

Non-technical summary
In some forms of incurable epilepsies, GABAergic interneurons, which physiologically mediate inhibition in the brain, are thought to mediate excitation. The presence of a specific form of electrical communication between these cells, which is mediated by structures called gap junctions, has been proposed to be involved in the generation of synchronized epileptiform discharges. In support of this hypothesis is the repeated finding in the literature that the drug carbenoxolone, which is an effective blocker of gap junction function, decreases epileptiform activity in models of epilepsy both in vivo and in vitro. Our work challenges this view and highlights additional side-effects of carbenoxolone, which are unrelated to gap junctions, but seem to contribute to its reported antiepileptic activity. A full knowledge of these additional mechanisms is important for the rational development of new molecules to be used in the therapy of epilepsy.
Abstract
Abstract
Synchronous bursting of cortical GABAergic interneurons is important in epilepsies associated with excitatory GABAergic signalling. If electrical coupling was critical for the generation of this pathological activity, then the development of selective blockers of connexin36-based interneuronal gap junctions could be of therapeutic value. We have addressed this issue in the 4-aminopyridine model of epilepsy in vitro by comparing GABAergic epileptiform currents and their sensitivity to gap junction blockers in wild-type vs. connexin36 knockout mice. Although electrical coupling was abolished in stratum lacunosum-moleculare interneurons from knockout animals, epileptiform currents were not eliminated. Furthermore, epileptiform currents propagated similarly across hippocampal layers in the two genotypic groups. Blockade of electrical coupling with carbenoxolone suppressed amplitude, frequency and half-width of the epileptiform currents both in wild-type and in knockout animals, whereas mefloquine had no effects. Carbenoxolone also depressed responses to exogenous and synaptic GABA application onto interneurons. We conclude that, in the 4-aminopyridine model of epilepsy in vitro, connexin36 is not critical for the generation of epileptiform discharges in GABAergic networks and that the observed antiepileptic effects of carbenoxolone are likely to be due to blockade of GABAA receptors and not of connexin36-based gap junctions. Lastly, because of its chemical structure and its effects on amplitude and kinetics of GABAergic currents, we tested the hypothesis that carbenoxolone acted via specific sites on GABAA receptors, such as the one mediating the effects of the neurosteroid pregnenolone sulfate, or the allosteric regulatory site of benzodiazepines/β-carbolines. Our results suggest that neither of these is involved.
Introduction
Synchronous firing of populations of cortical GABAergic interneurons (Galarreta & Hestrin, 2001) generating depolarizing responses (Staley, 2004) has been implicated in epileptiform discharges in vivo and in vitro (Avoli, 1996). Studies on tissue obtained after surgery from patients suffering from intractable epilepsy have identified depolarizing GABAergic signalling as a mechanism sustaining electroencephalographic spikes in temporal lobe epilepsy (Cohen et al. 2002; Huberfeld et al. 2007), ictal-interictal transitions in Taylor's type focal cortical dysplasia (D'Antuono et al. 2004), and possibly seizures in autosomal dominant nocturnal frontal lobe epilepsy (Klaassen et al. 2006). Developmentally regulated depolarizing vs. hyperpolarizing GABAergic signalling in cortical vs. subcortical networks has been proposed as a mechanism of electroclinical uncoupling of neonatal seizures (Glykys et al. 2009), further highlighting the importance of synchronized activity of cortical interneurons for some forms of epilepsy.
GABAergic interneurons are electrically coupled by gap junctions (Galarreta & Hestrin, 2001), which depend primarily on connexin36 (Venance et al. 2000; Deans et al. 2001; Hormuzdi et al. 2001). Direct paired recordings have shown that coupling between interneurons promotes synchronous firing under specific conditions (Mancilla et al. 2007), and propagates GABAergic excitatory potentials within interneuronal networks (Zsiros et al. 2007). If these mechanisms were operational in the aforementioned forms of epilepsies, then a strong rationale could be made for the development of connexin36-selective gap junction blockers as antiepileptic drugs (Margineanu & Klitgaard, 2001; Meldrum & Rogawski, 2007).
Several studies have taken advantage of the convulsive properties of the potassium channel blocker 4-aminopyridine (4-AP) to investigate seizures in vivo (Szente & Pongrácz, 1979) and epileptiform synchronization in vitro (Rutecki et al. 1987, see review by Avoli et al. 2002). In particular, 4-AP is very effective in triggering synchronized discharges in cortical interneurons (Lamsa & Kaila, 1997; Ziburkus et al. 2006; Zsiros et al. 2007). This activity depends on depolarizing GABAergic transmission (Lamsa & Taira, 2003; Kantrowitz et al. 2005; Zsiros et al. 2007) and can be conveniently isolated in vitro following blockade of ionotropic glutamate receptors (Perrault & Avoli, 1992). Application of gap junction blockers such as carbenoxolone, has been consistently reported to reduce 4-AP-dependent synchronization both in vivo (Szente et al. 2002; Gajda et al. 2003, 2005, 2006; Medina-Ceja et al. 2008) and in vitro (Ross et al. 2000; Yang & Michelson, 2001; Traub et al. 2001; Lamsa & Taira, 2003; Gigout et al. 2006; Zsiros et al. 2007). Unfortunately, currently available blockers (reviewed by Juszczak & Swiergiel, 2009) bind to several targets other than gap junctions, making a straightforward interpretation of pharmacological experiments problematic (Tovar et al. 2009). Furthermore, only a limited number of studies have compared 4-AP-induced activity in wild-type vs. connexin36 knockout mice (Cx36 KO), and contrasting observations have been reported (see Maier et al. 2002, but also Buhl et al. 2003 and Pais et al. 2003). We have re-examined this issue by taking advantage of recordings from interneurons in wild-type and Cx36 KO mice. Our results indicate that carbenoxolone possesses antiepileptic activity independent of its blockade of connexin36-based gap junctions.
Methods
Ethical approval
All experimental procedures described in the present work were in accordance with the National Institutes of Health (NIH) Guidelines for the Care and Use of Laboratory Animals, followed Northwestern University Institutional Animal Care and Use Committee (IACUC) approved protocols, and complied with the policies of The Journal of Physiology (Drummond, 2009) and UK regulations on animal experimentation.
Slice preparation
Slices were prepared from young (P14–P21) C57/B6 wild-type (n = 58) and C57BL6/129sv connexin36 knockout mice (n = 47, generous gift of Prof. David Paul, Harvard University) generated as described in Deans et al. (2001). Animals were first deeply anaesthetized using isoflurane in an induction chamber, in compliance with the guidelines provided by the IACUC of Northwestern University and the NIH. The level of anaesthesia was assessed by monitoring the pedal withdrawal reflex and by pinching the tail and ears. Following deep anaesthesia, mice were quickly decapitated and the brain removed from the skull in a small container filled with chilled solution of the following composition (in mm): 130 NaCl, 24 NaHCO3, 3.5 KCl, 1.25 NaH2PO4, 1 CaCl2, 2 MgCl2, 10 glucose saturated with 95% O2–5% CO2 at pH 7.4. Both hemispheres of the brain were glued on the stage of a vibrating microtome (Leica, VT1000S) and submerged in chilled artificial cerebrospinal fluid (ACSF), and sections of 400 μm were cut and stored in an incubation chamber at 34–35°C for at least 30 min, then stored at room temperature until use. Throughout the study, polymerase chain reaction testing was used (Center for Comparative Medicine, Northwestern University via Transnetyx, Cordova, TN, USA) to confirm the genotype of connexin36 knockout mice. Thirty-nine animals were tested and genotype was confirmed in 100% of them.
Whole-cell recordings
Conventional patch-clamp recordings were performed. Slices were superfused with preheated ACSF maintained at a constant temperature (32–35°C). ACSF was of the following composition (in mm): 130 NaCl, 24 NaHCO3, 3.5 KCl, 1.25 NaH2PO4, 2 CaCl2, 1 MgSO4, 10 glucose saturated with 95% O2–5% CO2 at pH 7.4. Cells were observed and selected for recording by means of 40× IR immersion DIC objective applied to a direct microscope (Zeiss, Germany) equipped with an infrared camera system (DAGE-MTI, Michigan City, IN, USA). Interneurons were selected in the CA1 stratum lacunosum-moleculare according to the same criteria as in Zsiros & Maccaferri (2005). Pipettes were pulled from borosilicate thin glass capillaries (MTW150F-3, WPI) and filled with the appropriate filtered intracellular solution to a ∼3 MΩ final resistance, as detailed below. Recordings were carried out using a Multiclamp 700 amplifier (Molecular Devices, Sunnyvale, CA, USA). Data were filtered at 3 kHz and digitized at 10–20 kHz using a Digidata A/D board and the Clampex 9 program suite (Molecular Devices). The holding potential for voltage-clamp experiments was set to −60 mV apart from a limited set of initial experiments where cells were held at −40 mV. GABA was puffed (>10 psi, 50 ms) on the recorded cells via a picospritzer device (TooheySpritzer, Toohey Company, Fairfield, NJ, USA).
The solution for current-clamp recordings was (in mm): 115 potassium methylsulfate, 4 ATP-Mg2, 30 NaCl, 0.3 GTP, 16 KHCO3 equilibrated with 95% O2–5% CO2 to give pH 7.3.
The solution for voltage-clamp recordings was (in mm): 125 KCl, 4 ATP-Mg2, 10 NaCl, 0.3 GTP, 16 KHCO3, 10 N-2(2,6-dimethylphenylcarbamoylmethyl) triethylammonium chloride (QX-314-Cl) equilibrated with 95% O2–5% CO2 to give pH 7.3. Estimated EGABA(A) was ∼0 mV (Zsiros et al. 2007). QX-314 was included in the intracellular solution at high concentration in order to block voltage-dependent conductances.
Evaluation of electrical coupling in paired current-clamp experiments
We tested for the presence of gap junctions between pairs by repetitively injecting a 500 ms current step of −50/−100 pA. Cells were considered coupled if they had a coupling coefficient (ratio of the voltage deflection in the non-injected cell to the voltage deflection in the injected cell measured during the last 50 ms of the stimulus) value higher than 0.005 and the shape of the electrotonic response was recognizable in the recording from the non-injected cell. For these experiments, synaptic blockers were added to ACSF (SR-95531 (gabazine; 12.5 μm), 1,2,3,4-tetrahydro-6-nitro-2,3-dioxo-benzo[f]quinoxaline-7-sulfonamide (NBQX; 20 μm), d(–)-2-amino-5-phosphonopentanoic acid (d-AP5; 50 μm), CGP55845 (5 μm)).
