2-02, are present in C. neoformans and both deletion mutants are in the large set of deletion mutants recently made publicly available (26). As part of that project, Liu et al. found that PKH2-02 was deficient for growth at 37°C, a virulence property for C. neoformans and, accordingly, showed a strong virulence defect in a mouse model of pulmonary cryptococcosis. We obtained both mutants from this collection and, consistent with the S. cerevisiae mutants, PKH2-02 is significantly more sensitive to KP-372-1 than wild type while PKH2-01 is slightly more sensitive than wild type at 30°C (Fig. 4C). Although not definitive, these chemical genetic studies strongly support the notion that KP-372-1 targets PDK1 orthologs in yeast as part of its mode of action as an antifungal molecule.
KP-372-1 inhibits phosphorylation of the Pkh substrate Pil1p
The genetic experiments presented above suggest that the antifungal properties of KP-372-1 are related to its activity as a PDK1 inhibitor. To further test this hypothesis, we took advantage of the fact that the PDK1 orthologs Pkh1/2 phosphorylate the eisosome component Pil1 while neither the Akt ortholog Sch9 nor the other downstream kinase targets of Pkh1/2 (Ypk1/2 & Pkc1) are involved in its phosphorylation (22, 23). Pil1p is a key component of eisosomes, punctate structures located beneath the plasma membrane that may play a role in endocytosis. Pkh1/2-mediated phosphorylation of Pil1 appears to be involved in eisosome regulation. Since Pkh1/2p-mediated phosphorylation of Pil1p generates a species with markedly decreased mobility by SDS-PAGE (23), this substrate provides an ideal system to test the hypothesis that KP-372-1 inhibits Pkh1/2p in the cell.
S. cerevisiae strains containing a chromosomally-integrated PIL1-GFP allele were transformed with a vector control or a plasmid expressing PKH2 under the control of a galactose-inducible promoter. The resulting strains were grown overnight in raffinose-containing medium to de-repress the galactose promoter and then shifted to galactose-containing medium to induce expression of PKH2 in the presence or absence of sub-inhibitory KP-372-1 (1/2 MIC). The phosphorylation status of Pil1-GFP was followed over a 3.5h time course by Western blot according a published protocol (23).
As shown in Fig. 5A, Pil1-GFP phosphorylation increases as the cells enter into logarithmic phase in untreated strains containing empty vector. Consistent with previously reported observations, galactose-induced, over-expression of PKH2 dramatically increases the proportion of phosphorylated Pil1-GFP compared to vector control (22). In the presence of KP-372-1, phosphorylation of Pil1-GFP is rapidly blocked in cells with endogenous Pkh1/2 as well as in cells over-expressing Pkh2. Deletion mutants of the Akt ortholog sch9Δ do not have defects in eisosome assembly (23) and, therefore, the ability of KP-372-1 to block Pil1-GFP phosphorylation cannot be due to its activity as an Akt inhibitor. Accordingly, this experiment indicates that KP-372-1 inhibits PDK1 orthologs in yeast. Our data also indicate that a substantial portion of the Pkh1/2 activity is inhibited in KP-372-1-treated cells since we can detect none of the Pkh1/2-phosphorylated form of Pil1; indeed, these blots are similar to those derived from pkh1ts pkh2Δ cells that have been shifted to the restrictive temperature (23). Since loss of PDK1 (Pkh1/2) activity in yeast is lethal and loss of Akt activity (Sch9) is not, these data also strongly support the notion that the antifungal activity of KP-372-1 is due in large part to its activity as a PDK1 inhibitor.
Figure 5.

KP-372-1 inhibits Pil1-GFP phosphorylation, eisosome assembly, and endocytosis in S. cerevisiae. a. S. cerevisiae strains containing a Pil1-GFP allele and either empty plasmid or a plasmid expressing PKH2 from a galactose-inducible promoter were shifted from raffinose to galactose medium and incubated in the presence or absence of sub-inhibitory KP-372-1 (1/2MIC). Cells were harvested at the indicated times and analyzed by Pil1-GFP immunoblotting. Pil1-P corresponds to the Pkh1/2-dependent phosphorylated Pil1 species. b. Pil1-GFP strains were treated for 3 h with KP-372-1 (1/2 MIC) or 1% DMSO and analyzed by fluorescence microscopy. c&d. S. cerevisiae cells were treated with either KP-372-1 (1/2 MIC) or 1% DMSO for 1h and then processed for Lucifer yellow (LY) uptake. C. Representative field of cells 15 min after addition of LY. D. Time course of LY uptake in the presence or absence of KP-372-1 (standard deviation for each point <10%).
