

Non-technical summary
Following a myocardial infarction, cardiac muscle becomes irreversibly damaged and over time this may lead to heart failure. Strategies to reduce ischaemic damage, enhance new vessel growth, and/or replace damaged heart muscle are currently being investigated. We have identified a novel non-angiogenic role for ephrinA1, a membrane-bound ligand receptor tyrosine kinase, in promoting myocardial tissue salvage after non reperfused myocardial infarction. Treating the heart with this protein at the time of injury reduced infarct size and overall damage, presumably by preventing cardiomyocyte loss after ischaemia. Further studies are in progress to determine the cellular mechanisms by which this occurs and the extent to which adverse remodelling is attenuated.
Abstract
Abstract
The purpose of this study was to investigate the role of intramyocardial administration of chimeric ephrinA1-Fc in modulating the extent of injury and inflammation in non reperfused myocardial infarction (MI). Our results show that intramyocardial injection of 6 μg ephrinA1-Fc into the border zone immediately after permanent coronary artery ligation in B6129s mice resulted in 50% reduction of infarct size, 64% less necrosis, 35% less chamber dilatation and 32% less left ventricular free wall thinning at 4 days post-MI. In the infarct zone, Ly6G+ neutrophil density was 57% reduced and CD45+ leukocyte density was 21% reduced. Myocyte damage was also reduced in ephrinA1-Fc-treated hearts, as evidenced by 54% reduced serum cardiac troponin I. Further, we observed decreased cleaved PARP, increased BAG-1 protein expression, increased phosphorylated AKT/total AKT protein, and reduced NF-κB protein with ephrinA1-Fc administration, indicating improved cellular survival. Of the eight EphA receptors known to be expressed in mice (A1–A8), RT-PCR revealed that A1–A4, A6 and A7 were expressed in the uninjured adult myocardium. Expression of EphA1–A3 and EphA7 were significantly increased following MI while EphA6 expression decreased. Treatment with ephrinA1-Fc further increased EphA1 and EphA2 gene expression and resulted in a 2-fold increase in EphA4. Upregulation and combinatorial activation of these receptors may promote tissue survival. We have identified a novel, beneficial role for ephrinA1-Fc administration at the time of MI, and propose this as a promising new target for infarct salvage in non reperfused MI. More experiments are in progress to identify receptor-expressing cell types as well as the functional implications of receptor activation.
Introduction
The heart lacks an endogenous regenerative capacity sufficient for repair after injury. Consequential left ventricular remodelling after myocardial infarction (MI) leads to left ventricle (LV) dilatation, ultimately leading to heart failure (Pfeffer & Braunwald, 1991; Gaudron et al. 1993; Goldstein et al. 1998; Holmes et al. 2005). To reduce this epidemiological and fiscal burden, it is imperative that strategies be developed to preserve cardiomyocyte survival, subsequently reducing myocardial infarct size, and reducing overall LV remodelling.
Immediately after coronary occlusion, ischaemic myocytes downstream from the occlusion become necrotic and/or undergo apoptosis (Cheng et al. 1996; MacLellan & Schneider, 1997; Freude et al. 1998) or autophagy (Nakai et al. 2007; Dorn & Diwan, 2008; Porrello & Delbridge, 2009). Cardiac troponin I is released, which can be measured in plasma and correlates to the size of injury (Bodor et al. 1995; Chapelle, 1999; Braunwald et al. 2002; Nageh et al. 2003; Oyama & Sisson, 2004; Jaffe, 2005). Neutrophils infiltrate the tissue immediately, while leukocytes, predominantly macrophages, arrive shortly thereafter and participate in digestion of necrotic cellular debris. Neutrophils in the ischaemic tissue can be toxic to the surrounding myocytes because they release reactive oxygen species and proteolytic enzymes which further injure the surrounding myocytes (Lefer & Granger, 2000; Frangogiannis et al. 2002; Frangogiannis, 2008; Lambert et al. 2008; Nah & Rhee, 2009). Once damage occurs, a hypocellular scar forms, leading to contractile dysfunction and heart failure (Fishbein et al. 1978; Frangogiannis et al. 2002; Virag & Murry, 2003; Dorn, 2009).
Since the discovery of the Eph (erythropoietin-producing hepatocellular carcinoma) receptor tyrosine kinase (RTK) in 1987 (Hirai et al. 1987), a great deal of effort has been focused on elucidating Eph RTK and ephrin ligand signalling in the context of numerous pathologies. A distinguishing characteristic of Eph–ephrin interactions is the ability to generate bidirectional signalling. ‘Forward’ signalling occurs in the direction of the receptor-expressing cell, while ‘reverse’ signalling occurs in the direction of the ligand-expressing cell (Bruckner et al. 1997; Mellitzer et al. 1999; Klein, 2001; Kullander & Klein, 2002). Upon ligand binding and receptor activation, endocytic internalization of the complex occurs (Pasquale, 2010), leading to downregulation of the protein. Intracellular cascades downstream of Eph/ephrin signalling are involved in cellular survival, growth, differentiation, and motility (Zhou, 1998; Kullander & Klein, 2002; Arvanitis & Davy, 2008; Pasquale, 2008, 2010). The EphA1 receptor has been linked to angiogenesis through endothelial cell migration. Like the ephrinA1 ligand, EphA1 is induced by tumour necrosis factor-α (TNF-α), vascular endothelial growth factor (VEGF) and interleukin-1β (IL-1β), leading to cellular adhesion via integrins and vessel destabilization (Pandey et al. 1995; Cheng et al. 2002a,b; Moon et al. 2007). Similarly, the EphA2 receptor, expressed on endothelial cells, is widely reported as a key player in angiogenesis, particularly in development and cancer (Ogawa et al. 2000; Brantley-Sieders et al. 2004, 2006; Wykosky et al. 2008).
Of the five ephrinA ligands, ephrinA1 is unique in that it is the only ligand which binds all eight EphA receptors known to be expressed in mice. Aside from its predominant characterization as a pro-angiogenic factor in adult mouse tumours, (Easty et al. 1999; Ogawa et al. 2000; Iida et al. 2005), ephrinA1 appears to be involved in inflammation and apoptosis, two very important facets of infarct progression. It was reported in 2006 that Eph receptors are differentially expressed at early and late stages of inflammation (Ivanov & Romanovsky, 2006). For example, at earlier stages of inflammation, EphA2 and ephrinB2 expression is predominantly localized to epithelial and endothelial cells, promoting disruption of the endothelial/epithelial barrier. However, as the inflammatory process progresses, expression of EphA1, EphA3, EphB3 and EphB4 on these cells decreases, allowing infiltrating leukocytes to adhere to endothelial cells by disrupting endothelial/epithelial barriers (Ivanov & Romanovsky, 2006). EphrinA1/EphA receptor expression changes also appear to be involved in regulating pathways involved with apoptosis. In 2006, Muñoz and colleagues reported that EphA4-deficient mice exhibited both defective T cell development and increased numbers of apoptotic cells (Muñoz et al. 2006). These two reports suggest a role for the EphA4 receptor in mediating cell death, and it is reasonable to suspect that activation of this receptor is anti-apoptotic, while inhibition or removal of this receptor is pro-apoptotic.
The present study was designed to characterize the expression of ephrinA1/EphA RTKs in the uninjured adult myocardium in response to ischaemia in non reperfused myocardium, and the role of exogenous ephrinA1 in limiting myocardial infarct injury. Specifically, we tested the hypothesis that intramyocardial administration of ephrinA1-Fc at the time of injury would promote myocyte survival and subsequently reduce infarct size and inflammatory cell infiltrate. Our results indicate a novel and robust cardioprotective role for ephrinA1-Fc in limiting excessive infarct injury in the non reperfused myocardium.
Methods
Ethical approval
All procedures were approved by the East Carolina University Institutional Animal Care and Use Committee and the investigation conforms to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996). The authors have read and complied with the guidelines in ‘Reporting ethical matters in The Journal of Physiology: standards and advice’ (Drummond, 2009).
Animals
Six-week-old B6/129S breeder pairs were obtained from Jackson Laboratories (Bar Harbor, ME, USA) to establish an in-house colony (strain no. 101045). Animals were housed in 12 h/12 h light/dark cycle conditions and received food and water ad libitum.
Surgical procedure
Male 8- to 10-week-old mice (22–28 g) were anaesthetized (20 μl g−1 Avertin i.p.), intubated and mechanically ventilated. The left anterior descending coronary artery was permanently ligated using an 8-0 suture. Sham controls, in which the suture was pulled through the heart but not ligated and either chimeric IgG-Fc or ephrinA1-Fc injected, were done to ensure that there was no injury caused by the injection (data not shown). Ischaemia was confirmed by blanching of the myocardium distal to the site of ligation. Immediately following coronary occlusion, using a Hamilton syringe with a sterile 30 gauge needle, animals received a single intramyocardial injection of either 6 μg IgG-Fc (R&D), or 6 μg ephrinA1-Fc (Sigma) resuspended in 6 μl sterile PBS at the peri-infarct zone. Injections occurred within 1 min of coronary ligation. This dose was chosen based on previous studies showing effective doses of intramyocardial injections of Tβ4 (Bock-Marquette et al. 2004) and intraperitoneal injection of ephrinB2-Fc (Mansson-Broberg et al. 2008). Additionally, this dose is within the therapeutic range (for humans) of the maximum recommended therapeutic dose of 0.00001 to 1000 mg (kg body wt)−1 day−1, as defined by the FDA (Contrera et al. 2004). Taking into account heart weight, the potential for efflux of the protein from the heart via the injection site, and that an average mouse left ventricle weighs approximately 150 mg, injecting 6 μg of protein intramyocardially is within this range (approximately 40 mg kg−1). The investigator performing the surgery was blinded as to the treatments, which were randomized by another investigator. Once the animals recovered, they were returned to the vivarium. The surgical procedure is described in more detail elsewhere (Virag & Murry, 2003; Virag et al. 2010).