Drug preparation and application in slices
All the following drugs were from Tocris Biosciences. Gabazine was used at 12.5 μm. The powder was dissolved in water as a stock solution at 25 mm, aliquoted in 100 μl vials, and frozen at −20°C. d-AP5 was dissolved in water, stocked frozen in 100 μl aliquots at 50 mm and used at a 50 μm. Stock solutions of NBQX were dissolved in DMSO at 100 mm, aliquoted in 40 μl vials and frozen at −20°C. The final concentration used was 20 μm. CGP55845 was dissolved in DMSO in stock solutions at 100 mm, then aliquoted and used at a final concentration of 5 μm. Carbenoxolone was obtained from Sigma as a disodium salt and was dissolved directly into the recording solution to a final concentration of 100 μm. 4-Aminopyridine was from Sigma and was also dissolved directly into the recording solution to its final concentration of 100 μm. Pregnenolone sulfate, mefloquine and flumazenil were also obtained from Sigma. Pregnenolone sulfate (sodium salt) and mefloquine hydrochloride were dissolved directly into the recording solution to a final concentration of 100 μm. Flumazenil was dissolved in DMSO, stored as frozen 1 ml aliquots at 25 mm, and used at a final concentration of 5 μm.
Analysis of network-driven events in slices exposed to 4-aminopyridine
Spontaneous epileptiform currents recorded from single neurons were analysed using the Clampfit 9.0 (Molecular Devices), OriginPro7.0 (OriginLab Corp., Northampton, MA, USA), and Microsoft Excel suites of programs. Events were first collected using the threshold-based analysis feature of Clampfit, reviewed by visual inspection, and aligned by their peaks. Analysis of the time course of the effect of various drugs (or stability) was performed first by averaging the amplitudes of spontaneous events within the same neuron in 1 min bins and then by averaging each bin across different cells. Cross-correlations were performed on traces digitally filtered at 30 Hz.
Digital summation of the effects of carbenoxolone and pregnenolone sulfate
The digital summation of the effects of the individual applications of carbenoxolone and pregnenolone sulfate was obtained by multiplying the averaged normalized values of peak and frequency for every bin during the time course of the effect (Fig. 14A) and the averaged normalized values during the last 5 min of the experiment in the presence of the drug (Fig. 14B).
Figure 14. The effects of pregnenolone sulfate and carbenoxolone on 4-AP-induced epileptiform events add linearly.
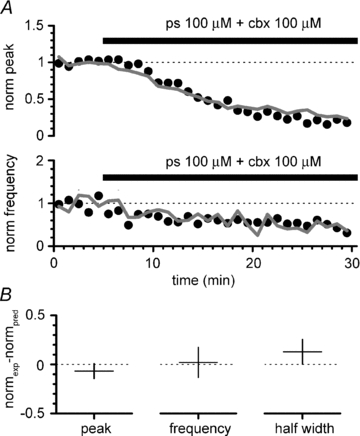
A, time course plot of the experimental effect of co-application of pregnenolone sulfate and carbenoxolone on amplitude and frequency of GABAergic currents (black circles, same data as Fig. 12B) compared to its predicted effect calculated from the linear summation of the effects of their individual applications (thick grey line). Notice the similarity of the effects. B, mean and 95% confidence intervals of the difference of the experimental effect of co-application (from left to right: on peak amplitude, event frequency and half-width) vs. the predicted effect if the two drugs act by purely independent mechanisms. Notice that all the confidence intervals of the difference (normexp– normpred) include a range of values indicating a very small difference between observed and predicted effects.
Statistical methods
Statistical analysis was performed using the following software packages: Clampfit 9.0, Excel, Origin, Prism (GraphPad Software Inc., La Jolla, CA, USA). Significance was set at 0.05. Fisher's exact test or Student's t test for paired or unpaired data was used as appropriate. Because multiple comparisons were often made, the Bonferroni correction was used (Motulsky, 2010). The Bonferroni correction was implemented by multiplying the extracted value of P by the m number of comparisons. The Bonferroni correction was also applied to multiple confidence intervals by using t0.025/m in place of t0.025 when constructing m 95% confidence intervals. Data are shown as means ± SEM unless stated otherwise in the legend.
Results
If the antiepileptic actions of carbenoxolone in vitro (Zsiros et al. 2007) depended on the blockade of connexin36 gap junction channels, then we would expect to see differences in 4-AP-induced epileptiform discharges recorded in hippocampal slices from wild-type vs. Cx36 KO mice. When glutamatergic transmission is blocked in this model of epilepsy in vitro, GABAergic epileptiform events are thought to be initiated in the stratum moleculare of the dentate gyrus (Gonzalez-Sulser et al. 2011) and propagate to the CA1 region starting from stratum lacunosum-moleculare (Sinha & Saggau, 2001; Perkins, 2002). Therefore, we decided to measure and compare 4-AP-induced epileptiform currents recorded from stratum lacunosum-moleculare interneurons, which are frequently electrically coupled (Price et al. 2005; Zsiros & Maccaferri, 2005).
However, as a preliminary step, we verified that electrical coupling between stratum lacunosum-moleculare interneurons was indeed abolished in slices from Cx36 animals. As shown in Figs 1 and 2, electrical coupling was present in pairs of interneurons from control slices, but absent in paired recordings from tissue obtained from Cx36 KO mice. The probability of finding connected pairs in wild-type animals was ∼30% (5 coupled pairs out of 15 tested), similar to what was previously reported for interneurons of the same layer in rat slices (Zsiros & Maccaferri, 2005). In contrast, no coupled pairs were found in slices from Cx36 KO mice (0 out of 25 pairs tested, P < 0.05). Therefore, we concluded that stratum lacunosum-moleculare interneurons of wild-type animals are electrically coupled via gap junctions that depend on connexin36, and that obvious compensatory mechanisms re-establishing coupling are absent in interneurons of Cx36 KO animals. It is also interesting to note that the membrane input resistance of interneurons from Cx36 KO mice was significantly higher from the one observed in control animals (372 ± 32 MΩ in wild-type, n = 30 vs. 520 ± 41 MΩ in Cx36 KO, n = 50, P < 0.05), consistent with the idea of the loss of a resting membrane conductance, and in agreement with what was previously reported in neocortical interneurons of the same mouse (Deans et al. 2001).
Figure 1. Electrical coupling between CA1 stratum lacunosum-moleculare interneurons in wild-type (WT, black traces) and connexin36 knockout (Cx36 KO, red traces) mice.
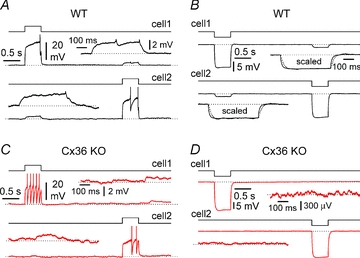
A, sequential injection of a depolarizing current step (90 pA, black steps) to interneurons of WT mice generates an attenuated response in the non-injected cell. Insets show the attenuated responses at enhanced magnification: notice the presence of spikelets. B, similar protocol as in A, but hyperpolarizing current steps of –50 pA (black steps) are injected. Notice the propagation of the voltage response to the non-injected neuron. Also notice, in the insets, after scaling, the slower kinetics of the propagated response (dotted line) vs. the original one (continuous trace). C and D illustrate the same type of experiments as A and B, respectively, performed on interneurons in a slice obtained from a Cx36 KO mouse. Notice the lack of a propagated response in the non-injected neuron. Current pulses were 50 pA in C (black steps) and –50 pA in D (black steps). Insets in C and D show the lack of a voltage response in the non-injected cell at a magnified scale. Gabazine (12.5 μm), NBQX (20 μm), d-AP5 (50 μm) and CGP55845 (1–5 μm) present throughout in A–D.
Figure 2. Network and membrane differences in interneurons of wild-type (WT) vs. connexin36 knockout (Cx36 KO) mouse.
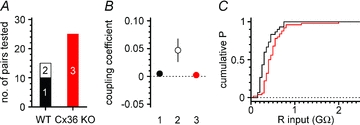
A, summary plot showing the different probability of finding electrically coupled pairs in the two types of animals. The white part of the bar (2) represents coupled pairs, in contrast to the black and red portions (1, 3), representing non-connected neurons in wild-type and Cx36 KO mice, respectively. B, summary graph showing the value of coupling coefficients for the uncoupled (1, black circle) and coupled (2, white circle) pairs of WT animals and for uncoupled (3, red circle) cells of Cx36 KO mice. C, cumulative probability distributions of resting membrane input resistances in WT (black line) vs. Cx36 KO animals (red line). Notice the shift towards higher values in the Cx36 KO mice.
Therefore, as mentioned above, if gap junctions played a critical role in the generation of epileptiform GABAergic activity, we would expect to see this reflected in a dramatic reduction of the amplitude and/or frequency of 4-AP-induced epileptiform currents in the animals lacking connexin36.
In the constant presence of blockers of ionotropic glutamatergic transmission and GABAB receptors (NBQX 20 μm, d-AP5 50 μm and CGP55845 5 μm), we recorded spontaneous epileptiform currents following exposure of the slices to 4-AP (100 μm, Fig. 3). Surprisingly, epileptiform events in Cx36 KO mice had a larger peak conductance than the ones observed in control animals (74 ± 8 nS in Cx36 KO, n = 37 vs. 52 ± 3 nS in wild-type mice, n = 66, P < 0.05), and no difference was found in their frequency (31 ± 1 mHz in Cx36 KO, n = 37 vs. 36 ± 1 mHz, n = 66, P > 0.05). The difference in peak conductance could not be explained by voltage-clamp errors due to different series resistance values for the recordings in the two datasets (series resistance was 18 ± 1 MΩ in Cx36 KO, n = 37 vs. 18 ± 1 MΩ, n = 66 in wild-type, P > 0.05). This result suggests that the expression of connexin36 and the presence of functional coupling between stratum lacunosum-moleculare interneurons are not an absolute requirement for the generation of GABAergic epileptiform discharges in vitro.
Figure 3. Properties of 4-AP-induced epileptiform currents and recording conditions in WT (black traces) vs. Cx36 KO mice (red traces).
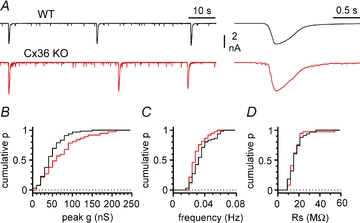
A, examples of voltage-clamp recording traces from stratum lacunosum-moleculare interneurons in WT and Cx36 KO mice shown at different temporal magnifications (left and right panels). B, cumulative probability plot of the peak conductance during epileptiform events in the two genotypes (black line, WT; red line, Cx36 KO). Notice the presence of larger conductance events in the Cx36 KO genotype. C, summary graph showing the similarity of the frequency of the epileptiform events in the two groups of animals. D, cumulative probability plot of the series resistance for the recordings in WT and Cx36 KO mice: notice the overlapping distributions. A–D, ionotropic glutamatergic synaptic transmission and GABAB receptors blocked throughout the experiments.
