

Non-technical summary
The neuromuscular junction (NMJ) is the synaptic connection between the nerve and the muscle. The neuromuscular synaptic transmission is highly reliable, as each nerve impulse results in the release of more neurotransmitter than is required for evoking an action potential in the muscle. This feature, often referred as the ‘safety factor’, ensures that a muscle contraction will occur in response to each nerve impulse under normal physiological conditions. Here we show that a small, integral membrane protein of synaptic vesicles, named synaptobrevin (Syb)/vesicle-associated membrane protein (VAMP), is required for optimum synaptic transmission at the NMJ. A genetic mutation in Syb1/VAMP1 in mice causes marked reduction of neurotransmitter release at the NMJ, suggesting an important role for Syb1/VAMP1 in maintaining the ‘safety factor’ of the NMJ.
Abstract
Abstract
Synaptobrevin (Syb)/vesicle-associated membrane protein (VAMP) is a small, integral membrane protein of synaptic vesicles. Two homologous isoforms of synaptobrevin, Syb1/VAMP1 and Syb2/VAMP2, exhibit distinct but partially overlapping patterns of expression in adult mammalian neurons: Syb1 is predominantly expressed in the spinal cord, especially in motor neurons and motor nerve terminals of the neuromuscular junction (NMJ), whereas Syb2 is primarily expressed in central synapses in the brain. Whereas many studies have focused on the function of Syb2 in the brain, few studies have examined the role of Syb1. Here we report that Syb1 plays a critical role in neuromuscular synaptic transmission. A null mutation of Syb1 resulting from a spontaneous, nonsense mutation in mice significantly impairs the function, but not the structure, of the NMJ. In particular, both spontaneous and evoked synaptic activities in Syb1 mutant mice are reduced significantly relative to control mice. Short-term synaptic plasticity in Syb1-deficient NMJs is markedly altered: paired-pulse facilitation is significantly enhanced, suggesting a reduction in the initial release probability of synaptic vesicles. Furthermore, Syb1-deficient NMJs display a pronounced asynchrony in neurotransmitter release. These impairments are not due to an alteration of the size of the readily releasable pool of vesicles, but are attributable to reduced sensitivity and cooperativity to calcium (Ca2+) due to the absence of Syb1. Our findings demonstrate that Syb1 plays an essential, non-redundant role in Ca2+-triggered vesicle exocytosis at the mouse NMJ.
Introduction
Soluble N-ethylmaleimide-sensitive factor attachment protein receptors (SNAREs) play a key role in synaptic transmission by forming the SNARE complex for Ca2+-triggered vesicle exocytosis, a membrane fusion process that results in neurotransmitter release (Rothman & Orci, 1992; Jahn & Scheller, 2006; Sudhof & Rothman, 2009). The core of the SNARE complex is a four-helix bundle (Sutton et al. 1998) composed of three SNARE proteins (Sollner et al. 1993), including synaptosomal-associated protein 25 kDa (SNAP-25) (Oyler et al. 1989), syntaxin1 (Bennett et al. 1992; Inoue et al. 1992) and Syb/VAMP, a class of small membrane proteins evolutionarily conserved in all eukaryotic cells (Ferro-Novick & Jahn, 1994; Hong, 2005). Two highly homologous isoforms of Syb/VAMP, Syb1/VAMP1 and Syb2/VAMP2 (thereafter referred to as Syb1 and Syb2, respectively), are abundantly expressed in neurons and exhibit distinct but partially overlapping expression patterns (Trimble et al. 1988; Baumert et al. 1989; Sudhof et al. 1989; Raptis et al. 2005). Syb2 is one of the most abundant synaptic vesicle proteins in the rat brain (Takamori et al. 2006), whereas Syb1 is predominantly expressed in the spinal cord (Elferink et al. 1989; Li et al. 1996; Jacobsson et al. 1998). Syb1 is also abundantly present in large myelinated axons and dorsal root ganglion (DRG) neurons, whereas Syb2 is detected in small- and medium-sized axons and small- and medium-sized DRG neurons (Li et al. 1996).
The role of Syb2 in synaptic function is well-studied. Syb2 plays critical roles in Ca2+-triggered vesicle exocytosis (Schoch et al. 2001; Deak et al. 2006) and in fast endocytosis in hippocampal synapses (Deak et al. 2004). In contrast, the role of Syb1 in synaptic function remains largely unexplored. Recent genetic studies demonstrate that Syb1 is essential for survival; a null mutation in Syb1 leads to the lethal-wasting (lew) phenotype and premature death in mice. In addition, these mutant (Syb1lew/lew) mice display profound motor impairments and become progressively immobile preceding death (Nystuen et al. 2007). This led us to hypothesize that Syb1 is required for proper functioning of the NMJ, the impairment of which often leads to motor defects and premature death. To test our hypothesis, we examined the formation and function of the NMJ in Syb1lew/lew mutant mice. We found that neuromuscular synapses were generated on schedule in mutant mice, but the NMJ function was severely compromised. Spontaneous synaptic activity was significantly reduced, as were evoked synaptic transmission and the initial release probability. Additionally, the loss of Syb1 led to a pronounced asynchrony in neurotransmitter release. These results demonstrate that Syb1 is essential for efficient Ca2+-triggered synaptic transmission in neuromuscular synapses.
Methods
Mice
We obtained Syb1+/lew heterozygous mice (C3H/HeDiSnJ-Vamp1lew/GrsrJ, stock number 004626) from The Jackson Laboratory (Bar Harbor, ME, USA). Although homozygous mutant mice die within 3 weeks of birth, heterozygous Syb1+/lew mice exhibit no morphological abnormalities and survive to adulthood. Breeder pairs of Syb1+/lew mice were mated to generate Syb1lew/lew mice, and the offspring were monitored daily. A total of 69 offspring (from 11 litters) were collected and 18 were identified as homozygous mutant mice (Syb1lew/lew). Analyses were carried out on homozygous mutants (Syb1lew/lew) and their littermate controls, which included both wild-type (WT) and heterozygous mice. To distinguish the WT and the mutant allele, we designed two primer sets and carried out two separate polymerase chain reactions (PCRs) using genomic DNA isolated from mouse tails. Because Syb1lew/lew mice carry a single point mutation at the Syb1 locus, both expected products were the same length (393 base pairs). Primers were as follows: for wild-type (WT) allele, primer 1 –AGA GGG ACC AGA AGT TGT CAG and primer 2 –TGG CTG CTG GGT GTC TGA GG; for mutant allele, primer 2 –TGG CTG CTG GGT GTC TGA GG and primer 3 –AGA GGG ACC AGA AGT TGT CAT. All experimental protocols followed National Institutes of Health (NIH) guidelines and were approved by the University of Texas Southwestern Institutional Animal Care and Use Committee.
Whole mount immunofluorescence staining
Labelling of neuromuscular synapses was performed as previously described (Liu et al. 2008). Briefly, diaphragm or triangularis sterni muscles (McArdle et al. 1981) from Syb1lew/lew mice and their littermate controls (P14) were fixed in 2% paraformaldehyde in 0.1 m phosphate buffer (pH 7.3) overnight at 4°C. Muscle samples then were incubated with Texas Red conjugated α-bungarotoxin (α-bgt) (2 nm, Invitrogen, Carlsbad, CA, USA) for 30 min, and then with primary antibodies overnight at 4°C. The following primary antibodies were used: neurofilament (NF150) (Chemicon, Temecula, CA, USA), synaptotagmin-2 (Syt-2) (I735) (Pang et al. 2006a), synaptobrevin1 (P938), and synaptobrevin2 (P939) (I735, P938 and P939 were generous gifts from Dr Thomas Südhof, Stanford University School of Medicine, Palo Alto, CA, USA). All primary antibodies were diluted by 1:1000 in antibody dilution buffer (500 mm NaCl, 0.01 m phosphate buffer, 3% BSA and 0.01% thimerosal). After extensive washes, muscle samples were incubated with fluorescein isothiocyanate (FITC)-conjugated goat anti-rabbit IgG (1:600, Jackson ImmunoResearch Laboratories, Inc., West Grove, PA, USA) overnight at 4°C. Muscle samples were then washed with phosphate-buffered saline and mounted in Vectashield mounting medium (H-1000, Vector Laboratories, Inc., Burlingame, CA, USA). Images were acquired using high numerical aperture (NA) objectives (either 40× oil NA 1.30 or 63× oil, NA 1.40) with a Zeiss LSM 510 confocal microscope. To avoid spectral bleed-through (crossover) artifacts in confocal imaging, we sequentially scanned wholemount muscle samples using two separate laser lines: an argon (488 nm) laser (LP 505 nm) for FITC (excitation 490 nm/emission 525 nm) and a He–Ne (543 nm) laser (BP 585–615 nm) for Texas Red (excitation 596/emission 615). The images are presented as maximum projections from Z-stack data acquisition serials.
Quantitative Western blot
Spinal cords and brains from Syb1lew/lew mice (n = 3) and their littermates (control, n = 3) (P14) were homogenized in Tris buffer containing 50 mm Tris (pH 7.4), 150 mm NaCl, 2 mm EDTA, 1 mm PMSF and protease inhibitor cocktail (Roche Applied Science, Indianapolis, IN, USA). Tissue homogenates were separated by SDS-PAGE and transferred to nitrocellulose membranes. The membranes were blocked in 5% milk in Tris-buffered saline and then incubated with primary antibodies against Syb1 (P938, 1:1000, rabbit polyclonal antibody), Syb2 (CL69.1, 1:5000, mouse monoclonal antibody, gift from Dr Reinhard Jahn, Max-Planck-Institute) (Edelmann et al. 1995), and α-tubulin (1:1000, Sigma-Aldrich, St Louis, MO, USA) at 4°C overnight. Membranes then were incubated with goat anti-mouse IRDye 680 and goat anti-rabbit IRDye 800 secondary antibodies (1:10000, Li-Cor Biosciences, Lincoln, NE, USA) at room temperature for 1 h. Membranes were scanned and analysed using the Odyssey infrared imaging system (Li-Cor Biosciences). Relative expression levels of either Syb1 or Syb2 between the control and Syb1lew/lew mice were determined by normalizing to that of α-tubulin, which served as a loading control.