Four days after surgery mice were given a 0.5 ml i.p. injection of 5-bromodeoxyuridine, (BrdU, 5 mg ml−1) to label proliferating endothelial cells, and anaesthetized 1 h later with an i.p. injection of 0.1 ml pentobarbital (390 mg ml−1) (Virag & Murry, 2003). The heart was arrested in diastole using cold KCl (30 mm), excised, rinsed in PBS, and immersed in zinc fixative with a segment of small intestine (used as a positive control for BrdU+ proliferating cells). Hearts were sectioned transversely into four slices of equal thickness and were processed and embedded in paraffin. Routine histological (haematoxylin and eosin) procedures and immunostaining were performed using 5 μm sections, as described below (Virag & Murry, 2003).
EphrinA1-Fc distribution in the myocardium
To determine the distribution pattern and duration of persistence of ephrinA1-Fc in the non reperfused myocardium, an anti-human IgG-Fc was used to immunolocalize the ephrinA1 chimera in hearts at 30 min, 4 h and 24 h post-injection (n = 3 per group). A representative image (Fig. 1) shows prominent epicardial and transmural staining at 30 min. Light staining was observed in 2 of 3 hearts at 4 h but none was observed at 24 h or 4 days post-injection in saline-injected hearts or in tissues incubated without the primary antibody (data not shown).
Figure 1. EphrinA1-Fc distribution in the infarcted myocardium.
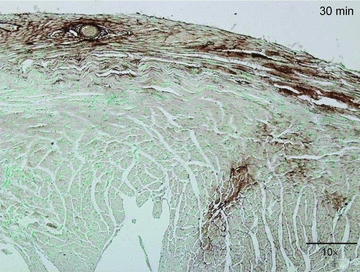
Anti-human IgG-Fc staining to detect exogenous ephrinA1-Fc in the myocardium 30 min after injection. This representative image shows an abundant concentration of ephrinA1-Fc on the epicardial surface, as well as transmural expression of the protein. To a lesser extent, ephrinA1-Fc was also detected 4 h post-injection, but could not be detected 24 h or 4 days post-injection. Scale bar, 200 μm.
Histology and morphometry
Images of four haematoxylin and eosin-stained sections per heart were taken at ×20 magnification using a DP70 digital camera. Two sections of infarct, approximately 1 mm apart (apical and closer to the ligation site) and two sections of base in non-infarcted regions, also 1 mm apart, were used. Scion imaging software (Scion Corporation, Frederick, MD, USA) was used to trace the cross sectional area of the left ventricular wall and chamber, as well as the infarct zone (necrosis + granulation tissue) and necrosis. Measurements from three to four complete, transverse profiles per heart were averaged. Septal and free wall thicknesses were also measured using the average of three radial measures in each of two sections containing infarct. The investigator was blinded as to the treatment while obtaining morphometric measurements. After determining that there was no significant difference between IgG-Fc-treated hearts and standard 4 day-infarcted hearts without any injection, we used 4 day MI hearts for protein and RNA analysis, and this experimental group is labelled as MI rather than IgG-Fc, which was the group used for histological and immunohistochemical analysis.
Immunostaining
Similarly to the histological analysis, two sections of infarct, approximately 1 mm apart (apical and closer to the ligation site) and two sections of base in non-infarcted regions, also 1 mm apart, were used. Tissue sections were deparaffinized in xylene and endogenous peroxidases quenched with 3% H2O2 in methanol. Slides were rinsed in PBS and incubated with anti-ephrinA1 (Zymed), CD45 (PharMingen; 1:2000) for leukocytes, Ly6G (PharMingen) for neutrophils, or CD31 (PharMingen) and anti-BrdU (Roche) for proliferating endothelial cells. Slides were incubated with appropriate biotinylated secondary antibodies and then with Avidin Biotin Complex (Vector Labs PK-6100). The reaction product was visualized with DAB (Vector, SK-4100), counterstained with methyl green, dehydrated in xylene, and slides were coverslipped. For the ephrinA1 staining, a second antibody, anti-ephrinA1 (Santa Cruz) was used to verify consistent staining pattern. Negative controls were performed in the same manner but without a primary antibody. For mast cell staining, slides were sent to Histo-Scientific Research Laboratories (Mount Jackson, VA, USA) for pinacyanol erthrosinate staining to identify mast cells (Murray et al. 2004). Leukocyte, neutrophil and mast cell density was measured in three fields per section of two sections of infarcted heart at ×400. Results were expressed as the number of cells per 0.1 mm2. For proliferating endothelial cells (BrdU++ CD31+), numbers are expressed as a percentage of 1000 endothelial cells (CD31+ only).
Cardiac troponin I (cTnI) measurements
Approximately 50–100 μl of whole blood was collected from mice pre-surgery and at the time of sacrifice by a submandibular bleed, stored in lithium–heparin-coated tubes on a rocker to prevent clotting, and analysed within 30 min of collection on an i-STAT Handheld Clinical Analyzer with cTnI cartridges (Abbott Labs). Values are expressed as nanograms per millilitre.
Protein isolation
Whole left ventricles were snap frozen in liquid nitrogen at the time of collection, and stored at −80°C until use. The whole LV was homogenized in a lysis buffer containing 50 mm Hepes, 10 mm EDTA, 100 mm NaF, 50 mm sodium pyrophosphate, and 1% each of protease and phosphatase inhibitors. Protein was quantified using the Bradford Assay.
Western blotting
Western blotting was performed on a 4–12% gradient Bis-Tris gel (BioRad) in 1× Mops running buffer. Fifty micrograms of sample was loaded per well, and the gel was run for 1 h at 155 V, and transferred for 55 min (for ephrinA1, B-cell lymphoma 2 (Bcl-2)-associated athanogene (BAG-1) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH)), 1 h 20 min (for nuclear factor κB (NF-κB)), or 1 h 30 min (for cleaved poly(ADP-ribose) polymerase (PARP), AKT and pAKT) onto pure nitrocellulose membranes (BioRad). The membrane was incubated with one of the following antibodies: cleaved PARP (89 kDa; Cell Signaling; 1:1000), ephrinA1 (28 kDa; Santa Cruz; 1:100), AKT (Cell Signaling, 1:1000), phospho-AKT (Cell Signaling; 1:2000), NF-κB p65 (Santa Cruz, 1:200) and GAPDH (37 kDa; Millipore; 1:100), followed by appropriate secondary antibodies. EphrinA1 and cleaved PARP were run on the same membrane, which was cut horizontally at 50 kDa, with the bottom half of the membrane used for the ephrinA1 blot and the top half used for the cleaved PARP blot. The ephrinA1 blot was then stripped/reprobed for anti-GAPDH to confirm equal protein loading. All blots were detected with Amersham ECL Advance (GE Healthcare) and imaged on a Typhoon Imager. Densitometry was performed using Image J software and the intensity of each protein was normalized to GAPDH. In the case of pAKT/AKT, the amount of phosphorylated AKT protein was normalized to total AKT.
RNA extraction and real-time RT-PCR
The Trizol method was used for RNA isolation, followed by the Qiagen RNeasy kit for additional purification. cDNA was synthesized using a high capacity cDNA kit. Real-time RT-PCR was conducted on an Applied Biosystems thermocycler. A reaction mixture of 10 μl containing 100 ng RNA was amplified using recommended conditions for TaqMan primers provided by Applied Biosciences. TaqMan primers and probes were obtained from Applied Biosciences (ephrinA1: Mm00438660_m1), EphA1: Mm00445804_m1, EphA2: Mm00438726_m1, EphA3: Mm00580743_m1, EphA4: Mm00433056_m1, EphA5: Mm00433074_m1, EphA6: Mm00433094_m1, EphA7: Mm00833876_m1, GAPDH: Mm99999915_g1). In each experiment, fluorescence data were analysed using the ΔΔCt method. Gene expression was normalized to the housekeeping gene GAPDH. ‘No template controls’ were included in each experiment, and all samples were run in triplicate.
Statistics
Student's t tests were used to test statistical significance between 4 day MI and ephrinA1-Fc-treated MI for RT-PCR, relative infarct size and necrosis. ANOVAs and Student–Newman–Keuls post hoc analyses were used to determine differences between control, 4 day MI and ephrinA1-Fc-treated MI for cTnI, inflammatory cell density, chamber area and left ventricular free wall thickness. The number of hearts analysed for each endpoint and significance levels have been specified for each experiment in the figure legends. Four animals were excluded from all experiments: two from each group, based on suboptimal cTnI and/or overall health of the animals.
Results
EphrinA1-Fc reduces infarct size, necrosis, chamber dilatation and left ventricular free wall thinning
EphrinA1-Fc or IgG-Fc was injected into the border zone of the infarct immediately after coronary ligation. Four days after surgery, tissue was collected and either fixed for histology and immunohistochemistry, or frozen for RNA and protein isolation. Overall survival for this study was 70%, and there was no difference in survival between experimental groups. Histological staining and morphometric analyses (Fig. 2) show that ephrinA1-Fc-treated mice had a 50% reduction in the size of the infarct (expressed as a percentage of the left ventricle), 64% less necrotic area, a 35% reduction in chamber dilatation, and 32% less thinning of the infarcted left ventricular free wall in comparison to IgG-Fc-treated mice. Of note, there was no significant difference in chamber area between uninjured control hearts and those treated with ephrinA1-Fc at day 4 post-MI.
Figure 2. EphrinA1-Fc administration reduces infarct size, chamber dilatation, necrosis, and thinning of the left ventricular free wall.
