

Abstract
Abstract
The transient receptor potential (TRP) family of ion channels is a large family of cation selective ion channels, which are expressed and functional in a variety of tissues. In this review we focus on the most recent results detailing the role of TRP channels in the cardiovascular system. The presented results underscore the role of TRP channels in cardiomyocytes, smooth cells and endothelium, and in disease states such as hypertension, cardiac conduction block and cardiac hypertrophy.
Rudi Vennekens (36) is at the Catholic University in Leuven, Belgium (KU Leuven). Being a Master in Biochemistry he was fortunate enough to join Bernd Nilius's lab at the Physiology Department of the KU Leuven in 1997, where he studied the likes of volume-regulated anion channels and transient receptor potential (TRP) cation channels from an electrophysiologist's point of view. After a post-doc with Profs Veit Flockerzi and Marc Freichel at the University of Saarland in Homburg (Germany) he rejoined the KU Leuven, where he received an independent position in 2008. He is now working in the Laboratory of Ion Channel Research (LICR). His current research is focused on the physiological role of TRP ion channels.
 |
The transient receptor potential (TRP) family is large group of ion channel genes, which are related to the Drosophila Trp gene. To date 28 mammalian Trp genes are known, which are divided into six subfamilies: TRPC, TRPV, TRPM, TRPA, TRPP and TRPML. All TRP proteins constitute cation channels, but they display a daunting diversity in gating mechanism and cation selectivity. TRP channels can open upon direct ligand binding, G-protein coupled signalling and membrane depolarization. Most TRP channels are Ca2+ permeable non-selective cation channels, but exceptions are common: TRPV5 and TRPV6 are highly selective Ca2+ channels, TRPM4 and TRPM5 are not Ca2+ permeable at all, and TRPM6 and TRPM7 are Mg2+ selective cation channels (for recent reviews see Flockerzi, 2007; Wu et al. 2010a).
TRPs in endothelium and vascular smooth muscle
A role for TRP channels in vascular endothelium and smooth muscle was extensively reviewed recently (Earley, 2006; Guibert et al. 2008; Watanabe et al. 2008; Di & Malik, 2010; Dietrich et al. 2010; Earley, 2010; Earley & Brayden, 2010; Gonzalez-Cobos & Trebak, 2010; Yang et al. 2010; Zholos, 2010). In short, TRP channels are involved in endothelial barrier function, release of vasoactive compounds such as nitric oxide (NO), hypoxia sensing and endothelial cell migration. Likewise, in vascular smooth cells various TRP channels have been implicated in Ca2+ induced smooth muscle cell proliferation and migration, contraction, hypoxia sensing by pulmonary smooth muscle and stretch or mechanical sensing by vascular smooth muscle cells.
Somewhat surprising, in a recent study TRPA1 has been implicated as a player in endothelium-induced vasorelaxation (Earley et al. 2009). Indeed, application of AITC (mustard oil) to precontracted cerebral artery rings induced endothelium-dependent vasorelaxation, not related to nitric oxide release. The mechanism apparently involves activation of K+ channels both in endothelium and smooth muscle cells, but how the coupling between both cell types occurs is unclear. It seems counterintuitive that the authors report endothelial expression of TRPA1, because TRPA1 expression until now was detected by several authors almost exclusively in sensory neurons and mechano-sensory epithelial cells of the inner ear (Story et al. 2003; Jordt et al. 2004; Kobayashi et al. 2005; Nagata et al. 2005; Garcia-Anoveros & Nagata, 2007). Whether sensory nerves that innervate endothelium or smooth muscle cells might be involved in mustard oil induced vasorelaxation remains to be determined. It should also be noted that the selectivity of mustard oil for TRPA1 is increasingly being disputed (M. Gees, K. Talavera et al. personal communication).
Regarding the function of TRP channels in smooth muscle cells, recent results highlight the role of TRPs in the myogenic response in blood vessels, or the so-called Bayliss effect. When high internal pressure stretches blood vessels, they don't inflate like a balloon, but instead develop an active tension, which restricts the increase of blood vessel diameter. This is the Bayliss effect, and it is essential to keep the blood flow through blood vessels relatively stable even when perfusion pressure fluctuates (Bayliss, 1902; Davis & Hill, 1999).
It is generally hypothesized that mechanosensitive ion channels in the vascular smooth muscle cells are essential for this mechanism and downregulation of TRPM4 and TRPC6 gene expression in rat cerebral artery smooth muscle cells led to a severe loss in pressure-induced vasoconstriction, leading to the hypothesis that both TRPM4 and TRPC6 constitute mechanosensitive ion channels in vascular smooth muscle cells (Welsh et al. 2002; Earley et al. 2004; Brayden et al. 2008; Inoue et al. 2009). However, for both proteins mechano-sensitivity has been questioned or was not reported by other authors (Nilius et al. 2003; Gottlieb et al. 2008). Moreover, in TRPM4 deficient mice the Bayliss effect in hind-limb resistance vessels is unaffected (Mathar et al. 2010) and in TRPC6 deficient mice the threshold for the Bayliss effect is even decreased, i.e. the vessel becomes apparently even more sensitive to pressure-induced stretch (Dietrich et al. 2005).