Electron microscopy
Diaphragm muscles (P14) from mutant (Syb1lew/lew, n = 5) and littermate controls (n = 5) were used for electron microscopy studies. Diaphragm muscles were dissected in Ringer solution and fixed with 1% glutaraldehyde in 0.1 m phosphate buffer (pH 7.4), and kept in the same fixative overnight at 4°C. Tissues were rinsed in 0.1 m phosphate buffer, trimmed to small pieces, and post-fixed with 1% osmium tetroxide for 3 h on ice. The tissues then were dehydrated in a graded series of ethanol, infiltrated and polymerized in Epon 812 (Polysciences, Warrington, PA, USA). Epon blocks were cut at 1 μm and stained with 1% toluidine blue for light microscopy observations. Ultrathin sections (70 nm) were prepared and mounted on Formvar-coated grids, then stained with uranyl acetate and lead citrate. Electron micrographs were acquired using a Tecnai electron microscope (the Netherlands) operated at 120 kV.
For quantitative analyses of ultrastructural parameters of the neuromuscular synapses, 50 nerve terminal profiles were obtained from four pairs of P14 control and Syb1lew/lew mice. The following measurements were made in each presynaptic nerve terminal profile using ImageJ software (NIH): nerve terminal area, perimeter length, synaptic contact length, total synaptic vesicle number, active zone number, and docked synaptic vesicle number. The synaptic contact was defined as the length of the presynaptic plasma membrane that was apposed to the postsynaptic muscle membrane. The active zone was defined as a cluster of synaptic vesicles at the electro-dense presynaptic membrane. A docked synaptic vesicle was defined as a presynaptic plasma membrane-attached synaptic vesicle. All synaptic vesicles were manually counted in each nerve terminal profile. Synaptic vesicle density was calculated as the number of synaptic vesicles per μm2 area.
Electrophysiology
Intracellular recordings were carried out in P14 diaphragm muscles from Syb1lew/lew mice (n = 5) and their littermate controls (n = 5). Diaphragm muscles with phrenic nerves attached were acutely isolated in oxygenated (95% O2, 5% CO2) Ringer solution (136.8 mm NaCl, 5 mm KCl, 12 mm NaHCO3, 1 mm NaH2PO4, 1 mm MgCl2, 2 mm CaCl2, and 11 mm d-glucose, pH 7.3) (Liley, 1956). End-plate regions identified under a water-immersion objective (Olympus BX51WI) were impaled with glass micropipettes (resistance 20–40 MΩ) filled with 2 m potassium citrate and 10 mm potassium chloride. Evoked end-plate potentials (EPPs) were elicited by supra-threshold stimulation (2 V, 0.1 ms) of the phrenic nerve via a suction electrode connected to an extracellular stimulator (SD9, Grass-Telefactor, West Warwick, RI, USA). To prevent muscle contraction, μ-conotoxin GIIIB (2 μm, Peptides International, Louisville, KY, USA) was added to the bath solution 30 min prior to recording. Miniature end-plate potentials (mEPPs) and EPPs were acquired using an intracellular amplifier (AxoClamp-2B) and digitized with Digidata 1332 (Molecular Devices, Sunnyvale, CA, USA). Data were analysed with pCLAMP 9.0 (Molecular Devices) and Mini Analysis Program (Synaptosoft, Inc., Decatur, GA, USA). Quantal content (the number of acetylcholine quanta released in response to a single nerve impulse) was estimated using the direct method: dividing the mean amplitude of EPPs by the mean amplitude of mEPPs of the same cell (Boyd & Martin, 1956; Hubbard et al. 1969; Wood & Slater, 2001).
Statistical analysis
All data are presented as means ± standard error of the mean (SEM). Student's t test was applied to assess statistical differences between the control and Syb1lew/lew mice. For quantitative analyses of ultrastructural parameters, the means of nerve terminal area, perimeter length, synaptic contact length, total synaptic vesicle number, vesicle density, active zone number and docked synaptic vesicle number were compared between the control (n = 50 nerve terminal, n = 5 mice) and Syb1lew/lew (n = 50 nerve terminal, n = 5 mice). For electrophysiological analyses, the means of mEPP frequency, mEPP amplitude, paired-pulse ratio and EPPn/EPP1 between control (n = 23 muscle fibres, n = 5 mice) and Syb1lew/lew (n = 29 muscle fibres, n = 5 mice) were assessed. In addition, the F test was applied to determine differences in the variance of the EPP amplitudes between control and Syb1lew/lew mice. The difference is considered statistically significant if the P value (P) is less than 0.05.
Results
Both Syb1 and Syb2 are expressed at the presynaptic nerve terminal in juvenile mice
Previous studies have shown that adult rodent motor endplates express Syb1, but not Syb2 (Li et al. 1996). To determine whether the expression of Syb1/Syb2 in juvenile mouse motor endplates is similar to that in adult motor endplates, we immunostained WT juvenile (P14) muscle tissues with either Syb1 or Syb2 antibodies. In addition, we co-labelled the muscle tissues with the postsynaptic marker α-bgt to identify the NMJ. We found that both Syb1 and Syb2 were expressed at the nerve terminal apposing the postsynaptic AChRs (Fig. 1A, merge). Next, we examined the expression of Syb1 and Syb2 in Syb1lew/lew mice. We found that Syb1 was absent from the nerve terminals of Syb1lew/lew mice, but Syb2 was present at the nerve terminals (Fig. 1B). Furthermore, quantitative immunoblot analysis confirmed that Syb1 was not detectable in Syb1lew/lew mice (Fig. 1C), and that the expression level of Syb2 in Syb1lew/lew mice was similar to that of their littermate controls (Fig. 1D). Thus, there is no compensatory increase in Syb2 expression in Syb1lew/lew mice, consistent with a previous report using quantitative RT-PCR (Nystuen et al. 2007).
Figure 1. Expression of Syb1 and Syb2 in WT and Syb1lew/lew mice.
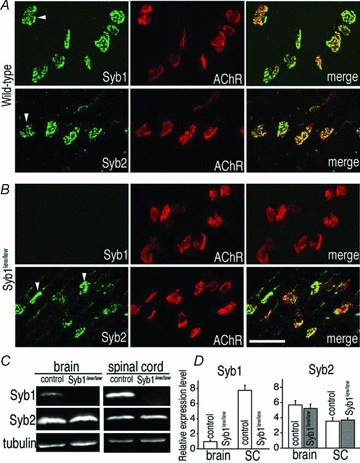
A and B, whole mounts of triangularis sterni (P14) muscles from WT mice (A) or Syb1lew/lew mutant mice (B) were immunostained with antibodies against Syb1 or Syb2, as well as Texas Red-conjugated α-bgt, which labels postsynaptic AChRs. Both Syb1 and Syb2 were detected at the WT nerve terminals (arrowheads in A). Syb1 was absent from Syb1lew/lew NMJs: only α-bgt staining was detected in the synaptic regions of the muscle (upper row in B). In contrast, Syb2 was detected in the nerve terminals (arrowheads in B) of Syb1lew/lew mutant mice. C, images of quantitative Western blot analysis of the brain and spinal cord (SC) from Syb1lew/lew and their littermate control mice (P14). D, comparison of relative levels of Syb1 or Syb2 between Syb1lew/lew mice (n = 3) and their littermates (control, n = 3). Relative expression levels of Syb1 and Syb2 were calculated by normalizing to that of α-tubulin (loading control). Syb1 was absent from Syb1lew/lew mice, but present in control mice, with significantly higher (P < 0.001) expression in the SC (7.73 ± 0.66, n = 3) compared to the brain (0.96 ± 0.21, n = 3) (normalized to α-tubulin level). In both Syb1lew/lew and control mice, Syb2 was more abundantly expressed in the brain compared to the SC (control: brain, 5.69 ± 0.57; SC, 3.54 ± 0.5, P < 0.05; Syb1lew/lew mutant: brain, 5.22 ± 0.57, SC, 3.69 ± 0.27, P < 0.05). In both the brain and SC, the relative levels of Syb2 remained similar between the Syb1lew/lew and control mice, P > 0.05. Scale bar in A and B, 50 μm.
Syb1 is not required for the formation of the NMJ
Using the Syb1lew/lew mouse as a model, we investigated the role of Syb1 in the formation of the NMJ. Among the 69 offspring (11 litters) derived from breeding the heterozygous (Syb1+/lew) mice, 18 were identified as homozygous mutant mice (Syb1lew/lew). These mutant mice appeared grossly normal during the first postnatal week. However, during the second postnatal week, the mutant mice exhibited noticeable motor defects, including the absence of the righting reflex and progressive immobility, consistent with a previous report (Nystuen et al. 2007). At P14, the mutant mice were noticeably smaller, with body weights less than 50% of their littermate controls (control: 8.91 ± 0.17 g, n = 16, Syb1lew/lew: 4.29 ± 0.09 g, n = 17, P < 0.001), indicating severe growth retardation in Syb1lew/lew mice.