KP-372-1 induces eisosome disassembly and blocks endocytosis in S. cerevisiae
The role of Pkh1/2-mediated phosphorylation of Pil1 in the regulation, assembly and turnover of eisosomes is controversial. Walther et al. reported that blocking Pkh1/2-mediated phosphorylation of Pil1-GFP by shifting a strain with a temperature sensitive allele of PKH1 (pkh1ts pkh2Δ) to the restrictive temperature (37°C) increased the number and intensity of Pil1-marked eisosomes (22), suggesting that Pil1 phosphorylation was involved in eisosome disassembly. Luo et al studied this process with essentially identical strains and, in contrast, found that eisosome number and intensity decreased upon shift to the restrictive temperature, suggesting that phosphorylation is required either for assembly or stabilization of eisosomes (23).
Since we observed significantly reduced Pil1-GFP phosphorylation at sub-lethal concentrations of KP-372-1, we hypothesized that the use of this inhibitor as a chemical probe of the role of Pkh1/2 phosphorylation might provide useful information regarding its role in eisosome assembly. Therefore, we treated S. cerevisiae cells containing Pil1 with a C-terminal GFP fusion with KP-372-1 and examined its effect on eisosome patterns by fluorescence microscopy. As shown in Fig. 5B, DMSO-treated cells show the typical pattern of eisosome distribution. However, within one hour of treatment, the number of peripheral eisosomes dramatically decreased. The micrographs of the KP-372-1-treated cells closely match those reported by Luo et al. (23) and, consequently, support a model in which Pil1 phosphorylation is required for eisosome assembly/stabilization.
Pkh kinases are also important for endocytosis in yeast (21). We, therefore, determined the effect of sub-lethal KP-372-1 on fluid-phase endocytosis using a Lucifer yellow (LY) uptake assay. LY binds to the plasma membrane and is transported to the vacuole (yeast lysosomal homolog) in PKH-dependent fashion. As shown in Fig. 5C, fewer KP-372-1-treated cells show vacuolar localization of the dye in comparison to untreated control cells. Scoring vacuolar localization over a 1 hr time course revealed that uptake is inhibited at early time points by KP-372-1 but that the proportion of cells with internalized LY approaches that of untreated cells at later time points (Fig. 5D). Importantly, substantial number of treated cells eventually internalized LY, indicating that KP-372-1 induced defects in endocytosis and eisosome assembly are unlikely to be due to cell death.
Antifungal activity of structurally distinct PDK1 inhibitors
Protein kinases have emerged as attractive drug targets for a variety of diseases including cancer, cardiovascular disease, diabetes and autoimmune disorders (1). Among the protein kinase inhibitors that have entered clinical development are two molecules with activity toward PDK1, UCN-01 (27) and OSU-03012 (28). In addition, BX-912 has been tested in animal models (29). These compounds are well tolerated and, in general, have low cytotoxicity toward human cells (13). Since these compounds are commercially available and have favorable pharmacological properties, we carried out a focused structure-activity study to determine whether PDK1 inhibitors based on other chemical scaffolds also display antifungal activity. Scaffolds with antifungal activity could then serve as starting points for further optimization of the antifungal activity of PDK1 inhibitors.
As summarized in Fig. 6A, both UCN-01 and OSU-03012 showed good antifungal activity against C. albicans and C. neoformans while BX-912 did not inhibit growth below 64 μg/mL. In addition, both OSU-03012 showed activity against C. albicans biofilms (Fig. 6C), although at higher concentrations than those active against planktonic cells. In contrast to KP-372-1 and OSU-03012, UCN-01 showed synergy with fluconazole (FIC = 0.3) but not with caspofungin (FIC = 1.0). UCN-01 is a modestly more selective derivative of the promiscuous protein kinase inhibitor staurosporine (30). Staurosporine has been shown to be synergistic with fluconazole (31) and we suspect that the synergy displayed by UCN-01 may be due to its structural similarity to staurosporine.
Figure 6.