An interesting alternative mechanism has been recently proposed, in which a mechanosensitive Gq-coupled receptor would initiate the downstream cell signalling of blood vessel stretch (Mederos y Schnitzler et al. 2008). Several TRP channels, including most TRPC channels and TRPM4, have been shown previously to be coupled with and activated by the Gq-PLC pathway, and could in this way be involved in the mechanosensitive current in vascular smooth muscle cells. In their study, Mederos y Schnitzler et al. convincingly show that TRPC6 does not meet the requirements for being a mechanosensitive ion channel in itself, but is clearly activated by a mechanical stimulus when co-expressed with the angiotensin receptor 1 (AT1R). In a smooth muscle cell line that does not exhibit mechanosensitive currents, overexpression of angiotensin receptors induces robust stretch activated current independent of receptor agonists. Agents that inhibit GPCRs or PLC inhibit stretch activation of the channel and the competitive AT1R antagonist losartan inhibits the Bayliss effect both in cerebral arteries and in isolated perfused kidney. Thus, it seems that mechanical activation of a G-protein coupled receptor might be the missing link between a mechanical stimulus and activation of TRPC channels. Further research will resolve how general this mechanism is (Earley et al. 2007; Gonzales et al. 2010).
Alternatively, very recently, two other TRP proteins, the polycystic kidney disease associated TRPP1 and TRPP2, were convincingly shown to be regulators of a stretch-activated ion channel (SAC) in smooth cells and regulators of myogenic tone (Sharif-Naeini et al. 2009). Indeed, SAC currents in smooth muscle cells are inhibited by TRPP2 expression, which is reversed by TRPP1 expression, indicating that the TRPP1/TRPP2 expression ratio regulates pressure-induced activation of SAC currents in smooth muscle cells. In mesenteric arteries, TRPP1 deletion in smooth muscle cells reduces SAC activity, and the arterial myogenic response. Inversely, depletion of TRPP2 in TRPP1-deficient arteries rescues both SAC activity and the myogenic response to intraluminal pressure (Sharif-Naeini et al. 2009). In this study the protein underlying the stretch-activated current remains to be identified.
Interesting in this regard, two new genes were presented at the TRP2010 Leuven meeting, Piezo-1 and Piezo-2, which are essential for very large and robust mechano-activated currents in the neuroblastoma cell line N2A (Coste et al. 2010). Whether these genes are expressed in smooth muscle cells (or endothelial cells) is unclear at this point. In light of the above-mentioned results, it might be interesting to consider the involvement of these proteins in the Bayliss effect, and whether they could interact with other TRP channels.
TRP channels in cardiac muscle
TRP channels are expressed in every cell type present in the heart, including cardiomyocytes, fibroblasts, endothelial cells and vascular smooth muscle cells (for a review see Nilius et al. 2007; Watanabe et al. 2008; Watanabe, 2009).
Recently, genetic analysis of human patients was used to shed light on the role of TRPM4 in the heart (Kruse et al. 2009; Liu et al. 2010). TRPM4 is a Ca2+ activated non-selective, Ca2+ impermeable cation channel that is expressed in atrial and ventricular tissue, in pacemaker cells, and in Purkinje fibres (Guinamard et al. 2004, 2006; Liu et al. 2010). Progressive familial heart block type I (PFHBI) is a progressive cardiac bundle branch disease in the His-Purkinje system that exhibits autosomal-dominant inheritance. In three branches of a large South African family with an autosomal-dominant form of PFHBI, Kruse et al. (2009) identified a specific mutation in the TRPM4 gene, which leads to an amino acid substitution E7K in the TRPM4 amino terminus. Overexpression studies showed that this specific mutation attenuates desumoylation of the TRPM4 channel in a cell line and thereby impairs endocytosis of the channel, leading to elevated TRPM4 channel density at the cell surface. In another study, one Lebanese family and two French families with autosomal dominant isolated cardiac conduction blocks were also used for linkage analysis (Liu et al. 2010). A heterozygous missense mutation of the TRPM4 gene was found in each family (p.Arg164Trp, p.Ala432Thr, and p.Gly844Asp), and all three mutations resulted again in an increased current density in an overexpression system, due to an elevated TRPM4 channel density at the cell surface secondary to deregulation of sumoylation and impaired endocytosis. It should, however, be noted that in the latter study, a penetrance value as low as 54% (in females) was reported, which is the proportion of individuals that carry a variation in the TRPM4 gene and exhibit also the associated trait. Also, TRPM4 mutations were found after sequencing 12 candidate genes, from the linked genomic interval of 4 megabase size that contains 300 genes. Thus, it cannot be ruled out completely that mutations in other genes also contribute to the syndrome.
Both studies imply that a gain-of-function of TRPM4 activity would lead to conduction block in the heart, but it is unclear how this would occur in detail. The increased membrane retention due to impaired desumoylation was for instance only shown in cell lines overexpressing mutant TRPM4, not in native cardiomyocytes. It is hypothesized that an excess depolarizing current through the TRPM4 channel might lead to conduction abnormalities in Purkinje fibres but also this remains to be shown.
TRP channels and blood-pressure regulation
Blood pressure is a critical haemodynamic parameter which results from a complex interplay of several organs (vasculature, the heart, kidneys and the central and autonomic nervous system) and external factors (stress, lifestyle, physical demand). Several TRPs have been implicated already in blood pressure regulation, but direct evidence for regulation of basal blood pressure by TRP channels is only available for three examples, TRPM4, TRPC1 and TRPC6 (Dietrich et al. 2005; Mathar et al. 2010).