Because the Syb1lew/lew mutant mice displayed motor deficiencies, we sought to determine whether their NMJs developed properly, as malformation of the NMJ may underlie motor defects. We found that the NMJ was established on schedule in Syb1lew/lew mice (Fig. 2D–F), similar to that of controls (Fig. 2A–C). Like the control mice, the nerve terminals in Syb1lew/lew mice (Fig. 2E) were intensely labelled by presynaptic markers, such as synaptotagmin2. In addition, the terminals were closely apposed to the postsynaptic AChRs, as revealed by α-bgt staining (Fig. 2F). In both control and Syb1lew/lew mice, 99% of the end-plates in P14 diaphragm muscles (control: 1026/1041 endplates, n = 3 mice; Syb1lew/lew: 1225/1242, n = 3 mice) were innervated by a single axon; 1% of end-plates (control: 15/1041 endplates, n = 3 mice; Syb1lew/lew: 17/1242, n = 3 mice) were innervated by two axons (Fig. 2G and H). Thus, there was no evidence of any delay in synapse elimination, a naturally occurring process that transforms multiply innervated NMJs into singly innervated NMJs during the first 2 weeks after birth in rodents (Brown et al. 1976; Thompson, 1983; Balice-Gordon & Lichtman, 1993; Walsh & Lichtman, 2003).
Figure 2. Formation of neuromuscular synapses in Syb1lew/lew mice.
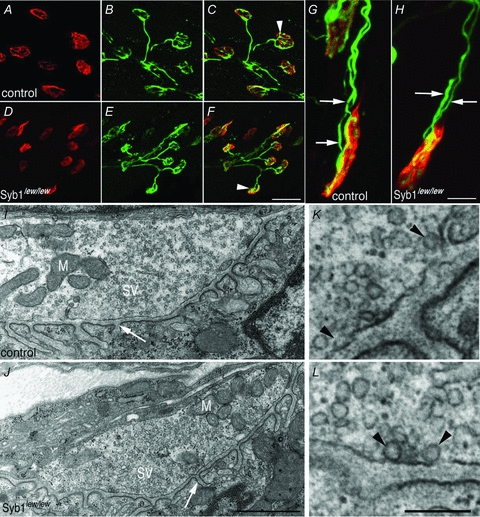
A–H, diaphragm muscles (P14) of control (A–C and G) and Syb1lew/lew mice (D–F and H) were doubly labelled with presynaptic markers (anti-synaptotagmin 2 and anti-neurofilament) (green) and postsynaptic markers (Texas Red-conjugated α-bgt) (red). In both control and Syb1lew/lew mice, nerve terminals (B and E) formed close apposition with postsynaptic AChRs (A and D), as shown in the merged images (white arrowheads in C and F). The majority (99%) of the end-plates were innervated by a single axon, except a small number (1%) of end-plates which were occupied by two axons in both control (G, white arrows point to two individual axons) and Syb1lew/lew mice (H, white arrows). I–L, electron micrographs of longitudinal sections of P14 diaphragm muscles. A typical nerve terminal from control (I) and Syb1lew/lew mice (J): both nerve terminals were packed with abundant synaptic vesicles (SV) and mitochondria (M). Postsynaptic junctional folds (white arrow in I and J) were morphologically normal in Syb1lew/lew mice compared with the control. K and L, examples of docked vesicles (black arrowheads) viewed under high magnification. K: control; L: Syb1lew/lew. Scale bars: A–F, 20 μm; G and H, 5 μm; I and J, 1 μm; K and L, 0.2 μm.
To determine whether the ultrastructure of the NMJ was affected in Syb1lew/lew mice, we carried out electron microscopy analyses. We examined 82 nerve terminals in Syb1lew/lew mutant mice (n = 5) and 62 nerve terminals in control mice (n = 5) and found that the neuromuscular ultrastructure of the Syb1lew/lew mutant mice was similar to that of controls. Motor nerve terminals contained an abundance of synaptic vesicles (SV) and mitochondria (M), and were closely apposed to the postsynaptic muscle membrane, in which characteristic postsynaptic junctional folds (arrows, Fig. 2I and J) were evident. Docked vesicles were present in the nerve terminals of both control and Syb1lew/lew mice (Fig. 2K and L). To further quantify the ultrastructural parameters, we measured 100 nerve terminals (50 control; 50 Syb1lew/lew) and measured the size of nerve terminals, the length of synaptic contacts, the number and density of synaptic vesicles, and the number of docked synaptic vesicles and active zones. All these parameters were similar between the control and Syb1lew/lew NMJs (Table 1). Together, these data demonstrated that neuromuscular synapses were established normally in Syb1lew/lew mice and that Syb1 is not required for the formation of the NMJ.
Table 1.
Ultrastructural parameters of motor nerve terminals in Syb1lew/lew and control mice
| Syb1lew/lew (n = 50) | Control (n = 50) | |
|---|---|---|
| Nerve terminal area (μm2) | 3.10 ± 0.37 | 3.34 ± 0.50 |
| Nerve terminal perimeter (μm) | 8.17 ± 0.56 | 8.50 ± 0.67 |
| Synaptic contact length (μm) | 3.76 ± 0.28 | 3.55 ± 0.36 |
| Synaptic vesicle number (per terminal) | 180 ± 21 | 173 ± 17 |
| Synaptic vesicle density (per μm2) | 66.3 ± 4.1 | 72.8 ± 6.5 |
| Active zone number (per terminal) | 1.2 ± 0.1 | 1.1 ± 0.1 |
| Docked synaptic vesicle number (per terminal) | 3.6 ± 0.3 | 3.4 ± 0.3 |
The data are presented as mean ± SEM. A total of 100 nerve terminals (50 control, 50 Syb1lew/lew) from 4 pairs of Syb1lew/lew and control mice were analysed. No statistical difference (Student's t test) was found between Syb1lew/lew and control mice.
Neuromuscular synaptic transmission is impaired in Syb1lew/lew mice
Given the apparently normal neuromuscular morphology but abnormal motor behaviour in Syb1lew/lew mice, we sought to determine whether neuromuscular synaptic transmission was affected in these mutant mice. We carried out intracellular recordings in the diaphragm muscles of control and Syb1lew/lew mice (P14). We observed similar resting membrane potentials in the control (−70.6 ± 1.1 mV, n = 21 muscle fibres, n = 5 mice) and Syb1lew/lew mice (−70.5 ± 1.2 mV, n = 25 muscle fibres, n = 4 mice), but marked impairments in neuromuscular synaptic transmission in Syb1lew/lew mice.
The mEPP frequency in Syb1lew/lew NMJs (3.2 ± 0.4 event min−1, n = 34) was reduced to less than 40% of that observed in control NMJs (8.9 ± 1.1 event min−1, n = 40) (Fig. 3A and B). The mEPP amplitude and rise slope for the Syb1lew/lew mutant mice were similar to those of controls (Fig. 3C and D), indicating that the quantal size, as measured by the size of the mEPP, was not affected by the absence of Syb1. These results suggest that the observed decreases in mEPP frequency in Syb1lew/lew were due to impairments of synaptic transmission at the presynaptic nerve terminals.
Figure 3. Reduction of spontaneous neurotransmitter release at the NMJ in Syb1lew/lew mice.
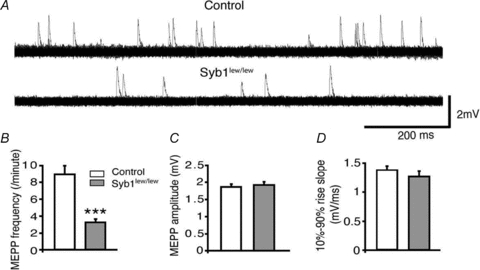
A, representative mEPP traces recorded from the diaphragm muscles (P14). Each trace represents a total of 2 min continuous recording from the same NMJ and is displayed as 120 superimposed 1 s sweeps. mEPP frequencies were markedly reduced in Syb1lew/lew mice compared with controls. B–D, quantitative analyses of mEPP frequencies, amplitudes and 10–90% rise slopes. The average mEPP frequencies were reduced to 3.2 ± 0.4 events min−1 in Syb1lew/lew mice, significantly lower than those in the controls (8.9 ± 1.1 events min−1) (B). mEPP amplitudes (C) of Syb1lew/lew mice (1.92 ± 0.09 mV) were similar to those of the controls (1.86 ± 0.08 mV). The 10–90% rise slopes (D) of Syb1lew/lew mice (1.3 ± 0.1 mV ms−1) and control mice (1.4 ± 0.1 mV ms−1) also were similar. Syb1lew/lew, n = 34 muscle fibres; control, n = 40 muscle fibres. ***P < 0.001, Student's t test.
Evoked synaptic activity was also markedly decreased in the NMJs of Syb1lew/lew mice. The average EPP amplitude in Syb1lew/lew NMJs was 15.5 ± 1.2 mV (n = 25 muscle fibres), 40% less than control NMJs (25.9 ± 1.3 mV, n = 22 muscle fibres) (Fig. 4B). As a result, quantal content was significantly reduced in Syb1lew/lew NMJs (8.6 ± 0.7, n = 25) compared with control NMJs (16.1 ± 0.6, n = 22 muscle fibres) (Fig. 4D). Additionally, EPPs in Syb1lew/lew mice were highly variable, in terms of changes in the amplitudes, rise slopes and decay slopes (Fig. 4A). These results demonstrate the unreliability of synaptic transmission at the NMJ of Syb1lew/lew mice. In contrast, EPPs in control NMJs exhibited constant amplitudes and slopes, which is characteristic of reliable synaptic transmission at the NMJ (Wood & Slater, 2001). For example, 10 repetitive, low frequency (0.2 Hz) stimuli to the nerve in control mice elicited EPPs within a narrow range of amplitudes (20–27 mV, Fig. 4A), consistent with previously reported EPPs from WT mouse diaphragm muscles (Hong & Chang, 1989). In contrast, the same stimuli to the nerve in Syb1lew/lew mice elicited EPPs over a broad range of amplitudes (2–17 mV, Fig. 4A). Additionally, the rise slopes in Syb1lew/lew mice were reduced to 60% of those observed in the control (Fig. 4C), suggesting an asynchrony of synaptic transmission at the NMJ of Syb1lew/lew mice.
Figure 4. Reduction of evoked neurotransmitter release at the NMJ in Syb1lew/lew mice.
