

Non-technical summary
Muscarinic receptors were recently shown to be modulated by membrane potential. Here, we show that membrane potential alters the binding of agonists in an agonist-specific manner. Moreover, agonist binding results in agonist-specific conformational changes in the muscarinic receptor, as measured by changes in the receptor's response to voltage. Voltage-dependent modulation of muscarinic receptors has important consequences for cellular signalling in excitable tissues and implications for cardiovascular drug development.
Abstract
Abstract
The ability to sense transmembrane voltage is a central feature of many membrane proteins, most notably voltage-gated ion channels. Gating current measurements provide valuable information on protein conformational changes induced by voltage. The recent observation that muscarinic G-protein-coupled receptors (GPCRs) generate gating currents confirms their intrinsic capacity to sense the membrane electrical field. Here, we studied the effect of voltage on agonist activation of M2 muscarinic receptors (M2R) in atrial myocytes and how agonist binding alters M2R gating currents. Membrane depolarization decreased the potency of acetylcholine (ACh), but increased the potency and efficacy of pilocarpine (Pilo), as measured by ACh-activated K+ current, IKACh. Voltage-induced conformational changes in M2R were modified in a ligand-selective manner: ACh reduced gating charge displacement while Pilo increased the amount of charge displaced. Thus, these ligands manifest opposite voltage-dependent IKACh modulation and exert opposite effects on M2R gating charge displacement. Finally, mutations in the putative ligand binding site perturbed the movement of the M2R voltage sensor. Our data suggest that changes in voltage induce conformational changes in the ligand binding site that alter the agonist–receptor interaction in a ligand-dependent manner. Voltage-dependent GPCR modulation has important implications for cellular signalling in excitable tissues. Gating current measurement allows for the tracking of subtle conformational changes in the receptor that accompany agonist binding and changes in membrane voltage.
Introduction
The ability to sense transmembrane potential is an inherent feature of all proteins that harness the energy of the membrane electrical field to perform a cellular function. Voltage-gated ion channels are prototypical proteins that undergo voltage-induced conformational changes that open or close the channel pore. Gating current measurements provide valuable information on conformational changes that precede opening of the pore (Bezanilla, 2000). Recent studies have uncovered voltage regulation of other membrane proteins not previously considered to be voltage sensitive. Membrane voltage was reported to modulate several G-protein-coupled receptors (GPCRs) (Itoh et al. 1992; Ben-Chaim et al. 2003; Martinez-Pinna et al. 2005; Ohana et al. 2006). GPCRs constitute the largest family of signalling proteins in the human genome and are classically activated by external stimuli including photons, hormones, neurotransmitters, odorants and chemokines. The observation that voltage regulates GPCR activity suggests an additional capacity to fine-tune cellular signalling in excitable tissues.
Voltage-dependent GPCR modulation is best characterized in muscarinic receptors where the affinity for acetylcholine (ACh) varies in a voltage-dependent manner. For example, membrane depolarization decreases the affinity of M2 muscarinic receptors (M2R) for ACh, while increasing that of M1 receptors (M1R) (Ben-Chaim et al. 2003). Voltage sensitivity was noted to be a property of the receptor itself and not of the downstream effectors (Ben-Chaim et al. 2003). The intrinsic capacity of muscarinic receptors to ‘sense’ transmembrane potential was confirmed by the recording of gating currents (Ben-Chaim et al. 2006) that reflect reorientation of charges within the receptor in response to changes in voltage. The voltage dependence of charge movement correlated with a calculated measure of the ‘low’-affinity M2R state. These authors concluded that depolarization-induced conformational changes in the receptor reduce coupling to downstream trimeric G-proteins, which promotes the low-affinity receptor state (Ben-Chaim et al. 2006; Parnas & Parnas, 2007). The rapid kinetics of gating charge displacement suggests that dynamic conformational changes in M2R may be induced during the time course of neuronal firing.
In the heart, parasympathetic (vagal) stimulation releases ACh which binds to M2R to trigger G-protein-mediated activation of the acetylcholine-activated K+ current (IKACh). Cardiac KACh channels are heteromultimers composed of two homologous G-protein inward rectifier K+ channel subunits, Kir 3.1 and Kir 3.4 (Corey et al. 1998). These subunits are highly expressed in sinoatrial (SA) node, atrium, atrioventricular node and purkinje fibres; however, the primary effect of ACh is exerted at the level of the SA node and atrium. As such, M2R activation of IKACh plays a crucial role in regulating heart rate variability and vulnerability to atrial arrhythmias (Wickman et al. 1998; Kovoor et al. 2001).
In this study, we characterized the effect of voltage on M2R activation of IKACh in atrial myocytes and on conformational changes in M2R measured by gating currents. The muscarinic cholinergic agonists ACh and pilocarpine (Pilo) manifested opposite voltage-dependent IKACh modulation and exerted distinct effects on gating charge displacement. Mutations in the putative ligand binding site perturbed the movement of the M2R voltage sensor. The observation that the affinity and/or efficacy of muscarinic agonists are differentially modulated by voltage has important implications for the design of therapeutic agents that target M2R. Analysis of gating currents provides a sensitive measure of receptor conformations induced by voltage and ligand binding that can be exploited to study novel therapeutic pathways.
Methods
Ethical approval
All animal studies were performed in accordance with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996) and after securing approval by the University of Utah and University of Colima Institutional Animal Care and Use Committees. All animal studies conform to the principles of UK regulations, as described in Drummond (2009). The experiments presented in this manuscript involved the isolation of left atrial myocytes from adult cats. The experimental protocol, including the use of sodium pentobarbitone as the anaesthetic agent, was approved by the Institutional Animal Care and Use Committee of the University of Colima. Animals were killed by excision of the heart en bloc while under anaesthesia.
Cell isolation, heterologous expression and molecular biology
Single feline left atrial myocytes were isolated using an enzymatic perfusion protocol as described previously (Navarro-Polanco & Sanchez-Chapula, 1997) and in accordance with the Public Health Service Policy on the Humane Care and Use of Laboratory Animals. Oocytes were isolated, maintained and injected with cRNA as per standard protocols in this lab (Ferrer et al. 2006). For gating current analysis, oocytes were injected with 10 ng M2R cRNA. HEK-293 cells were transiently transfected with M2R (2 μg), Kir 3.1 (0.5 μg), Kir 3.4 (0.5 μg) and green fluorescent protein (GFP, 0.6 μg) using Lipofectamine (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions. Mutations were introduced into M2R using the QuikChange site-directed mutagenesis kit and manufacturer's protocol (Stratagene, La Jolla, CA, USA).