Next, we verified that epileptiform events in the interneurons of the two types of animals were sustained by GABAA receptors and similarly sensitive to gabazine (Fig. 4), as previously reported for 4-AP-induced epileptiform activity in wild-type rats (Zsiros et al. 2007). As expected, application of gabazine (12.5 μm) blocked epileptiform currents in both wild-type (from 100 ± 11 nS in control to 5 ± 1 nS in the presence of the drug, n = 3, P < 0.05) and Cx36 KO mice (from 66 ± 9 nS in control to 1 ± 1 nS in the presence of gabazine, n = 3, P < 0.05) to a similar degree (residual currents in the presence of gabazine were 5 ± 1% of control values in wild-type and 2 ± 1% of control values in Cx36 KO).
Figure 4. Pharmacological demonstration that 4-AP-induced epileptiform currents are mediated by GABAA receptors in both WT (black traces) and Cx36 KO mice (red traces).
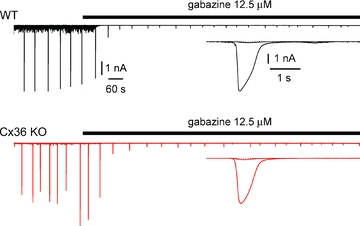
Application of the GABAA receptor blocker gabazine (12.5 μm) powerfully reduces epileptiform currents in interneurons recorded in slices obtained from the two different genotypes. Upper traces show wild-type and lower traces connexin36 knockout. Insets show superimposed averaged events in control and in the presence of the drug.
Despite the fact that the peak of the epileptiform events did not appear reduced in Cx36 KO mice, we thought that the absence of functional gap junctions might nevertheless impact their propagation to other hippocampal layers. Previous work by Perrault & Avoli (1992) has indeed shown that the physical separation of different hippocampal layers via surgical cuts does not stop the occurrence of epileptiform GABAergic events in slices, but impairs their synchronization. Therefore, if the surgical cuts of Perrault & Avoli (1992) prevented synchronization of the different hippocampal layers by physically separating GABAergic networks connected via gap-junctions, we would expect to observe non-synchronized events in different hippocampal layers of slices from the Cx36 KO mice. We tested this possibility by performing simultaneous double recordings from interneurons located in stratum lacunosum-moleculare and stratum oriens of both wild-type animals and Cx36 KO mice (Fig. 5). Contrary to our expectations, synchronized epileptiform currents were observed in the two layers in slices obtained from both wild-type and Cx36 KO animals. Neither the peak (peak cross correlation was 0.76 ± 0.03 in wild-type, n = 10 double recordings vs. 0.74 ± 0.05 in Cx36 KO, n = 10 double recordings, P > 0.05) nor the position of the peak (+78 ± 16 ms in wild-type, n = 10 double recordings, +40 ± 19 ms, n = 10 ms in Cx36 KO, P > 0.05) of the cross correlations were different in the two types of animals. This result confirms the reports of Sinha & Saggau (2001) and Perkins (2002) that, in the CA1 subfield, these experimental conditions trigger GABAergic epileptiform activity initiated in stratum lacunosum-moleculare. However, it shows that connexin36 and electrical coupling between interneurons are not required for propagation across layers.
Figure 5. Propagation of 4-AP-induced epileptiform currents across hippocampal layers in WT (black traces) vs. Cx36 KO animals (red traces).
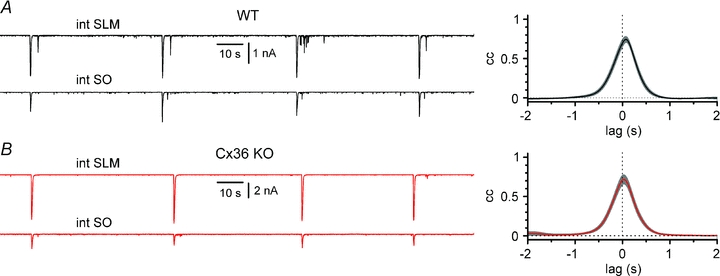
A, left panel, simultaneous recordings from a stratum lacunosum-moleculare and a stratum oriens interneuron from a WT mouse (int SLM and int SO, respectively). Notice the presence of synchronous events. Right panel, summary cross-correlogram of the activity in 10 SLM-SO WT interneuron pairs. Grey bands indicate ±SEM. B, identical experiments and analysis as in A, but performed in 10 pairs of interneurons from Cx36 KO animals. Notice the similarity of the results. NBQX (20 μm), d-AP5 (50 μm) and CGP55845 (5 μm) present throughout in A and B.
Taken together, the observations presented so far suggest that the reported effects of carbenoxolone on GABAergic discharges induced by 4-AP (Ross et al. 2000; Yang & Michelson, 2001; Traub et al. 2001; Lamsa & Kaila, 1997; Gigout et al. 2006; Zsiros et al. 2007) are not due to the blockade of connexin36-dependent gap junctions between interneurons, but are likely to rely on different mechanism(s).
We decided to test this hypothesis directly by comparing the effect of carbenoxolone on epileptiform currents in wild-type vs. Cx36 KO animals. However, as a control experiment, we first verified that the absence of connexin36 in the KO animals does not impair the stability of epileptiform activity in prolonged recordings (Rouach et al. 2008). The amplitude and frequency of synchronized GABAergic currents were not different at the beginning compared to the end of 30 min recordings either in wild-type or in Cx36 KO mice, (Fig. 6). For wild-type animals the amplitude and frequency of the events was 3928 ± 454 pA and 29 ± 2 mHz calculated for the initial 5 min of the recording vs. 3376 ± 268 pA and 28 ± 2 mHz in the last 5 min (P > 0.05, n = 13). Similarly, neither the amplitude nor the frequency of the epileptiform events changed in slices from Cx36 KO mice (4189 ± 734 pA and 22 ± 1 mHz at the beginning vs. 3884 ± 663 pA and 26 ± 2 mHz at the end of the recording, P > 0.05, n = 8).
Figure 6. 4-AP-induced epileptiform activity can be maintained for long recording times in both WT (black circles and traces) and Cx36 KO mice (red circles and traces).
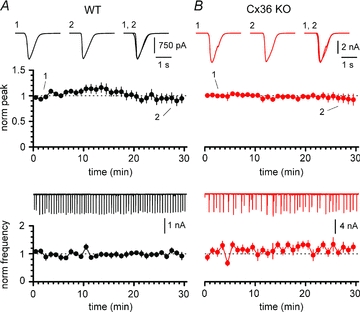
A, notice the stability of the amplitude (upper panel) and frequency (lower panel) of the events. Insets in the upper panel represent averaged events during the first (1) and last (2) 5 min of recording, and superimposed (1, 2). Inset in the lower panel shows the continuous experimental recording. B, as in A, but experiments were performed in slices from Cx36 KO animals. Note that epileptiform activity is similarly maintained for 30 min in both genotypes. NBQX (20 μm), d-AP5 (50 μm) and CGP55845 (5 μm) present throughout in A and B.
When the effect of carbenoxolone was tested (Fig. 7), we observed that epileptiform currents were reduced in wild-type animals from 3099 ± 395 pA to 1434 ± 171 pA (first 5 min in control vs. last 5 min in carbenoxolone, n = 16) and from 5016 ± 457 pA to 2457 ± 351 pA (n = 22, P < 0.05) in Cx36 KOs. The frequency of the events was also slightly reduced in both cases (from 35 ± 3 mHz to 21 ± 3 mHz in wild-type (n = 16, P < 0.05), and from 28 ± 2 mHz to 22 ± 2 mHz in Cx36 KO mice (n = 22, P < 0.05)). In conclusion, carbenoxolone reduced the amplitudes of epileptiform currents to 43 ± 4% in wild-type animals and to 48 ± 5% in Cx36 KO mice, and simultaneously decreased their frequency to 70 ± 7% and 76 ± 6%, respectively. When we examined the kinetics of epileptiform currents (Fig. 8), we also noticed that carbenoxolone reduced the duration of the events. In particular, the half-width of the events was reduced from 339 ± 21 ms to 226 ± 26 ms in wild-type animals (n = 16, P < 0.05) and from 350 ± 20 ms to 239 ± 19 ms in Cx36 KO mice (n = 22, P < 0.05). Overall, these results show that carbenoxolone has similar effects on epileptiform events recorded in tissue from wild-type and Cx36 KO mice, thus suggesting that blockade of connexin36 is unlikely to mediate its observed antiepileptic actions. As an alternative, direct suppression of GABAA-mediated synaptic transmission could potentially explain our results. Data in the literature, however, appear contradictory. Although Traub et al. (2001) found that carbenoxolone at 100 μm had no effect on pharmacologically isolated depolarizing GABAergic potentials evoked by tetanic stimulation in slices, Tovar and colleagues (2009) have recently reported that carbenoxolone, at the same concentration, reduces the amplitude of IPSC evoked in autaptic cultures by ∼50%.
Figure 7. Carbenoxolone depresses GABAergic epileptiform events in both WT (black circles and traces) and Cx36 KO animals (red circles and traces).
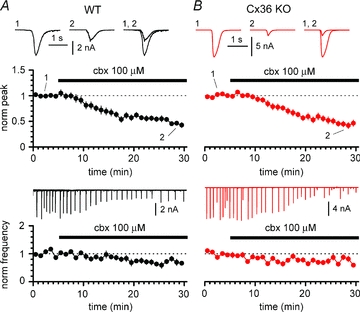
A, time course of normalized peak (upper panel) and frequency (lower panel) during application of carbenoxolone (cbx, black bar) to slices from WT mice. Notice the strong decrease in the amplitude of the events and also the reduction in their frequency. Insets in the upper panel are averaged events in control (1), in the presence of carbenoxolone (2), and superimposed (1, 2). Inset in the lower panel shows the continuous experimental recording. B, as in A, but experiments were performed on Cx36 KO animals. Notice that carbenoxolone also has an effect on both amplitude and frequency in this genotype. Ionotropic glutamatergic synaptic transmission and GABAB receptors blocked throughout the experiments both in A and B.
Figure 8. Carbenoxolone reduces the half-width of GABAergic epileptiform events in both WT (black circles and traces) and Cx36 KO animals (red circles and traces).