TRPM4 is expressed in several organs implicated in blood-pressure regulation, including the kidneys, the heart (see above), vascular endothelium and smooth muscle and the adrenal glands (Nilius et al. 2003, 2007; Nilius & Vennekens, 2006; Vennekens & Nilius, 2007). Trpm4 deficient mice display increased blood pressure (Mathar et al. 2010). Indeed, after a recovery period from implantation of a blood pressure transmitter, TRPM4 mice have on average about a 10mmHg increase in blood pressure compared to wild-type mice. This hypertension remains present during resting and active periods of mouse behaviour and is not due to changes in locomotor activity and heart rate. Mathar et al. present a detailed analysis of this phenotype, including kidney function, the renin–angiotensin system and the myogenic response and agonist induced changes in contractility of hindlimb resistance levels. In short, in identical experimental conditions none of these parameters is essentially changed in Trpm4−/− mice, and could account for the hypertension. Also, cardiac output, ejection fraction and cardiac contractility of the heart are not changed in Trpm4−/− mice, at least in basal conditions. Strikingly, however, a non-specific ganglion blocker, hexamethonium, can abolish the difference in blood pressure between WT and Trpm4−/− mice. This results in a fall in blood pressure to the same level in WT and Trpm4−/− animals, suggesting that the there is a differential regulation of blood pressure by the autonomic nervous system in WT and Trpm4−/− mice. Indeed, Trpm4−/− mice show increased plasma adrenaline levels and increased urinary excretion of the catecholamine breakdown products metanephrin and vanillyl mandelic acid, suggesting that Trpm4−/− mice display an increased sympathetic tone. The increased plasma catecholamine level can account for the increased mean arterial blood pressure through its well-known effect on vessel tone and cardiac output (Guyenet, 2006).
To further delineate this, it should be clear that the primary source of adrenaline in the body is the chromaffin cells in the medulla of the adrenal gland (Ungar & Phillips, 1983), which are innervated by preganglionic fibres of the sympathetic nervous system. When Mathar and colleagues analysed the function of the chromaffin cells it became clear that Trpm4−/− chromaffin display more exocytotic release events (as determined by amperometry), as compared to wild-type cells, when excited with a similar dose of acetylcholine. Strikingly, this result is independent of the global intracellular Ca2+ signal in the cell, which triggers the exocytosis. These results raise the intriguing suggestion that TRPM4 might play an unexpected role in the exocytosis machinery of chromaffin cells. It apparently serves as a negative regulator of vesicle release, though it is completely unclear how it could play this role. Taken together, these data clearly indicate that TRPM4 is a regulator of sympathetic tone and in this way has a profound impact on the regulation of blood pressure.
Another murine model in which blood pressure was probed is the Trpc6−/− deficient mouse. Here an elevation of about 7 mmHg in basal mean arterial blood pressure was detected in conscious mice (Dietrich et al. 2005). This seems somewhat counterintuitive, since it was shown previously that vascular smooth muscle contraction activated by α1-adrenergic agonists can be blocked by suppressing Trpc6 expression, and that vasopressin stimulation of A7r5 smooth muscle cells leads to TRPC6 activation. However, Trpc6−/− mice display higher agonist induced contractility in isolated tracheal and aortic rings. These apparently conflicting results can, however, be explained by the unexpected and compensatory overexpression of TRPC3, which was reported in Trpc6−/− mice in the same study. TRPC3 is a constitutively active non-selective cation channel, and its overexpression leads to enhanced basal and agonist-induced Ca2+ entry into smooth cells, both through the TRPC3 channel and as a result of increased depolarization and activation of voltage-gated Ca2+ channels, which eventually leads to enhanced contractility of smooth muscle cells (Dietrich et al. 2005, 2010).
Finally, in Trpc1−/− mice endothelium-derived hyperpolarizing factor-dependent vasorelaxation seems to be augmented, while NO mediated vasorelaxation is unchanged. TRPC1 is a non-selective cation channel, activated upon phospholipase C activation, although exact details remain unclear. The data suggest that TRPC1 contributes to the depolarization of the endothelial cell after stimulation with acetylcholine, thereby counteracting the hyperpolarisation mediated by activation of Ca2+-dependent K+ channels. A lack of TRPC1 then leads to stronger hyperpolarisation, extensive influx of Ca2+ and augmented release of endothelium derived hyperpolarizing factor (EDHF). Concurrently with these data, blood pressure in TRPC1 deficient mice is moderately decreased.
Also in TRPV1 and TRPV4 KO mice, blood pressure was studied in freely moving mice. In neither of them could a change in basal mean arterial pressure be detected (Suzuki et al. 2003; Pacher et al. 2004; Zhang et al. 2009). However, a TRPV1-specific agonist, capsaicin, induced a marked drop in blood pressure, which is not present in TRPV1 deficient mice (Pacher et al. 2004). Analogously, a TRPV4-specific agonist, GSK1016790A, induces a dose-dependent reduction of blood pressure followed by circulatory collapse, whereas Trpv4−/− show no acute cardiovascular effects in response to the same compound. The drop in blood pressure seems to be due to a potent endothelial and NO-dependent relaxation of vasculature. The circulatory collapse is associated with TRPV4-dependent vascular leakage and tissue haemorrhage in the lung, intestine and kidney (Willette et al. 2008).
TRP channels and hypertrophy
Cardiac hypertrophy is a thickening of the heart muscle, which results in a decrease in size of the chambers of the heart, including the left and right ventricles. A common cause of cardiac hypertrophy is high blood pressure (hypertension) and heart valve stenosis (Heineke & Molkentin, 2006; Molkentin, 2006). A role for especially TRPC channels has been suggested in several studies already (Watanabe et al. 2008).