Cut-open oocyte voltage-clamp
Gating currents were measured using a CA-1B amplifier (Dagan Corp, Minneapolis, MN, USA) at room temperature (22–24°C) as described previously (Piper et al. 2003, 2005; Ferrer et al. 2006). Voltage commands were generated using pCLAMP 9.0 software (Molecular Devices, Sunnyvale, CA, USA), a personal computer and a Digidata 1320 interface (Molecular Devices). Signals were digitized at 50 kHz and filtered at 10 kHz with an eight-pole Bessel filter. Microelectrodes were pulled from borosilicate glass capillary tubes to obtain resistances of 0.2 MΩ when filled with 3 m KCl. The extracellular solution consisted of (in mm): 120 NMG (N-methyl-d-glucamine)-MES, 2 Ca-MES, 10 Hepes, pH 7.6; and the intracellular solution was the same but without Ca-MES. Electrical internal access to the oocyte was obtained by permeabilization with 0.1–0.3% saponin for 2 min. Linear leak and capacitance currents were compensated by analog circuitry and subtracted on-line using a P/8 protocol from a holding potential (Vh) of +60 mV. In preliminary experiments, no differences in gating charge displacement were observed using a Vh of +60 or +80 mV. In general, a P/8 protocol from a Vh of +60 mV was better tolerated by the oocytes. For the W99A M2R mutant, a P/–8 leak subtraction protocol from Vh−120 mV was performed. Qualitatively similar gating currents were recorded in NMG- and TEA-containing solutions, although NMG was most effective at eliminating residual endogenous currents.
IKACh recording in isolated myocytes and transfected cells
Macroscopic currents were recorded in the whole-cell configuration of the patch-clamp technique by means of an Axopatch-200B amplifier (Molecular Devices). All electrophysiological experiments were performed at room temperature (22–24°C) using a rapid perfusion system (Warner Instruments, Hamden, CT, USA) for application and removal of agonists. Data acquisition and command potentials were controlled by pCLAMP 10.0 software (Molecular Devices). Cells were voltage-clamped to a negative (−100 mV) and positive (+50 mV) holding potential, resulting in inward and outward currents, respectively. Patch pipettes with a resistance of 1.5–3 MΩ were made from borosilicate capillary glass (WPI, Sarasota, FL, USA). The bath was grounded using an agar–KCl bridge. The external solution contained (in mm): 120 NaCl, 20 KCl, 1.0 MgCl2, 10 Hepes-Na+, 0.5 CaCl2, 2.0 CoCl2 and 11 glucose; pH 7.4. The internal solution contained (in mm): 80 potassium aspartate, 10 KH2PO4, 1.0 MgSO4, 20 KCl, 5 Hepes, 5 K4BAPTA, 0.2 GTP-Na and 3 ATP-Na2; pH 7.25. Recordings from atrial myocytes were performed in the presence of 3 μm E-4031, 50 μm chromanol 293B and 10 μm glibenclamide to block IKr, IKs and IKATP, respectively. External cobalt (2 mm) and internal BAPTA (5 mm) were included to block L-type calcium and calcium-activated currents, respectively.
Sequential concentration–response curves
After obtaining the whole-cell configuration, the cell was clamped to −100 or +50 mV (randomly) and perfused with increasing concentrations of agonist to activate IKACh and a washout period between each concentration. As it is well described, repeated application of muscarinic agonists can result in either decreased current (desensitization) or increased current (potentiation) depending upon the duration of agonist exposure and the experimental protocol (Honjo et al. 1992; Wang & Lipsius, 1995). Using a rapid switcher device, the duration of agonist exposure in our experiments was 8–12 s for low and intermediate agonist concentrations and 4–6 s for highest agonist concentrations (1 mm Pilo and 10 μm ACh), which was sufficiently brief that neither desensitization nor potentiation was observed with repetitive applications of agonist (Supplemental Fig. S1). After assaying several agonist concentrations at a given voltage, the cell was maintained in a resting condition (I = 0) for 2–3 min, before performing the same agonist application procedure at the other testing voltage. In all concentration–response experiments, the IKACh amplitude, measured as the peak response, was normalized to that obtained with a maximal ACh concentration (10 μm).
Single oocyte chemiluminescence assay
Hemagglutinin (HA) tag was inserted into the extracellular portion of the M2R N-terminus. This construct generated gating currents and activated IKACh similar to wild-type (WT) M2R (data not shown). Cell surface expression of HA-tagged M2R WT and various mutants were estimated using the single oocyte chemiluminescence technique as described previously (Chen et al. 2000). Briefly, oocytes were blocked with ND96 and 1% bovine serum albumin, labelled with anti-HA antibody and horseradish peroxidase-conjugated secondary antibody, then washed with ND96 solution. Oocytes were immersed in 80 μl of SuperSignal enzyme-linked immunosorbent assay substrate (Thermo Scientific, Rockford, IL, USA). Relative light units (RLU) were counted for 4 s with an MLX microtitre plate luminometer.
Drugs
ACh and Pilo (Sigma-Aldrich, St Louis, MO, USA) were dissolved directly in the external solution at the desired concentrations and prepared fresh daily. Tertiapin Q and chromanol 293B were obtained from Tocris Bioscience (Ellisville, MO, USA). E-4031, glibenclamide, atropine and all other reagents employed were from Sigma-Aldrich.
Data analysis
Concentration–response curve fitting
Concentration–response curves, performed as the normalized IKAChversus agonist concentration, were fitted according to the Hill equation:
| (1) |
where X represents agonist concentration, Emax the maximal asymptotic value, EC50 the half-maximal effect and nH the Hill coefficient.
Affinity and efficacy estimates of pilocarpine
The comparative method (Barlow et al. 1967) was used to analyse the partial agonist Pilo according to the operational model-fitting methodology described by Leff et al. (1990). This method compares the partial agonist (Pilo) with a reference full agonist (ACh) assayed in the same study tissue (in our case in the same myocyte) such that an estimate of affinity (KA) and efficacy (τ) of Pilo can be obtained. The equation describing drug action through the operational model (Black & Leff, 1983) is given as:
| (2) |
in which KA, τ, Em and n are the equilibrium dissociation constant, efficacy, the maximum possible effect and the slope factor parameter in the receptor system, respectively. For agonist of high efficacy (such as ACh), eqn (2) reduces to an equation of the logistic form:
| (3) |
Em and n were estimated by fitting eqn (3) to the ACh (full agonist) concentration–response curve data. The values of Em and n were used to calculate KA and τ for Pilo using eqn (2) and Pilo concentration–response data.
Gating currents
The voltage dependence of charge movement was determined by integrating IgON or IgOFF at each test potential and plotting the values of Q versus test potential. The resulting data were normalized to the maximum charge moved by fitting the data to a Boltzmann function:
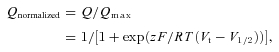 |
(4) |
where Qmax is the maximum charge moved, Vt is the test potential, V1/2 is the half activation voltage, z is the effective valence, F is Faraday's constant, R is the ideal gas constant, and T is temperature in degrees Kelvin.
Statistics
All data are expressed as mean ± SEM. When appropriate, Student's paired or unpaired t tests were used for evaluating statistical difference. Significance was assumed for P < 0.05.