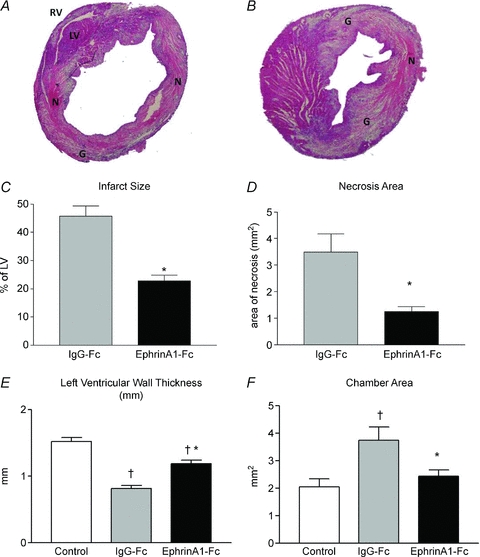
Representative histological images shown are of IgG-Fc-treated (A) and ephrinA1-Fc-treated (B) hearts 4 days post-MI. There was a 50% reduction in infarct size (C), 64% less necrosis (D), 35% less chamber dilatation (E), and 32% less thinning of the left ventricular free wall (F) in ephrinA1-Fc-treated hearts compared to IgG-Fc-treated hearts. n = 7 control, n = 9 IgG-Fc, n = 9 ephrinA1-Fc. P < 0.05: †different from control; *different from IgG-Fc. LV, left ventricle; RV, right ventricle; N, necrosis; G, granulation tissue.
Cardiac troponin I levels reduced with ephrinA1-Fc administration
In the present study, serum cTnI levels were measured prior to surgery and at the time of sacrifice (4 days post-MI) in the same animals. There was an 89% increase in cTnI levels following MI in vehicle-treated hearts. However, cTnI levels in ephrinA1-Fc-treated hearts were 54% lower than those from vehicle-treated animals (Fig. 3A). Interestingly, there was no significant difference between pre-surgery levels and those of ephrinA1-Fc-treated animals 4 days post-surgery.
Figure 3. Intramyocardial ephrinA1-Fc administration reduces tissue injury.
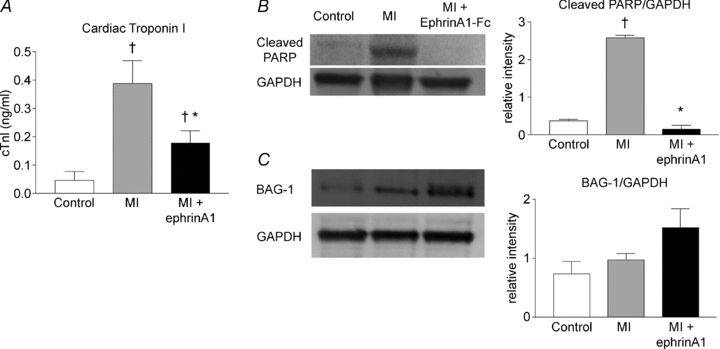
Administration of ephrinA1-Fc resulted in a 54% reduction in cTnI serum levels 4 days after MI (A), (n = 8 control, n = 13 vehicle, n = 11 ephrinA1-Fc). Cleaved PARP expression was reduced with ephrinA1-Fc administration (B). BAG-1 protein (C) increased with ephrinA1-Fc administration by 36% when normalized to GAPDH. P < 0.05: †different from control, *different from MI.
EphrinA1-Fc-treated hearts show diminished cleaved PARP expression and increased BAG-1 expression
In the present study, cleaved PARP, the main target of caspase-3 and an indicator of increased apoptosis (Nicholson et al. 1995; Tewari et al. 1995; Oliver et al. 1998), increased by approximately 88% in response to MI, but diminished with ephrinA1-Fc treatment (Fig. 3B) below control levels. Although we did not observe a change in the level of anti-apoptotic Bcl-2 protein expression with ephrinA1-Fc treatment (data not shown), we did observe a change in Bcl-2-associated athanogene-1 (BAG-1). BAG-1 is a protein that enhances the anti-apoptotic effects of Bcl-2 and has also been identified as a cardioprotective protein through interactions with heat shock proteins (Doong et al. 2002; Townsend et al. 2004). We report here that ephrinA1-Fc administration upregulated the expression of the BAG-1 protein by approximately 54% (Fig. 3C).
EphrinA1-Fc treatment reduces inflammatory cell infiltration to infarcted myocardium
Our results indicate a 57% reduction in neutrophil density (Fig. 4A) and a 21% reduction in leukocyte density in ephrinA1-Fc-treated versus IgG-Fc-treated hearts at 4 days post-MI (Fig. 4B), indicating that ephrinA1-Fc attenuates the inflammatory response. We observed no statistical differences in the numbers of mast cells between ephrinA1-Fc and vehicle-treated hearts, with only a few (1–6 per section of LV) mast cells per heart (data not shown).
Figure 4. EphrinA1-Fc reduces inflammatory cell infiltration.
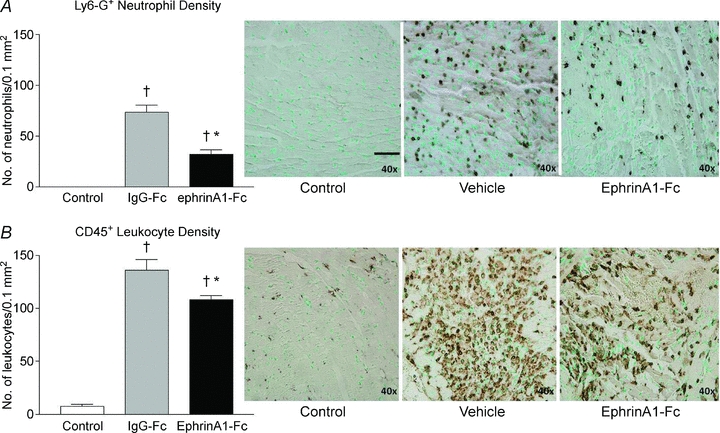
EphrinA1-Fc administration significantly reduced infiltration of neutrophils (A) and leukocytes (B) at 4 days. n = 3 control, n = 9 IgG-Fc, n = 9 ephrinA1-Fc. P < 0.05: †different from control, *different from MI. Representative images of Ly6G+ neutrophil infiltration (top panels) and CD45+ pan-leukocyte infiltration (bottom panels) are shown in control (left), vehicle-treated (middle) and ephrinA1-Fc-treated (right) hearts. Scale bar in A (Control) represents 50 μm.
EphrinA1-Fc treatment does not influence the angiogenic response to MI
No differences were seen in endothelial cell proliferation (5.0 ± 1%vs. 6.1 ± 1.3%; n = 3 vehicle, n = 5 ephrinA1-Fc) or capillary density (111 ± 26.4 vs. 111 ± 26.0 vessels per ×40 high power field, n = 4 per group) between vehicle- and ephrinA1-Fc-treated hearts, respectively.
EphrinA1 and EphA receptor gene expression in response to ephrinA1-Fc treatment
EphrinA1 gene expression was quantified using qRT-PCR. mRNA levels decrease significantly by 35% following MI, and remain unchanged with ephrinA1-Fc treatment (Fig. 5). Of the eight receptors, EphA1, A2, A3 and A7 were all significantly upregulated 4 days after MI (5-fold, 2-fold, 5-fold and 28%, respectively); EphA1 and A2 were further upregulated with ephrinA1-Fc treatment (10-fold and 3-fold, respectively, from control). Despite not changing in response to MI, EphA4 was significantly upregulated 2-fold with ephrinA1-Fc treatment. EphA6 was detected in control hearts, but significantly decreased in response to MI, and expression in the ephrinA1-Fc-treated group was unchanged relative to the untreated MI group (Fig. 5). Ligands ephrinA2–A5 and ephrinB3 (the only B ligand known to bind to an EphA receptor, specifically, EphA4) were also detected in the heart, but their expression did not change in response to MI or ephrinA1-Fc administration (data not shown).
Figure 5. Altered gene expression of ephrinA1 and EphA receptors in response to MI and MI + ephrinA1-Fc.
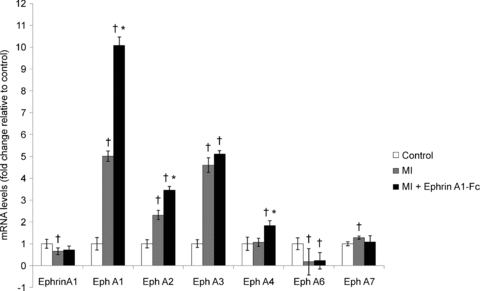
Following MI, ephrinA1 gene expression was significantly reduced (grey bars), and remained relatively unchanged in response to ephrinA1-Fc administration (black bars). Receptors A1, A2, A3 and A7 were significantly upregulated in response to MI, by 5-fold, 2-fold, 5-fold and 28%, respectively, while EphA4 remained unchanged. EphA6 was detected in control hearts but dropped significantly following MI, and expression was not recovered with ephrinA1-Fc administration. In response to ephrinA1-Fc administration, receptors A1 and A2 were further upregulated, by approximately 2-fold each, and A4 was also upregulated by almost 2-fold. Values were calculated using the Ct method, normalized to GAPDH, and presented here as fold changes relative to uninjured control (white bars). n = 8 control, n = 8 MI, n = 8 ephrinA1-Fc. P < 0.05: †different from control, *different from MI.