In an early study, TRPC3 and TRPC6 were implicated in angiotensin II-induced nuclear factor of activated T-cells (NFAT) activation in isolated cardiomyocytes (Onohara et al. 2006), which is an essential step of cardiac hypertrophy development in the whole heart. Mechanistic data were presented showing that they are apparently essential components of the Ang-II induced signalling pathway, which leads to production of diacylglycerol and TRPC3 and TRPC6 mediated Ca2+ influx (Onohara et al. 2006). Studies that followed tried to correlate these data with animal models either overexpressing TRP channels or with decreased TRP channel expression.
When mRNA and protein expression of several TRP channel subunits were evaluated using hearts from abdominal aortic-banded (AAB) rats (a common technique to induce cardiac hypertrophic growth), TRPC1 expression was significantly increased in the hearts of AAB rats compared to sham-operated rats. Using primary cultures of neonatal rat cardiomyocytes, it was shown that expression of TRPC1, brain natriuretic peptide (BNP), and atrial natriuretic factor (ANF), as well as store-operated Ca2+ entry (SOCE) and cell surface area, was increased following endothelin-1 (ET-1) treatment. Silencing of the TRPC1 gene via small interfering RNA (siRNA) could attenuate SOCE and prevented ET-1-, angiotensin II- and phenylephrine-induced cardiac hypertrophy (Ohba et al. 2007). In addition to this, a recent study using TRPC1 knockout mice, showed that they are resistant to the induction of cardiac hypertrophy, either in response to haemodynamic stress (through aortic banding) or neurohormonal excess (through chronic angiotensin infusion) (Seth et al. 2009).
Overexpression of TRPC3 in mouse cardiac myocytes elicited increased store-operated Ca2+ entry, increased calcineurin-NFAT activation in vivo, cardiomyopathy, and increased hypertrophy after neurohormonal excess or hemodynamic stress stimulation. In effect, this study suggests that enhanced store-operated Ca2+ entry in the heart can regulate calcineurin-NFAT signalling in vivo, which secondarily impacts the hypertrophic response and cardiomyopathy (Nakayama et al. 2006). In parallel, another study showed that TRPC6 was upregulated in mouse hearts in response to activated calcineurin and pressure overload, as well as in failing human hearts. Two conserved NFAT consensus sites in the promoter of the TRPC6 gene conferred responsiveness to cardiac stress. Cardiac-specific overexpression of TRPC6 in transgenic mice resulted in heightened sensitivity to stress, a propensity for lethal cardiac growth and heart failure, and an increase in NFAT-dependent expression of β-myosin heavy chain, a sensitive marker for pathological hypertrophy. These findings implicate TRPC6 as a positive regulator of calcineurin-NFAT signalling and a key component of a calcium-dependent regulatory loop that drives pathological cardiac remodelling (Kuwahara et al. 2006). In a follow-up study, functional data suggest that TRPC6 blockade might be a promising tool to prevent the development of cardiac hypertrophy upon excessive calcineurin-NFAT signalling (Kinoshita et al. 2010).
In a recent report, it was shown that in transgenic mice expressing, specifically in cardiac myocytes, a dominant-negative (dn) TRPC3, dnTRPC6, or dnTRPC4 construct the cardiac hypertrophic response following either neuroendocrine agonist infusion or pressure-overload stimulation is attenuated. dnTRPC transgenic mice also were partially protected from loss of cardiac functional performance following long-term pressure-overload stimulation. They show less of a reduction of fractional shortening in the ECG, they were protected from lung oedema, which are characteristic for heart failure and showed less ventricular fibrosis than WT controls. Importantly, adult myocytes isolated from hypertrophic WT hearts showed a Ca2+ influx activity under store-depleted conditions that was not observed in myocytes from hypertrophied dnTRPC3, dnTRPC6, or dnTRPC4 hearts. Moreover, dnTRPC4 inhibited the activity of the TRPC3/6/7 subfamily in the heart, suggesting that these two subfamilies function in coordinated complexes. Mechanistically, it is suggested that inhibition of TRPC channels in transgenic mice or in cultured neonatal myocytes leads to significantly reduced activity of the transcription factor NFAT, thereby attenuating cardiac remodelling (Wu et al. 2010b).
Finally, data presented at the Leuven TRP2010 meeting suggest that in Trpc1/Trpc4−/− mice the isoproterenol-induced hypertrophy is reduced, whereas the angiotensin II-induced hypertrophic response is increased in Trpc3/Trpc6−/− mice (Camacho-Londono et al.). Especially the latter data are somewhat surprising considering the above-mentioned elements that both TRPC3 and TRPC6 activity seem essential for hypertrophic signalling, but further details are lacking at the time of writing.