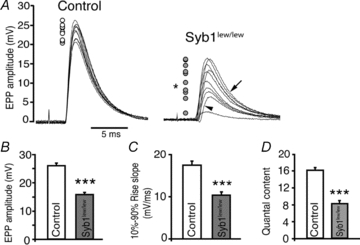
A, 10 consecutive EPP traces recorded from P14 diaphragm muscles (evoked by 0.2 Hz stimulation of the phrenic nerves) are superimposed. EPPs in the control mice were highly reproducible; the peaks of all 10 EPPs, as indicated by circles located to left of the traces, all fall within a range of 7 mV (between 20 and 27 mV). In contrast, EPPs in Syb1lew/lew mice were highly variable, fluctuating over a range of 15 mV (between 2 and 17 mV, filled circles). An F test demonstrated that the variances in Syb1lew/lew mice were significantly (*P < 0.05) increased compared to those of the controls. Similarly, EPP rising phases (arrowhead) and decay phases (arrow) were also highly variable in Syb1lew/lew mice compared with control mice. B, quantification of EPP amplitudes between control and Syb1lew/lew mice: EPP amplitudes were significantly reduced in Syb1lew/lew mice (15.5 ± 1.2 mV), compared with those in the control mice (25.9 ± 1.3 mV). C, the EPP rise-slopes (10–90%) in Syb1lew/lew mice (10.3 ± 0.8 mV ms−1) were significantly reduced compared to those in the controls (17.3 ± 0.9 mV ms−1). D, quantal contents were significantly reduced in Syb1lew/lew mice (8.6 ± 0.7) compared to controls (16.1 ± 0.6). Syb1lew/lew, n = 25 muscle fibres; control, n = 22 muscle fibres. ***P < 0.001, Student's t test.
Abnormal short-term synaptic plasticity at the NMJs in Syb1lew/lew mice
To further investigate the impact of a Syb1-null on neuromuscular synaptic transmission, we examined short-term synaptic plasticity in Syb1lew/lew mice. We applied repetitive stimulation to the phrenic nerves, either paired-pulse or train stimulation. These frequencies are within the normal physiological range of the phrenic motor neuron firing rate in rodents (Kong & Berger, 1986; Road et al. 1995). We then measured the resulting successive postsynaptic EPPs in the diaphragm muscles; an increase or decrease in EPPs reflects synaptic facilitation or depression, respectively. This method indirectly measures the initial release probability of synaptic vesicles (Zucker & Regehr, 2002). Using paired-pulse stimulation with interpulse intervals ranging from 20 to 50 ms, control muscles exhibited a slight (5–7%) increase in postsynaptic potential following the preceding EPP (Fig. 5); this suggests mild paired-pulse facilitation (PPF) consistent with previous studies in WT diaphragm muscles (Hong & Chang, 1989). In contrast, the same paired-pulse stimulation protocol evoked markedly enhanced PPF in Syb1lew/lew mice. For example, at an inter-pulse interval of 20 ms, the paired-pulse ratio was 1.33 ± 0.05 (n = 21) in Syb1lew/lew NMJs and 1.05 ± 0.02 (n = 18) in control NMJs (Fig. 5). The impairments of PPF in Syb1lew/lew NMJs indicate a reduction of the initial release probability.
Figure 5. Impairment of paired-pulse facilitation at the NMJ in Syb1lew/lew mice.
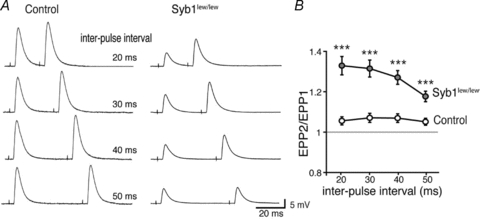
A, sample EPP traces recorded from P14 diaphragm muscles following twin-pulse stimulation of the phrenic nerves at various interpulse intervals (20–50 ms). B, paired pulse ratio (EPP2/EPP1) plotted as a function of the inter-pulse-interval. Syb1lew/lew NMJs exhibited enhanced facilitation compared with control NMJs. Number of NMJs: n = 21, Syb1lew/lew; n = 18, control. ***P < 0.001, Student's t test.
Next, we measured the ratio of each subsequent EPP to the first EPP in response to a train of repetitive stimulation at increasing frequency (30, 50 and 70 Hz). Consistent with previous studies of EPPs evoked by high-frequency stimulation of the phrenic nerve in WT diaphragm muscles (Hong & Chang, 1989), EPP amplitudes in the control mice were rapidly reduced to a plateau, demonstrating a run-down of EPP resulted from synaptic depression under high-frequency stimulation (Fig. 6). In contrast, EPP amplitudes in Syb1lew/lew mice were increased at 30 and 50 Hz, suggesting synaptic facilitation (Fig. 6B and D); this is consistent with the enhanced synaptic facilitation observed during PPF (Fig. 5). Interestingly, at 70 Hz, the enhanced facilitation in Syb1lew/lew mice was observed only during the first few pulses and was then followed by a run-down of the EPP (Fig. 6F). Nevertheless, the rate of EPP run-down was still significantly reduced in Syb1lew/lew mice compared with that of the control mice (Fig. 6F). These results demonstrate that short-term synaptic plasticity was markedly altered at the NMJs of Syb1lew/lew mice.
Figure 6. Altered short-term neuromuscular synaptic plasticity in Syb1lew/lew mice.
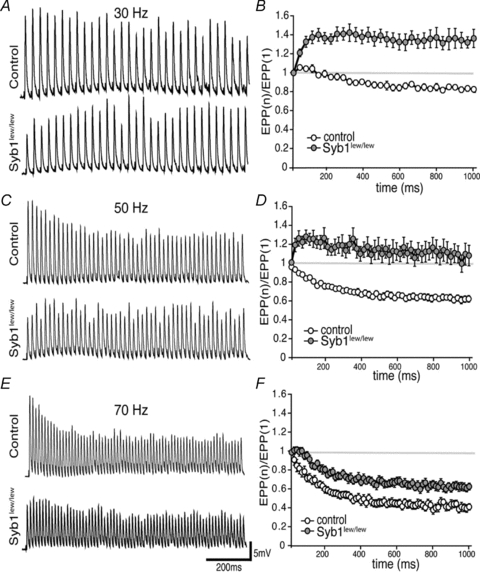
Left (A, C and E), representative EPP traces responded to 30 Hz (A), 50 Hz (C) or 70 Hz (E) train stimulation (1 s train). Right (B, D and F), EPP run-down calculated by the ratio of EPP amplitude to the first EPP amplitude during 30, 50 and 70 Hz stimulation. In the control NMJ, EPP ratios were progressively decreased during the course of train stimulation at 30, 50, and 70 Hz, with more pronounced depression at higher frequency (70 Hz > 50 Hz > 30 Hz). In contrast, EPP amplitudes in Syb1lew/lew NMJs were facilitated during 30 Hz and 50 Hz stimulation, as well as during the first few traces of 70 Hz stimulation (F). At all three frequencies (30, 50 and 70 Hz), the rate of EPP run-down in Syb1lew/lew NMJs (n = 19) was significantly reduced compared to the control (n = 14) (P < 0.001, Student's t test).
The impairment of short-term plasticity in Syb1lew/lew NMJs described above suggest a possibility of altered size of readily releasable neurotransmitter quanta, or readily releasable pool (RRP) (Rosenmund & Stevens, 1996). To test this possibility, we analysed EPP amplitudes under high-frequency repetitive stimulation (70 Hz) as shown in Fig. 6E and F. The cumulative EPP amplitudes in both control and Syb1lew/lew NMJs were then fitted by linear regression and back-extrapolated to time zero to give rise to an estimated number of quanta (Schneggenburger et al. 1999). As shown in Fig. 7, there was no significant difference in the estimated number of quanta between the control (57.2 ± 8.9, n = 8) and Syb1lew/lew (50.4 ± 5.9, n = 9; P > 0.05) (Fig. 7). These analyses suggest that the size of the RRP is normal in Syb1lew/lew NMJs (see also Discussion).
Figure 7. Normal size of readily releasable pool (RRP) at the NMJ in Syb1lew/lew mice.
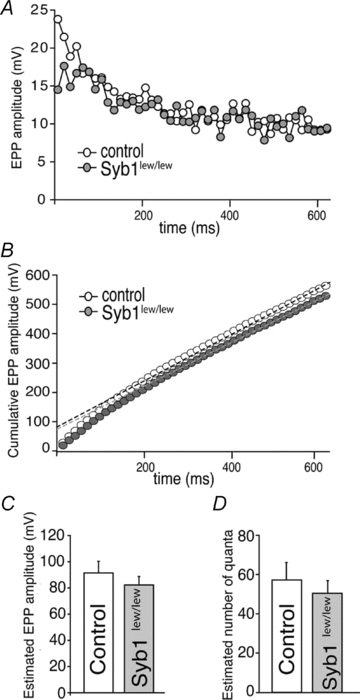
A, analyses of EPP amplitudes in the diaphragm muscle (P14) evoked by a high frequency repetitive stimulation (70 Hz) of the phrenic nerve. The peak of EPP amplitudes reaches a steady state after 40–44 stimuli in both control and Syb1lew/lew NMJs. The first 44 EPPs are shown in this plot. B, cumulative EPP amplitudes as shown in A. Five data points from 40th to 44th stimulus were fitted by linear regression and back-extrapolated to time zero (control: black dashed line; Syb1lew/lew: grey dashed line). C, estimated EPP amplitudes at time zero. D, estimated number of quanta obtained by dividing the estimated EPP amplitudes at time zero in C with mEPP amplitude of the same cell. The resulting estimated number of quanta is similar between the control (57.2 ± 8.9, n = 8) and Syb1lew/lew (50.4 ± 5.9, n = 9; P > 0.05), suggesting the size of the RRP is normal in Syb1lew/lew NMJs.