Endogenous ephrinA1 tissue expression pattern post-MI and in response to ephrinA1-Fc treatment
In uninjured control hearts, endogenous ephrinA1 protein expression appeared to be expressed at a low, basal level on cardiac myocytes throughout the myocardium (Fig. 6A and D). Four days after MI, ephrinA1 protein expression was expressed in cardiomyocytes throughout the uninjured regions of the hearts and was also localized to the spared cardiac myocytes on both the epicardial and endocardial surfaces of the myocardium, at the border zones of the infarct (Fig. 6B and E). In the ephrinA1-Fc-treated hearts at 4 days post-MI, endogenous ephrinA1 protein expression appeared to be localized not only to the cardiomyocytes, but also to infiltrating granulation tissue cells throughout the infarct zone (Fig. 6C and F).
Figure 6. EphrinA1 protein distribution in the myocardium.
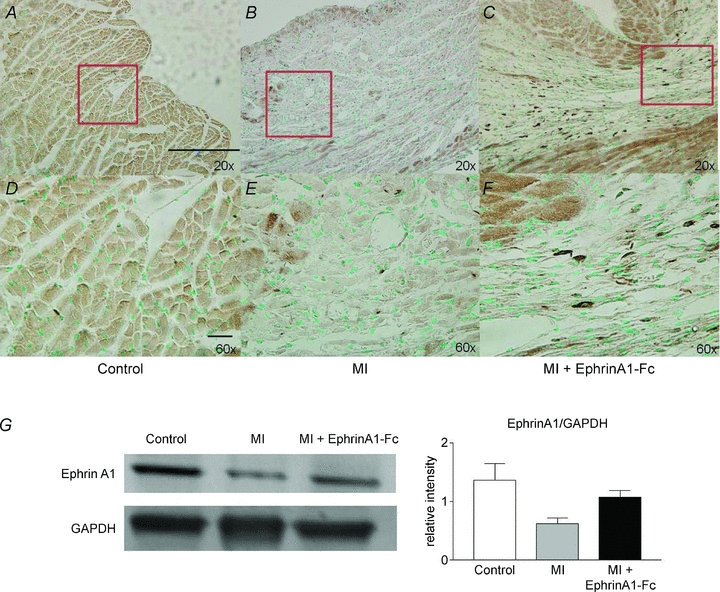
Representative immunostaining for ephrinA1 protein showed a low basal expression of ephrinA1 in cardiomyocytes of control hearts (A and D), intense staining in endo- and epicardial myocytes following 4 days non reperfused MI (B and E), and more intense staining in myocytes as well as numerous granulation tissue cells in the infarct zone following ephrinA1-Fc treatment at 4 days post-MI (C and F). EphrinA1 total protein expression (G) was reduced by 50% in response to MI, but only reduced 36% in response to ephrinA1-Fc administration (normalized to GAPDH). Scale bars: 100 μm in A, 20 μm in D.
EphrinA1 protein expression post-MI and in response to ephrinA1-Fc treatment
Western blotting was used to quantify endogenous ephrinA1 expression. Since anti-IgG-Fc immunostaining (Fig. 1) shows that expression of the chimeric protein was greatly reduced by 4 h post-injection, and completely abolished by 24 h, ephrinA1 protein expression detected at 4 days is only the endogenous protein. In addition, the molecular mass for the chimera is 42 kDa (not observed), vs. 28 kDa for the native protein. Endogenous ephrinA1 protein expression decreased 50% with MI, but was only diminished by approximately 36% with ephrinA1-Fc treatment (Fig. 6G).
EphrinA1-Fc administration increases pAKT/total AKT ratio
Total and phosphorylated AKT protein was measured using Western blotting. While total AKT remained unchanged in the three groups (Control, 4 day MI, and ephrinA1-Fc treated), phosphorylated AKT levels increased with ephrinA1-Fc treatment following MI (Fig. 7A). The pAKT/AKT ratio was significantly higher in the MI group compared to control, and further increased in ephrinA1-Fc-treated hearts compared to both control and MI.
Figure 7. EphrinA1-Fc increases pAKT/AKT.
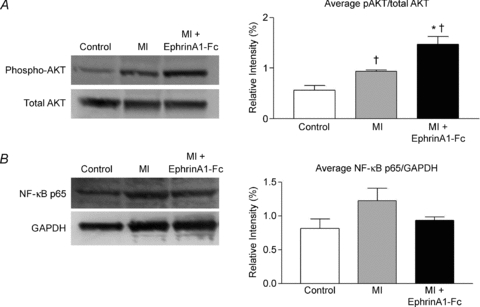
A, representative blot of phosphorylated and total AKT, with the average densitometric analysis of three repeated blots. n = 3 control, n = 3 MI, n = 3 ephrinA1-Fc. P < 0.05: †different from control, *different from MI. B, representative blot of NF-κB p65 protein expression, normalized to GAPDH. n = 3 control, n = 3 MI, n = 3 ephrinA1-Fc.
NF-κB p65 protein reduced with ephrinA1-Fc administration
Protein levels of nuclear factor κ light-chain enhancer of activated B cells (NF-κB) were measured with Western blotting, and a trend for reduced expression of this immune-modulatory protein with ephrinA1-Fc administration was observed (Fig. 7B).
Discussion
We report here that ephrinA1, and several of its receptors, are expressed in the adult myocardium, and their expression profile is altered in response to ischaemia in the non reperfused myocardium. In addition, we have identified a novel and protective role for intramyocardial administration of ephrinA1-Fc at the time of MI, leading to reduced infarct size, necrosis, chamber dilatation, and wall thinning, as well as less inflammatory cell infiltration. A significant decrease in cardiac troponin I levels, coupled with reduced cleaved PARP and increased BAG-1 protein expression, indicates less overall cell death. This is further supported by the fact that ephrinA1-Fc administration leads to increased phosphorylated AKT protein, a known modulator of myocyte survival (Matsui et al. 2003; Latronico et al. 2004; Matsui & Rosenzweig, 2005; Hausenloy & Yellon, 2006; Miyamoto et al. 2009). In addition, we observed reduced levels of NF-κB with administration of ephrinA1-Fc. This protein is a downstream target of AKT and is involved in mediating the inflammatory response (Frangogiannis, 2008).
The heart lacks significant regenerative capacity to overcome myocardial injury. Therefore, there has been much investigation into a number of cell-, gene- and protein-based therapeutic strategies aimed at augmenting the cardiac regenerative potential and promoting tissue salvage (Urbich et al. 2006; Laflamme et al. 2007; Dorn & Diwan, 2008; van Rooij et al. 2008; Abbate et al. 2009; Bartunek et al. 2010; Hwang & Kloner, 2010). To effectively reduce injury and limit the progression of remodelling and dysfunction, it is necessary to reduce inflammation and cell death, and promote revascularization. Optimization of the mode of delivery, timing and bioavailability of proteins and growth factors has been an attractive target for infarct salvage and regeneration. In 2002, Edelberg et al. reported that pre-treatment of an infarcted, non reperfused rat heart with platelet-derived growth factor-AB resulted in a ∼50% reduction in infarct size. However, treatment at the time of coronary occlusion did not alter infarct size (Edelberg et al. 2002). There has also been interest in using the thymosin β4 peptide for myocardial salvage. A 2004 report by Bock-Marquette et al. concluded that thymosin β4 was crucial for myocyte survival, migration and repair (Bock-Marquette et al. 2004). In that study, intramyocardial (400 ng in 10 μl collagen) and intraperitoneal (150 μg in 300 μl PBS) delivery of the thymosin β4 peptide immediately after permanent coronary ligation in mice reduced infarct volume by 50% and improved contractile performance. The authors identified AKT activation as a potential mechanism of Tβ4-mediated protection. More work is needed to identify the factor(s) and their mechanisms of action that will promote optimal therapeutic efficacy (Segers & Lee, 2010).
The literature is currently limited in the number of reports involving ephrin and/or Eph expression and signalling in the adult heart. In 2008, Mansson-Broberg and colleagues demonstrated a protective role for ephrinB2/EphB4 signalling in the repair process after MI (Mansson-Broberg et al. 2008). An intraperitoneal injection of 100 μg ephrinB2-Fc 1 week after ligation of the left anterior descending coronary artery in mice resulted in increased capillary density. This study suggests a role for Eph/ephrin signalling in the infarcted heart and its effect on capillary density, but did not investigate how these interactions influence infarct size or cell behaviour, including that of cardiac myocytes, infiltrating inflammatory cells and fibroblasts. Additionally, the authors reported that ephrinB2-Fc treatment of cultured human aortic endothelial cells induced proliferation and that ephrinB2-Fc also induced increased sprouting in murine aortic ring studies, demonstrating a pro-angiogenic role for the EphB4/ephrinB2 signalling cascade (Mansson-Broberg et al. 2008). More recently, it has been proposed that downregulation of angiogenic factors in early injury impairs performance and this can be remediated by administration of an angiogen (Siddiqui et al. 2010). In accordance with this notion, the results of our study demonstrate decreased protein expression of the angiogenic factor ephrinA1 from control levels to 4 day post-MI levels, yet implicate a non-angiogenic role for exogenous chimeric ephrinA1-Fc-induced signalling in the context of acute MI that has not been previously reported. Administration of this chimera ‘rescued’ the loss of ephrinA1 protein expression observed at 4 days. Further, Mansson-Broberg and colleagues reported that Eph receptors A1–A4, A6, B1 and B4, as well as ephrin ligands A1, A2, A5, B2 and B3 were expressed in adult hearts. We have also detected the EphA receptors mentioned above, in addition to the EphA7 receptor. The EphA6 and EphA7 receptors have been implicated in angiogenesis, both are expressed on vascular endothelium (Shaut et al. 2007) and EphA7 is also expressed on mural cells (Stadler et al. 2001), making it an attractive target to modulate vessel integrity via cell adhesion. Pathologically, increased expression of EphA7 correlates with increased severity of disease outcome in glioblastoma patients (Wang et al. 2008). Although we did not observe an angiogenic effect in the present study, long-term studies are underway to examine later time points, since involvement of these receptors could potentially mediate vessel persistence and/or revascularization of the infarcted heart.