Thus, it is now extensively proposed that TRPC channels are necessary components of pathological cardiac hypertrophy, apparently predominantly by being an essential part of the signalling pathway leading to calcineurin-NFAT activation. The exact choreography of how these channels each influences the development of cardiac hypertrophy remains, however, a matter of study. Notably, what seems to be lacking in this field of study is a systematic analysis of cardiac hypertrophy development of TRPC knockout mice, and in extenso cardiac specific knockout mice. Especially considering the repeated use of cardiac-specific overexpression constructs (either WT or ‘specific’ dominant-negative genes), it seems essential to delineate TRPC involvement in cardiac hypertrophy to cardiac myocytes. Interestingly in this regard, it was recently suggested that another transient receptor potential channel (TRPM7) is the molecular basis of a major Ca2+ permeable channel in human atrial fibroblasts. Knocking down TRPM7 by small hairpin RNA largely eliminates TRPM7 current in atrial fibroblasts. More importantly, atrial fibroblasts from atrial fibrillation patients show a striking upregulation of TRPM7 mediated Ca2+ influx and are more prone to myofibroblast differentiation. Cardiac fibrosis contributes to pathogenesis of atrial fibrillation (AF), and although it has been suggested that Ca2+ signals are involved in fibrosis promotion, the molecular basis of Ca2+ signalling mechanisms and how Ca2+ signals contribute to fibrogenesis remained largely unknown. TRPM7 gene knockdown markedly reduced basal AF fibroblast differentiation, and aparrently transforming growth factor (TGF)-β1, the major stimulator of atrial fibrosis, requires TRPM7-mediated Ca2+ influx for its effect on fibroblast proliferation and differentiation (Du et al. 2010). Thus it is clear that TRP channels are not only functional in cardiac myocytes but also influence Ca2+ signalling in cardiac fibroblasts. This obviously has important consequences for the pathogenesis of atrial fibrillation, but could also have important implications for the development of cardiac hypertrophy. Indeed, it has been shown extensively that cardiac fibroblasts play an important role in the development of this condition (Porter & Turner, 2009). However, in this cell type hardly anything is known about the role of TRP channels other than TRPM7.
Conclusion
Figure 1 summarizes the role of TRP channels in cells of the cardiovascular system. A role for TRP channels has now been extensively shown in several cell types of the cardiovascular system, including endothelium, smooth muscle and cardiac myocytes. Accordingly they play an essential role in diverse functions such as blood pressure regulation, vascular myogenic tone, cardiac hypertrophy and cardiac fibrosis. However, it is also clear that how TRP channels function in these different cell types remains largely to be determined in detail. There are clear indications for the potential of TRP channel targeting pharmaca in the treatment of cardiovascular disease, but a more detailed knowledge of the cellular physiology of TRP channels will increase our understanding of disease development and could open even brighter perspectives for early detection and prevention.
Figure 1. Schematic summary of the role of TRP channels in cells of the cardiovascular system.
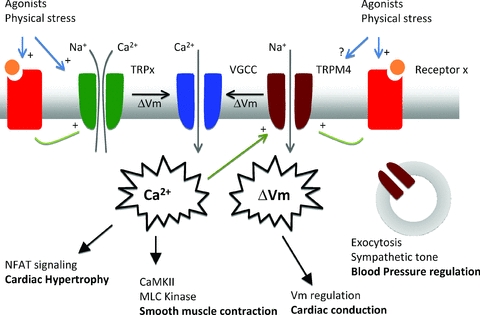
Roughly TRP channels can be subdivided in two groups: Ca2+ permeable and Ca2+ impermeable cation channels. Both will have an effect on intracellular Ca2+ dynamics, either directly by providing a Ca2+ influx pathway, or indirectly through membrane depolarisation, activation of voltage-gated Ca2+ channels and/or influencing the driving force for Ca2+ entry. TRP channels might be activated directly by agonists, or indirectly through G-protein coupled receptors. TRPC channels are Ca2+ permeable channels activated by G-protein coupled receptors (GPCR), through Gq and phospholipase C. The resulting Ca2+ influx apparently regulates specifically NFAT signalling and development of cardiac hypertrophy, without changing the Ca2+ transient during normal heart cycle. Ca2+ influx either through TRP channels or voltage-gated Ca2+ channel activation after TRP channel mediated depolarization is linked with myosin light chain kinase phosphorylation and smooth muscle contraction during the development of myogenic tone. A mechanically activated GPCR is linked with activation of TRPC6 and possibly TRPM4. Membrane potential regulation by TRPM4 in cardicac Purkinje cells might be important for proper cardiac conduction of the action potential through the heart. Through an unknown mechanism, TRPM4 also plays a role in the exocytosis of adrenaline from chromaffin cells, regulating in this way the sympathetic control of blood pressure. For more details and references, see the text.