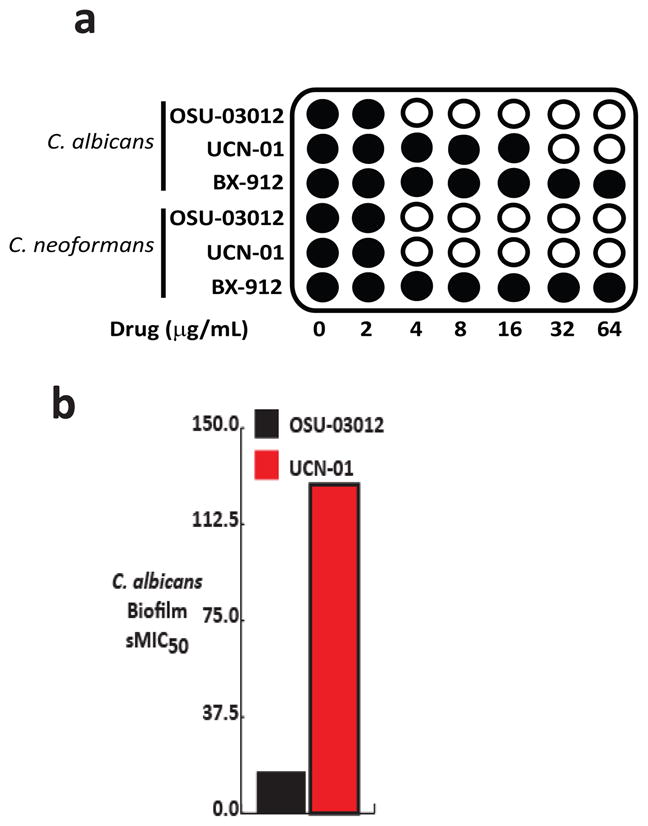
Structurally diverse PDK1 inhibitors display antifungal activity a. Schematic representation of the minimum inhibitory concentration (MIC) of KP-372-1 against C. albicans and C. neoformans. Filled circles indicate growth and clear circles indicate no detectable turbidity. The lowest concentration of drug resulting in no growth is defined as the MIC. c. sMIC50 of OSU-03012 and UCN-01 toward C. albicans biofilms as determined by XTT assay (17).
Only one new mechanistic class of antifungal drugs has been introduced into clinical use in the last thirty years (6). One strategy to increase the rate of new antifungal development is to identify compounds with antifungal activity within classes of molecules that have been developed for other purposes. A very large number of PKIs have been generated in recent years (1). To discover PKIs with antifungal activity, we designed a screening strategy to identify PKIs that both cause yeast cell lysis and target the cell wall stress response. Through this approach, we have discovered that mammalian PDK1 inhibitors display potent antifungal activity toward Candida spp., C. neoformans, and fungal biofilms.
Mechanistic characterization of our lead compound, KP-372-1, indicates that it targets fungal PDK1 orthologs as part of its mechanism of action. Although KP-372-1 also has well characterized activity against the PDK1 target Akt in human cells, it is unlikely that this activity accounts for its antifungal activity because the yeast Akt ortholog, Sch9, is not essential in either S. cerevisiae or C. albicans (16). However, it is important to note that very few PKIs are completely specific and we cannot exclude the possibility that at least a portion of the antifungal activity of these molecules is due to the inhibition of closely related protein kinases. Indeed, it is possible that inhibition of Sch9 by KP-372-1 contributes partially to its antifungal effects. Of the other ACG family protein kinases that PDK1 inhibitors could target in yeast, PKC1, the protein kinase C ortholog, seems the most likely because it is also involved in the regulation of cell wall integrity (9, 30). Although PKC1 orthologs are essential in S. cerevisiae (9) and C. neoformans (32), pkc1Δ/Δ mutants are viable in C. albicans (33) and KP-372-1 is as active against this mutant as it is toward wild type cells (L. DiDone, unpublished results). This suggests that, in C. albicans, the majority of the antifungal activity of KP-372-1 is through its effect on kinases other than Pkc1. Our biochemical and cell biological results indicate that KP-372-1 inhibits the phosphorylation of a substrate of the yeast PDK1 orthologs Pkh1/2 and inhibits cellular processes dependent on these kinases. Since Pkh1/2 are essential kinases, these data strongly support the conclusion that a substantial portion of the antifungal activity of KP-372-1 is due to its activity as a PDK1 inhibitor and suggest that PDK1 orthologs are promising antifungal drug targets.