As in other RTKs, activation of Eph receptors by their ephrin ligands results in autophosphorylation of the receptors, and endocytic internalization and degradation of the ligand–receptor complex (Pasquale, 2010), which would result in reduced protein expression. In our study, we identified increased mRNA expression of several receptors (A1, A2 and A4) following ephrinA1-Fc administration. This is probably due to a compensatory increase in mRNA following internalization and degradation of the receptors. Of particular interest was the significant upregulation of EphA4 receptor expression following ephrinA1-Fc administration, since expression of this receptor was unaffected by MI alone. EphA4 and EphA1 are both expressed on T cells. EphrinA1 stimulation of EphA4-expressing T cells resulted in cell migration (Aasheim et al. 2005; Holen et al. 2010), so it is possible that activation of this receptor in our model muted the inflammatory response, reducing necrotic debris and tissue damage, with an overall reduced inflammatory cell population at 4 days post-MI. This is further supported by a recent report showing that a small subset of T cells, which express the angiotensin AT2R, are non-cytotoxic compared to other T cells, and their transplantation into the ischaemic myocardium increased expression of the protective cytokine IL-10, thus reducing injury (Curato et al. 2010). EphA4 is also involved in apoptosis. Furne and colleagues reported that removal of the ephrinB3 ligand from EphA4 resulted in caspase-dependent cell death (Furne et al. 2009). We did not see changes in ephrinB3 mRNA expression, but it is plausible that ephrinA1-Fc stimulation of EphA4 in our model reduced, or inhibited, apoptotic cell death. A 2006 study by Muñoz and colleagues showed that EphA4-deficient mice had increased numbers of apoptotic cells, again suggesting a role for EphA4 forward signalling in the inhibition of cellular apoptosis (Muñoz et al. 2006). More studies are needed to determine the cell-specific expression of EphA4 and its level of activation in response to ephrinA1-Fc.
There is evidence that ephrinA reverse signalling results in AKT phosphorylation and inhibition of apoptosis (Holen et al. 2008). Since ephrinA1-Fc administration increased the endogenous protein expression of ephrinA1 in the myocardium (Fig. 6), this may also play a role in the observed protection. The EphA1 and EphA2 receptors have mainly been characterized in the setting of tumour angiogenesis (Wykosky et al. 2005; Wykosky & Debinski, 2008; Giaginis et al. 2010; Chen et al. 2010). Further investigation into the mechanism of salvage afforded by forward signalling by each receptor and reverse signalling by the ligand in addition to the role that activation of these pathways may play in promoting vessel stability and/or angiogenesis in the infarcted heart will be determined in future investigations by in vitro studies and examination of later time points.
In the early stages following myocardial infarction, cardiomyocytes are lost through regulated cell death, or apoptosis, as well as unregulated death or necrosis. Protein expression of cleaved PARP, a marker of cellular apoptosis, was substantially reduced with ephrinA1-Fc administration, as shown in Fig. 3. Self digestion, or autophagy, can also occur in cardiomyocytes during MI, and may be involved in survival mechanisms, as well as cell death (Whelan et al. 2010). Autophagy is activated in hibernating myocardium, an adaptive feature of cardiomyocytes to survive limited flow and depleted oxygen supply (Slezak et al. 2009). In addition to enhancing the anti-apoptotic effects of the Bcl-2 protein (Reed et al. 1996; Tang, 2002), it has been previously reported that the BAG-1 protein can induce autophagy in a rat model of ischaemia–reperfusion by linking heat shock proteins Hsc70/Hsp70 with the proteasome (Gurusamy et al. 2009), leading to cardioprotection. In accordance with these findings, in this study, we observed increased BAG-1 expression coupled with the significant reduction in myocardial injury following ephrinA1-Fc administration. Although we did not observe a change in Bcl-2 protein expression, we hypothesize that BAG-1 expression is leading to increased cellular survival through myocyte autophagy and studies in our laboratory are currently underway to specifically explore the cellular mechanism by which increased BAG-1 expression affords protection in ephrinA1-Fc-treated hearts (Terman & Brunk, 2005).
Cardiac troponin I (cTnI) is a highly sensitive, specific and reliable serum biomarker for cardiac injury in the clinical setting (Chapelle, 1999; Nageh et al. 2003; Oyama & Sisson, 2004), and there is a proportional relationship of the extent of myocardial injury with the measured level of cTnI (Bodor et al. 1995; Braunwald et al. 2002; Jaffe, 2005). In our study, cTnI levels were reduced by approximately 55% (P < 0.05) with ephrinA1-Fc administration. Combined with the reduced cleaved PARP and increased BAG-1 protein expression, these data suggest reduced cardiomyocyte injury as the mechanism for the observed salvage.
Moreover, our data demonstrate that ephrinA1-Fc administration post-infarction leads to phosphorylation of AKT, a protein involved in cellular survival. Our findings are in agreement with similar studies, including a recent paper which showed that administration of nerve growth factor induced neovascularization and improved cardiac function in a permanent coronary occlusion model, which was coincident with a significant increase in phosphorylated AKT 3 days post-MI (Meloni et al 2010). In another study by the same group, inhibition of PI3K signalling led to reduced pAKT/AKT, increased cardiomyocyte apoptosis in vitro and reduced infarct size in mice 14 days following permanent coronary occlusion (Siragusa et al. 2010). Treating swine for 7 days with subcutaneous injections of granulocyte colony-stimulating factor (G-CSF) beginning 24 h after MI led to reduced infarct size, increased VEGF expression, and increased pAKT/AKT (Iwanaga et al. 2004). Clearly, AKT activation is involved in cellular survival and favours cardiac salvage. While several other groups have identified AKT activation as a survival mechanism in the setting of non reperfused MI (Patten & Karas, 2006; Haider et al. 2008; Shujia et al. 2008), the downstream targets for AKT in this setting are not fully understood. It has been proposed that three main methods of AKT cardioprotection involve anti-apoptotic factors, promotion of cell growth, and promotion of survival and improved function of dysfunctional cardiomyocytes (Matsui & Rosenzweig, 2005). Studies are currently underway to investigate this signalling process.
NF-κB is a downstream target of AKT, and we also observed reduced expression of this protein with ephrinA1-Fc administration. Additionally, it has been reported that PARP-1 may serve as a coactivator of NF-κB (Hassa & Hottiger, 2002), which is in agreement with our observation that ephrinA1-Fc administration reduces cleaved PARP expression. Increased expression has been associated with increased cardiac fibrosis and apoptosis (Feuerstein, 1999; Hamid et al. 2011). In heart failure, NF-κB is activated in cardiac myocytes, and its expression was further increased with an angiotensin-converting enzyme inhibitor (ACE) inhibitor (Frantz et al. 2003). Thus, downregulation of this protein in our model appears to be associated with cardioprotection.
In summary, our data provide the first evidence that intramyocardial administration of recombinant ephrinA1-Fc promotes myocardial tissue salvage. Modulating ephrinA1/EphA signalling may play a significant role in governing the repair process following myocardial infarction, and so exogenous ephrinA1-Fc may prove to be an attractive therapeutic target. Although reperfusion has been the clinical standard for post-MI therapy, approximately 25% of these patients still have an infarct size greater than 75% of the ischaemic zone, which is associated with an even greater incidence of mortality and poor outcome (Miura & Miki, 2008). In addition, reperfusion should be initiated within 2 h of the onset of MI for the greatest success in salvaging ischaemic tissue (Milavetz et al. 1998). Thus, reperfusion after MI may not always be feasible, so investigating the time-dependency of ephrinA1-Fc administration may provide new insight into a promising treatment option. Our laboratory is currently working to elucidate the mechanism for the considerable degree of salvage observed by examining the effects of ephrinA1-Fc on cell-specific receptor expression patterns, signalling cascades activated, and isolated cell behaviour and metabolism. Additionally, we are examining the long-term impact that ephrinA1-Fc administration has on remodelling and cardiac function, as well as the timing, frequency and route of administration required to elicit protection.
Acknowledgments
This work was supported by funding from the Division of Research and Graduate Studies at East Carolina University (ECU). The authors wish to thank Anita Coburn and Robin Alligood (ECU Department of Comparative Medicine) for their assistance with blood collection for cTnI measurements. We would also like to acknowledge Dr Robert Carroll for his thorough review of this manuscript. The authors have no conflicts of interest to disclose.
Glossary
Abbreviations
- BAG-1
B-cell lymphoma 2-associated athanogene
- Bcl-2
B-cell lymphoma 2
- BrdU
5-bromodeoxyuridine
- cTnI
cardiac troponin I
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- LV
left ventricle
- MI
myocardial infarction
- PARP
poly(ADP-ribose) polymerase
- RTK
receptor tyrosine kinase
Author contributions
J.L.D. and J.A.I.V. contributed to the conception, design, drafting and revision of the manuscript. J.A.I.V. performed the surgical procedure, and J.L.D. and S.D.K. collected and analysed the data. All authors have read and approved the final manuscript. All experiments were performed in the Department of Physiology, Brody School of Medicine at East Carolina University in Greenville, NC, USA.