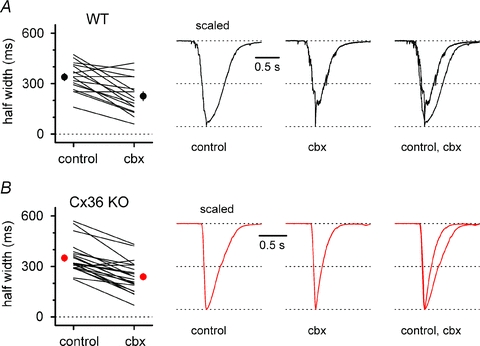
A, left, summary graph comparing half-widths in control and in the presence of carbenoxolone (cbx) for individual experiments (lines) and for their overall average (circle). Traces in control, in the presence of carbenoxolone, and superimposed are shown in the right panel. B, as in A, but experiments were performed on slices from Cx36 KO animals. Notice that carbenoxolone has a similar effect irrespective of the genotype. NBQX (20 μm), d-AP5 (50 μm) and CGP55845 (5 μm) present throughout in A and B.
We designed an experiment to test the blocking effect of carbenoxolone on both exogenous and endogenous GABAergic events recorded from stratum lacunosum-moleculare interneurons (bathed in standard ACSF plus blockers of glutamatergic ionotropic synaptic transmission and GABAB receptor antagonists, but no 4-AP added). In order to completely exclude effects due to blockade of connexin36, slices were prepared from Cx36 KO mice. While interneurons were held in voltage-clamp at −60 mV, we first puffed GABA (50 μm, 50 ms duration) onto their soma, and then, after a delay of 42 s, an inhibitory postsynaptic current was evoked on the same cell by extracellular stimulation. As shown in Fig. 9, carbenoxolone depressed responses to exogenous GABA application and reduced inhibitory postsynaptic currents in 9 out of 10 cells tested. Currents generated by exogenous GABA application decreased from 645 ± 152 pA to 237 ± 59 pA (n = 9, P < 0.05), and inhibitory postsynaptic currents from 1020 ± 211 pA to 582 ± 181 pA (n = 9, P < 0.05). The relative effects of carbenoxolone on GABA puffs and synaptic currents were similar (residual GABA puff and synaptic current in the presence of the drug were 43 ± 9% and 54 ± 10%, respectively), thus suggesting that, under our experimental conditions, the major impact of carbenoxolone is on postsynaptic GABAA receptors and not on presynaptic mechanisms. If this was the case, an additional prediction would be that carbenoxolone should also affect the kinetics of GABA puffs and evoked synaptic transmission (Fig. 10). The half-width of the GABA puff was reduced from 829 ± 73 ms in control conditions to 639 ± 55 ms in the presence of the drug (n = 9, P < 0.05), and, similarly, the half-width of the evoked synaptic current decreased from 60 ± 9 ms to 46 ± 6 ms (n = 9, P < 0.05). Thus, our data suggest that direct blockade of GABAA receptors, and not blockade of connexin36-based gap junctions, is the major mechanism underlying the antiepileptic effect of carbenoxolone in this model in vitro.
Figure 9. Carbenoxolone reduces direct responses both to exogenous and synaptic GABA applications.
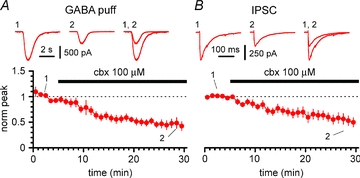
A, summary plot of the time course of the effect of carbenoxolone (cbx, black bar) on currents evoked by GABA puffs onto stratum lacunosum-moleculare interneurons of the Cx36 KO mouse. Insets show averaged responses to GABA application in control (1), in the presence of carbenoxolone (2) and superimposed (1, 2). B, time course summary graph for the effect of carbenoxolone (black bar) on evoked inhibitory postsynaptic currents (IPSCs) recorded from the same cells in A. Notice that IPSCs are also depressed by carbenoxolone, similarly to responses to exogenous GABA. All experiments in A and B were performed on slices from Cx36 KO animals and ionotropic glutamatergic synaptic transmission and GABAB receptors were pharmacologically blocked.
Figure 10. Effect of carbenoxolone on the half-width of responses to exogenous GABAergic applications and of inhibitory postsynaptic potentials in interneurons from Cx36 KO animals.
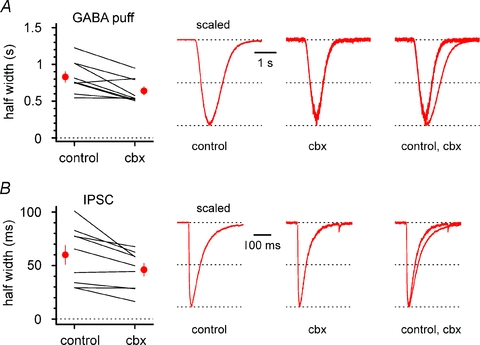
A, left, summary graph comparing half-widths of voltage-clamp responses to GABA puffs in control and in the presence of carbenoxolone (cbx) for individual experiments (lines) and for their overall average (red circles). Traces in control, in the presence of carbenoxolone and superimposed are shown in the right panel. B, as in A, but data and analysis regards evoked inhibitory postsynaptic currents (IPSCs). Notice that carbenoxolone has a similar effect on GABA puffs and IPSCs. NBQX (20 μm), d-AP5 (50 μm) and CGP55845 (5 μm) present throughout in A and B.
We decided to further confirm this interpretation by measuring the effect of a different gap junction blocker, mefloquine, on GABAergic epileptiform currents. The advantage of mefloquine is that this compound has been reported to lack non-specific effects on GABAergic synaptic transmission (Cruikshank et al. 2004). As shown in Fig. 11, application of mefloquine did not affect either the amplitude or the frequency of synchronized currents recorded from either wild-type or Cx36 KO mice. The peak of epileptiform currents in wild-type animals was 3971 ± 383 pA during the first 5 min in control conditions vs. 3545 ± 421 pA during the last 5 min in mefloquine (n = 11, P > 0.05). In Cx36 KOs, the amplitude of the events was 4112 ± 857 pA in control and 4248 ± 745 pA (n = 5, P > 0.05) in the presence of the drug. The frequency of the epileptiform currents did not change in either case (from 15 ± 1 mHz to 14 ± 2 mHz in wild-type (n = 11, P > 0.05), and from 17 ± 3 mHz to 15 ± 1 mHz in Cx36 KO mice (n = 5, P > 0.05)). No changes either were observed on the kinetics of the events as indicated by half-width measurements (from 473 ± 28 ms to 543 ± 32 ms in wild-type (n = 11, P > 0.05), and from 376 ± 23 ms to 466 ± 28 ms in Cx36 KO mice (n = 5, P > 0.05)).
Figure 11. Mefloquine does not affect GABAergic epileptiform events in either WT (black circles and traces) or Cx36 KO animals (red circles and traces).
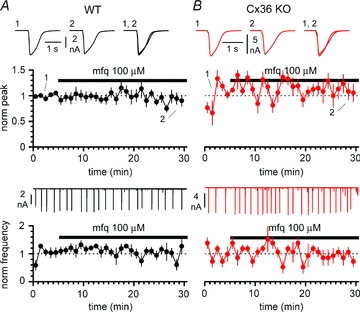
A, time course of normalized peak (upper panel) and frequency (lower panel) during application of mefloquine (mfq, black bar) to slices from WT mice. Notice the lack of effect both on the amplitude and on the frequency of the events. Insets in the upper panel are averaged events in control (1), in the presence of mefloquine (2), and superimposed (1, 2). Inset in the lower panel shows the continuous experimental recording. B, as in A, but experiments were performed on Cx36 KO animals. Notice that mefloquine does not affect epileptiform activity in this genotype either. Ionotropic glutamatergic synaptic transmission and GABAB receptors blocked throughout the experiments in both A and B.
Taken together, these experiments exclude connexin36 as the molecular target of carbenoxolone mediating the decrease in GABAergic epileptiform activity, and support the hypothesis that a direct effect on GABAA receptors is involved.
GABAA receptors are the target of several drugs and endogenous modulators. In particular, endogenous neurosteroids can affect both the amplitude and the duration of GABAA receptor-mediated currents (Belelli & Lambert, 2005). Because carbenoxolone (Pubchem CID 636403, Davidson & Baumgarten, 1988) shares some structural similarities with steroids, we next tested the possibility that the observed negative regulation of GABAA receptor activity following exposure to carbenoxolone shared a common mechanism with one of the best characterized GABAA receptor negative modulating neurosteroids, pregnenolone sulfate (Majewska et al. 1988; Mienville & Vicini, 1989; Shen et al. 2000; Eisenman et al. 2003). This hypothesis would predict that pregnenolone sulfate should reduce the amplitude, frequency and half-width of GABAergic epileptiform currents. As shown in Figs 12 and 13, application of pregnenolone sulfate (100 μm) on slices from wild-type animals fulfilled these predictions. The amplitude of epileptiform currents was reduced from a value of 2584 ± 330 pA (measured during the initial 5 min in the absence of the drug) to 1372 ± 148 pA (during the last 5 min in the presence of pregnenolone sulfate, n = 8, P < 0.05). Similarly, the frequency of the events decreased from 22 ± 2 mHz to 14 ± 2 mHz (n = 8, P < 0.05). Lastly, as expected, the measured half-width of the collected events was reduced by pregnenolone sulfate from 419 ± 42 ms to 204 ± 27 ms (n = 8, P < 0.05). These results indicate that carbenoxolone and pregnenolone sulfate affect GABAergic epileptiform currents in a similar way, although this could still be achieved by independent sites. If the sites and mechanism(s) underlying carbenoxolone- and pregnenolone sulfate-dependent modulation of epileptiform currents were truly independent, then co-application of the two drugs should summate algebraically. In contrast, if the effect of the two molecules shared sites and/or mechanism, co-application should reveal a sublinear summation. The IC50 of pregnenolone sulfate for GABAA receptors varies with the status of activation of the receptor, but typical values are in the high nanomolar (Zaman et al. 1992) to low micromolar range (4.9 μm, see Neelands et al. 1998; 7.2 μm, see Park-Chung et al. 1999; 3–10 μm, see Shen et al. 2000). Furthermore, the IC50 for pregnenolone sulfate decreases in the presence of elevated GABA concentrations, and inhibition of multiquantal GABAergic responses has been shown to be especially efficient (Eisenman et al. 2003). Therefore, 100 μm applied to epileptiform GABAergic currents may be considered a high dose, potentially close to saturation (Zaman et al. 1992).
Figure 12. Effects of the application of pregnenolone sulfate on 4-AP-induced epileptiform currents and of its co-application with carbenoxolone in slices from WT animals.