Reduced Ca2+ sensitivity and cooperativity of transmitter release in Syb1lew/lew NMJs
We next asked whether neuromuscular synaptic transmission impairments in Syb1lew/lew mice were due to reduced sensitivity to Ca2+. We measured EPP amplitude and quantal content at various external [Ca2+] (0.5, 1, 2 and 4 mm). As [Ca2+] increased, EPP amplitude and quantal contents increased in both control and Syb1lew/lew NMJs (Fig. 8A and B). However, the increase was greater in the control than in the Syb1lew/lew NMJs. In Syb1lew/lew NMJs, the EC50 for EPP amplitude and quantal content (2.3 mm for EPP, 2.1 mm for quantal content, n = 29) were significantly higher than those measured for the control NMJs (1.3 mm for EPP, 1.4 mm for quantal content, n = 23). In addition, the Hill slopes were significantly reduced in Syb1lew/lew NMJs (1.1 for EPP, 1.3 for quantal content, n = 29) compared with the control NMJs (2.0 for EPP, 2.0 for quantal content, n = 23). These results demonstrated a marked reduction in both Ca2+ sensitivity and cooperativity in Syb1lew/lew NMJs (Fig. 8C and D).
Figure 8. Reduced Ca2+ sensitivity and cooperativity of neuromuscular synaptic transmission in Syb1lew/lew mice.
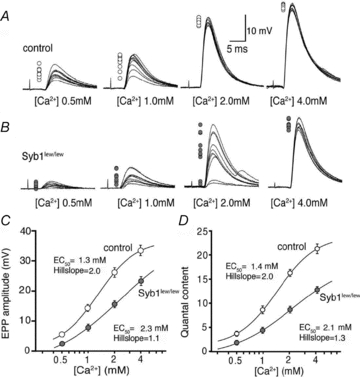
A, sample EPP traces recorded at various external [Ca2+] (0.5, 1.0, 2.0 and 4.0 mm). Each panel represents 10 superimposed EPP traces, with peaks indicated by open circles on the left side. In both control (A) and Syb1lew/lew mice (B), EPP amplitudes decrease and variability increases as external [Ca2+] is reduced. C and D, dose–response curve of average EPP amplitude (C) and quantal content (D), as a function of external [Ca2+], from recordings of P14 diaphragm muscles from Syb1lew/lew and control mice. The curve is fitted by a sigmoidal dose–response equation: 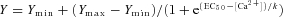 , where Y is the value of the EPP amplitude or the quantal content at the indicated [Ca2+], Ymax and Ymin are the maximum and minimum EPP amplitude or quantal content, respectively, EC50 is the [Ca2+] when Y is 50% of Ymax, and k is the Hill coefficient (Hill slope), which reflects the steepness of the curve. The sigmoidal dose–response curve of EPP amplitude and quantal content as a function of external [Ca2+] showed that the EC50 was markedly increased and the Hill slope was markedly reduced in Syb1lew/lew NMJs (n = 29) compared with the control NMJs (n = 23).
, where Y is the value of the EPP amplitude or the quantal content at the indicated [Ca2+], Ymax and Ymin are the maximum and minimum EPP amplitude or quantal content, respectively, EC50 is the [Ca2+] when Y is 50% of Ymax, and k is the Hill coefficient (Hill slope), which reflects the steepness of the curve. The sigmoidal dose–response curve of EPP amplitude and quantal content as a function of external [Ca2+] showed that the EC50 was markedly increased and the Hill slope was markedly reduced in Syb1lew/lew NMJs (n = 29) compared with the control NMJs (n = 23).
These observations led us to hypothesize that impairments in neuromuscular synaptic transmission in Syb1lew/lew mice could be ameliorated by elevating external [Ca2+]. We compared the EPPs in Syb1lew/lew NMJs at 4 mm[Ca2+] to those in controls at 2 mm[Ca2+]. Indeed, we found that the variability of EPPs displayed in Syb1lew/lew NMJs was eliminated when the external [Ca2+] was increased to 4 mm. For example, at 2 mm external [Ca2+], the EPPs were constant in control mice (Fig. 8A, third trace from the left), but were highly variable in Syb1lew/lew mice (Fig. 8B, third trace from the left). When the external [Ca2+] was raised to 4 mm, the variability of the EPPs in Syb1lew/lew mice was prevented, and the EPPs in Syb1lew/lew mice became constant (Fig. 8B, fourth trace from the left), similar to those of control mice at 2 mm[Ca2+] (Fig. 8A, third trace from the left). EPP amplitudes of Syb1lew/lew NMJs recorded at 4 mm[Ca2+] were in the range of 21.8–26.3 mV with a mean of 23.5 ± 1.5 mV (n = 16), similar to EPP amplitudes in control NMJs recorded at 2 mm[Ca2+], which were in a range of 22.4–27.1 mV with a mean of 25.4 ± 1.9 mV (n = 16). Furthermore, EPP run-down responses evoked by a repetitive stimulation (30 Hz) in the Syb1lew/lew NMJs at 4 mm[Ca2+] (Fig. 9) were similar to those of the control NMJs at 2 mm[Ca2+], reversing the facilitation seen in Syb1lew/lew NMJs at 2 mm[Ca2+] (Fig. 9). Thus, elevating external [Ca2+] restores short-term plasticity of neuromuscular synapses in Syb1lew/lew mice.
Figure 9. Elevating external [Ca2+] restores short-term plasticity of Syb1lew/lew NMJs.
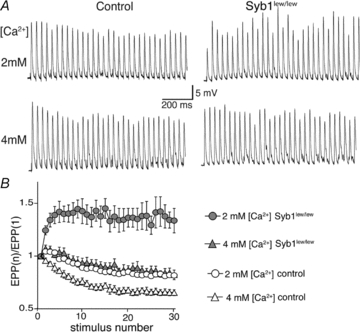
A, sample EPP traces of control and Syb1lew/lew NMJs (P14 diaphragm muscle) evoked by 30 Hz, 1 s train stimulation of the phrenic nerve at 2 mm[Ca2+] and 4 mm[Ca2+]. B, quantification of the EPP amplitudes shown in A. EPP amplitudes were facilitated during 30 Hz stimulation in Syb1lew/lew NMJs (n = 29) at 2 mm[Ca2+] (filled circles), and this facilitation was eliminated when [Ca2+] was increased to 4 mm (filled triangles), approaching the normal EPP run-down typically exhibited in control NMJs (n = 24) at 2 mm[Ca2+] (open circles).
Discussion
Syb1 contributes to the ‘safety factor’ of the NMJ and is required for survival
One of the remarkable features of the vertebrate NMJ is its reliability in synaptic transmission as each nerve impulse normally results in the release of more neurotransmitter than is required for evoking an action potential in the muscle. This feature, often referred as the ‘safety factor’, ensures that a muscle contraction will occur in response to each nerve impulse under normal physiological conditions (Wood & Slater, 2001).
In this study, we investigated the role of Syb1 in the formation and function of the NMJ. A spontaneous, nonsense mutation in Syb1 in mice leads to a null Syb1 mutant (Syb1lew/lew). Mutant mice die within 3 weeks of birth, indicating that Syb1 is essential for survival. In the absence of Syb1, the structure of the NMJ in Syb1lew/lew mutant mice is normal: the presynaptic nerve terminals contain normal number of total synaptic vesicles and docked vesicles, and the postsynaptic junctional folds develop normally, suggesting that Syb1 is not required for the formation of the NMJ. However, synaptic transmission in Syb1lew/lew NMJs is markedly impaired, with a significant reduction in both spontaneous and evoked neurotransmitter release and impairment in short-term synaptic plasticity. A supra-threshold stimulation of the nerve in Syb1lew/lew mutant mice elicits EPPs with a broad range of amplitudes (2–17 mV), the value of which would sometimes fall below the threshold (11–25 mV) required for generating action potentials in rodent muscles (Wood & Slater, 1995, 1997). This reduction in neurotransmitter release is probably responsible, at least in part, for the premature death of Syb1lew/lew mutant mice, as sub-threshold EPPs would not be sufficient to elicit muscle action potentials and muscle contractions required for the body's normal functioning such as breathing activity. Therefore, the ‘safety factor’ of the NMJ in Syb1lew/lew mutant mice is compromised due to a marked reduction of neurotransmitter release. However, it must also be noted that Syb1 is expressed in select regions in the brain, including the thalamus, subthalamus, hippocampus and cerebral cortex (Raptis et al. 2005; Nystuen et al. 2007). It is possible that the absence of Syb1 from these brain regions also contributes to the lethality in Syb1lew/lew mutant mice.
The overall levels of Sybs are essential for efficient neuromuscular synaptic transmission in vivo
While the defects of synaptic transmission in Syb1lew/lew mice are consistent with those reported in previous studies of other Syb mutants in mice (Schoch et al. 2001; Deak et al. 2004, 2006), Drosophila (DiAntonio et al. 1993; Deitcher et al. 1998; Bhattacharya et al. 2002) and Caenorhabditis elegans (Nonet et al. 1998), the synaptic transmission in Syb1lew/lew mice appears to be less severely affected. For example, in hippocampal synapses in Syb2−/− mice, or in the NMJ in neuronal-synaptobrevin (n-syb) null Drosophila, action potential evoked release is completely abolished (Deitcher et al. 1998; Schoch et al. 2001). In contrast, evoked release is reduced by approximately 50% in Syb1lew/lew NMJs. One plausible contributing factor for this different degree of severity in synaptic transmission defect is the expression level of Syb isoforms in specific synapses. For example, Syb2 and n-Syb are the predominant isoforms expressed in mouse hippocampal synapses (Schoch et al. 2001) and Drosophila NMJs (Deitcher et al. 1998), respectively. However, both Syb1 and Syb2 are expressed in the NMJ in mice. Thus, the absence of Syb1 can be partially compensated by the presence of Syb2, and this may explain why both spontaneous and evoked synaptic transmissions are detected in Syb1lew/lew NMJs, albeit to a reduced extent. Nevertheless, because the overall expression levels of Syb2 in the spinal cord are far less than that of Syb1 (Elferink et al. 1989) (see also Fig. 1), and because no compensatory increase in the expression of other SNARE proteins occurs in Syb1lew/lew mutant mice (Nystuen et al. 2007), neuromuscular synaptic transmission fails to sustain at the level required for survival. These results suggest that the overall levels of Sybs are essential for efficient neuromuscular synaptic transmission.