Results
Opposite voltage-dependent activation of atrial IKACh by ACh and Pilo
Consistent with a previous study of heterologously expressed M2R–Kir3.1/3.4 in oocytes (Ben-Chaim et al. 2003), we found that ACh activation of IKACh in single, isolated feline atrial myocytes was modified by membrane voltage (Fig. 1A and B). The EC50 for IKACh activation by ACh was ∼4-fold less when measured at Vh−100 mV compared to +50 mV (EC50 45 ± 10 nm and 176 ± 39 nm, respectively). Unexpectedly, we found the opposite effect of voltage for IKACh activation by the muscarinic agonist (Pilo). The EC50 for IKACh activation by Pilo was 3-fold greater when measured at Vh−100 mV compared to +50 mV (EC50 12 ± 3 μm and 4 ± 1 μm, respectively; Fig. 1A and B). Additionally, membrane potential modified the maximal asymptote of the response induced by Pilo from 0.68 ± 0.04 at −100 mV to 0.84 ± 0.03 at +50 mV, suggesting an increase in the efficacy.
Figure 1. Opposite voltage-dependent effects of acetylcholine (ACh) and pilocarpine (Pilo) on M2R activation of IKACh recorded in atrial myocytes.
A, the effects of increasing concentrations of ACh and Pilo on IKACh recorded from feline atrial myocytes at a holding potential (Vh) of +50 mV (top traces) and −100 mV (bottom traces). Horizontal bars indicate the duration of ligand application. The dashed line indicates the zero current level. B, concentration–response curves for ACh (open symbols) and Pilo (filled symbols) activation of IKACh at Vh+50 mV (circles) and −100 mV (squares). Data were normalized to current elicited by a saturating concentration of ACh (10 μm) and plotted as a function of ligand concentration. The lines represent data fits to a Hill equation. The EC50 and Hill coefficient for ACh were 45 ± 10 nm and 0.8 ± 0.1 (Vh−100 mV) and 176 ± 39 nm and 1.0 ± 0.2 (Vh+50 mV). The EC50 and Hill coefficient for Pilo were 12 ± 3 μm and 0.9 ± 0.2 (Vh−100 mV) and 4 ± 1 μm and 0.8 ± 0.1 (Vh+50 mV). n = 8–10 cells.
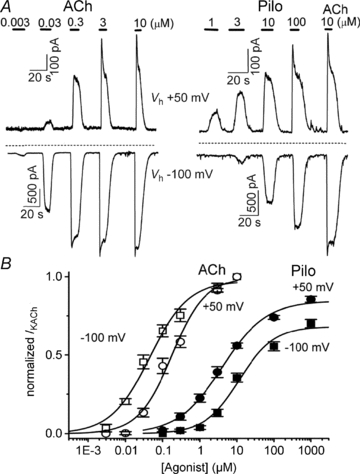
We considered several possible sources of error that could complicate the results obtained above. First, Pilo might activate a cardiac current other than IKACh. However, the experiments were performed in the presence of blockers of L-type calcium current, IKr, IKs and IKATP, as well as chelation of intracellular calcium (see Methods). To further characterize the nature of current activated by Pilo in atrial myocytes, a voltage ramp was applied during brief applications of Pilo and ACh. Pilo (10 and 100 μm) activated an inwardly rectifying K+ current that was similar in magnitude to current activated by ACh (30 nm and 3 μm, Fig. 2). Next, we repeated the experimental protocol in Fig. 1 in a heterologous system. In untransfected HEK-293 cells, no current was activated by the application of ACh or Pilo (data not shown). However, in HEK-293 cells transfected with components of the muscarinic/IKACh pathway (M2R, Kir3.1 and Kir3.4), ACh and Pilo activated current with opposite voltage dependence, similar to that obtained in atrial myocytes (Supplemental Fig. S2).
Figure 2. Inward rectifying nature of IKACh activated by Pilo and ACh in feline atrial myocytes.
IKACh activation by Pilo and ACh at Vh of −100 (A, top panel) and +50 mV (B, top panel). Rapid vertical deflections represent changes in membrane current induced by 50 ms voltage ramps from −120 to + 50 mV. Current–voltage curves of ACh- and Pilo-induced currents at Vh−100 mV (A, bottom) and +50 mV (B, bottom). I–V relationships were determined by subtracting currents recorded in the absence of agonist from currents in the presence of agonist in response to linear voltage ramps from −120 mV to +50 mV. Note that for all I–V curves the reversal potential is close to EK∼−50 mV in external K+ concentration of 20 mm.
Changes in the EC50 induced by voltage may be a consequence of alterations in the affinity and/or the efficacy of the response. Depolarization was previously shown to reduce M2R affinity for ACh using a radio-labelled binding assay (Ben-Chaim et al. 2003). To obtain an estimate of the affinity and efficacy values of Pilo relative to voltage, the data were analysed using the comparative method (Barlow et al. 1967) (see Methods and Supplemental Fig. S3). Average estimates from several individual analyses confirmed a greater efficacy (τ) for Pilo at +50 mV (3.9 ± 0.8) than at −100 mV (2.0 ± 0.3). Moreover, membrane depolarization increased the affinity (KA) of Pilo (28.6 ± 5.4 and 15.2 ± 4.6 μm at −100 and +50 mV, respectively; n = 10). These findings are not compatible with the notion that depolarization favours a low-affinity receptor state by reduced G-protein coupling, as one would expect the affinity for all agonists to decrease with depolarization. Instead, we propose that membrane voltage induces a conformational change within the ligand binding site to influence agonist affinity in a ligand-selective fashion.
ACh and Pilo exert distinct effects on M2R gating charge displacement
To address the disparate ligand-specific M2R voltage dependency, we measured M2R gating charge displacement using the cut-open voltage-clamp technique (COVC) (Stefani & Bezanilla, 1998). Robust gating currents were generated by test voltages of 20 ms duration between −200 mV and +60 mV from a Vh of −70 mV (Fig. 3). Depolarization and hyperpolarization from a Vh of −70 mV elicited gating currents that decayed with single or bi-exponential kinetics, qualitatively similar to that reported by Ben-Chaim et al. (2006). Membrane repolarization to −70 mV elicited return gating currents that also showed bi-exponential kinetics. The complex kinetic behaviour of M2R gating currents implies that the receptor undergoes multiple transitions in response to changes in membrane potential. The integral of charge moved during the test pulse (QON) provides a measure of the charge moved for a given step in voltage. M2R charge displacement occurred over a wide range of voltages and saturated at voltages near −200 and +60 mV (Fig. 3B). The half-point of maximal charge movement (V1/2) was −67 ± 3 mV with a valence (z, the amount of charge moved per receptor) of 0.55 ± 0.02 eo (n = 10). The V1/2 was more negative than that reported (−44 mV) by Ben-Chaim et al. (2006), probably because we assayed charge movement over a wider range of voltages. In voltage-gated ion channels, prolonged depolarization induces conformational changes in the voltage-sensing domain that shift the voltage dependence of charge movement, the so-called gating charge conversion (Olcese et al. 1997). By contrast, varying the Vh did not markedly influence the voltage dependence of gating charge displacement in M2R. The QON–V relationship obtained from a Vh of +60 mV was similar to Vh−70 mV (V1/2−69 mV; z 0.6). Likewise, V1/2 and z were not appreciably altered by measuring the QON–V relationship at Vh of −140 mV and −120 mV (data not shown). Since Vh did not markedly influence M2R gating charge displacement, we selected a Vh of −70 mV to perform all subsequent experiments as the potential was well-tolerated by the cells and allowed us to perform step hyperpolarizations as negative as −200 mV.