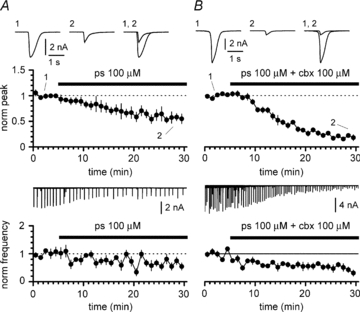
A, time course of normalized peak (upper panel) and frequency (lower panel) during application of pregnenolone sulfate (ps, black bar). Notice the reduction in both amplitude and frequency of the events. Insets in the upper panel are averaged events in control (1), in the presence of pregnenolone sulfate (2), and superimposed (1, 2). Inset in the lower panel shows the continuous experimental recording. B, as in A, but pregnenolone sulfate and carbenoxolone (ps+cbx) were co-applied. Ionotropic glutamatergic synaptic transmission and GABAB receptors blocked throughout the experiments both in A and B.
Figure 13. Kinetic changes of 4-AP-induced epileptiform currents following either application of pregnenolone sulfate or its co-application with carbenoxolone in slices from WT animals.
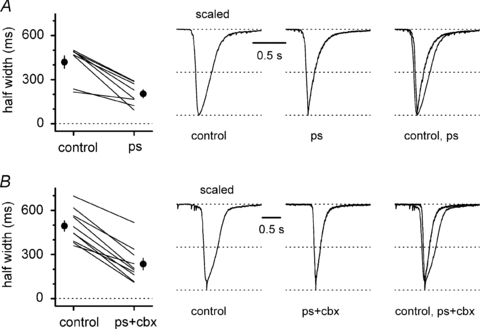
A, left, summary graph comparing half-widths in control and in the presence of pregnenolone sulfate (ps) for individual experiments (lines) and for their overall average (black circles). Traces in control, in the presence of pregnenolone sulfate and superimposed are shown in the right panel. B, as in A, but pregnenolone sulfate and carbenoxolone were co-applied. Ionotropic glutamatergic synaptic transmission and GABAB receptors were pharmacologically blocked in A and B.
Co-application of carbenoxolone and pregnenolone sulfate (Fig. 13) powerfully decreased the peak amplitude of GABAergic epileptiform events from a control value of 3186 ± 455 pA to 610 ± 85 pA (n = 10, P < 0.05), and their frequency from 32 ± 4 mHz to 16 ± 4 mHz (n = 10, P < 0.05). In addition, the half-width of the recorded epileptiform currents was also strongly reduced from 494 ± 35 ms to 236 ± 39 ms (n = 10, P < 0.05).
In order to establish whether the experimental co-application of the two drugs resulted in a linear or non-linear summation of the individual effects, we digitally added (see Methods for details) the data previously obtained with separate applications of carbenoxolone and pregnenolone sulfate (Figs 7–8 and 12–13, respectively). As shown in Fig. 14, the time course of the effect of the experimental co-application of carbenoxolone and pregnenolone sulfate on amplitude and frequency of epileptiform events was very similar to one predicted by the digital summation of the individual effects. This would suggest that carbenoxolone and pregnenolone sulfate have independent sites of action. To further assess this possibility we calculated the confidence intervals of the difference between the experimentally measured (n = 10) and predicted effects (expressed as normalized values in the last 5 min of the co-application of the drugs) for amplitude, frequency and half-width of the epileptiform currents. The confidence intervals of the difference between measured and predicted effects were –0.07 ± 0.08 for the amplitude of the events, 0.02 ± 0.15 for their frequency, and 0.13 ± 0.13 for their half-width. Thus, the pharmacological profile of the combination of pregnenolone sulfate and carbenoxolone indicates that these two compounds are very unlikely to share the same site of action on GABAA receptors.
Therefore, we next decided to test the possibility that carbenoxolone acted as an inverse agonist at the allosteric regulatory benzodiazepine/β-carboline binding site (Sieghart, 1995) by measuring its effect on epileptiform currents recorded in the constant presence of the specific high affinity competitive antagonist flumazenil. Experiments were performed on slices obtained from Cx36 KO mice in order to exclude effects due to blockade of gap junctions between interneurons. As shown in Figs 15 and 16, the constant presence of flumazenil did not prevent the reduction of epileptiform activity induced by carbenoxolone. GABAergic events were reduced in amplitude from 3142 ± 261 pA to 1809 ± 227 pA (first 5 min in flumazenil vs. last 5 min in flumazenil and carbenoxolone, n = 12, P < 0.05) and their frequency decreased from 26 ± 4 mHz to 14 ± 3 mHz (n = 12, P < 0.05). Also, the half-width of the epileptiform currents was shortened from 387 ± 41 ms in the presence of flumazenil alone to 292 ± 41 ms in the presence of flumazenil and carbenoxolone (Fig. 16, n = 12, P < 0.05). We directly ruled out the possibility that the lack of block of carbenoxolone-induced reduction of epileptiform activity was due to a subnanomolar affinity of carbenoxolone (as an inverse agonist) for the flumazenil binding site (affinity of flumazenil is in the low nanomolar range, see Hunkeler et al. 1981). If this was the case, carbenoxolone should begin to considerably affect epileptiform activity at nanomolar, or even lower concentrations. Yet, when we tested carbenoxolone at 5 μm, no effect was observed. As illustrated in Fig. 15, the peak of epileptiform events was 5148 ± 1283 pA in control vs. 5844 ± 1322 pA in the presence of the drug (first 5 min in control vs. last 5 min in carbenoxolone, n = 5, P > 0.05) and their frequencies were 29 ± 6 mHz and 28 ± 6 mHz, respectively (n = 5, P > 0.05). Lastly, the half-width of the epileptiform currents did not change and was 348 ± 37 ms in control conditions and 392 ± 49 ms in the presence of 5 μm carbenoxolone (n = 5, P < 0.05).
Figure 15. Carbenoxolone does not act via the allosteric regulatory site of benzodiazepines.

A, time course of normalized peak (upper panel) and frequency (lower panel) during the application of carbenoxolone (cbx, black bar) in the constant presence of the competitive antagonist flumazenil: notice the similarity of these results (red circles) to those obtained in the absence of flumazenil (thick grey lines, same data as in Fig. 7B). Insets in the upper panel are averaged events in flumazenil (1), in the presence of flumazenil and carbenoxolone (2), and superimposed (1, 2). Inset in the lower panel shows the continuous experimental recording. B, carbenoxolone does not change GABAergic epileptiform currents at low concentrations. Notice the lack of effects on both amplitude and frequency. Insets in the upper panel are averaged events in control (1), in the presence of low concentrations of carbenoxolone (2), and superimposed (1, 2). Inset in the lower panel shows the continuous experimental recording. Experiments shown both in A and B were performed in slices obtained from Cx36 KO mice and in the constant additional presence of NBQX (20 μm), d-AP5 (50 μm) and CGP55845 (5 μm).
Figure 16. Kinetic changes of 4-AP-induced epileptiform currents following application of carbenoxolone in the constant presence of flumazenil.
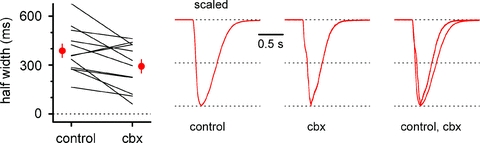
Left, summary graph comparing half-widths in flumazenil (5 μm, control) and in the added presence of carbenoxolone (100 μm, cbx) for individual experiments (lines) and for their overall average (red circles). Scaled traces in flumazenil, in the presence of flumazenil and carbenoxolone, and superimposed are shown in the right panel. Notice that flumazenil does not prevent carbenoxolone-induced kinetic changes.
In conclusion, these results indicate that carbenoxolone does not act as an inverse agonist at the benzodiazepine allosteric regulatory site.
Discussion
This study provides direct evidence that connexin36 is not a critical requirement for 4-AP-induced epileptiform synchronization of interneurons. We also demonstrate that the antiepileptic effect of carbenoxolone on 4-AP-induced GABAergic epileptiform currents is not due to the blockade of connexin36-based gap junctions, but, most likely, to a direct action on postsynaptic GABAA receptors. These results are important for a reinterpretation of previous studies claiming a link between gap junction blockade and antiepileptic activity of carbenoxolone in the 4-AP and, possibly, other models of epilepsy (see for example Fujiwara-Tsukamoto et al. 2010).
Antiepileptic effect of carbenoxolone in the 4-AP model of epilepsy
At least three main different cellular populations may form electrically coupled networks in cortical circuits: glial cells (Kettenmann & Ransom, 1988; Meme et al. 2009), principal neurons (MacVicar & Dudek, 1981; Mercer et al. 2006; Wang et al. 2010, see review by Bennett & Pereda, 2006) and GABAergic interneurons (Galarreta & Hestrin, 1999; Gibson et al. 1999). Therefore, the antiepileptic effect of carbenoxolone in wild-type and Cx36 KO mice may simply suggest that other connexins expressed by glial cells or pyramidal neurons (Yamamoto et al. 1990; Belluardo et al. 2000; Nagy et al. 2004; Weickert et al. 2005) are the real molecular targets. However, this possibility seems unlikely for the following reasons. First, the complete elimination of electrical coupling between astroglial cells by knocking out both connexin43 and connexin30 has resulted in animals with reduced spatial buffering and enhanced susceptibility to epileptiform activity in vitro (Wallraff et al. 2006). Similarly, reduced spatial buffering was observed in mice lacking connexin32 and connexin47, which is expressed by oligodendrocytes (Menichella et al. 2006; Odermatt et al. 2003). Therefore, blockade of glial gap junctions by carbenoxolone would be predicted to increase, and not to reduce, epileptiform activity. Second, our experiments were performed in the presence of ionotropic glutamate receptor antagonists. These experimental conditions should severely limit the impact of pyramidal cell activity in the network. In fact, epileptiform activity under our experimental conditions appeared fundamentally dependent on depolarizing GABAA receptor mediated signalling (see Fig. 4, Kantrowitz et al. 2005, Zsiros et al. 2007, and the review by Avoli et al. 2002). Third, epileptiform activity was not changed by a different gap junction blocker, mefloquine, which has been reported to lack non-specific effects on GABAergic synaptic transmission (Cruikshank et al. 2004). In conclusion, the most parsimonious explanation for our results is that the antiepileptic effect of carbenoxolone on GABAergic currents in the 4-AP model of epilepsy in vitro is due to direct reduction of GABAA receptor activity, and not to the blockade of connexin36-dependent electrical coupling between interneurons. However, it needs to be acknowledged that our work was performed on slices obtained from mice. Therefore, the possibility that (when other animal species were used) species-related differences may explain some of the inconsistencies with previously published literature cannot be excluded. As mentioned earlier in the Introduction, some forms of epilepsies are thought to be sustained by depolarizing GABAergic signalling. Our conclusion that blockade of electrical coupling in interneurons does not seem to be the mechanism involved in the antiepileptic effect of carbenoxolone on GABAergic synchronous discharges is therefore important and potentially clinically relevant. We would predict that the development of a new therapeutic agent selectively targeting connexin36 would have little clinical impact in those specific forms of epilepsy.