The role of Syb1 in the maintenance of short-term synaptic plasticity at the NMJ
Our electrophysiology analyses indicate that the initial release probability is reduced in Syb1lew/lew NMJs, as PPF is markedly increased compared to the controls. This reduction in the initial release probability is unlikely to be due to changes in RRP size. The estimated RRP size is similar between the control and Syb1lew/lew NMJs. This estimation of RRP size, based upon the strategies previously described in studies of calyx synapses (Schneggenburger et al. 1999), however, is likely to underestimate the size of the RRP at the NMJ. This is because neuromuscular synaptic transmission remains remarkably sustainable even under high-frequency repetitive stimulation. In contrast, synaptic transmission at the calyx synapses becomes rapidly depressed in response to high-frequency repetitive stimulation (Borst et al. 1995; von Gersdorff et al. 1997; Wang & Kaczmarek, 1998; Schneggenburger et al. 1999). Nevertheless, the RRP size is similar between the control and Syb1lew/lew NMJs, and this is consistent with our ultrastructural analyses demonstrating that the total synaptic vesicle number, vesicle density and the number of docked vesicles are similar between the control and Syb1lew/lew NMJs. Together, these results suggest that the loss of Syb1 does not affect RRP size at the presynaptic nerve terminal and that the impairment in short-term synaptic plasticity in Syb1lew/lew NMJs is not likely to be due to an alteration of RRP size.
The role of Syb1 in Ca2+-triggered exocytosis
Upon the arrival of an action potential, Ca2+-influx via presynaptic Ca2+ channels (Tsien et al. 1988) triggers exocytosis of synaptic vesicles resulting in the release of neurotransmitter (Katz, 1969; Augustine et al. 1987; Pang & Sudhof, 2010). An important feature of the Ca2+ action in transmitter release is its non-linear, cooperativity (Dodge & Rahamimoff, 1967). Our data indicate that Syb1 plays an important role in Ca2+ cooperativity in neurotransmitter release. In the absence of Syb1, Ca2+-triggered EPP amplitudes and rise slopes are markedly reduced and become highly variable. This is attributable to reduced Ca2+ sensitivity and cooperativity in Syb1-deficient NMJs, because the variability of EPPs could be ameliorated by elevating the external [Ca2+]. The reduction in Ca2+ sensitivity and cooperativity decreases the release probability and quantal content and increases the asynchrony of neurotransmitter release. This is consistent with studies by Stewart et al. (2000), who found that reduced levels of SNAREs in Drosophila lead to reduced Ca2+ cooperativity, and by Hu et al. (2002), who found that Sybs are the rate-limiting factors in Ca2+-triggered membrane fusion.
Ca2+ triggers the formation of the SNARE complex and vesicle fusion (Chen et al. 1999; Hu et al. 2002; Rizo et al. 2006), but SNAREs do not appear to function directly as Ca2+ sensors (Chen et al. 2005). A large body of evidence has demonstrated that synaptotagmin (Syt), a synaptic vesicle protein that binds to phospholipid (Perin et al. 1990; Davletov & Sudhof, 1993; Chapman & Davis, 1998; Arac et al. 2006), functions as a Ca2+ sensor for action potential evoked neurotransmitter release (reviewed in Pang & Sudhof, 2010). In addition, Syt also plays an important role in positioning synaptic vesicles for synchronous release (Young & Neher, 2009). Both Syt1 (Perin et al. 1991) and Syt2 (Geppert et al. 1991; Wendland et al. 1991) are expressed in mouse NMJs (Pang et al. 2006a), and the loss-of-function of Syt2 leads to marked impairments in neuromuscular synaptic transmission, including a significant reduction in EPP amplitude and quantal content, and alternation of short-term plasticity (Pang et al. 2006a). These defects are similar to those displayed by Syb1lew/lew mutant mice. However, Syt2 mutants and Syb1 mutants differ in terms of the defect in spontaneous release. For example, mEPP frequencies are significantly increased in Syt2 point mutants, which exhibit reduced Syt2 expression (Pang et al. 2006b), and in Syt2 null mutant mice (Pang et al. 2006a). In contrast, mEPP frequencies are significantly reduced in Syb1lew/lew mutant mice. This can be attributed to the specific roles that Syt2 and Syb1 play during synaptic transmission – that is, Syt2 as a Ca2+ sensor (Sun et al. 2007), and Syb1 as an essential SNARE protein for Ca2+-triggered membrane fusion.
Synaptic transmission in Syb1-deficient NMJs remains Ca2+ dependent, but exhibits a marked reduction in Ca2+ sensitivity and cooperativity. The mechanism underlying this alteration of Ca2+ sensitivity and cooperativity in Syb1-deficient NMJs remains to be further elucidated. The simplest explanation is a reduction of the Syb level, as discussed above, in Syb1-deficient NMJs. As Syb1 is the predominant Syb in the spinal cords, its absence would drastically decrease the number of Syb molecules available for each synaptic vesicle. This may result in a decreased stoichiometry of SNAREs and hence reduces the rate of vesicle fusion.
Another possibility is that the loss of Syb1 may impair the coupling between the Ca2+ channels at the presynaptic nerve terminal and the synaptic vesicles, a rate-limiting step for fast neurotransmitter release (Wadel et al. 2007). Studies with clostridial neurotoxins such as botulinum neurotoxins (BoNT) and tetanus toxin (TeNT) in the central synapses elegantly demonstrate that the kinetics and Ca2+ dependency in neurotransmitter release differ distinctly between synapses poisoned with BoNT, which cleaves SNAP-25 and/or syntaxin, and synapses poisoned with TeNT that cleaves Syb (Capogna et al. 1997; Sakaba et al. 2005; Young, 2005). These studies indicate that the cleavage of Syb may specifically impede the coupling between the Ca2+ channels and synaptic vesicles (Sakaba et al. 2005). Therefore, it is entirely possible that in the absence of Syb1, the coupling between the Ca2+ channels and synaptic vesicles is impaired, resulting in asynchrony in neurotransmitter release.
A third possibility is that Syb1, or SNARE complex containing Syb1, may be required for interacting with the calcium sensor (e.g. Syt, the major coordinator of Ca2+-triggered synchronous release) to achieve optimum Ca2+ efficiency. Previous studies have shown that Syt may confer the Ca2+ sensitivity to SNAREs via its interaction with Snapin (Hilfiker et al. 1999), which binds to SNAREs (Ilardi et al. 1999). Consistent with this notion, increased asynchrony in Syb1lew/lew mice is reminiscent of that displayed in mutant mice deficient in the P/Q-type Ca2+ channel (Urbano et al. 2003; Depetris et al. 2008) or in mutant mice deficient in Snapin (Pan et al. 2009). In this scenario, spontaneous and evoked synaptic transmission at the NMJ would occur without Syb1, but it would be much less efficient and would require a greater [Ca2+] for consistent neurotransmission. Indeed, our results indicated that doubling the external [Ca2+] resulted in WT-like EPPs in Syb1lew/lew NMJs.
In conclusion, by analysing the formation and function of the NMJ in mutant mice deficient in Syb1, a SNARE protein co-expressed with Syb2 at the presynaptic motor nerve terminals, we have demonstrated that Syb1 is not required for the formation of the NMJ, but is essential for maintaining normal levels of neurotransmitter release at the NMJ. Syb1-null NMJs display a pronounced asynchrony in neurotransmitter release attributable to reduced sensitivity and cooperativity to Ca2+. The mechanism underlying this reduced Ca2+ sensitivity and cooperativity remains to be further elucidated. It may be attributable to decreased levels of Syb, defects in the coupling between Ca2+ channels and synaptic vesicles, or defects in the interaction between SNARE complex the calcium sensor. Together, our findings demonstrate that Syb1 plays an essential, non-redundant role in Ca2+-triggered vesicle exocytosis at the mouse NMJ.
Acknowledgments
We thank Drs Thomas Südhof, Jane Johnson, Joseph McArdle, Kimberly Huber and Ege Kavalali for critical reading and commenting on the manuscript. We are indebted to Drs George Augustine and Tsung-Chang Sung for valuable suggestions on electrophysiology and molecular biology, and Ms Lori Nguyen for assistance in animal care. This study was supported by grants (to W.L.) from the NIH/NINDS (NS 055028) and from the Edward Mallinckrodt, Jr Scholar Program.
Glossary
Abbreviations
- α-bgt
α-bungarotoxin
- EPP
end-plate potential
- NMJ
neuromuscular junction
- SNARE
soluble N-ethylmaleimide-sensitive factor attachment protein receptor
- Syb
synaptobrevin
- VAMP
vesicle-associated membrane protein
Author contributions
Y.L. carried out electrophysiology, immunofluorescence staining and quantitative Western blotting; Y.S. performed electron microscopy. Y.L. and Y.S. collected and analysed the data. W.L. supervised the design of the experiments, and collection, analysis and interpretation of the data. Y.L. and W.L. co-wrote the paper with critical input from Y.S.