References
- Aasheim HC, Delabie J, Finne EF. Ephrin-A1 binding to CD4+ T lymphocytes stimulates migration and induces tyrosine phosphorylation of PYK2. Blood. 2005;105:2869–2876. doi: 10.1182/blood-2004-08-2981. [DOI] [PubMed] [Google Scholar]
- Abbate A, Biondi-Zoccai GG, Van Tassell BW, Baldi A. Cellular preservation therapy in acute myocardial infarction. Am J Physiol Heart Circ Physiol. 2009;296:H563–H565. doi: 10.1152/ajpheart.00066.2009. [DOI] [PubMed] [Google Scholar]
- Arvanitis D, Davy A. Eph/ephrin signaling: networks. Genes Dev. 2008;22:416–429. doi: 10.1101/gad.1630408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartunek J, Vanderheyden M, Hill J, Terzic A. Cells as biologics for cardiac repair in ischaemic heart failure. Heart. 2010;96:792–800. doi: 10.1136/hrt.2007.139394. [DOI] [PubMed] [Google Scholar]
- Bock-Marquette I, Saxena A, White MD, Dimaio JM, Srivastava D. Thymosin beta4 activates integrin-linked kinase and promotes cardiac cell migration, survival and cardiac repair. Nature. 2004;432:466–472. doi: 10.1038/nature03000. [DOI] [PubMed] [Google Scholar]
- Bodor GS, Porterfield D, Voss EM, Smith S, Apple FS. Cardiac troponin-I is not expressed in fetal and healthy or diseased adult human skeletal muscle tissue. Clin Chem. 1995;41:1710–1715. [PubMed] [Google Scholar]
- Brantley-Sieders D, Schmidt S, Parker M, Chen J. Eph receptor tyrosine kinases in tumor and tumor microenvironment. Curr Pharm Des. 2004;10:3431–3442. doi: 10.2174/1381612043383160. [DOI] [PubMed] [Google Scholar]
- Brantley-Sieders DM, Fang WB, Hwang Y, Hicks D, Chen J. Ephrin-A1 facilitates mammary tumor metastasis through an angiogenesis-dependent mechanism mediated by EphA receptor and vascular endothelial growth factor in mice. Cancer Res. 2006;66:10315–10324. doi: 10.1158/0008-5472.CAN-06-1560. [DOI] [PubMed] [Google Scholar]
- Braunwald E, Antman EM, Beasley JW, Califf RM, Cheitlin MD, Hochman JS, Jones RH, Kereiakes D, Kupersmith J, Levin TN, Pepine CJ, Schaeffer JW, Smith EE, 3rd, Steward DE, Theroux P, Gibbons RJ, Alpert JS, Faxon DP, Fuster V, Gregoratos G, Hiratzka LF, Jacobs AK, Smith SC., Jr ACC/AHA guideline update for the management of patients with unstable angina and non-ST-segment elevation myocardial infarction–2002: summary article: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Committee on the Management of Patients With Unstable Angina) Circulation. 2002;106:1893–1900. doi: 10.1161/01.cir.0000037106.76139.53. [DOI] [PubMed] [Google Scholar]
- Bruckner K, Pasquale EB, Klein R. Tyrosine phosphorylation of transmembrane ligands for Eph receptors. Science. 1997;275:1640–1643. doi: 10.1126/science.275.5306.1640. [DOI] [PubMed] [Google Scholar]
- Chapelle JP. Cardiac troponin I and troponin T: recent players in the field of myocardial markers. Clin Chem Lab Med. 1999;37:11–20. doi: 10.1515/CCLM.1999.002. [DOI] [PubMed] [Google Scholar]
- Chen G, Wang Y, Zhou M, Shi H, Yu Z, Zhu Y, Yu F. EphA1 receptor silencing by small interfering RNA has antiangiogenic and antitumor efficacy in hepatocellular carcinoma. Oncol Rep. 2010;23:563–570. [PubMed] [Google Scholar]
- Cheng N, Brantley DM, Chen J. The ephrins and Eph receptors in angiogenesis. Cytokine Growth Factor Rev. 2002a;13:75–85. doi: 10.1016/s1359-6101(01)00031-4. [DOI] [PubMed] [Google Scholar]
- Cheng N, Brantley DM, Liu H, Lin Q, Enriquez M, Gale N, Yancopoulos G, Cerretti DP, Daniel TO, Chen J. Blockade of EphA receptor tyrosine kinase activation inhibits vascular endothelial cell growth factor-induced angiogenesis. Mol Cancer Res. 2002b;1:2–11. [PubMed] [Google Scholar]
- Cheng W, Kajstura J, Nitahara JA, Li B, Reiss K, Liu Y, Clark WA, Krajewski S, Reed JC, Olivetti G, Anversa P. Programmed myocyte cell death affects the viable myocardium after infarction in rats. Exp Cell Res. 1996;226:316–327. doi: 10.1006/excr.1996.0232. [DOI] [PubMed] [Google Scholar]
- Contrera JF, Matthews EJ, Kruhlak NL, Benz RD. Estimating the safe starting dose in phase I clinical trials and no observed effect level based on QSAR modeling of the human maximum recommended daily dose. Regul Toxicol Pharmacol. 2004;40:185–206. doi: 10.1016/j.yrtph.2004.08.004. [DOI] [PubMed] [Google Scholar]
- Curato C, Slavic S, Dong J, Skorska A, Altarche-Xifro W, Miteva K, Kaschina E, Thiel A, Imboden H, Wang J, Steckelings U, Steinhoff G, Unger T, Li J. Identification of noncytotoxic and IL-10-producing CD8+AT2R+ T cell population in response to ischemic heart injury. J Immunol. 2010;185:6286–6293. doi: 10.4049/jimmunol.0903681. [DOI] [PubMed] [Google Scholar]
- Doong H, Vrailas A, Kohn EC. What's in the ‘BAG’?–A functional domain analysis of the BAG-family proteins. Cancer Lett. 2002;188:25–32. doi: 10.1016/s0304-3835(02)00456-1. [DOI] [PubMed] [Google Scholar]
- Dorn GW., 2nd Novel pharmacotherapies to abrogate postinfarction ventricular remodeling. Nat Rev Cardiol. 2009;6:283–291. doi: 10.1038/nrcardio.2009.12. [DOI] [PubMed] [Google Scholar]
- Dorn GW, 2nd, Diwan A. The rationale for cardiomyocyte resuscitation in myocardial salvage. J Mol Med. 2008;86:1085–1095. doi: 10.1007/s00109-008-0362-y. [DOI] [PubMed] [Google Scholar]
- Drummond GB. Reporting ethical matters in The Journal of Physiology: standards and advice. J Physiol. 2009;587:713–719. doi: 10.1113/jphysiol.2008.167387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Easty DJ, Hill SP, Hsu MY, Fallowfield ME, Florenes VA, Herlyn M, Bennett DC. Up-regulation of ephrin-A1 during melanoma progression. Int J Cancer. 1999;84:494–501. doi: 10.1002/(sici)1097-0215(19991022)84:5<494::aid-ijc8>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- Edelberg JM, Lee SH, Kaur M, Tang L, Feirt NM, McCabe S, Bramwell O, Wong SC, Hong MK. Platelet-derived growth factor-AB limits the extent of myocardial infarction in a rat model: feasibility of restoring impaired angiogenic capacity in the aging heart. Circulation. 2002;105:608–613. doi: 10.1161/hc0502.103672. [DOI] [PubMed] [Google Scholar]
- Feuerstein GZ. Apoptosis in cardiac diseases – new opportunities for novel therapeutics for heart diseases. Cardiovasc Drugs Ther. 1999;13:289–294. doi: 10.1023/a:1007735413477. [DOI] [PubMed] [Google Scholar]
- Fishbein MC, Maclean D, Maroko PR. Experimental myocardial infarction in the rat: qualitative and quantitative changes during pathologic evolution. Am J Pathol. 1978;90:57–70. [PMC free article] [PubMed] [Google Scholar]
- Frangogiannis NG. The immune system and cardiac repair. Pharmacol Res. 2008;58:88–111. doi: 10.1016/j.phrs.2008.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frangogiannis NG, Smith CW, Entman ML. The inflammatory response in myocardial infarction. Cardiovasc Res. 2002;53:31–47. doi: 10.1016/s0008-6363(01)00434-5. [DOI] [PubMed] [Google Scholar]
- Frantz S, Fraccarollo D, Wagner H, Behr TM, Jung P, Angermann CE, Ertl G, Bauersachs J. Sustained activation of nuclear factor kappa B and activator protein 1 in chronic heart failure. Cardiovasc Res. 2003;57:749–756. doi: 10.1016/s0008-6363(02)00723-x. [DOI] [PubMed] [Google Scholar]
- Freude B, Masters TN, Kostin S, Robicsek F, Schaper J. Cardiomyocyte apoptosis in acute and chronic conditions. Basic Res Cardiol. 1998;93:85–89. doi: 10.1007/s003950050066. [DOI] [PubMed] [Google Scholar]
- Furne C, Ricard J, Cabrera JR, Pays L, Bethea JR, Mehlen P, Liebl DJ. EphrinB3 is an anti-apoptotic ligand that inhibits the dependence receptor functions of EphA4 receptors during adult neurogenesis. Biochim Biophys Acta. 2009;1793:231–238. doi: 10.1016/j.bbamcr.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudron P, Eilles C, Kugler I, Ertl G. Progressive left ventricular dysfunction and remodeling after myocardial infarction. Potential mechanisms and early predictors. Circulation. 1993;87:755–763. doi: 10.1161/01.cir.87.3.755. [DOI] [PubMed] [Google Scholar]
- Giaginis C, Tsourouflis G, Zizi-Serbetzoglou A, Kouraklis G, Chatzopoulou E, Dimakopoulou K, Theocharis SE. Clinical significance of ephrin (eph)-A1, -A2, -a4, -a5 and -a7 receptors in pancreatic ductal adenocarcinoma. Pathol Oncol Res. 2010;16:267–276. doi: 10.1007/s12253-009-9221-6. [DOI] [PubMed] [Google Scholar]