In addition to being promising antifungal drug candidates, PDK1 inhibitors also appear to be useful mechanistic probes for the study of the function of PDK1 orthologs in yeast. Pkh kinases are essential in S. cerevisiae (4, 24) and based on our results appear to be similarly essential in pathogenic yeast. Genetic studies of Pkh function in S. cerevisiae have utilized temperature sensitive mutants of pkh1 generated in a pkh2Δ background. In other systems, chemical genetic studies with PKIs have proven to be useful approaches to studying kinases (34) and, thus, we utilized KP-372-1 to examine the role of Pil1 in eisosome function under conditions where the cells remain viable. We found that chemical inhibition of Pil1 phosphorylation leads to decreased numbers of eisosomes and an increase in cytosolic Pil1-GFP. These observations are consistent with the findings of Luo et al. (23) and support a model whereby Pkh promotes eisosome assembly or stabilization through phosphorylation of Pil1. At this point nature of the discrepancy between the observations of Luo et al. (23) and Walther et al. (22) remains unclear and further work will be required to conclusively characterize the role of Pil1 phosphorylation in eisosome regulation. KP-372-1 should also prove useful as a probe in studies of the function of PDK1 orthologs in pathogenic yeast with less tractable genetics.
PDK1 inhibitors have been extensively studied as targeted anti-cancer agents because they show favorable patterns of cytotoxicity toward normal cells. Our results indicate that fungal PDK1 orthologs are a promising antifungal drug target and that at least three structural classes of molecules that inhibit mammalian PDK1 have antifungal activity. Since a number of other structural classes of PDK1 inhibitors have also been reported (34), it seems that a systematic evaluation and optimization of the antifungal properties of PDK1 inhibitors represents an attractive approach to new antifungal drug development.
METHODS
Yeast strains, plasmids, culture media and reagents
A complete list of yeast strains, genotypes, and sources is provided in Supplementary Table 1. The reporter plasmid pRLM1-lacZ was a generous gift of David Levin (15). pGAL-PKH2 was a gift of Eric Phizicky (35). The InhibitorSelect library of protein kinase inhibitors was obtained from EMD Chemicals USA. Compounds for re-testing were obtained from separate lots or from alternative suppliers. All compounds and reagents were used as received.
Adenylate kinase (AK) assay
AK assays were performed according to a recently published protocol (11) using the 96-well plate version using the Toxi-Light Assay kit (Lonza). Luminescence was measured using a SpectraMax plate reader (Molecular Devices). Screening hits were defined as molecules inducing >3-fold increase in RLU relative to wells containing cells treated with 1% dimethylsulfoxide.
Antifungal susceptibility assays
The antifungal activity of protein kinase inhibitors was determined using the Clinical and Laboratory Science Institute microdilution protocol M-27A2 (12). Biofilm antifungal activity was determined using the protocol of Pierce et al. and is reported as MIC50S (17). Disk diffusion assays were performed as described previously (24).
β-Galactosidase reporter assays
Logarithmic phase S. cerevisiae (BY4741) cells harboring the pRLM1-lacZ plasmid were transferred to a 96-well plate, and treated with Calcofluor white (25 μg/mL) +/− protein kinase inhibitor at sub-inhibitory concentrations (1/2MIC). The cells were incubated at room temperature for 5 h and processed for β-galactosidase activity using the ThermoScientific Yeast β-galactosidase kit according to the manufacturer’s instructions (Pierce). β-galactosidase activity was determined by measuring OD420 using a SpectraMax Plate reader (Molecular Devices) and expressed as fold change in Miller units (nmole/mg/min) relative to untreated cells. Each experiment was performed in duplicate with three independent isolates.
Pil1 Immunobloting
Western blot analysis of Pil1-GFP was performed essentially as described by Luo et al (23). Briefly, Pil1-GFP containing cells were harvested and lysed using the SDS-PAGE sample buffer method. Extracts corresponding to equivalent numbers of cells were fractionated by SDS-PAGE electrophoresis on 7% gels, transferred to nitrocellulose and blocked overnight in 50 mM Tris pH 7.5/150 mM NaCl/0.05% (v/v) Tween-20+5% (w/v) non-fat skim milk. Pil1-GFP was detected using mouse anti-GFP (1:10,000 dilution, Clontech Living Colors) as primary and goat anti-mouse antibodies conjugated with horse-radish peroxidase (1:10,000) followed by visualization with ECL-Plus reagents (Amersham).