Direct effect of carbenoxolone on GABAA receptors
Our work indicates that the most likely site of action of carbenoxolone in suppressing GABAergic epileptiform currents is directly located on postsynaptic GABAA receptors. Indeed, in addition to depressing GABAergic events and IPSCs, carbenoxolone was able to reduce direct responses to exogenous GABA application. The degree of block of all these events was roughly similar, suggesting that modulation of GABA release from presynaptic terminals is not a majorly contributing mechanism. These results are consistent with the observation of Tovar et al. (2009) in neuronal cultures, that carbenoxolone reduces the amplitude of autaptic IPSCs. The precise site mediating the effect of carbenoxolone on GABAA receptors, however, remains to be identified. Indeed, despite the relative similarity of both the chemical structures of carbenoxolone and pregnenolone sulfate and their actions on GABAergic epileptiform activity, their effects appeared to summate algebraically, thus indicating that they are likely to act through independent sites. Neither did pharmacological blockade of the allosteric benzodiazepine regulatory site prevent the effect of carbenoxolone, thus making it unlikely that it acts as an inverse agonists at this site. Further studies in artificial expression systems will clarify both the detailed molecular mechanism(s) of action of carbenoxolone and their potential dependence on specific GABAA receptor subunits.
Although GABAA receptor blockade could suppress excitatory depolarizing GABAergic signalling occurring at the epileptic focus, the lack of spatial selectivity of this approach would promote hyperexcitability and possibly epileptiform activity in healthy areas outside the focus. The advantage of our present study was to have addressed the effects of carbenoxolone in a simplified model of network activity, where the contribution of pyramidal cells was drastically reduced because of the pharmacological blockade of ionotropic glutamate receptors and epileptiform activity was essentially driven by GABAA receptors. However, we propose that the antiepileptic effects of carbenoxolone observed in vivo and in vitro (in more complex scenarios: i.e. with ionotropic glutamatergic transmission intact) are due to its lack of specificity and to its coordinated effects on several targets. Several studies have found that carbenoxolone reduces neuronal membrane excitability (Rekling et al. 2000; Rouach et al. 2003; Vessey et al. 2004; Tovar et al. 2009, but see also Schmitz et al. 2001 and Jahromi et al. 2002 for no effects and increased excitability, respectively). In addition carbenoxolone has been shown to reduce glutamatergic transmission by a presynaptic mechanism (Tovar et al. 2009) and to block postsynaptic NMDA receptors directly (Chepkova et al. 2008). These latter effects could compensate the potential hyperexcitability due to GABAA receptor blockade, and maintain the balance between excitation and inhibition below the threshold for epileptiform activity.
In conclusion, the simultaneous effects on cell membrane excitability and glutamatergic and GABAergic synaptic transmission may explain the efficacy of carbenoxolone in preventing pathological activity driven by either glutamatergic or GABAergic inputs.
Epileptiform synchronization in the Cx36 KO mouse
Although several studies have highlighted the importance of connexin36 for electrical coupling between cortical interneurons, their anatomical and functional heterogeneity (Freund & Buzsaki, 1996; Maccaferri & Lacaille, 2003) prevents extending results obtained in a specific subtype to the entire population. Here, we have shown for the first time that interneurons of CA1 hippocampal stratum lacunosum-moleculare require connexin36 for the expression of a detectable electrical coupling. Similarly to what was shown by Deans et al. (2001) in various neocortical interneurons, the lack of connexin36 was associated to an increased membrane input resistance. This predicts a larger voltage response to the same synaptic current, and hence might easily explain the initially unexpected larger amplitude of the epileptiform GABAergic events in Cx36 KO mice.
In addition, connexin36 does not appear critical for the propagation of GABAergic epileptiform currents across interneuronal network of different hippocampal layers, supporting the proposal by Avoli et al. (1996) that propagation of this type of epileptiform activity is critically mediated by transients in the external potassium concentration. This consideration also suggests the possibility that blockade of glial gap junctions by carbenoxolone might actually promote the generation and propagation of GABAergic events by impairing potassium spatial buffering. Nevertheless, this effect is overcome by the direct blocking action on GABA receptors.
Alternative mechanisms
The general conclusion that, under our experimental conditions, connexin36 does not play a significant role for the generation and propagation of GABAergic epileptiform events could imply different detailed scenarios. One possibility is simply that, during GABAergic epileptiform events, cells are already synchronized by potassium transients and their membranes undergo a significant decrease of input resistance. Both conditions would strongly attenuate the propagation of voltage signals via gap junctions, which is prominent when the membrane potentials of coupled cells are different and when membrane input resistance is high (Bennett, 1977). However, alternative mechanisms can also be envisaged. For example, a potentially alternative explanation is that bursting of interneurons during GABAergic epileptiform currents triggers some type of metabotropic signalling that negatively modulates the connexin36 gap junction channels. Connexin36 is known to be phosphorylated in vitro by several kinases (Urschel et al. 2006), which are likely to influence either the functional properties of the gap junction channels or, potentially, their trafficking at the gap junction site in vivo (see Flores et al. 2010 for connexin35, the fish orthologue of connexin36). Unfortunately, a direct experimental test of our hypothesis is challenging because, in the presence of 4-AP, interneurons receive a powerful barrage of synaptic currents that makes the direct determination of the modulation of coupling in paired cells problematic. Future studies addressing the response of connexin36-based gap junctions to various signalling pathways will be needed to clarify this issue.
Conclusions
Our work shows that connexin36 is not critically involved in shaping 4-AP-induced GABAergic epileptiform events in vitro and underscores the importance of a very cautious interpretation of the antiepileptic effects of carbenoxolone. Suppression of epileptiform activity by this drug should not be taken as unequivocal evidence for the involvement of gap junctions.
Acknowledgments
We would like to thank Dr Kwang-Youn Kim and Dr Marco Martina (Northwestern University) for comments on a previous version of the manuscript and discussion. We are especially grateful to Prof. David L. Paul (Harvard University) for his generous gift of the Cx36 KO mice. This work was supported by NIH Grants NS057445 to G.M. and GM37751 and EY014127 to D.L.P.
Author contributions
Conception and design of the experiments: G.M. Collection, analysis and interpretation of the data: M.B. and G.M. Drafting the article or revising it critically for important intellectual content: M.B. and G.M. Both authors approved the final version of the manuscript.
References
- Avoli M. GABA-mediated synchronous potentials and seizure generation. Epilepsia. 1996;37:1035–1042. doi: 10.1111/j.1528-1157.1996.tb01022.x. [DOI] [PubMed] [Google Scholar]
- Avoli M, Barbarosie M, Lücke A, Nagao T, Lopantsev V, Köhling R. Synchronous GABA-mediated potentials and epileptiform discharges in the rat limbic system in vitro. J Neurosci. 1996;16:3912–3924. doi: 10.1523/JNEUROSCI.16-12-03912.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avoli M, D'Antuono, Louvel J, Kohling R, Biagini G, Pumain R, D'Arcangelo G, Tancredi V. Network and pharmacological mechanisms leading to epileptiform synchronization in the limbic system in vitro. Prog Neurobiol. 2002;68:167–207. doi: 10.1016/s0301-0082(02)00077-1. [DOI] [PubMed] [Google Scholar]
- Belelli D, Lambert JJ. Neurosteroids: endogenous regulators of the GABAA receptor. Nat Rev Neurosci. 2005;6:565–575. doi: 10.1038/nrn1703. [DOI] [PubMed] [Google Scholar]
- Bennett MVL. Electrical transmission: a functional analysis and comparison to chemical transmission. In: Kandel EK, editor. Handbook of Physiology, section 1, The Nervous System, vol. I, Cellular Biology of Neurons. Baltimore: Williams & Wilkins; 1977. pp. 357–416. [Google Scholar]
- Bennett MV, Pereda A. Pyramid power: principal cells of the hippocampus unite! Brain Cell Biol. 2006;35:5–11. doi: 10.1007/s11068-006-9004-x. [DOI] [PubMed] [Google Scholar]
- Belluardo N, Mudò G, Trovato-Salinaro A, Le Gurun S, Charollais A, Serre Beinier V, Amato G, Haefliger JA, Meda P, Condorelli DF. Expression of connexin36 in the adult and developing rat brain. Brain Res. 2000;865:121–138. doi: 10.1016/s0006-8993(00)02300-3. [DOI] [PubMed] [Google Scholar]
- Buhl DL, Harris KD, Hormuzdi SG, Monyer H, Buzsáki G. Selective impairment of hippocampal gamma oscillations in connexin-36 knock-out mouse in vivo. J Neurosci. 2003;23:1013–1018. doi: 10.1523/JNEUROSCI.23-03-01013.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chepkova AN, Sergeeva OA, Haas HL. Carbenoxolone impairs LTP and blocks NMDA receptors in murine hippocampus. Neuropharmacology. 2008;55:139–147. doi: 10.1016/j.neuropharm.2008.05.001. [DOI] [PubMed] [Google Scholar]