- Goldstein S, Ali AS, Sabbah H. Ventricular remodeling. Mechanisms and prevention. Cardiol Clin. 1998;16:623–632. vii–viii. doi: 10.1016/s0733-8651(05)70039-4. [DOI] [PubMed] [Google Scholar]
- Gurusamy N, Lekli I, Gorbunov NV, Gherghiceanu M, Popescu LM, Das DK. Cardioprotection by adaptation to ischaemia augments autophagy in association with BAG-1 protein. J Cell Mol Med. 2009;13:373–387. doi: 10.1111/j.1582-4934.2008.00495.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haider H, Jiang S, Idris NM, Ashraf M. IGF-1-overexpressing mesenchymal stem cells accelerate bone marrow stem cell mobilization via paracrine activation of SDF-1α/CXCR4 signaling to promote myocardial repair. Circ Res. 2008;103:1300–1308. doi: 10.1161/CIRCRESAHA.108.186742. [DOI] [PubMed] [Google Scholar]
- Hamid T, Guo SZ, Kingery JR, Xiang X, Dawn B, Prabhu SD. Cardiomyocyte NF-κB p65 promotes adverse remodelling, apoptosis, and endoplasmic reticulum stress in heart failure. Cardiovasc Res. 2011;89:129–138. doi: 10.1093/cvr/cvq274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassa PO, Hottiger MO. The functional role of poly(ADP-ribose)polymerase 1 as novel coactivator of NF-κB in inflammatory disorders. Cell Mol Life Sci. 2002;59:1534–1553. doi: 10.1007/s00018-002-8527-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hausenloy DJ, Yellon DM. Survival kinases in ischemic preconditioning and postconditioning. Cardiovasc Res. 2006;70:240–253. doi: 10.1016/j.cardiores.2006.01.017. [DOI] [PubMed] [Google Scholar]
- Hirai H, Maru Y, Hagiwara K, Nishida J, Takaku F. A novel putative tyrosine kinase receptor encoded by the eph gene. Science. 1987;238:1717–1720. doi: 10.1126/science.2825356. [DOI] [PubMed] [Google Scholar]
- Holen HL, Nustad K, Aasheim HC. Activation of EphA receptors on CD4+CD45RO+ memory cells stimulates migration. J Leukoc Biol. 2010;87:1059–1068. doi: 10.1189/jlb.0709497. [DOI] [PubMed] [Google Scholar]
- Holen HL, Shadidi M, Narvhus K, Kjosnes O, Tierens A, Aasheim HC. Signaling through ephrin-A ligand leads to activation of Src-family kinases, Akt phosphorylation, and inhibition of antigen receptor-induced apoptosis. J Leukoc Biol. 2008;84:1183–1191. doi: 10.1189/jlb.1207829. [DOI] [PubMed] [Google Scholar]
- Holmes JW, Borg TK, Covell JW. Structure and mechanics of healing myocardial infarcts. Annu Rev Biomed Eng. 2005;7:223–253. doi: 10.1146/annurev.bioeng.7.060804.100453. [DOI] [PubMed] [Google Scholar]
- Hwang H, Kloner RA. Improving regenerating potential of the heart after myocardial infarction: factor-based approach. Life Sci. 2010;86:461–472. doi: 10.1016/j.lfs.2010.01.004. [DOI] [PubMed] [Google Scholar]
- Iida H, Honda M, Kawai HF, Yamashita T, Shirota Y, Wang BC, Miao H, Kaneko S. Ephrin-A1 expression contributes to the malignant characteristics of α-fetoprotein producing hepatocellular carcinoma. Gut. 2005;54:843–851. doi: 10.1136/gut.2004.049486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov AI, Romanovsky AA. Putative dual role of ephrin-Eph receptor interactions in inflammation. IUBMB Life. 2006;58:389–394. doi: 10.1080/15216540600756004. [DOI] [PubMed] [Google Scholar]
- Iwanaga K, Takano H, Ohtsuka M, Hasegawa H, Zou Y, Qin Y, Odaka K, Hiroshima K, Tadokoro H, Komuro I. Effects of G-CSF on cardiac remodeling after acute myocardial infarction in swine. Biochem Biophys Res Commun. 2004;325:1353–1359. doi: 10.1016/j.bbrc.2004.10.149. [DOI] [PubMed] [Google Scholar]
- Jaffe AS. Use of biomarkers in the emergency department and chest pain unit. Cardiol Clin. 2005;23:453–465. vi. doi: 10.1016/j.ccl.2005.08.013. [DOI] [PubMed] [Google Scholar]
- Klein R. Excitatory Eph receptors and adhesive ephrin ligands. Curr Opin Cell Biol. 2001;13:196–203. doi: 10.1016/s0955-0674(00)00197-6. [DOI] [PubMed] [Google Scholar]
- Kullander K, Klein R. Mechanisms and functions of Eph and ephrin signalling. Nat Rev Mol Cell Biol. 2002;3:475–486. doi: 10.1038/nrm856. [DOI] [PubMed] [Google Scholar]
- Laflamme MA, Zbinden S, Epstein SE, Murry CE. Cell-based therapy for myocardial ischemia and infarction: pathophysiological mechanisms. Annu Rev Pathol. 2007;2:307–339. doi: 10.1146/annurev.pathol.2.010506.092038. [DOI] [PubMed] [Google Scholar]
- Lambert JM, Lopez EF, Lindsey ML. Macrophage roles following myocardial infarction. Int J Cardiol. 2008;130:147–158. doi: 10.1016/j.ijcard.2008.04.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latronico MV, Costinean S, Lavitrano ML, Peschle C, Condorelli G. Regulation of cell size and contractile function by AKT in cardiomyocytes. Ann N Y Acad Sci. 2004;1015:250–260. doi: 10.1196/annals.1302.021. [DOI] [PubMed] [Google Scholar]
- Lefer DJ, Granger DN. Oxidative stress and cardiac disease. Am J Med. 2000;109:315–323. doi: 10.1016/s0002-9343(00)00467-8. [DOI] [PubMed] [Google Scholar]
- MacLellan WR, Schneider MD. Death by design. Programmed cell death in cardiovascular biology and disease. Circ Res. 1997;81:137–144. doi: 10.1161/01.res.81.2.137. [DOI] [PubMed] [Google Scholar]
- Mansson-Broberg A, Siddiqui AJ, Genander M, Grinnemo KH, Hao X, Andersson AB, Wardell E, Sylven C, Corbascio M. Modulation of ephrinB2 leads to increased angiogenesis in ischemic myocardium and endothelial cell proliferation. Biochem Biophys Res Commun. 2008;373:355–359. doi: 10.1016/j.bbrc.2008.06.036. [DOI] [PubMed] [Google Scholar]
- Matsui T, Nagoshi T, Rosenzweig A. Akt and PI 3-kinase signaling in cardiomyocyte hypertrophy and survival. Cell Cycle. 2003;2:220–223. [PubMed] [Google Scholar]
- Matsui T, Rosenzweig A. Convergent signal transduction pathways controlling cardiomyocyte survival and function: the role of PI 3-kinase and Akt. J Mol Cell Cardiol. 2005;38:63–71. doi: 10.1016/j.yjmcc.2004.11.005. [DOI] [PubMed] [Google Scholar]
- Mellitzer G, Xu Q, Wilkinson DG. Eph receptors and ephrins restrict cell intermingling and communication. Nature. 1999;400:77–81. doi: 10.1038/21907. [DOI] [PubMed] [Google Scholar]
- Meloni M, Caporali A, Graiani G, Lagrasta C, Katare R, Van Linthout S, Spillmann F, Campesi I, Madeddu P, Quaini F, Emanueli C. Nerve growth factor promotes cardiac repair following myocardial infarction. Circ Res. 2010;106:1275–1284. doi: 10.1161/CIRCRESAHA.109.210088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milavetz JJ, Giebel DW, Christian TF, Schwartz RS, Holmes DR, Jr, Gibbons RJ. Time to therapy and salvage in myocardial infarction. J Am Coll Cardiol. 1998;31:1246–1251. doi: 10.1016/s0735-1097(98)00088-6. [DOI] [PubMed] [Google Scholar]
- Miura T, Miki T. Limitation of myocardial infarct size in the clinical setting: current status and challenges in translating animal experiments into clinical therapy. Basic Res Cardiol. 2008;103:501–513. doi: 10.1007/s00395-008-0743-y. [DOI] [PubMed] [Google Scholar]
- Miyamoto S, Murphy AN, Brown JH. Akt mediated mitochondrial protection in the heart: metabolic and survival pathways to the rescue. J Bioenerg Biomembr. 2009;41:169–180. doi: 10.1007/s10863-009-9205-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon JJ, Lee SH, West JL. Synthetic biomimetic hydrogels incorporated with ephrin-A1 for therapeutic angiogenesis. Biomacromolecules. 2007;8:42–49. doi: 10.1021/bm060452p. [DOI] [PubMed] [Google Scholar]
- Muñoz JJ, Alfaro D, García-Ceca J, Alonso-C LM, Jiménez E, Zapata A. Thymic alterations in EphA4-deficient mice. J Immunol. 2006;177:804–813. doi: 10.4049/jimmunol.177.2.804. [DOI] [PubMed] [Google Scholar]
- Murray DB, Gardner JD, Brower GL, Janicki JS. Endothelin-1 mediates cardiac mast cell degranulation, matrix metalloproteinase activation, and myocardial remodeling in rats. Am J Physiol Heart Circ Physiol. 2004;287:H2295–H2299. doi: 10.1152/ajpheart.00048.2004. [DOI] [PubMed] [Google Scholar]
- Nageh T, Sherwood RA, Harris BM, Byrne JA, Thomas MR. Cardiac troponin T and I and creatine kinase-MB as markers of myocardial injury and predictors of outcome following percutaneous coronary intervention. Int J Cardiol. 2003;92:285–293. doi: 10.1016/s0167-5273(03)00105-0. [DOI] [PubMed] [Google Scholar]