Microscopy
Light and fluorescence microscopy was performed using a Nikon ES80 epi-fluorescence microscope equipped with a CoolSnap CCD camera. Images were collected using NIS-Elements Software and processed in PhotoShop. All images were collected with identical exposure settings and equally processed with respect to tone and contrast.
Lucifer yellow (LY) endocytosis assays
LY uptake assays were performed as described by Dulic et al. (36) using LY obtained from Sigma. Briefly, yeast cells were grown to logarithmic phase, treated with either 10 μM KP-372-1 or 1% DMSO and incubated for 1 h. Cells were then exposed to LY and aliquots were removed at 15 min intervals. Endocytosis was stopped by the addition of sodium azide/succinate and the percentage of cells with vacuolar LY staining was determined by fluorescence microscopy.
Supplementary Material
Acknowledgments
We thank A. Kumar, D. Levin, A. Mitchell, J. Heitman and E. Phizicky for providing strains and plasmids. D.O. was a summer undergraduate research student supported by National Science Foundation grant 0849892. This work was supported by a grant from the National Institute of Allergy and Infectious Diseases 1R01AI075033 (D.J.K).
Footnotes
Supplementary Information Available: This material is available free of charge via the internet.
References
- 1.Eglen RM, Reisine T. The current status of drug discovery against the human kinome. Assay Drug Dev Technol. 2009;7:22–43. doi: 10.1089/adt.2008.164. [DOI] [PubMed] [Google Scholar]
- 2.Wash CT, Fischbach MA. Repurposing libraries of eukaryotic protein kinase inhibitors for antibiotic discovery. Proc Natl Acad Sci USA. 2009;106:1689–1690. doi: 10.1073/pnas.0813405106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen RE, Thorner J. Function and regulation of MAPK signaling pathways: lessons learned from the yeast Saccharomyces cerevisiae. Biochim Biophys Acta. 2007;1773:1311–340. doi: 10.1016/j.bbamcr.2007.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Casamyayor A, Torrance PD, Kobayashi T, Thorner J, Alessi DR. Functional counterparts of mammalian protein kinase PDK1 and SGK in budding yeast. Curr Biol. 1999;9:186–197. doi: 10.1016/s0960-9822(99)80088-8. [DOI] [PubMed] [Google Scholar]
- 5.Pfaller MA, Diekma DJ. Epidemiology of invasive candidiasis: persistent public health problem. Clin Microbiol Rev. 2007;20:133–163. doi: 10.1128/CMR.00029-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Denning DW, Hope WW. Therapy for fungal diseases: opportunities and priorities. Trends Microbiol. 2010;18:195–204. doi: 10.1016/j.tim.2010.02.004. [DOI] [PubMed] [Google Scholar]
- 7.Sussman A, Huss K, Chio LC, Heidler S, Shaw M, Ma D, Zhu G, Campbell RM, Park TS, Palaniappan K, Scott JE, Carpenter JW, Strege MA, Belvo MD, Swartling JR, Fischl A, Yeh WK, Ye XS. Discovery of cercosporamide, a known antifungal natural product as a selective Pkc1 kinase inhibitor through high-throughput screening. Eukaryot Cell. 2004;3:932–943. doi: 10.1128/EC.3.4.932-943.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim HJ, Park JE, Jin S, Kim JH, Song K. An isoquinolinium derivative selectively inhibits MAPK Spc1 of the stress-activated MAPK cascade of Schizosaccharomyces pombe. Chem Biol. 2006;13:881–889. doi: 10.1016/j.chembiol.2006.06.012. [DOI] [PubMed] [Google Scholar]
- 9.Levin D. Cell wall integrity signaling in S. cerevisiae. Microbiol Mol Biol Rev. 2005;69:262–291. doi: 10.1128/MMBR.69.2.262-291.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Heinisch JJ. Baker’s yeast as a tool for the development of antifungal kinase inhibitors-targeting protein kinase C and the cell integrity pathway. Biochim Biophys Acta. 2005;1754:171–182. doi: 10.1016/j.bbapap.2005.07.032. [DOI] [PubMed] [Google Scholar]