References
- Arac D, Chen X, Khant HA, Ubach J, Ludtke SJ, Kikkawa M, Johnson AE, Chiu W, Sudhof TC, Rizo J. Close membrane-membrane proximity induced by Ca2+-dependent multivalent binding of synaptotagmin-1 to phospholipids. Nat Struct Mol Biol. 2006;13:209–217. doi: 10.1038/nsmb1056. [DOI] [PubMed] [Google Scholar]
- Augustine GJ, Charlton MP, Smith SJ. Calcium action in synaptic transmitter release. Annu Rev Neurosci. 1987;10:633–693. doi: 10.1146/annurev.ne.10.030187.003221. [DOI] [PubMed] [Google Scholar]
- Balice-Gordon RJ, Lichtman JW. In vivo observations of pre- and postsynaptic changes during the transition from multiple to single innervation at developing neuromuscular junctions. J Neurosci. 1993;13:834–855. doi: 10.1523/JNEUROSCI.13-02-00834.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumert M, Maycox PR, Navone F, De Camilli P, Jahn R. Synaptobrevin: an integral membrane protein of 18,000 daltons present in small synaptic vesicles of rat brain. EMBO J. 1989;8:379–384. doi: 10.1002/j.1460-2075.1989.tb03388.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett MK, Calakos N, Scheller RH. Syntaxin: a synaptic protein implicated in docking of synaptic vesicles at presynaptic active zones. Science. 1992;257:255–259. doi: 10.1126/science.1321498. [DOI] [PubMed] [Google Scholar]
- Bhattacharya S, Stewart BA, Niemeyer BA, Burgess RW, McCabe BD, Lin P, Boulianne G, O'Kane CJ, Schwarz TL. Members of the synaptobrevin/vesicle-associated membrane protein (VAMP) family in Drosophila are functionally interchangeable in vivo for neurotransmitter release and cell viability. Proc Natl Acad Sci U S A. 2002;99:13867–13872. doi: 10.1073/pnas.202335999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borst JG, Helmchen F, Sakmann B. Pre- and postsynaptic whole-cell recordings in the medial nucleus of the trapezoid body of the rat. J Physiol. 1995;489:825–840. doi: 10.1113/jphysiol.1995.sp021095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd IA, Martin AR. The end-plate potential in mammalian muscle. J Physiol. 1956;132:74–91. doi: 10.1113/jphysiol.1956.sp005503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MC, Jansen JK, Van Essen D. Polyneuronal innervation of skeletal muscle in new-born rats and its elimination during maturation. J Physiol. 1976;261:387–422. doi: 10.1113/jphysiol.1976.sp011565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capogna M, McKinney RA, O'Connor V, Gahwiler BH, Thompson SM. Ca2+ or Sr2+ partially rescues synaptic transmission in hippocampal cultures treated with botulinum toxin A and C, but not tetanus toxin. J Neurosci. 1997;17:7190–7202. doi: 10.1523/JNEUROSCI.17-19-07190.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman ER, Davis AF. Direct interaction of a Ca2+-binding loop of synaptotagmin with lipid bilayers. J Biol Chem. 1998;273:13995–14001. doi: 10.1074/jbc.273.22.13995. [DOI] [PubMed] [Google Scholar]
- Chen X, Tang J, Sudhof TC, Rizo J. Are neuronal SNARE proteins Ca2+ sensors? J Mol Biol. 2005;347:145–158. doi: 10.1016/j.jmb.2005.01.024. [DOI] [PubMed] [Google Scholar]
- Chen YA, Scales SJ, Patel SM, Doung YC, Scheller RH. SNARE complex formation is triggered by Ca2+ and drives membrane fusion. Cell. 1999;97:165–174. doi: 10.1016/s0092-8674(00)80727-8. [DOI] [PubMed] [Google Scholar]
- Davletov BA, Sudhof TC. A single C2 domain from synaptotagmin I is sufficient for high affinity Ca2+/phospholipid binding. J Biol Chem. 1993;268:26386–26390. [PubMed] [Google Scholar]
- Deak F, Schoch S, Liu X, Sudhof TC, Kavalali ET. Synaptobrevin is essential for fast synaptic-vesicle endocytosis. Nat Cell Biol. 2004;6:1102–1108. doi: 10.1038/ncb1185. [DOI] [PubMed] [Google Scholar]
- Deak F, Shin OH, Kavalali ET, Sudhof TC. Structural determinants of synaptobrevin 2 function in synaptic vesicle fusion. J Neurosci. 2006;26:6668–6676. doi: 10.1523/JNEUROSCI.5272-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deitcher DL, Ueda A, Stewart BA, Burgess RW, Kidokoro Y, Schwarz TL. Distinct requirements for evoked and spontaneous release of neurotransmitter are revealed by mutations in the Drosophila gene neuronal-synaptobrevin. J Neurosci. 1998;18:2028–2039. doi: 10.1523/JNEUROSCI.18-06-02028.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depetris RS, Nudler SI, Uchitel OD, Urbano FJ. Altered synaptic synchrony in motor nerve terminals lacking P/Q-calcium channels. Synapse. 2008;62:466–471. doi: 10.1002/syn.20516. [DOI] [PubMed] [Google Scholar]
- DiAntonio A, Burgess RW, Chin AC, Deitcher DL, Scheller RH, Schwarz TL. Identification and characterization of Drosophila genes for synaptic vesicle proteins. J Neurosci. 1993;13:4924–4935. doi: 10.1523/JNEUROSCI.13-11-04924.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodge FA, Jr, Rahamimoff R. Co-operative action a calcium ions in transmitter release at the neuromuscular junction. J Physiol. 1967;193:419–432. doi: 10.1113/jphysiol.1967.sp008367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelmann L, Hanson PI, Chapman ER, Jahn R. Synaptobrevin binding to synaptophysin: a potential mechanism for controlling the exocytotic fusion machine. EMBO J. 1995;14:224–231. doi: 10.1002/j.1460-2075.1995.tb06995.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elferink LA, Trimble WS, Scheller RH. Two vesicle-associated membrane protein genes are differentially expressed in the rat central nervous system. J Biol Chem. 1989;264:11061–11064. [PubMed] [Google Scholar]
- Ferro-Novick S, Jahn R. Vesicle fusion from yeast to man. Nature. 1994;370:191–193. doi: 10.1038/370191a0. [DOI] [PubMed] [Google Scholar]
- Geppert M, Archer BT, 3rd, Sudhof TC. Synaptotagmin II. A novel differentially distributed form of synaptotagmin. J Biol Chem. 1991;266:13548–13552. [PubMed] [Google Scholar]
- Hilfiker S, Greengard P, Augustine GJ. Coupling calcium to SNARE-mediated synaptic vesicle fusion. Nat Neurosci. 1999;2:104–106. doi: 10.1038/5659. [DOI] [PubMed] [Google Scholar]
- Hong SJ, Chang CC. Use of geographutoxin II (mu-conotoxin) for the study of neuromuscular transmission in mouse. Br J Pharmacol. 1989;97:934–940. doi: 10.1111/j.1476-5381.1989.tb12034.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong W. SNAREs and traffic. Biochim Biophys Acta. 2005;1744:120–144. doi: 10.1016/j.bbamcr.2005.03.014. [DOI] [PubMed] [Google Scholar]
- Hu K, Carroll J, Fedorovich S, Rickman C, Sukhodub A, Davletov B. Vesicular restriction of synaptobrevin suggests a role for calcium in membrane fusion. Nature. 2002;415:646–650. doi: 10.1038/415646a. [DOI] [PubMed] [Google Scholar]
- Hubbard JI, Llinas R, Quastel DMJ. In: Investigation of Presynaptic Function. Davson H, editor. Vol. 19. Baltimore: The Williams & Wilkins Company; 1969. pp. 112–173. Monographs of the Physiological Society. [Google Scholar]
- Ilardi JM, Mochida S, Sheng ZH. Snapin: a SNARE-associated protein implicated in synaptic transmission. Nat Neurosci. 1999;2:119–124. doi: 10.1038/5673. [DOI] [PubMed] [Google Scholar]
- Inoue A, Obata K, Akagawa K. Cloning and sequence analysis of cDNA for a neuronal cell membrane antigen, HPC-1. J Biol Chem. 1992;267:10613–10619. [PubMed] [Google Scholar]
- Jacobsson G, Piehl F, Meister B. VAMP-1 and VAMP-2 gene expression in rat spinal motoneurones: differential regulation after neuronal injury. Eur J Neurosci. 1998;10:301–316. doi: 10.1046/j.1460-9568.1998.00050.x. [DOI] [PubMed] [Google Scholar]
- Jahn R, Scheller RH. SNAREs – engines for membrane fusion. Nat Rev Mol Cell Biol. 2006;7:631–643. doi: 10.1038/nrm2002. [DOI] [PubMed] [Google Scholar]
- Katz B. The Release of Neural Transmitter Substances. Liverpool: Liverpool University Press; 1969. [Google Scholar]
- Kong FJ, Berger AJ. Firing properties and hypercapnic responses of single phrenic motor axons in the rat. J Appl Physiol. 1986;61:1999–2004. doi: 10.1152/jappl.1986.61.6.1999. [DOI] [PubMed] [Google Scholar]
- Li JY, Edelmann L, Jahn R, Dahlstrom A. Axonal transport and distribution of synaptobrevin I and II in the rat peripheral nervous system. J Neurosci. 1996;16:137–147. doi: 10.1523/JNEUROSCI.16-01-00137.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liley AW. An investigation of spontaneous activity at the neuromuscular junction of the rat. J Physiol. 1956;132:650–666. doi: 10.1113/jphysiol.1956.sp005555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Padgett D, Takahashi M, Li H, Sayeed A, Teichert RW, Olivera BM, McArdle JJ, Green WN, Lin W. Essential roles of the acetylcholine receptor γ-subunit in neuromuscular synaptic patterning. Development. 2008;135:1957–1967. doi: 10.1242/dev.018119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McArdle JJ, Angaut-Petit D, Mallart A, Bournaud R, Faille L, Brigant JL. Advantages of the triangularis sterni muscle of the mouse for investigations of synaptic phenomena. J Neurosci Methods. 1981;4:109–115. doi: 10.1016/0165-0270(81)90044-3. [DOI] [PubMed] [Google Scholar]