Figure 3. M2R gating charge displacement as measured by the cut-open oocyte voltage-clamp technique.
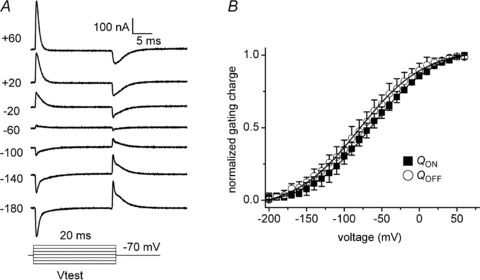
A, representative M2R gating currents elicited by 20 ms voltage steps between −180 and +60 mV from a Vh of −70 mV. Capacitive currents were subtracted using a P/8 subtraction protocol from a Vh of +60 mV. B, the integral of gating currents elicited during the variable voltage pulses (QON) and following return to Vh−70 mV (QOFF) were plotted versus voltage and fitted to a Boltzmann function. For clarity of presentation, the extrapolated charge at the most hyperpolarized potential was assigned the value 0, such that the range of normalized charge (y-axis) was between 0 and 1. The V1/2 and z for the QON–V relationship was −67 ± 3 mV and 0.54 ± 0.02 eo, respectively (n = 10). The V1/2 and z for the QOFF–V relationship was −73 ± 3 mV and 0.53 ± 0.03 eo, respectively (n = 10).
Given the opposite effects of voltage on IKACh activation by ACh and Pilo, we wondered whether these ligands might selectively alter the conformation of the receptor, as measured by gating charge displacement. Indeed, ACh and Pilo exerted disparate effects on M2R gating currents. ACh caused a dose-dependent decrease in M2R gating current magnitude (Figs 4 and 5). Application of 1 μm ACh decreased peak M2R gating to 0.52 ± 0.06 of control values at +60 mV, while 10 μm ACh nearly eliminated M2R gating current (Figs 4 and 5). By contrast, Pilo slowed the kinetics of gating current decay and overall increased the total amount of charge displaced (1.47 ± 0.09-fold increase compared to control values at +60 mV, Fig. 4). The rate of IgON decay in the presence of Pilo was 1.9 ± 0.2 ms at +60 mV, compared to 0.60 ± 0.04 ms in control conditions and 0.89 ± 0.05 ms in ACh.
Figure 4. Opposite effects of ACh and Pilo on M2R gating charge displacement.
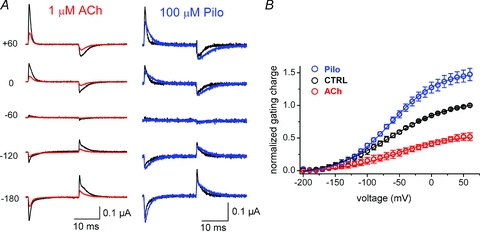
A, effect of ACh and Pilo on M2R gating currents at various membrane potentials. 20 ms step depolarizations or hyperpolarizations were performed from a Vh of −70 mV, followed by return to −70 mV (P/8 leak subtraction at +60 mV). Gating currents in the presence of indicated concentration of agonist (red, ACh; blue, Pilo) are superimposed on control gating currents (black) at the indicated membrane potential. B, M2R QON–V relationships under control conditions (black) and after external application of 100 μm Pilo (blue) or 1 μm ACh (red). n = 4–10 cells. The V1/2 and z for the QON–V relationship during ACh was −55 ± 3mV and 0.58 ± 0.03 eo, respectively, and −63 ± 3 mV and 0.69 ± 0.02 eo, respectively, during Pilo.
Figure 5. Concentration–response for ACh-induced reduction in M2R gating current.
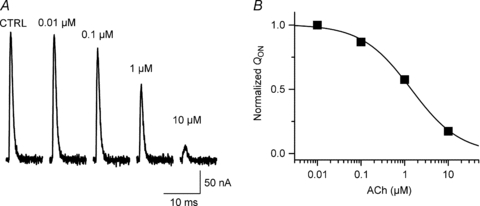
A, effect of indicated ACh concentration on M2R gating currents elicited by 20 ms step to +60 mV from Vh−70 mV, recorded in the cut-open oocyte voltage-clamp system. For clarity, only IgON is shown. B, normalized QONversus ACh concentration. Line represents data fit to Hill equation (concentration that caused 50% reduction in QON= 1.3 μm, Hill coefficient 0.8).
Mutations in the ligand binding site perturb M2R gating charge displacement
Given that membrane voltage influences the potency of specific ligands for M2R and that ligand binding, in turn, influences gating charge displacement, we wondered whether mutations in the putative ligand binding site might also affect gating currents. The agonist binding site of rhodopsin-like GPCRs is located within the outer third of a helical bundle formed by the seven transmembrane (TM) helices (Fig. 6). In muscarinic receptors, the binding pocket is formed by a highly conserved Asp residue (D103) and W99, Y104, S107 in TM3, plus Y403 in TM6 (Lu et al. 2002; Pedretti et al. 2006). Mutation of D103 to Ala (D103A) resulted in receptors that exhibited robust gating currents, but the voltage dependence of gating charge displacement was shifted −67 mV compared to WT M2R (V1/2–134 ± 4 mV, n = 5; Fig. 7A). Y104A and Y403A mutant receptors also shifted the voltage dependence of charge displacement in the hyperpolarizing direction to variable degrees (Fig. 7A). By contrast, W99A caused a +82 mV depolarizing shift in the V1/2 of the QON–V relationship (Fig. 7A) and increased the steepness of the relationship (V1/2=+15 ± 1 mV, z = 0.8 ± 0.02; n = 8). Finally, mutation of the putative binding site residue (S107A) did not appreciably alter the voltage dependence of gating charge displacement (Fig. 7A). In summary, mutation of 4 of 5 residues within the putative binding pocket perturbed M2R charge movement by shifting the voltage dependence of the Q–V relationship, but did not appreciably alter the effective valence (Table 1). Thus, these charge-perturbing residues do not function as the primary voltage sensor, but rather allosterically influence voltage sensor movement.