- 11.DiDone L, Scrimale TS, Baxter BK, Krysan DJ. A high-throughput assay of yeast lysis for drug discovery and genetic analysis. Nat Protocol. 2010;5:1107–1114. doi: 10.1038/nprot.2010.47. [DOI] [PubMed] [Google Scholar]
- 12.National Committee for Clinical Laboratory Standards. Reference method for broth dilution antifungal susceptibility testing of yeasts. Approved standard M27-A2; National Committee for Clinical Laboratory Standards; Wayne, PA. 2002. [Google Scholar]
- 13.Granville CA, Memmott RM, Gillis JJ, Dennis PA. Handicapping the race to develop inhibitors of the phosphoinositide-3-kinase/Akt/mammalian target of rapamycin pathway. Clin Cancer Res. 2006;12:679–689. doi: 10.1158/1078-0432.CCR-05-1654. [DOI] [PubMed] [Google Scholar]
- 14.Weinblatt ME, Kavanaugh A, Genovese MC, Musser TK, Grosshard EB, Magilavy DB. An oral spleen tyrosine kinase (Syk) inhibitor for rheumatoid arthritis. N Engl J Med. 2010;363:1303–1312. doi: 10.1056/NEJMoa1000500. [DOI] [PubMed] [Google Scholar]
- 15.Jung US, Sobering AK, Romo MJ, Levin DE. Regulation of Rlm1 transcription factor by the Mpk1 cell wall integrity MAP kinase. Mol Microbiol. 2002;46:781–789. doi: 10.1046/j.1365-2958.2002.03198.x. [DOI] [PubMed] [Google Scholar]
- 16.Zeng Z, Samudio IJ, Zhang W, Estrov Z, Pelicano H, Harris D, Frolova O, Hail N, Jr, Chen W, Kornblau SM, Haung P, Lu Y, Mills GB, Andreeff M, Konopleva M. Simultaneous inhibition of PDK1/Akt and Fms-like tyrosine kinase 3 signaling by a small-molecule KP372–1 induces mitochondrial dysfunction and apoptosis in acute myelogenous leukemia. Cancer Res. 2006;66:3737–3746. doi: 10.1158/0008-5472.CAN-05-1278. [DOI] [PubMed] [Google Scholar]
- 17.Pierce CG, Uppuluri P, Tristan AR, Wormley FL, Jr, Mowat E, Ramage G, Lopez-Ribot JL. A simple and reproducible 96-well plate based method for the formation of fungal biofilms and its application to antifungal susceptibility testing. Nat Protoc. 2008;3:1494–1500. doi: 10.1038/nport.2008.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu W, Zhao J, Li X, Li Y, Jiang L. The protein kinase Sch9p is required for the cell growth, filamentation, and virulence in the human fungal pathogen Candida albicans. FEMS Yeast Res. 2010;10:462–470. doi: 10.1111/j.1567-1364.2010.00617.x. [DOI] [PubMed] [Google Scholar]
- 19.Inagaki M, Schmelzle T, Yamaguchi K, Irie K, Hall MN, Matsumoto K. PDK1 homologs activate Pkc1-mitogen-activated protein kinase pathway in yeast. Mol Cell Biol. 1999;19:8344–8352. doi: 10.1128/mcb.19.12.8344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roelants FM, Baltz AG, Trott AE, Thorner J. A protein kinase network regulates the function of aminophospholipid flippases. Proc Natl Acad Sci USA. 2010;107:34–39. doi: 10.1073/pnas.0912497106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Friant S, Lombardi R, Schmelzle T, Hall MN, Riezman H. Sphingoid base signaling via Pkh kinases is required for endocytosis in yeast. EMBO J. 2001;20:6783–6792. doi: 10.1093/emboj/20.23.6783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Walther TC, Aguilar PS, Frolich F, Chu F, Moreira K, Burlingame, Walter P. Pkh-kinases control eisosome assembly and organization. EMBO J. 2007;26:4946–4955. doi: 10.1038/sj.emboj.7601933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Luo G, Gruhler A, Liu Y, Jensen ON, Dickson RC. The sphingolipid long chain base-Pkh1/2-Ypk1/2 signaling pathway regulates eisosome assembly and turnover. J Biol Chem. 2008;283:10433–10444. doi: 10.1074/jbc.M709972200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roelants FM, Torrance PD, Thorner J. Differential roles of PDK1- and PDK2-phosphorylation sites in the yeast AGC kinases Ypk1, Pkc1, and Sch9. Microbiology. 2004;150:3289–3304. doi: 10.1099/mic.0.27286-0. [DOI] [PubMed] [Google Scholar]