- Nah DY, Rhee MY. The inflammatory response and cardiac repair after myocardial infarction. Korean Circ J. 2009;39:393–398. doi: 10.4070/kcj.2009.39.10.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakai A, Yamaguchi O, Takeda T, Higuchi Y, Hikoso S, Taniike M, Omiya S, Mizote I, Matsumura Y, Asahi M, Nishida K, Hori M, Mizushima N, Otsu K. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med. 2007;13:619–624. doi: 10.1038/nm1574. [DOI] [PubMed] [Google Scholar]
- Nicholson DW, Ali A, Thornberry NA, Vaillancourt JP, Ding CK, Gallant M, Gareau Y, Griffin PR, Labelle M, Lazebnik YA, et al. Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature. 1995;376:37–43. doi: 10.1038/376037a0. [DOI] [PubMed] [Google Scholar]
- Ogawa K, Pasqualini R, Lindberg RA, Kain R, Freeman AL, Pasquale EB. The ephrin-A1 ligand and its receptor, EphA2, are expressed during tumor neovascularization. Oncogene. 2000;19:6043–6052. doi: 10.1038/sj.onc.1204004. [DOI] [PubMed] [Google Scholar]
- Oliver FJ, de la Rubia G, Rolli V, Ruiz-Ruiz MC, de Murcia G, Murcia JM. Importance of poly(ADP-ribose) polymerase and its cleavage in apoptosis. Lesson from an uncleavable mutant. J Biol Chem. 1998;273:33533–33539. doi: 10.1074/jbc.273.50.33533. [DOI] [PubMed] [Google Scholar]
- Oyama MA, Sisson DD. Cardiac troponin-I concentration in dogs with cardiac disease. J Vet Intern Med. 2004;18:831–839. doi: 10.1892/0891-6640(2004)18<831:ctcidw>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- Pandey A, Shao H, Marks RM, Polverini PJ, Dixit VM. Role of B61, the ligand for the Eck receptor tyrosine kinase, in TNF-alpha-induced angiogenesis. Science. 1995;268:567–569. doi: 10.1126/science.7536959. [DOI] [PubMed] [Google Scholar]
- Pasquale EB. Eph-ephrin bidirectional signaling in physiology and disease. Cell. 2008;133:38–52. doi: 10.1016/j.cell.2008.03.011. [DOI] [PubMed] [Google Scholar]
- Pasquale EB. Eph receptors and ephrins in cancer: bidirectional signalling and beyond. Nat Rev Cancer. 2010;10:165–180. doi: 10.1038/nrc2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patten RD, Karas RH. Estrogen replacement and cardiomyocyte protection. Trends Cardiovasc Med. 2006;16:69–75. doi: 10.1016/j.tcm.2006.01.002. [DOI] [PubMed] [Google Scholar]
- Pfeffer MA, Braunwald E. Ventricular enlargement following infarction is a modifiable process. Am J Cardiol. 1991;68:127D–131D. doi: 10.1016/0002-9149(91)90270-u. [DOI] [PubMed] [Google Scholar]
- Porrello ER, Delbridge LM. Cardiomyocyte autophagy is regulated by angiotensin II type 1 and type 2 receptors. Autophagy. 2009;5:1215–1216. doi: 10.4161/auto.5.8.10153. [DOI] [PubMed] [Google Scholar]
- Reed JC, Zha H, Aime-Sempe C, Takayama S, Wang HG. Structure-function analysis of Bcl-2 family proteins. Regulators of programmed cell death. Adv Exp Med Biol. 1996;406:99–112. [PubMed] [Google Scholar]
- Segers VF, Lee RT. Protein therapeutics for cardiac regeneration after myocardial infarction. J Cardiovasc Transl Res. 2010;3:469–477. doi: 10.1007/s12265-010-9207-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaut CA, Saneyoshi C, Morgan EA, Knosp WM, Sexton DR, Stadler HS. HOXA13 directly regulates EphA6 and EphA7 expression in the genital tubercle vascular endothelia. Dev Dyn. 2007;236:951–960. doi: 10.1002/dvdy.21077. [DOI] [PubMed] [Google Scholar]
- Shujia J, Haider HK, Idris NM, Lu G, Ashraf M. Stable therapeutic effects of mesenchymal stem cell-based multiple gene delivery for cardiac repair. Cardiovasc Res. 2008;77:525–533. doi: 10.1093/cvr/cvm077. [DOI] [PubMed] [Google Scholar]
- Siddiqui AJ, Fischer H, Widegren U, Grinnemo KH, Hao X, Mansson-Broberg A, Sylvén C, Gustafsson T. Depressed expression of angiogenic growth factors in the subacute phase of myocardial ischemia: a mechanism behind the remodeling plateau? Coron Artery Dis. 2010;21:65–71. doi: 10.1097/MCA.0b013e3283349cbb. [DOI] [PubMed] [Google Scholar]
- Siragusa M, Katare R, Meloni M, Damilano F, Hirsch E, Emanueli C, Madeddu P. Involvement of phosphoinositide 3-kinase γ in angiogenesis and healing of experimental myocardial infarction in mice. Circ Res. 2010;106:757–768. doi: 10.1161/CIRCRESAHA.109.207449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slezak J, Tribulova N, Okruhlicova L, Dhingra R, Bajaj A, Freed D, Singal P. Hibernating myocardium: pathophysiology, diagnosis, and treatment. Can J Physiol Pharmacol. 2009;87:252–265. doi: 10.1139/Y09-011. [DOI] [PubMed] [Google Scholar]
- Stadler HS, Higgins KM, Capecchi MR. Loss of Eph-receptor expression correlates with loss of cell adhesion and chondrogenic capacity in Hoxa13 mutant limbs. Development. 2001;128:4177–4188. doi: 10.1242/dev.128.21.4177. [DOI] [PubMed] [Google Scholar]
- Tang SC. BAG-1, an anti-apoptotic tumour marker. IUBMB Life. 2002;53:99–105. doi: 10.1080/15216540211473. [DOI] [PubMed] [Google Scholar]
- Terman A, Brunk UT. Autophagy in cardiac myocyte homeostasis, aging, and pathology. Cardiovasc Res. 2005;68:355–365. doi: 10.1016/j.cardiores.2005.08.014. [DOI] [PubMed] [Google Scholar]
- Tewari M, Quan LT, O'Rourke K, Desnoyers S, Zeng Z, Beidler DR, Poirier GG, Salvesen GS, Dixit VM. Yama/CPP32β, a mammalian homolog of CED-3, is a CrmA-inhibitable protease that cleaves the death substrate poly(ADP-ribose) polymerase. Cell. 1995;81:801–809. doi: 10.1016/0092-8674(95)90541-3. [DOI] [PubMed] [Google Scholar]
- Townsend PA, Cutress RI, Carroll CJ, Lawrence KM, Scarabelli TM, Packham G, Stephanou A, Latchman DS. BAG-1 proteins protect cardiac myocytes from simulated ischemia/reperfusion-induced apoptosis via an alternate mechanism of cell survival independent of the proteasome. J Biol Chem. 2004;279:20723–20728. doi: 10.1074/jbc.M400399200. [DOI] [PubMed] [Google Scholar]
- Urbich C, Rossig L, Dimmeler S. Restoration of cardiac function with progenitor cells. Novartis Found Symp. 2006;274:214–223. Discussion 223–217, 272–216. [PubMed] [Google Scholar]
- van Rooij E, Marshall WS, Olson EN. Toward microRNA-based therapeutics for heart disease: the sense in antisense. Circ Res. 2008;103:919–928. doi: 10.1161/CIRCRESAHA.108.183426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virag JI, Dries JL, Easton PR, Friesland AM, DeAntonio JH, Chintalgattu V, Cozzi E, Lehman BD, Ding JM, Lust RM. Attenuation of myocardial injury in mice with functional deletion of the circadian rhythm gene mPer2. Am J Physiol Heart Circ Physiol. 2010;298:H1088–H1095. doi: 10.1152/ajpheart.01280.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virag JI, Murry CE. Myofibroblast and endothelial cell proliferation during murine myocardial infarct repair. Am J Pathol. 2003;163:2433–2440. doi: 10.1016/S0002-9440(10)63598-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang LF, Fokas E, Juricko J, You A, Rose F, Pagenstecher A, Engenhart-Cabillic R, An HX. Increased expression of EphA7 correlates with adverse outcome in primary and recurrent glioblastoma multiforme patients. BMC Cancer. 2008;8:79. doi: 10.1186/1471-2407-8-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whelan RS, Kaplinskiy V, Kitsis RN. Cell death in the pathogenesis of heart disease: mechanisms and significance. Annu Rev Physiol. 2010;72:19–44. doi: 10.1146/annurev.physiol.010908.163111. [DOI] [PubMed] [Google Scholar]
- Wykosky J, Debinski W. The EphA2 receptor and ephrinA1 ligand in solid tumors: function and therapeutic targeting. Mol Cancer Res. 2008;6:1795–1806. doi: 10.1158/1541-7786.MCR-08-0244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wykosky J, Gibo DM, Stanton C, Debinski W. EphA2 as a novel molecular marker and target in glioblastoma multiforme. Mol Cancer Res. 2005;3:541–551. doi: 10.1158/1541-7786.MCR-05-0056. [DOI] [PubMed] [Google Scholar]
- Wykosky J, Palma E, Gibo DM, Ringler S, Turner CP, Debinski W. Soluble monomeric EphrinA1 is released from tumor cells and is a functional ligand for the EphA2 receptor. Oncogene. 2008;27:7260–7273. doi: 10.1038/onc.2008.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou R. The Eph family receptors and ligands. Pharmacol Ther. 1998;77:151–181. doi: 10.1016/s0163-7258(97)00112-5. [DOI] [PubMed] [Google Scholar]