Figure 6. M2R structure and topology.
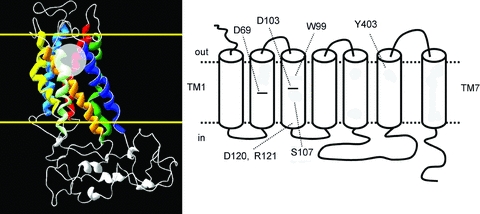
Left, homology model of M2R based on the rhodopsin crystal structure (Pedretti et al. 2006) showing the helical bundle formed by the 7 transmembrane helices. Border of cell membrane is delineated by the yellow lines. Orthosteric binding site is depicted by transparent circle. Right, topology of M2R showing positions of key amino acid residues that were mutated in this study.
Figure 7. Mutations in residues forming the putative ligand binding pocket alter the voltage dependence of M2R gating charge displacement.
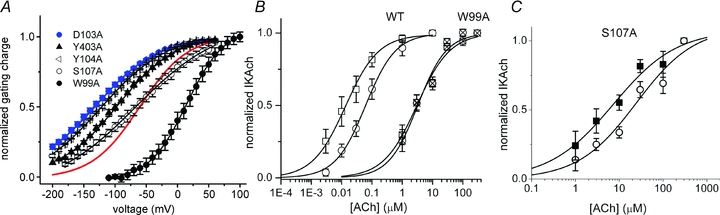
A, QON–V relationships for M2R mutants heterologously expressed in oocytes. Red line denotes WT M2R QON–V relationship. B, concentration–response for IKACh activation in HEK-293 cells heterologously expressing WT or mutant M2R and Kir3.1/3.4 channels. Concentration–response curves for WT M2R-mediated IKACh activation at Vh−100 mV (open squares) and +50 mV (open circles). The EC50 and Hill coefficient were 14 ± 3 nm and 0.8 ± 0.1 (Vh−100 mV) and 62 ± 6 nm and 0.9 ± 0.1 (Vh+50 mV). Concentration–response curves for W99A M2R-mediated IKACh activation at Vh−100 mV (hatched squares) and +30 mV (hatched circles). The EC50 and Hill coefficient were 3.0 ± 0.4 μm and 0.8 ± 0.1 (Vh−100 mV) and 3.2 ± 0.5 μm and 0.9 ± 0.1 (Vh+30 mV). Data points represent mean ± SEM of 4–8 cells. C, the potency of S107 M2R for ACh varies with membrane voltage. Concentration–response curves for S107A M2R-mediated IKACh activation in HEK-293 cells heterologously expressing S107A mutant M2R and Kir3.1/3.4 channels at Vh−100 mV (squares) and +50 mV (circles). The EC50 and Hill coefficient were 5.4 ± 1.0 μm and 0.7 ± 0.2 (Vh−100 mV) and 20.6 ± 4.6 μm and 0.7 ± 0.2 (Vh+50 mV). Data points represent mean ± SEM of 4–7 cells.
Table 1.
Voltage-dependent parameters for Q–V relationships of WT and mutant M2R
| V1/2 (mV) | SEM | z | SEM | n | |
|---|---|---|---|---|---|
| WT | −67.0 | 3.0 | 0.55 | 0.02 | 10 |
| D103A | −133.9 | 4.1 | 0.50 | 0.01 | 5 |
| W99A | +15 | 1 | 0.8 | 0.02 | 8 |
| Y104A | −119.8 | 1.4 | 0.54 | 0.02 | 4 |
| S107A | −66.1 | 8.2 | 0.49 | 0.02 | 4 |
| Y403 | −99.2 | 2.9 | 0.57 | 0.02 | 5 |
| D120N–R121N | −100.0 | 4.1 | 0.52 | 0.04 | 3 |
To determine if alterations in gating charge displacement are associated with changes in the voltage dependence of IKACh activation, we studied the effect of the charge-perturbing mutation W99A on M2R activation of IKACh. In HEK-293 cells heterologously expressing M2R and Kir3.1/3.4 channels, depolarization caused a 4-fold increase in the EC50 of WT M2R activation of IKACh (Supplemental Fig. S2 and Fig. 7B), similar to our findings in native atrial tissue. By contrast, the effect of ACh on W99A M2R activation of IKACh did not vary with depolarization (EC50 3.0 ± 0.4 μm and 3.2 ± 0.5 μm for Vh−100 mV and +30 mV, respectively) (Fig. 7B). Unlike W99A, S107A M2R (a binding pocket mutant that did not alter the Q–V relationship for gating charge displacement) demonstrated a 4-fold decrease in the potency for ACh with depolarization (Fig. 7C). These findings suggest that perturbations in gating charge displacement are accompanied by changes in the voltage dependence of IKACh activation.
Effect of charge neutralization on M2R gating currents and cell surface expression
Unlike the S4 domain of voltage-gated ion channels, there is a paucity of charged amino acids within the M2R TM helices. D69 and D103 constitute the only charged amino acids predicted to lie within the membrane electrical field and both residues are highly conserved across the rhodopsin-like GPCR family. We already excluded D103 as a voltage-sensing residue as neutralization of this amino acid generated gating currents with a similar effective valence as WT (Fig. 7A). Injection of D69A RNA failed to elicit measurable gating currents, even with extreme voltage steps to +200 or −200 mV (data not shown). To determine whether the absence of detectable gating current was a consequence of reduced protein expression, we estimated surface expression of HA epitope-tagged WT and mutant M2R using single oocyte chemiluminescence. The HA epitope was inserted into the M2R extracellular N-terminus (M2R–HA) as described in Methods. The insertion of the HA epitope at this location did not alter M2R gating current properties or IKACh activation (data not shown). The chemiluminescence signal (in RLU) of oocytes expressing WT M2R–HA was approximately 150-fold greater than the signal from H2O-injected oocytes (Fig. 8A). By contrast, the chemiluminescence signal of D69A M2R-HA was only 7-fold greater than that of H2O-injected oocytes (Fig. 8A). The magnitude of D69A M2R–HA chemiluminescence represented only 5% of the signal generated by WT M2R–HA (14 ± 2 × 103versus 310 ± 58 × 103 RLU for D69A and WT, respectively; n = 16 each). Thus, the inability to measure D69A M2R gating current is probably a consequence of markedly reduced protein surface expression.
Figure 8. Membrane surface expression of WT and mutant M2R as measured by oocyte chemiluminescence.