References
- Bayliss WM. On the local reactions of the arterial wall to changes of internal pressure. J Physiol. 1902;28:220–231. doi: 10.1113/jphysiol.1902.sp000911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brayden JE, Earley S, Nelson MT, Reading S. Transient receptor potential (TRP) channels, vascular tone and autoregulation of cerebral blood flow. Clin Exp Pharmacol Physiol. 2008;35:1116–1120. doi: 10.1111/j.1440-1681.2007.04855.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coste B, Mathur J, Schmidt M, Earley TJ, Ranade S, Petrus MJ, Dubin AE, Patapoutian A. Piezo1 and Piezo2 are essential components of distinct mechanically activated cation channels. Science. 2010;330:55–60. doi: 10.1126/science.1193270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis MJ, Hill MA. Signaling mechanisms underlying the vascular myogenic response. Physiol Rev. 1999;79:387–423. doi: 10.1152/physrev.1999.79.2.387. [DOI] [PubMed] [Google Scholar]
- Di A, Malik AB. TRP channels and the control of vascular function. Curr Opin Pharmacol. 2010;10:127–132. doi: 10.1016/j.coph.2009.11.010. [DOI] [PubMed] [Google Scholar]
- Dietrich A, Kalwa H, Gudermann T. TRPC channels in vascular cell function. Thromb Haemost. 2010;103:262–270. doi: 10.1160/TH09-08-0517. [DOI] [PubMed] [Google Scholar]
- Dietrich A, Mederos YSM, Gollasch M, Gross V, Storch U, Dubrovska G, Obst M, Yildirim E, Salanova B, Kalwa H, Essin K, Pinkenburg O, Luft FC, Gudermann T, Birnbaumer L. Increased vascular smooth muscle contractility in TRPC6– mice. Mol Cell Biol. 2005;25:6980–6989. doi: 10.1128/MCB.25.16.6980-6989.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Xie J, Zhang Z, Tsujikawa H, Fusco D, Silverman D, Liang B, Yue L. TRPM7-mediated Ca2+ signals confer fibrogenesis in human atrial fibrillation. Circ Res. 2010;106:992–1003. doi: 10.1161/CIRCRESAHA.109.206771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earley S. Molecular diversity of receptor operated channels in vascular smooth muscle: a role for heteromultimeric TRP channels? Circ Res. 2006;98:1462–1464. doi: 10.1161/01.RES.0000231255.32630.df. [DOI] [PubMed] [Google Scholar]
- Earley S. Vanilloid and melastatin transient receptor potential channels in vascular smooth muscle. Microcirculation. 2010;17:237–249. doi: 10.1111/j.1549-8719.2010.00026.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earley S, Brayden JE. Transient receptor potential channels and vascular function. Clin Sci (Lond) 2010;119:19–36. doi: 10.1042/CS20090641. [DOI] [PubMed] [Google Scholar]
- Earley S, Gonzales AL, Crnich R. Endothelium- dependent cerebral artery dilation mediated by TRPA1 and Ca2+-activated K+ channels. Circ Res. 2009;104:987–994. doi: 10.1161/CIRCRESAHA.108.189530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earley S, Straub SV, Brayden JE. Protein kinase C regulates vascular myogenic tone through activation of TRPM4. Am J Physiol Heart Circ Physiol. 2007;292:H2613–2622. doi: 10.1152/ajpheart.01286.2006. [DOI] [PubMed] [Google Scholar]
- Earley S, Waldron BJ, Brayden JE. Critical role for transient receptor potential channel TRPM4 in myogenic constriction of cerebral arteries. Circ Res. 2004;95:922–929. doi: 10.1161/01.RES.0000147311.54833.03. [DOI] [PubMed] [Google Scholar]
- Flockerzi V. An introduction on TRP channels. Handb Exp Pharmacol. 2007:1–19. doi: 10.1007/978-3-540-34891-7_1. [DOI] [PubMed] [Google Scholar]
- Garcia-Anoveros J, Nagata K. Trpa1. Handb Exp Pharmacol. 2007:347–362. doi: 10.1007/978-3-540-34891-7_21. [DOI] [PubMed] [Google Scholar]
- Gonzales AL, Amberg GC, Earley S. Ca2+ release from the sarcoplasmic reticulum is required for sustained TRPM4 activity in cerebral artery smooth muscle cells. Am J Physiol Cell Physiol. 2010;299:C279–288. doi: 10.1152/ajpcell.00550.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Cobos JC, Trebak M. TRPC channels in smooth muscle cells. Front Biosci. 2010;15:1023–1039. doi: 10.2741/3660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlieb P, Folgering J, Maroto R, Raso A, Wood TG, Kurosky A, Bowman C, Bichet D, Patel A, Sachs F, Martinac B, Hamill OP, Honore E. Revisiting TRPC1 and TRPC6 mechanosensitivity. Pflugers Arch. 2008;455:1097–1103. doi: 10.1007/s00424-007-0359-3. [DOI] [PubMed] [Google Scholar]
- Guibert C, Ducret T, Savineau JP. Voltage-independent calcium influx in smooth muscle. Prog Biophys Mol Biol. 2008;98:10–23. doi: 10.1016/j.pbiomolbio.2008.05.001. [DOI] [PubMed] [Google Scholar]
- Guinamard R, Chatelier A, Demion M, Potreau D, Patri S, Rahmati M, Bois P. Functional characterization of a Ca2+-activated non-selective cation channel in human atrial cardiomyocytes. J Physiol. 2004;558:75–83. doi: 10.1113/jphysiol.2004.063974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guinamard R, Demion M, Magaud C, Potreau D, Bois P. Functional expression of the TRPM4 cationic current in ventricular cardiomyocytes from spontaneously hypertensive rats. Hypertension. 2006;48:587–594. doi: 10.1161/01.HYP.0000237864.65019.a5. [DOI] [PubMed] [Google Scholar]