- Nonet ML, Saifee O, Zhao H, Rand JB, Wei L. Synaptic transmission deficits in Caenorhabditis elegans synaptobrevin mutants. J Neurosci. 1998;18:70–80. doi: 10.1523/JNEUROSCI.18-01-00070.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nystuen AM, Schwendinger JK, Sachs AJ, Yang AW, Haider NB. A null mutation in VAMP1/synaptobrevin is associated with neurological defects and prewean mortality in the lethal-wasting mouse mutant. Neurogenetics. 2007;8:1–10. doi: 10.1007/s10048-006-0068-7. [DOI] [PubMed] [Google Scholar]
- Oyler GA, Higgins GA, Hart RA, Battenberg E, Billingsley M, Bloom FE, Wilson MC. The identification of a novel synaptosomal-associated protein, SNAP-25, differentially expressed by neuronal subpopulations. J Cell Biol. 1989;109:3039–3052. doi: 10.1083/jcb.109.6.3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan PY, Tian JH, Sheng ZH. Snapin facilitates the synchronization of synaptic vesicle fusion. Neuron. 2009;61:412–424. doi: 10.1016/j.neuron.2008.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang ZP, Melicoff E, Padgett D, Liu Y, Teich AF, Dickey BF, Lin W, Adachi R, Sudhof TC. Synaptotagmin-2 is essential for survival and contributes to Ca2+ triggering of neurotransmitter release in central and neuromuscular synapses. J Neurosci. 2006a;26:13493–13504. doi: 10.1523/JNEUROSCI.3519-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang ZP, Sudhof TC. Cell biology of Ca2+-triggered exocytosis. Curr Opin Cell Biol. 2010;22:496–505. doi: 10.1016/j.ceb.2010.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang ZP, Sun J, Rizo J, Maximov A, Sudhof TC. Genetic analysis of synaptotagmin 2 in spontaneous and Ca2+-triggered neurotransmitter release. EMBO J. 2006b;25:2039–2050. doi: 10.1038/sj.emboj.7601103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perin MS, Brose N, Jahn R, Sudhof TC. Domain structure of synaptotagmin (p65) J Biol Chem. 1991;266:623–629. [PubMed] [Google Scholar]
- Perin MS, Fried VA, Mignery GA, Jahn R, Sudhof TC. Phospholipid binding by a synaptic vesicle protein homologous to the regulatory region of protein kinase C. Nature. 1990;345:260–263. doi: 10.1038/345260a0. [DOI] [PubMed] [Google Scholar]
- Raptis A, Torrejon-Escribano B, Gomez de Aranda I, Blasi J. Distribution of synaptobrevin/VAMP 1 and 2 in rat brain. J Chem Neuroanat. 2005;30:201–211. doi: 10.1016/j.jchemneu.2005.08.002. [DOI] [PubMed] [Google Scholar]
- Rizo J, Chen X, Arac D. Unraveling the mechanisms of synaptotagmin and SNARE function in neurotransmitter release. Trends Cell Biol. 2006;16:339–350. doi: 10.1016/j.tcb.2006.04.006. [DOI] [PubMed] [Google Scholar]
- Road J, Osborne S, Cairns A. Phrenic motoneuron firing rates during brief inspiratory resistive loads. J Appl Physiol. 1995;79:1540–1545. doi: 10.1152/jappl.1995.79.5.1540. [DOI] [PubMed] [Google Scholar]
- Rosenmund C, Stevens CF. Definition of the readily releasable pool of vesicles at hippocampal synapses. Neuron. 1996;16:1197–1207. doi: 10.1016/s0896-6273(00)80146-4. [DOI] [PubMed] [Google Scholar]
- Rothman JE, Orci L. Molecular dissection of the secretory pathway. Nature. 1992;355:409–415. doi: 10.1038/355409a0. [DOI] [PubMed] [Google Scholar]
- Sakaba T, Stein A, Jahn R, Neher E. Distinct kinetic changes in neurotransmitter release after SNARE protein cleavage. Science. 2005;309:491–494. doi: 10.1126/science.1112645. [DOI] [PubMed] [Google Scholar]
- Schneggenburger R, Meyer AC, Neher E. Released fraction and total size of a pool of immediately available transmitter quanta at a calyx synapse. Neuron. 1999;23:399–409. doi: 10.1016/s0896-6273(00)80789-8. [DOI] [PubMed] [Google Scholar]
- Schoch S, Deak F, Konigstorfer A, Mozhayeva M, Sara Y, Sudhof TC, Kavalali ET. SNARE function analyzed in synaptobrevin/VAMP knockout mice. Science. 2001;294:1117–1122. doi: 10.1126/science.1064335. [DOI] [PubMed] [Google Scholar]
- Sollner T, Whiteheart SW, Brunner M, Erdjument-Bromage H, Geromanos S, Tempst P, Rothman JE. SNAP receptors implicated in vesicle targeting and fusion. Nature. 1993;362:318–324. doi: 10.1038/362318a0. [DOI] [PubMed] [Google Scholar]
- Stewart BA, Mohtashami M, Trimble WS, Boulianne GL. SNARE proteins contribute to calcium cooperativity of synaptic transmission. Proc Natl Acad Sci U S A. 2000;97:13955–13960. doi: 10.1073/pnas.250491397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudhof TC, Baumert M, Perin MS, Jahn R. A synaptic vesicle membrane protein is conserved from mammals to Drosophila. Neuron. 1989;2:1475–1481. doi: 10.1016/0896-6273(89)90193-1. [DOI] [PubMed] [Google Scholar]
- Sudhof TC, Rothman JE. Membrane fusion: grappling with SNARE and SM proteins. Science. 2009;323:474–477. doi: 10.1126/science.1161748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Pang ZP, Qin D, Fahim AT, Adachi R, Sudhof TC. A dual-Ca2+-sensor model for neurotransmitter release in a central synapse. Nature. 2007;450:676–682. doi: 10.1038/nature06308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton RB, Fasshauer D, Jahn R, Brunger AT. Crystal structure of a SNARE complex involved in synaptic exocytosis at 2.4 A resolution. Nature. 1998;395:347–353. doi: 10.1038/26412. [DOI] [PubMed] [Google Scholar]
- Takamori S, Holt M, Stenius K, Lemke EA, Gronborg M, Riedel D, Urlaub H, Schenck S, Brugger B, Ringler P, Muller SA, Rammner B, Grater F, Hub JS, De Groot BL, Mieskes G, Moriyama Y, Klingauf J, Grubmuller H, Heuser J, Wieland F, Jahn R. Molecular anatomy of a trafficking organelle. Cell. 2006;127:831–846. doi: 10.1016/j.cell.2006.10.030. [DOI] [PubMed] [Google Scholar]
- Thompson W. Synapse elimination in neonatal rat muscle is sensitive to pattern of muscle use. Nature. 1983;302:614–616. doi: 10.1038/302614a0. [DOI] [PubMed] [Google Scholar]
- Trimble WS, Cowan DM, Scheller RH. VAMP-1: a synaptic vesicle-associated integral membrane protein. Proc Natl Acad Sci U S A. 1988;85:4538–4542. doi: 10.1073/pnas.85.12.4538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsien RW, Lipscombe D, Madison DV, Bley KR, Fox AP. Multiple types of neuronal calcium channels and their selective modulation. Trends Neurosci. 1988;11:431–438. doi: 10.1016/0166-2236(88)90194-4. [DOI] [PubMed] [Google Scholar]
- Urbano FJ, Piedras-Renteria ES, Jun K, Shin HS, Uchitel OD, Tsien RW. Altered properties of quantal neurotransmitter release at endplates of mice lacking P/Q-type Ca2+ channels. Proc Natl Acad Sci U S A. 2003;100:3491–3496. doi: 10.1073/pnas.0437991100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Gersdorff H, Schneggenburger R, Weis S, Neher E. Presynaptic depression at a calyx synapse: the small contribution of metabotropic glutamate receptors. J Neurosci. 1997;17:8137–8146. doi: 10.1523/JNEUROSCI.17-21-08137.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadel K, Neher E, Sakaba T. The coupling between synaptic vesicles and Ca2+ channels determines fast neurotransmitter release. Neuron. 2007;53:563–575. doi: 10.1016/j.neuron.2007.01.021. [DOI] [PubMed] [Google Scholar]
- Walsh MK, Lichtman JW. In vivo time-lapse imaging of synaptic takeover associated with naturally occurring synapse elimination. Neuron. 2003;37:67–73. doi: 10.1016/s0896-6273(02)01142-x. [DOI] [PubMed] [Google Scholar]
- Wang LY, Kaczmarek LK. High-frequency firing helps replenish the readily releasable pool of synaptic vesicles. Nature. 1998;394:384–388. doi: 10.1038/28645. [DOI] [PubMed] [Google Scholar]
- Wendland B, Miller KG, Schilling J, Scheller RH. Differential expression of the p65 gene family. Neuron. 1991;6:993–1007. doi: 10.1016/0896-6273(91)90239-v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood SJ, Slater CR. Action potential generation in rat slow- and fast-twitch muscles. J Physiol. 1995;486:401–410. doi: 10.1113/jphysiol.1995.sp020821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood SJ, Slater CR. The contribution of postsynaptic folds to the safety factor for neuromuscular transmission in rat fast- and slow-twitch muscles. J Physiol. 1997;500:165–176. doi: 10.1113/jphysiol.1997.sp022007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood SJ, Slater CR. Safety factor at the neuromuscular junction. Prog Neurobiol. 2001;64:393–429. doi: 10.1016/s0301-0082(00)00055-1. [DOI] [PubMed] [Google Scholar]
- Young SM., Jr Proteolysis of SNARE proteins alters facilitation and depression in a specific way. Proc Natl Acad Sci U S A. 2005;102:2614–2619. doi: 10.1073/pnas.0409656102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young SM, Jr, Neher E. Synaptotagmin has an essential function in synaptic vesicle positioning for synchronous release in addition to its role as a calcium sensor. Neuron. 2009;63:482–496. doi: 10.1016/j.neuron.2009.07.028. [DOI] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]