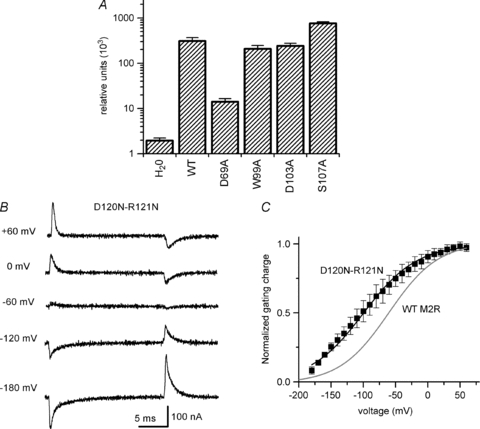
A, relative chemiluminescence of HA-tagged M2R (relative light units) expressed in oocytes. H2O represents signal from water-injected oocytes. The signal of oocytes expressing WT M2R-HA was approximately 150-fold greater than that of water-injected oocytes. The magnitude of D69A M2R-HA chemiluminescence represented only 5% of the signal generated by WT M2R-HA, indicating a marked reduction in mutant receptor cell surface expression. Data represent mean ± SEM, n = 10–16 oocytes. B, representative family of D120N-R121N M2R gating currents elicited by 20 ms voltage steps between −180 and +60 mV from a Vh of −70 mV using a P/–8 leak subtraction protocol at +60 mV. C, QON–V relationship for D120N-R121N M2R:V1/2−100 ± 4 mV, z 0.52 ± 0.04, n = 3. For comparison, the Boltzmann fit of the QON–V relationship for WT M2R is shown as a grey line.
Neutralization of two charged residues located outside the membrane electrical field (D120N–R121N) was reported to abolish M2R gating currents, suggesting that these residues may contribute to the voltage sensor (Ben-Chaim et al. 2006). However, in our hands D120N–R121N M2R generated measurable gating currents (Fig. 8B). While the voltage dependence of gating charge displacement was shifted −33 mV compared to WT M2R (V1/2–100 ± 4 mV, n = 3), the effective valence was similar to WT (z 0.52 ± 0.04, n = 3). Thus, these residues do not function as the principal M2R voltage sensor.
Discussion
GPCRs are classically activated by external stimuli, such as photons, hormones, neurotransmitters and odorants. The recent observation that membrane voltage modulates GPCR activity (Itoh et al. 1992; Ben-Chaim et al. 2003; Martinez-Pinna et al. 2005; Ohana et al. 2006) was surprising and implied an intrinsic capacity to sense membrane potential. Indeed, the inherent ability to sense membrane potential was confirmed by the recording of M2R gating currents (Ben-Chaim et al. 2006), which reflect rearrangement of dipoles within the receptor in response to changes in voltage. In this study, we used gating current measurements to probe conformational changes in the receptor induced by voltage and ligand binding.
Ben-Chaim and colleagues (2003) demonstrated that voltage directly influenced the affinity of M2R for ACh, with depolarization decreasing ACh affinity. Interestingly, M1R and M2R manifest opposite voltage-dependent changes in ACh affinity (Ben-Chaim et al. 2003). Analysis of M1R–M2R chimeric receptors revealed that the intracellular loop between helices V and VI was critical for imparting receptor-specific voltage-dependent ACh affinity (Ben-Chaim et al. 2006). While this finding explains the muscarinic receptor-specific voltage sensitivity, we discovered that a single muscarinic receptor displayed opposite voltage sensitivity in a ligand-specific manner. That is, depolarization decreased the potency of M2R for ACh while increasing that of Pilo (by increasing the affinity and efficacy of the latter), as measured by IKACh in atrial myocytes.
How does voltage induce opposite changes in agonist–receptor interaction (affinity and/or efficacy)? The agonist binding pocket is probably formed by the principal binding residue D103 in TM3, together with a series of aromatic residues in neighbouring helices that form a cage surrounding the ligand (Hibert et al. 1991; Lu et al. 2001; Goodwin et al. 2007). According to classic receptor theory, receptors exist in low- and high-affinity states and the equilibrium between these states is modulated by association with trimeric G-proteins. Thus, voltage-induced conformational changes in the receptor might alter the association with G-proteins, thereby favouring the high-affinity state (Ben-Chaim et al. 2006; Parnas & Parnas, 2007). If true, one would expect that the affinity for ACh and Pilo would vary in the same direction (e.g. hyperpolarization increasing the affinity for both agonists). Based on our findings that voltage exerts opposite effects on M2R affinity for Pilo compared to that found for ACh, we propose that voltage-induced conformational changes are transmitted directly to the agonist binding pocket, independent of coupling to downstream proteins. In this scenario, rearrangements in the voltage sensor induce subtle reorientations of the binding site side chains, thereby favouring binding of one agonist over the other. Alternatively, voltage sensor rearrangements might influence the accessibility of the agonist to the binding site. Either hypothesis is consistent with a recent study demonstrating that agonist-specific voltage sensitivity in the D2 dopamine receptor was related to agonist–receptor interactions and not to downstream effectors (Sahlholm et al. 2008).
Gating current measurement in voltage-gated ion channels has provided important insight into protein conformational changes that precede channel opening (Bezanilla & Stefani, 1998; Bezanilla, 2008). Likewise, analysis of M2R gating currents offers insight into receptor conformational changes induced by voltage and ligand binding. Different ligands produced markedly distinct changes in the magnitude and kinetics of M2R gating charge displacement. Binding of ACh appears to immobilize or ‘lock’ the voltage sensor, while Pilo binding augments translocation of gating charge. It is possible that ACh directly screens a voltage-sensing charge, while Pilo does not. Alternatively, protein rearrangements induced by ACh binding could move voltage-sensing residues outside of the membrane electrical field, restrict the movement of the voltage sensor or alter the local electrical field. Protein rearrangements associated with Pilo binding might recruit voltage-sensing residues into the electrical field or augment their movement across the electrical field. In any case, our data reveal that binding of distinct ligands uniquely alters the conformation of the muscarinic receptor and that gating current measurements offer a window into these conformational changes. The notion that agonists induce distinct receptor conformational changes is supported by fluorescent lifetime experiments in the β2-adrenergic receptor (Ghanouni et al. 2001). There is an increasing consensus that ligands induce unique, ligand-specific receptor conformations that can differentially activate signalling pathways associated with a particular receptor (Kenakin, 2002). Various terminologies have been used to describe this phenomenon: functional selectivity, agonist-directed trafficking and biased agonism. Our data indicate that membrane voltage, per se, is able to evoke distinct receptor conformations to influence not only how tightly an agonist binds to its receptor (affinity), but also the subsequent molecular rearrangements that occur after binding (efficacy). These findings add a new dimension to the phenomenon of functional selectivity, suggesting that the membrane voltage might dynamically modulate a signalling pathway.
This study does not define the fundamental nature of the voltage sensor in GPCRs. GPCRs are unique among voltage-sensing proteins in that robust gating currents are measurable and yet there is a paucity of charged amino acids located within the transmembrane domains. Neutralization of the highly conserved binding site residue D103 did not reduce the effective valence of charge movement. Neutralization of another charged and highly conserved residue (D69A) markedly reduced cell surface protein expression. The absence of measurable gating currents is probably a consequence of decreased protein expression; however, we cannot exclude the possibility that D69 senses voltage. While mutation of two charged residues located outside the membrane electrical field (D120N–R121N) was reported to abolish gating currents (Ben-Chaim et al. 2006), it now appears that these residues do not function as the primary voltage sensor. Mutation of several residues that form the ligand binding pocket influenced movement of the voltage sensor, causing hyper- or depolarizing shifts in the voltage dependence of charge movement, but did not affect the effective valence. Thus, the precise nature of the M2R voltage sensor remains elusive.