- Guyenet PG. The sympathetic control of blood pressure. Nat Rev Neurosci. 2006;7:335–346. doi: 10.1038/nrn1902. [DOI] [PubMed] [Google Scholar]
- Heineke J, Molkentin JD. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat Rev Mol Cell Biol. 2006;7:589–600. doi: 10.1038/nrm1983. [DOI] [PubMed] [Google Scholar]
- Inoue R, Jian Z, Kawarabayashi Y. Mechanosensitive TRP channels in cardiovascular pathophysiology. Pharmacol Ther. 2009;123:371–385. doi: 10.1016/j.pharmthera.2009.05.009. [DOI] [PubMed] [Google Scholar]
- Jordt SE, Bautista DM, Chuang HH, McKemy DD, Zygmunt PM, Hogestatt ED, Meng ID, Julius D. Mustard oils and cannabinoids excite sensory nerve fibres through the TRP channel ANKTM1. Nature. 2004;427:260–265. doi: 10.1038/nature02282. [DOI] [PubMed] [Google Scholar]
- Kinoshita H, Kuwahara K, Nishida M, Jian Z, Rong X, Kiyonaka S, Kuwabara Y, Kurose H, Inoue R, Mori Y, Li Y, Nakagawa Y, Usami S, Fujiwara M, Yamada Y, Minami T, Ueshima K, Nakao K. Inhibition of TRPC6 channel activity contributes to the antihypertrophic effects of natriuretic peptides-guanylyl cyclase-A signaling in the heart. Circ Res. 2010;106:1849–1860. doi: 10.1161/CIRCRESAHA.109.208314. [DOI] [PubMed] [Google Scholar]
- Kobayashi K, Fukuoka T, Obata K, Yamanaka H, Dai Y, Tokunaga A, Noguchi K. Distinct expression of TRPM8, TRPA1, and TRPV1 mRNAs in rat primary afferent neurons with aδ/c-fibers and colocalization with trk receptors. J Comp Neurol. 2005;493:596–606. doi: 10.1002/cne.20794. [DOI] [PubMed] [Google Scholar]
- Kruse M, Schulze-Bahr E, Corfield V, Beckmann A, Stallmeyer B, Kurtbay G, Ohmert I, Schulze-Bahr E, Brink P, Pongs O. Impaired endocytosis of the ion channel TRPM4 is associated with human progressive familial heart block type I. J Clin Invest. 2009;119:2737–2744. doi: 10.1172/JCI38292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwahara K, Wang Y, McAnally J, Richardson JA, Bassel-Duby R, Hill JA, Olson EN. TRPC6 fulfills a calcineurin signaling circuit during pathologic cardiac remodeling. J Clin Invest. 2006;116:3114–3126. doi: 10.1172/JCI27702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, El Zein L, Kruse M, Guinamard R, Beckmann A, Bozio A, Kurtbay G, Megarbane A, Ohmert I, Blaysat G, Villain E, Pongs O, Bouvagnet P. Gain-of-function mutations in TRPM4 cause autosomal dominant isolated cardiac conduction disease. Circ Cardiovasc Genet. 2010;3:374–385. doi: 10.1161/CIRCGENETICS.109.930867. [DOI] [PubMed] [Google Scholar]
- Mathar I, Vennekens R, Meissner M, Kees F, Van Der Mieren G, Camacho Londono JE, Uhl S, Voets T, Hummel B, Van Den Bergh A, Herijgers P, Nilius B, Flockerzi V, Schweda F, Freichel M. Increased catecholamine secretion contributes to hypertension in TRPM4-deficient mice. J Clin Invest. 2010;120:3267–3279. doi: 10.1172/JCI41348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mederos y Schnitzler M, Storch U, Meibers S, Nurwakagari P, Breit A, Essin K, Gollasch M, Gudermann T. Gq-coupled receptors as mechanosensors mediating myogenic vasoconstriction. EMBO J. 2008;27:3092–3103. doi: 10.1038/emboj.2008.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molkentin JD. Dichotomy of Ca2+ in the heart: contraction versus intracellular signaling. J Clin Invest. 2006;116:623–626. doi: 10.1172/JCI27824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagata K, Duggan A, Kumar G, Garcia-Anoveros J. Nociceptor and hair cell transducer properties of TRPA1, a channel for pain and hearing. J Neurosci. 2005;25:4052–4061. doi: 10.1523/JNEUROSCI.0013-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama H, Wilkin BJ, Bodi I, Molkentin JD. Calcineurin-dependent cardiomyopathy is activated by TRPC in the adult mouse heart. FASEB J. 2006;20:1660–1670. doi: 10.1096/fj.05-5560com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilius B, Owsianik G, Voets T, Peters JA. Transient receptor potential cation channels in disease. Physiol Rev. 2007;87:165–217. doi: 10.1152/physrev.00021.2006. [DOI] [PubMed] [Google Scholar]
- Nilius B, Prenen J, Droogmans G, Voets T, Vennekens R, Freichel M, Wissenbach U, Flockerzi V. Voltage dependence of the Ca2+-activated cation channel TRPM4. J Biol Chem. 2003;278:30813–30820. doi: 10.1074/jbc.M305127200. [DOI] [PubMed] [Google Scholar]
- Nilius B, Vennekens R. From cardiac cation channels to the molecular dissection of the transient receptor potential channel TRPM4. Pflugers Arch. 2006;453:313–321. doi: 10.1007/s00424-006-0088-z. [DOI] [PubMed] [Google Scholar]