- Cohen I, Navarro C, Clemenceau S, Baulac M, Miles R. On the origin of interictal activity in human temporal lobe epilepsy in vitro. Science. 2002;298:1418–1421. doi: 10.1126/science.1076510. [DOI] [PubMed] [Google Scholar]
- Cruikshank SJ, Hopperstad M, Younger M, Connors BW, Spray DC, Srinivas M. Potent block of Cx36 and Cx50 gap junction channels by mefloquine. Proc Natl Acad Sci U S A. 2004;101:12364–12369. doi: 10.1073/pnas.0402044101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Antuono M, Louvel J, Kohling R, Mattia D, Bernasconi A, Olivier A, Turak B, Devaux A, Pumain R, Avoli M. GABAA receptor-dependent synchronization leads to ictogenesis in the human dysplastic cortex. Brain. 2004;127:1626–1640. doi: 10.1093/brain/awh181. [DOI] [PubMed] [Google Scholar]
- Davidson JS, Baumgarten IM. Glycyrrhetinic acid derivatives: a novel class of inhibitors of gap-junctional intercellular communication. Structure-activity relationships. J Pharmacol Exp Ther. 1988;246:1104–1107. [PubMed] [Google Scholar]
- Deans MR, Gibson JR, Sellitto C, Connors BW, Paul DL. Synchronous activity of inhibitory networks in neocortex requires electrical synapses containing connexin36. Neuron. 2001;31:477–485. doi: 10.1016/s0896-6273(01)00373-7. [DOI] [PubMed] [Google Scholar]
- Drummond GB. Reporting ethical matters in The Journal of Physiology: standards and advice. J Physiol. 2009;587:713–719. doi: 10.1113/jphysiol.2008.167387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenman LN, He Y, Fields C, Zorumski CF, Mennerick S. Activation-dependent properties of pregnenolone sulfate inhibition of GABAA receptor-mediated current. J Physiol. 2003;550:679–691. doi: 10.1113/jphysiol.2003.043810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores CE, Cachope R, Nannapaneni S, Ene S, Nairn AC, Pereda AE. Variability of distribution of Ca2+/calmodulin-dependent kinase II at mixed synapses on the mauthner cell: colocalization and association with connexin 35. J Neurosci. 2010;30:9488–9499. doi: 10.1523/JNEUROSCI.4466-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freund TF, Buzsaki G. Interneurons of the Hippocampus. Hippocampus. 1996;6:347–470. doi: 10.1002/(SICI)1098-1063(1996)6:4<347::AID-HIPO1>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Fujiwara-Tsukamoto Y, Isomura Y, Imanishi M, Ninomiya T, Tsukada M, Yanagawa Y, Fukai T, Takada M. Prototypic seizure activity driven by mature hippocampal fast-spiking interneurons. J Neurosci. 2010;30:13679–13689. doi: 10.1523/JNEUROSCI.1523-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gajda Z, Gyengesi E, Hermesz E, Ali KS, Szente M. Involvement of gap junctions in the manifestation and control of the duration of seizures in rats in vivo. Epilepsia. 2003;44:1596–1600. doi: 10.1111/j.0013-9580.2003.25803.x. [DOI] [PubMed] [Google Scholar]
- Gajda Z, Szupera Z, Blazso G, Szente M. Quinine, a blocker of neuronal Cx36 chennels, suppresses seizure activity in rat neocortex in vivo. Epilepsia. 2005;46:1581–1591. doi: 10.1111/j.1528-1167.2005.00254.x. [DOI] [PubMed] [Google Scholar]
- Gajda Z, Hermesz E, Gyengesi E, Szupera Z, Szente M. The functional significance of gap junction channels in the epileptogenicity and seizure susceptibility of juvenile rats. Epilepsia. 2006;47:1009–1022. doi: 10.1111/j.1528-1167.2006.00573.x. [DOI] [PubMed] [Google Scholar]
- Galarreta M, Hestrin S. A network of fast-spiking cells in the neocortex connected by electrical synapses. Nature. 1999;402:72–75. doi: 10.1038/47029. [DOI] [PubMed] [Google Scholar]
- Galarreta M, Hestrin S. Electrical synapses between GABA-releasing interneurons. Nat Rev Neurosci. 2001;2:425–433. doi: 10.1038/35077566. [DOI] [PubMed] [Google Scholar]
- Gibson JR, Beierlein M, Connors BW. Two networks of electrically coupled inhibitory neurons in neocortex. Nature. 1999;402:75–79. doi: 10.1038/47035. [DOI] [PubMed] [Google Scholar]
- Gigout S, Louvel J, Kawasaki H, D'Antuono M, Armand V, Kurcewicz I, Olivier A, Laschet J, Turak B, Devaux B, Pumain R, Avoli M. Effects of gap junction blockers on human neocortical synchronization. Neurobiol Dis. 2006;22:496–508. doi: 10.1016/j.nbd.2005.12.011. [DOI] [PubMed] [Google Scholar]
- Glykys J, Dzhala VI, Kuchibhotla KV, Feng G, Kuner T, Augustine G, Bacskai BJ, Staley KJ. Differences in cortical versus subcortical GABAergic signaling: a candidate mechanism of electroclinical uncoupling of neonatal seizures. Neuron. 2009;63:657–672. doi: 10.1016/j.neuron.2009.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Sulser A, Wang J, Motamedi GK, Avoli M, Vicini S, Dzakpasu R. The 4-aminopyridine in vitro epilepsy model analyzed with a perforated multi-electrode array. Neuropharmacology. 2011 doi: 10.1016/j.neuropharm.2010.10.007. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hormuzdi SG, Pais I, LeBeau FE, Towers SK, Rozov A, Buhl EH, Whittington MA, Monyer H. Impaired electrical signaling disrupts gamma frequency oscillations in connexin36-deficient mice. Neuron. 2001;31:487–495. doi: 10.1016/s0896-6273(01)00387-7. [DOI] [PubMed] [Google Scholar]
- Huberfeld G, Wittner L, Clemenceau S, Baulac M, Kaila K, Miles R, Rivera C. Perturbed chloride homeostasis and GABAergic signaling in human temporal lobe epilepsy. J Neurosci. 2007;27:9866–9873. doi: 10.1523/JNEUROSCI.2761-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunkeler W, Möhler H, Pieri L, Polc P, Bonetti EP, Cumin R, Schaffner R, Haefely W. Selective antagonists of benzodiazepines. Nature. 1981;290:514–516. doi: 10.1038/290514a0. [DOI] [PubMed] [Google Scholar]
- Jahromi SS, Wentlandt K, Piran S, Carlen PL. Anticonvulsant actions of gap junctional blockers in an in vitro seizure model. J Neurophysiol. 2002;88:1893–1902. doi: 10.1152/jn.2002.88.4.1893. [DOI] [PubMed] [Google Scholar]
- Juszczak GR, Swiergiel AH. Properties of gap junction blockers and their behavioural, cognitive and electrophysiological effects: animal and human studies. Prog Neuropsychopharmacol Biol Psychiatry. 2009;33:181–198. doi: 10.1016/j.pnpbp.2008.12.014. [DOI] [PubMed] [Google Scholar]
- Kantrowitz JT, Francis NN, Salah A, Perkins KL. Synaptic depolarizing GABA response in adults is excitatory and proconvulsive when GABAB receptors are blocked. J Neurophysiol. 2005;93:2656–2667. doi: 10.1152/jn.01026.2004. [DOI] [PubMed] [Google Scholar]
- Kettenmann H, Ransom BR. Electrical coupling between astrocytes and between oligodendrocytes studied in mammalian cell cultures. Glia. 1988;1:64–73. doi: 10.1002/glia.440010108. [DOI] [PubMed] [Google Scholar]
- Klaassen A, Glykys J, Maguire J, Labarca C, Mody I, Boulter J. Seizures and enhanced cortical GABAergic inhibition in two mouse models of human autosomal dominant nocturnal frontal lobe epilepsy. Proc Natl Acad Sci U S A. 2006;103:19152–19157. doi: 10.1073/pnas.0608215103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamsa K, Kaila K. Ionic mechanisms of spontaneous GABAergic events in rat hippocampal slices exposed to 4-aminopyridine. J Neurophysiol. 1997;78:2582–2591. doi: 10.1152/jn.1997.78.5.2582. [DOI] [PubMed] [Google Scholar]
- Lamsa K, Taira T. Use-dependent shift from inhibitory to excitatory GABAA receptor action in SP-O interneurons in the rat hippocampal CA3 area. J Neurophsiol. 2003;90:1983–1995. doi: 10.1152/jn.00060.2003. [DOI] [PubMed] [Google Scholar]
- Maccaferri G, Lacaille JC. Interneuron Diversity series: Hippocampal interneuron classifications – making things as simple as possible, not simpler. Trends Neurosci. 2003;26:564–571. doi: 10.1016/j.tins.2003.08.002. [DOI] [PubMed] [Google Scholar]
- MacVicar BA, Dudek FE. Electrotonic coupling between pyramidal cells: a direct demonstration in rat hippocampal slices. Science. 1981;213:782–785. doi: 10.1126/science.6266013. [DOI] [PubMed] [Google Scholar]
- Maier N, Güldenagel M, Söhl G, Siegmund H, Willecke K, Draguhn A. Reduction of high-frequency network oscillations (ripples) and pathological network discharges in hippocampal slices from connexin36-deficient mice. J Physiol. 2002;541:521–528. doi: 10.1113/jphysiol.2002.017624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majewska MD, Mienville JM, Vicini S. Neurosteroid pregnenolone sulfate antagonizes electrophysiological responses to GABA in neurons. Neurosci Lett. 1988;90:279–284. doi: 10.1016/0304-3940(88)90202-9. [DOI] [PubMed] [Google Scholar]
- Mancilla JG, Lewis TJ, Pinto DJ, Rinzel J, Connors BW. Synchronization of electrically coupled pairs of inhibitory interneurons in neocortex. J Neurosci. 2007;27:2058–2073. doi: 10.1523/JNEUROSCI.2715-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margineanu DG, Klitgaard H. Can gap-junction blockade preferentially inhibit neuronal hypersynchrony vs. excitability? Neuropharmacology. 2001;41:377–383. doi: 10.1016/s0028-3908(01)00080-6. [DOI] [PubMed] [Google Scholar]
- Medina-Ceja L, Cordero-Romero A, Morales-Villagrán A. Antiepileptic effect of carbenoxolone on seizures induced by 4-aminopyridine: a study in the rat hippocampus and entorhinal cortex. Brain Res. 2008;1187:74–81. doi: 10.1016/j.brainres.2007.10.040. [DOI] [PubMed] [Google Scholar]
- Meldrum BS, Rogawski MA. Molecular targets for antiepileptic drug development. Neurotherapeutics. 2007;4:18–61. doi: 10.1016/j.nurt.2006.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meme W, Vandecasteele M, Giaume C, Venance L. Electrical coupling between hippocampal astrocytes in rat brain slices. Neurosci Res. 2009;63:236–243. doi: 10.1016/j.neures.2008.12.008. [DOI] [PubMed] [Google Scholar]