In the heart, voltage-dependent M2R modulation has important implications for understanding the role of parasympathetic regulation of basal heart rate. For decades, the contribution of IKACh to the modest chronotropic effects of ‘physiological’ or low-dose ACh has been debated (Shibata et al. 1985; DiFrancesco et al. 1989; Boyett et al. 1995). While low concentrations of ACh exert a negative chronotropic effect, membrane hyperpolarization (as would be expected from IKACh activation) was not observed (Bywater et al. 1989; Campbell et al. 1989). However, subsequent experiments by Boyett and colleagues demonstrated an important role for IKACh in mediating the chronotropic response of low-dose ACh (Boyett et al. 1995). Our experiments and those of Ben-Chaim et al. (2003) imply that low ACh concentrations will exert a greater effect during diastolic (hyperpolarized) membrane potentials, compared to the plateau (depolarized) potentials. These findings predict that weak vagal stimulation preferentially slows the pacemaker rate with minimal effect on action potential duration. Thus, voltage-dependent M2R modulation provides a mechanistic basis to explain the participation of IKACh in the modest chronotropic effects induced by resting vagal tone. Moreover, the observation that the affinity and efficacy of muscarinic agonists are differentially modulated by voltage has important implications for the design of therapeutic agents that target GPCRs in excitable tissues, such as the nervous system and heart.
Acknowledgments
This work was supported by ConsejoNacional de Ciencia y Tecnología, México (CONACyT-0545777 to R.A.N.-P.). We recognize the support of the Nora Eccles Treadwell Foundation. The authors wish to thank Miguel Angel Flores for technical assistance.
Glossary
Abbreviations
- ACh
acetylcholine
- GPCR
G-protein-coupled receptor
- IKACh
acetylcholine-activated K+ current
- M2R
muscarinic receptor type-2
- Pilo
pilocarpine
- TM
transmembrane
Author contributions
R.A.N.-P and E.G.M.-G. contributed equally to this work. R.A.N.-P., E.G.M.-G., J.A.S.-C. and M.T.-F. contributed to the conception and design of the study; R.A.N.-P, E.G.M.-G., T.F.-V. and M.T.-F. performed the experimental work; J.R.R. and M.A. contributed new reagents/analytic tools; R.A.N.-P, E.G.M.-G. and M.T.-F. participated in the analysis of results, interpretation of data and writing of the manuscript. All authors have approved the version to be published.
Supplementary material
Suppl. Figure 1
Suppl. Figure 2
Suppl. Figure 3
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer-reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors
References
- Barlow HB, Scott NC, Stephenson RP. The affinity and efficacy of onium salts on the frog rectus abdominis. Br J Pharmacol Chemother. 1967;31:188–196. doi: 10.1111/j.1476-5381.1967.tb01989.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Chaim Y, Chanda B, Dascal N, Bezanilla F, Parnas I, Parnas H. Movement of ‘gating charge’ is coupled to ligand binding in a G-protein-coupled receptor. Nature. 2006;444:106–109. doi: 10.1038/nature05259. [DOI] [PubMed] [Google Scholar]
- Ben-Chaim Y, Tour O, Dascal N, Parnas I, Parnas H. The M2 muscarinic G-protein-coupled receptor is voltage-sensitive. J Biol Chem. 2003;278:22482–22491. doi: 10.1074/jbc.M301146200. [DOI] [PubMed] [Google Scholar]
- Bezanilla F. The voltage sensor in voltage-dependent ion channels. Physiol Rev. 2000;80:555–592. doi: 10.1152/physrev.2000.80.2.555. [DOI] [PubMed] [Google Scholar]
- Bezanilla F. How membrane proteins sense voltage. Nat Rev. 2008;9:323–332. doi: 10.1038/nrm2376. [DOI] [PubMed] [Google Scholar]
- Bezanilla F, Stefani E. Gating currents. Methods Enzymol. 1998;293:331–352. doi: 10.1016/s0076-6879(98)93022-1. [DOI] [PubMed] [Google Scholar]
- Black JW, Leff P. Operational models of pharmacological agonism. Proc R Soc Lond B Biol Soc. 1983;220:141–162. doi: 10.1098/rspb.1983.0093. [DOI] [PubMed] [Google Scholar]
- Boyett MR, Kodama I, Honjo H, Arai A, Suzuki R, Toyama J. Ionic basis of the chronotropic effect of acetylcholine on the rabbit sinoatrial node. Cardiovasc Res. 1995;29:867–878. [PubMed] [Google Scholar]
- Bywater RA, Campbell G, Edwards FR, Hirst GD, O'Shea JE. The effects of vagal stimulation and applied acetylcholine on the sinus venosus of the toad. J Physiol. 1989;415:35–56. doi: 10.1113/jphysiol.1989.sp017710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell GD, Edwards FR, Hirst GD, O'Shea JE. Effects of vagal stimulation and applied acetylcholine on pacemaker potentials in the guinea-pig heart. J Physiol. 1989;415:57–68. doi: 10.1113/jphysiol.1989.sp017711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Mitcheson JS, Lin M, Sanguinetti MC. Functional roles of charged residues in the putative voltage sensor of the HCN2 pacemaker channel. J Biol Chem. 2000;275:36465–36471. doi: 10.1074/jbc.M007034200. [DOI] [PubMed] [Google Scholar]
- Corey S, Krapivinsky G, Krapivinsky L, Clapham DE. Number and stoichiometry of subunits in the native atrial G-protein-gated K+ channel, IKACh. J Biol Chem. 1998;273:5271–5278. doi: 10.1074/jbc.273.9.5271. [DOI] [PubMed] [Google Scholar]
- DiFrancesco D, Ducouret P, Robinson RB. Muscarinic modulation of cardiac rate at low acetylcholine concentrations. Science. 1989;243:669–671. doi: 10.1126/science.2916119. [DOI] [PubMed] [Google Scholar]
- Drummond GB. Reporting ethical matters in The Journal of Physiology: standards and advice. J Physiol. 2009;587:713–719. doi: 10.1113/jphysiol.2008.167387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer T, Rupp J, Piper DR, Tristani-Firouzi M. The S4-S5 linker directly couples voltage sensor movement to the activation gate in the human ether-a’-go-go-related gene (hERG) K+ channel. J Biol Chem. 2006;281:12858–12864. doi: 10.1074/jbc.M513518200. [DOI] [PubMed] [Google Scholar]