- Ohba T, Watanabe H, Murakami M, Takahashi Y, Iino K, Kuromitsu S, Mori Y, Ono K, Iijima T, Ito H. Upregulation of TRPC1 in the development of cardiac hypertrophy. J Mol Cell Cardiol. 2007;42:498–507. doi: 10.1016/j.yjmcc.2006.10.020. [DOI] [PubMed] [Google Scholar]
- Onohara N, Nishida M, Inoue R, Kobayashi H, Sumimoto H, Sato Y, Mori Y, Nagao T, Kurose H. TRPC3 and TRPC6 are essential for angiotensin II-induced cardiac hypertrophy. EMBO J. 2006;25:5305–5316. doi: 10.1038/sj.emboj.7601417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacher P, Batkai S, Kunos G. Haemodynamic profile and responsiveness to anandamide of TRPV1 receptor knock-out mice. J Physiol. 2004;558:647–657. doi: 10.1113/jphysiol.2004.064824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter KE, Turner NA. Cardiac fibroblasts: at the heart of myocardial remodeling. Pharmacol Ther. 2009;123:255–278. doi: 10.1016/j.pharmthera.2009.05.002. [DOI] [PubMed] [Google Scholar]
- Seth M, Zhang ZS, Mao L, Graham V, Burch J, Stiber J, Tsiokas L, Winn M, Abramowitz J, Rockman HA, Birnbaumer L, Rosenberg P. TRPC1 channels are critical for hypertrophic signaling in the heart. Circ Res. 2009;105:1023–1030. doi: 10.1161/CIRCRESAHA.109.206581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharif-Naeini R, Folgering JH, Bichet D, Duprat F, Lauritzen I, Arhatte M, Jodar M, Dedman A, Chatelain FC, Schulte U, Retailleau K, Loufrani L, Patel A, Sachs F, Delmas P, Peters DJ, Honore E. Polycystin-1 and -2 dosage regulates pressure sensing. Cell. 2009;139:587–596. doi: 10.1016/j.cell.2009.08.045. [DOI] [PubMed] [Google Scholar]
- Story GM, Peier AM, Reeve AJ, Eid SR, Mosbacher J, Hricik TR, Earley TJ, Hergarden AC, Andersson DA, Hwang SW, McIntyre P, Jegla T, Bevan S, Patapoutian A. ANKTM1, a TRP-like channel expressed in nociceptive neurons, is activated by cold temperatures. Cell. 2003;112:819–829. doi: 10.1016/s0092-8674(03)00158-2. [DOI] [PubMed] [Google Scholar]
- Suzuki M, Mizuno A, Kodaira K, Imai M. Impaired pressure sensation in mice lacking TRPV4. J Biol Chem. 2003;278:22664–22668. doi: 10.1074/jbc.M302561200. [DOI] [PubMed] [Google Scholar]
- Ungar A, Phillips JH. Regulation of the adrenal medulla. Physiol Rev. 1983;63:787–843. doi: 10.1152/physrev.1983.63.3.787. [DOI] [PubMed] [Google Scholar]
- Vennekens R, Nilius B. Insights into TRPM4 function, regulation and physiological role. Handb Exp Pharmacol. 2007:269–285. doi: 10.1007/978-3-540-34891-7_16. [DOI] [PubMed] [Google Scholar]
- Watanabe H. Pathological role of TRP channels in cardiovascular and respiratory diseases. Nippon Yakurigaku Zasshi. 2009;134:127–130. doi: 10.1254/fpj.134.127. [DOI] [PubMed] [Google Scholar]
- Watanabe H, Murakami M, Ohba T, Takahashi Y, Ito H. TRP channel and cardiovascular disease. Pharmacol Ther. 2008;118:337–351. doi: 10.1016/j.pharmthera.2008.03.008. [DOI] [PubMed] [Google Scholar]
- Welsh DG, Morielli AD, Nelson MT, Brayden JE. Transient receptor potential channels regulate myogenic tone of resistance arteries. Circ Res. 2002;90:248–250. doi: 10.1161/hh0302.105662. [DOI] [PubMed] [Google Scholar]
- Willette RN, Bao W, Nerurkar S, Yue TL, Doe CP, Stankus G, Turner GH, Ju H, Thomas H, Fishman CE, Sulpizio A, Behm DJ, Hoffman S, Lin Z, Lozinskaya I, Casillas LN, Lin M, Trout RE, Votta BJ, Thorneloe K, Lashinger ES, Figueroa DJ, Marquis R, Xu X. Systemic activation of the transient receptor potential vanilloid subtype 4 channel causes endothelial failure and circulatory collapse: Part 2. J Pharmacol Exp Ther. 2008;326:443–452. doi: 10.1124/jpet.107.134551. [DOI] [PubMed] [Google Scholar]
- Wu LJ, Sweet TB, Clapham DE. International Union of Basic and Clinical Pharmacology. LXXVI. Current progress in the mammalian TRP ion channel family. Pharmacol Rev. 2010a;62:381–404. doi: 10.1124/pr.110.002725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Eder P, Chang B, Molkentin JD. TRPC channels are necessary mediators of pathologic cardiac hypertrophy. Proc Natl Acad Sci U S A. 2010b;107:7000–7005. doi: 10.1073/pnas.1001825107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang XR, Lin MJ, Sham JS. Physiological functions of transient receptor potential channels in pulmonary arterial smooth muscle cells. Adv Exp Med Biol. 2010;661:109–122. doi: 10.1007/978-1-60761-500-2_7. [DOI] [PubMed] [Google Scholar]
- Zhang DX, Mendoza SA, Bubolz AH, Mizuno A, Ge ZD, Li R, Warltier DC, Suzuki M, Gutterman DD. Transient receptor potential vanilloid type 4-deficient mice exhibit impaired endothelium-dependent relaxation induced by acetylcholine in vitro and in vivo. Hypertension. 2009;53:532–538. doi: 10.1161/HYPERTENSIONAHA.108.127100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zholos A. Pharmacology of transient receptor potential melastatin channels in the vasculature. Br J Pharmacol. 2010;159:1559–1571. doi: 10.1111/j.1476-5381.2010.00649.x. [DOI] [PMC free article] [PubMed] [Google Scholar]