- Menichella DM, Majdan M, Awatramani R, Goodenough DA, Sirkowski E, Scherer SS, Paul DL. Genetic and physiological evidence that oligodendrocyte gap junctions contribute to spatial buffering of potassium released during neuronal activity. J Neurosci. 2006;26:10984–10991. doi: 10.1523/JNEUROSCI.0304-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer A, Bannister AP, Thomson AM. Electrical coupling between pyramidal cells in adult cortical regions. Brain Cell Biol. 2006;35:13–27. doi: 10.1007/s11068-006-9005-9. [DOI] [PubMed] [Google Scholar]
- Mienville JM, Vicini S. Pregnenolone sulfate antagonizes GABAA receptor-mediated currents via a reduction of channel opening frequency. Brain Res. 1989;489:190–194. doi: 10.1016/0006-8993(89)90024-3. [DOI] [PubMed] [Google Scholar]
- Motulsky H. Intuitive Biostatistic. 2nd edn. New York: Oxford University Press; 2010. [Google Scholar]
- Nagy JI, Dudek FE, Rash JE. Update on connexins and gap junctions in neurons and glia in the mammalian nervous system. Brain Res Brain Res Rev. 2004;47:191–215. doi: 10.1016/j.brainresrev.2004.05.005. [DOI] [PubMed] [Google Scholar]
- Neelands TR, Greenfield LJ, Jr, Zhang J, Turner RS, Macdonald RL. GABAA receptor pharmacology and subtype mRNA expression in human neuronal NT2-N cells. J Neurosci. 1998;18:4993–5007. doi: 10.1523/JNEUROSCI.18-13-04993.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odermatt B, Wellershaus K, Wallraff A, Seifert G, Degen J, Euwens C, Fuss B, Büssow H, Schilling K, Steinhäuser C, Willecke K. Connexin 47 (Cx47)-deficient mice with enhanced green fluorescent protein reporter gene reveal predominant oligodendrocytic expression of Cx47 and display vacuolized myelin in the CNS. J Neurosci. 2003;23:4549–4559. doi: 10.1523/JNEUROSCI.23-11-04549.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pais I, Hormuzdi SG, Monyer H, Traub RD, Wood IC, Buhl EH, Whittington MA, LeBeau FE. Sharp wave-like activity in the hippocampus in vitro in mice lacking the gap junction protein connexin36. J Neurophysiol. 2003;89:2046–2054. doi: 10.1152/jn.00549.2002. [DOI] [PubMed] [Google Scholar]
- Park-Chung M, Malayev A, Purdy RH, Gibbs TT, Farb DH. Sulfated and unsulfated steroids modulate γ-aminobutyric acidA receptor function through distinct sites. Brain Res. 1999;830:72–87. doi: 10.1016/s0006-8993(99)01381-5. [DOI] [PubMed] [Google Scholar]
- Perkins KL. GABA application to hippocampal CA3 or CA1 stratum lacunosum-moleculare excites and interneuron network. J Neurophysiol. 2002;87:1404–1414. doi: 10.1152/jn.00430.2001. [DOI] [PubMed] [Google Scholar]
- Perrault P, Avoli M. 4-Aminopyridine-induced epileptiform activity and a GABA-mediated long-lasting depolarization in the rat hippocampus. J Neurosci. 1992;12:104–115. doi: 10.1523/JNEUROSCI.12-01-00104.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price CJ, Cauli B, Kovacs ER, Kulik A, Lambolez B, Shigemoto R, Capogna M. Neurogliaform neurons form a novel inhibitory network in the hippocampal CA1 area. J Neurosci. 2005;25:6775–6786. doi: 10.1523/JNEUROSCI.1135-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross FM, Gwyn P, Spanswick D, Davies SN. Carbenoxolone depresses spontaneous epileptiform activity in the CA1 region of rat hippocampal slices. Neuroscience. 2000;100:789–796. doi: 10.1016/s0306-4522(00)00346-8. [DOI] [PubMed] [Google Scholar]
- Rekling JC, Shao XM, Feldman JL. Electrical coupling and excitatory synaptic transmission between rhythmogenic respiratory neurons in the preBötzinger complex. J Neurosci. 2000;20:RC113. doi: 10.1523/JNEUROSCI.20-23-j0003.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouach N, Segal M, Koulakoff A, Giaume C, Avignone E. Carbenoxolone blockade of neuronal network activity in culture is not mediated by an action on gap junctions. J Physiol. 2003;553:729–745. doi: 10.1113/jphysiol.2003.053439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouach N, Koulakoff A, Abudara V, Willecke K, Giaume C. Astroglial metabolic networks sustain hippocampal synaptic transmission. Science. 2008;322:1551–1551. doi: 10.1126/science.1164022. [DOI] [PubMed] [Google Scholar]
- Rutecki PA, Lebeda FJ, Johnston D. 4-Aminopyridine produces epileptiform activity in hippocampus and enhances synaptic excitation and inhibition. J Neurophysiol. 1987;57:1911–1924. doi: 10.1152/jn.1987.57.6.1911. [DOI] [PubMed] [Google Scholar]
- Schmitz D, Schuchmann S, Fisahn A, Draguhn A, Buhl EH, Petrasch-Parwez E, Dermietzel R, Heinemann U, Traub RD. Axo-axonal coupling. A novel mechanism for ultrafast neuronal communication. Neuron. 2001;31:831–840. doi: 10.1016/s0896-6273(01)00410-x. [DOI] [PubMed] [Google Scholar]
- Shen W, Mennerick S, Covey DF, Zorumski CF. Pregnenolone sulfate modulates inhibitory synaptic transmission by enhancing GABAA receptor desensitization. J Neurosci. 2000;20:3571–3579. doi: 10.1523/JNEUROSCI.20-10-03571.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieghart W. Structure and pharmacology of γ-aminobutyric acidA receptor subtypes. Pharmacol Rev. 1995;47:181–234. [PubMed] [Google Scholar]
- Sinha SR, Saggau P. Imaging of 4-AP-induced GABAA-dependent spontaneous synchronized activity mediated by the hippocampal interneuron network. J Neurophysiol. 2001;86:381–391. doi: 10.1152/jn.2001.86.1.381. [DOI] [PubMed] [Google Scholar]
- Staley KJ. Role of the depolarizing GABA response in epilepsy. Adv Exp Med Biol. 2004;548:104–109. doi: 10.1007/978-1-4757-6376-8_8. [DOI] [PubMed] [Google Scholar]
- Szente M, Pongrácz F. Aminopyridine-induced seizure activity. Electroencephalogr Clin Neurophysiol. 1979;46:605–608. doi: 10.1016/0013-4694(79)90014-2. [DOI] [PubMed] [Google Scholar]
- Szente M, Gajda Z, Said Ali K, Hermesz E. Involvement of electrical coupling in the in vivo ictal epileptiform activity induced by 4-aminopyridine in the neocortex. Neuroscience. 2002;115:1067–1078. doi: 10.1016/s0306-4522(02)00533-x. [DOI] [PubMed] [Google Scholar]
- Tovar KR, Maher BJ, Westbrook GL. Direct actions of carbenoxolone on synaptic transmission and neuronal membrane properties. J Neurophysiol. 2009;102:974–978. doi: 10.1152/jn.00060.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traub RD, Bibbig R, Piechotta A, Draguhn R, Schmitz D. Synaptic and nonsynaptic contributions to giant IPSPs and ectopic spikes induced by 4-aminopyridine in the hippocampus in vitro. J Neurophysiol. 2001;85:1246–1256. doi: 10.1152/jn.2001.85.3.1246. [DOI] [PubMed] [Google Scholar]
- Urschel S, Honer T, Schubert T, Alev C, Sohl G, Worsdorfer P, Asahara T, Dermietzel R, Weiler R, Willecke K. Protein kinase A-mediated phosphorylation of connexin36 in mouse retina results in decreased gap junctional communication between AII amacrine cells. J Biol Chem. 2006;281:33163–33171. doi: 10.1074/jbc.M606396200. [DOI] [PubMed] [Google Scholar]
- Venance L, Rozov A, Blatow M, Burnashev N, Feldmayer D, Monyer H. Connexin expression in electrically coupled postnatal rat brain neurons. Proc Natl Acad Sci U S A. 2000;97:10260–10265. doi: 10.1073/pnas.160037097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vessey JP, Lalonde MR, Mizan HA, Welch NC, Kelly ME, Barnes S. Carbenoxolone inhibitionof voltage-gated Ca channels and synaptic transmission in the retina. J Neurophysiol. 2004;92:1252–1256. doi: 10.1152/jn.00148.2004. [DOI] [PubMed] [Google Scholar]
- Wallraff A, Köhling R, Heinemann U, Theis M, Willecke K, Steinhäuser C. The impact of astrocytic gap junctional coupling on potassium buffering in the hippocampus. J Neurosci. 2006;26:5438–5447. doi: 10.1523/JNEUROSCI.0037-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Barakat A, Zhou H. Electrotonic coupling between pyramidal neurons in the neocortex. PLoS One. 2010;5:e10253. doi: 10.1371/journal.pone.0010253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weickert S, Ray A, Zoidl G, Dermietzel R. Expression of neural connexins and pannexin1 in the hippocampus and inferior olive: a quantitative approach. Brain Res Mol Brain Res. 2005;133:102–109. doi: 10.1016/j.molbrainres.2004.09.026. [DOI] [PubMed] [Google Scholar]
- Yamamoto T, Ochalski A, Hertzberg EL, Nagy JI. On the organization of astrocytic gap junctions in rat brain as suggested by LM and EM immunohistochemistry of connexin43 expression. J Comp Neurol. 1990;302:853–883. doi: 10.1002/cne.903020414. [DOI] [PubMed] [Google Scholar]
- Yang Q, Michelson HB. Gap junctions synchronize the firing of inhibitory interneurons in guinea pig hippocampus. Brain Res. 2001;907:139–143. doi: 10.1016/s0006-8993(01)02582-3. [DOI] [PubMed] [Google Scholar]
- Zaman SH, Shingai R, Harvey RJ, Darlison MG, Barnard EA. Effects of subunit types of the recombinant GABAA receptor on the response to a neurosteroid. Eur J Pharmacol. 1992;225:321–330. doi: 10.1016/0922-4106(92)90106-6. [DOI] [PubMed] [Google Scholar]
- Ziburkus J, Cressman JR, Barreto E, Schiff SJ. Interneuron and pyramidal cell interplay during in vitro seizure-like events. J Neurophysiol. 2006;95:3948–3954. doi: 10.1152/jn.01378.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zsiros V, Maccaferri G. Electrical coupling between interneurons with different excitable properties in stratum lacunosum-moleculare of the juvenile CA1 rat hippocampus. J Neurosci. 2005;25:8686–8695. doi: 10.1523/JNEUROSCI.2810-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zsiros V, Aradi I, Maccaferri G. Propagation of postsynaptic currents and potentials via gap junctions in GABAergic networks of the rat hippocampus. J Physiol. 2007;578:527–544. doi: 10.1113/jphysiol.2006.123463. [DOI] [PMC free article] [PubMed] [Google Scholar]