- 25.Giaver G, Shoemaker DD, Jones TW, Liang H, Winzeler EA, Astromoff A, Davis RW. Genomic profiling of drug sensitivities via induced haploinsufficiency. Nat Genet. 1995;21:278–283. doi: 10.1038/6791. [DOI] [PubMed] [Google Scholar]
- 26.Liu OW, Chun CD, Chow ED, Chen C, Madhani HD, Noble SM. Systematic genetic analysis of virulence in the human fungal pathogen Cryptococcus neoformans. Cell. 2008;135:174–188. doi: 10.1016/j.cell.2008.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hotte SJ, Oza A, Winquist EW, Moore M, Chen EX, Brown S, Pond GR, Dancey JE, Hirte HW. Phase I clinical trial of UCN-01 in combination with topotecan in patients with advanced solid cancers: a Princess Margaret Hospital phase II consortium study. Ann Oncol. 2006;17:334–340. doi: 10.1093/annonc/mdj076. [DOI] [PubMed] [Google Scholar]
- 28.Zhu J, Huang JW, Tseng PH, Yang YZ, Fowble J, Shiau CW, Shaw YJ, Kulp SK, Chen CS. From the cyclooxygenase-2 inhibitor celecoxib to a novel class of 3-phosphoinositide-dependent kinase-1 inhibitors. Cancer Res. 2004;64:4309–4318. doi: 10.1158/0008-5472.CAN-03-4063. [DOI] [PubMed] [Google Scholar]
- 29.Peifer C, Alessi DR. Small-molecule inhibitors of PDK1. Chem MedChem. 2008;3:1810–1838. doi: 10.1002/cmdc.200800195. [DOI] [PubMed] [Google Scholar]
- 30.Komander D, Kular GS, Bain J, Elliot M, Alessi DR, van Aalten DMF. Structural basis for UCN-01 (7-hydroxystaurosporine) specificity and PDK1 (3-phosphoinositide-dependent protein kinase-1) inhibition. Biochem J. 2003;375:255–262. doi: 10.1042/BJ20031119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.LaFayette SL, Collins C, Zaas AK, Schell WA, Betancourt-Quiroz M, Guanatilaka AA, Perfect JR, Cowen LE. Pkc signaling regulates drug resistance of the fungal pathogen Candida albicans via circuitry comprised of Mkc1, calcineurin, and Hsp90. PLoS Path. 2010;6:e1001069. doi: 10.1371/journal.ppat.1001069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gerik KJ, Bhimireddy SR, Ryerse JS, Specht CA, Lodge JK. Pkc1 is essential for protection against both oxidative and nitrosative stresses, cell integrity, and normal manifestations of virulence factors in the pathogenic fungus Cryptococcus neoformans. Eukaryot Cell. 2008;7:1685–1698. doi: 10.1128/EC.00146-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pavavincini G, Mendoza A, Antonsson B, Cooper M, Losberger C, Payton MA. The Candida albicans PKC1 gene encodes a protein kinase C homolog necessary for cellular integrity but not dimorphism. Yeast. 1996;12:741–756. doi: 10.1002/(sici)1097-0061(19960630)12:8<741::aid-yea967>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 34.Knight ZA, Shokat KM. Chemical genetics: where genetics and pharmacology meet. Cell. 2007;128:425–430. doi: 10.1016/j.cell.2007.01.021. [DOI] [PubMed] [Google Scholar]
- 35.Gelperin DM, White MA, Wilkinson ML, Kon Y, Kung LA, Wise KJ, Lopez-Hoyo N, Jiang L, Piccirillo S, Yu H, Gerstein M, Dumont ME, Phizicky EM, Snyder M, Grayhack EJ. Biochemical and genetic analysis of the yeast proteome with a movable ORF collection. Genes Dev. 2005;19:2816–2826. doi: 10.1101/gad.1362105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dulic V, Egerton M, Elguindi I, Rath S, Singer B, Reizman H. Yeast endocytosis assays. Methods Enzymol. 1991;194:697–710. doi: 10.1016/0076-6879(91)94051-d. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.