- Ghanouni P, Gryczynski Z, Steenhuis JJ, Lee TW, Farrens DL, Lakowicz JR, Kobilka BK. Functionally different agonists induce distinct conformations in the G protein coupling domain of the β2 adrenergic receptor. J Biol Chem. 2001;276:24433–24436. doi: 10.1074/jbc.C100162200. [DOI] [PubMed] [Google Scholar]
- Goodwin JA, Hulme EC, Langmead CJ, Tehan BG. Roof and floor of the muscarinic binding pocket: variations in the binding modes of orthosteric ligands. Mol Pharmacol. 2007;72:1484–1496. doi: 10.1124/mol.107.038265. [DOI] [PubMed] [Google Scholar]
- Hibert MF, Trumpp-Kallmeyer S, Bruinvels A, Hoflack J. Three-dimensional models of neurotransmitter G-binding protein-coupled receptors. Mol Pharmacol. 1991;40:8–15. [PubMed] [Google Scholar]
- Honjo H, Kodama I, Zang WJ, Boyett MR. Desensitization to acetylcholine in single sinoatrial node cells isolated from rabbit hearts. Am J Physiol. 1992;263:H1779–H1789. doi: 10.1152/ajpheart.1992.263.6.H1779. [DOI] [PubMed] [Google Scholar]
- Itoh T, Seki N, Suzuki S, Ito S, Kajikuri J, Kuriyama H. Membrane hyperpolarization inhibits agonist-induced synthesis of inositol 1,4,5-trisphosphate in rabbit mesenteric artery. J Physiol. 1992;451:307–328. doi: 10.1113/jphysiol.1992.sp019166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenakin T. Efficacy at G-protein-coupled receptors. Nat Rev Drug Discov. 2002;1:103–110. doi: 10.1038/nrd722. [DOI] [PubMed] [Google Scholar]
- Kovoor P, Wickman K, Maguire CT, Pu W, Gehrmann J, Berul CI, Clapham DE. Evaluation of the role of IKACh in atrial fibrillation using a mouse knockout model. J Am Coll Cardiol. 2001;37:2136–2143. doi: 10.1016/s0735-1097(01)01304-3. [DOI] [PubMed] [Google Scholar]
- Leff P, Prentice DJ, Giles H, Martin GR, Wood J. Estimation of agonist affinity and efficacy by direct, operational model-fitting. J Pharmacol Methods. 1990;23:225–237. doi: 10.1016/0160-5402(90)90066-t. [DOI] [PubMed] [Google Scholar]
- Lu ZL, Saldanha JW, Hulme EC. Transmembrane domains 4 and 7 of the M1 muscarinic acetylcholine receptor are critical for ligand binding and the receptor activation switch. J Biol Chem. 2001;276:34098–34104. doi: 10.1074/jbc.M104217200. [DOI] [PubMed] [Google Scholar]
- Lu ZL, Saldanha JW, Hulme EC. Seven-transmembrane receptors: crystals clarify. Trends Pharmacol Sci. 2002;23:140–146. doi: 10.1016/S0165-6147(00)01973-8. [DOI] [PubMed] [Google Scholar]
- Martinez-Pinna J, Gurung IS, Vial C, Leon C, Gachet C, Evans RJ, Mahaut-Smith MP. Direct voltage control of signaling via P2Y1 and other Gaq-coupled receptors. J Biol Chem. 2005;280:1490–1498. doi: 10.1074/jbc.M407783200. [DOI] [PubMed] [Google Scholar]
- Navarro-Polanco RA, Sanchez-Chapula JA. 4-Aminopyridine activates potassium currents by activation of a muscarinic receptor in feline atrial myocytes. J Physiol. 1997;498:663–678. doi: 10.1113/jphysiol.1997.sp021891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohana L, Barchad O, Parnas I, Parnas H. The metabotropic glutamate G-protein-coupled receptors mGluR3 and mGluR1a are voltage-sensitive. J Biol Chem. 2006;281:24204–24215. doi: 10.1074/jbc.M513447200. [DOI] [PubMed] [Google Scholar]
- Olcese R, Latorre R, Toro L, Bezanilla F, Stefani E. Correlation between charge movement and ionic current during slow inactivation in Shaker K+ channels. J Gen Physiol. 1997;110:579–589. doi: 10.1085/jgp.110.5.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parnas H, Parnas I. The chemical synapse goes electric: Ca2+- and voltage-sensitive GPCRs control neurotransmitter release. Trends Neurosci. 2007;30:54–61. doi: 10.1016/j.tins.2006.12.001. [DOI] [PubMed] [Google Scholar]
- Pedretti A, Vistoli G, Marconi C, Testa B. Muscarinic receptors: a comparative analysis of structural features and binding modes through homology modelling and molecular docking. Chem Biodivers. 2006;3:481–501. doi: 10.1002/cbdv.200690052. [DOI] [PubMed] [Google Scholar]
- Piper DR, Hinz WA, Tallurri CK, Sanguinetti MC, Tristani-Firouzi M. Regional specificity of human ether-a’-go-go-related gene channel activation and inactivation gating. J Biol Chem. 2005;280:7206–7217. doi: 10.1074/jbc.M411042200. [DOI] [PubMed] [Google Scholar]
- Piper DR, Varghese A, Sanguinetti MC, Tristani-Firouzi M. Gating currents associated with intramembrane charge displacement in HERG potassium channels. Proc Natl Acad Sci U S A. 2003;100:10534–10539. doi: 10.1073/pnas.1832721100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahlholm K, Marcellino D, Nilsson J, Fuxe K, Arhem P. Voltage-sensitivity at the human dopamine D2S receptor is agonist-specific. Biochem Biophys Res Commun. 2008;377:1216–1221. doi: 10.1016/j.bbrc.2008.10.117. [DOI] [PubMed] [Google Scholar]
- Shibata EF, Giles W, Pollack GH. Threshold effects of acetylcholine on primary pacemaker cells of the rabbit sino-atrial node. Proc R Soc Lond B Biol Sci. 1985;223:355–378. doi: 10.1098/rspb.1985.0006. [DOI] [PubMed] [Google Scholar]
- Stefani E, Bezanilla F. Cut-open oocyte voltage-clamp technique. Methods Enzymol. 1998;293:300–318. doi: 10.1016/s0076-6879(98)93020-8. [DOI] [PubMed] [Google Scholar]
- Wang YG, Lipsius SL. Acetylcholine potentiates acetylcholine-induced increases in K+ current in cat atrial myocytes. Am J Physiol Heart Circ Physiol. 1995;268:H1313–H1321. doi: 10.1152/ajpheart.1995.268.3.H1313. [DOI] [PubMed] [Google Scholar]
- Wickman K, Nemec J, Gendler SJ, Clapham DE. Abnormal heart rate regulation in GIRK4 knockout mice. Neuron. 1998;20:103–114. doi: 10.1016/s0896-6273(00)80438-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.