

Non-technical summary
Normal heart rate variability is critically dependent upon the G-protein-coupled, acetylcholine (ACh)-activated inward rectifier K+ current, IKACh. A unique feature of IKACh is the so-called ‘relaxation’ gating property that contributes to increased current at hyperpolarized membrane potentials. Here, we consider a novel explanation for IKACh relaxation based upon the recent finding that G-protein-coupled receptors are intrinsically voltage sensitive and that the muscarinic agonists acetylcholine and pilocarpine manifest opposite voltage-dependent IKACh modulation. Based on experimental and computational findings, we propose that IKACh relaxation represents a voltage-dependent change in agonist affinity as a consequence of a voltage-dependent conformational change in the muscarinic receptor.
Abstract
Abstract
Normal heart rate variability is critically dependent upon the G-protein-coupled, acetylcholine (ACh)-activated inward rectifier K+ current, IKACh. A unique feature of IKACh is the so-called ‘relaxation’ gating property that contributes to increased current at hyperpolarized membrane potentials. IKACh relaxation refers to a slow decrease or increase in current magnitude with depolarization or hyperpolarization, respectively. The molecular mechanism underlying this perplexing gating behaviour remains unclear. Here, we consider a novel explanation for IKACh relaxation based upon the recent finding that G-protein-coupled receptors (GPCRs) are intrinsically voltage sensitive and that the muscarinic agonists acetylcholine (ACh) and pilocarpine (Pilo) manifest opposite voltage-dependent IKACh modulation. We show that Pilo activation of IKACh displays relaxation characteristics opposite to that of ACh. We explain the opposite effects of ACh and Pilo using Markov models of IKACh that incorporate ligand-specific, voltage-dependent parameters. Based on experimental and computational findings, we propose a novel molecular mechanism to describe the enigmatic relaxation gating process: IKACh relaxation represents a voltage-dependent change in agonist affinity as a consequence of a voltage-dependent conformational change in the muscarinic receptor.
Introduction
Normal heart rate variability is critically dependent upon the G-protein-coupled, acetylcholine (ACh)-activated inward rectifier K+ current, IKACh (Wickman et al. 1998). Cardiac KACh channels are heteromultimers composed of two homologous G-protein-coupled inward rectifier K+ channel subunits, Kir 3.1 and Kir 3.4 (Corey et al. 1998). In the heart, parasympathetic stimulation releases ACh, which binds to the muscarinic type-2 receptor (M2R) to initiate activation of IKACh through a G-protein-coupled mechanism (Breitwieser & Szabo, 1985; Pfaffinger et al. 1985). IKACh activation augments repolarizing current throughout phases 3 and 4 of the cardiac action potential to shorten action potential duration, increase the maximum diastolic potential and retard spontaneous diastolic depolarization. While there is no controversy as to the cardiac effects of strong vagal stimulation, the participation of IKACh in mediating the purely chronotropic effects of low (or ‘physiological’) ACh concentrations has been debated over the past several decades (DiFrancesco et al. 1989; Boyett et al. 1995). In part, the debate concerns the degree to which low ACh concentrations preferentially activate IKACh at hyperpolarized membrane potentials (during diastole) compared to depolarized potentials (during systole). A unique feature of IKACh that contributes to increased current at hyperpolarized membrane potentials is the so-called ‘relaxation’ gating property. IKACh relaxation refers to a slow decrease or increase in current magnitude with depolarization or hyperpolarization, respectively. This time-dependent change in current is also agonist concentration dependent, present at low, but not high agonist concentrations (Noma & Trautwein, 1978; Ishii et al. 2001).
Since the initial description of IKACh relaxation in the late 1970s, the molecular mechanism underlying this perplexing gating behaviour has remained unknown. Recently, a role for regulators of G-protein signalling (RGS) proteins in mediating IKACh relaxation has been postulated. Membrane depolarization increases intracellular calcium which is proposed to activate RGS proteins and decrease the activity of IKACh channels. IKACh relaxation reflects the reverse process: a decrease in intracellular Ca2+ associated with decreased RGS activity and increased open probability of KACh channels (Fujita et al. 2000; Ishii et al. 2001).
In this manuscript, we consider a novel explanation for IKACh relaxation based upon the recent finding that G-protein-coupled receptors (GPCRs) are intrinsically voltage sensitive. Membrane depolarization was recently shown to decrease the affinity of M2R for ACh and this voltage sensitivity was noted to be a property of the receptor itself (Ben-Chaim et al. 2003). The intrinsic capacity of muscarinic receptors to ‘sense’ transmembrane potential was confirmed by the recording of M2R gating currents (Ben-Chaim et al. 2006). In the preceding manuscript, we studied the effects of ACh and pilocarpine (Pilo) on voltage-dependent M2R activation of IKACh and on M2R gating currents. ACh and Pilo manifested opposite voltage-dependent IKACh modulation and exerted opposite effects on the magnitude of gating charge displacement. We propose that depolarization induces a conformational change in the M2R agonist binding pocket that decreases the affinity for ACh while increasing that for Pilo. In this context, we predict that Pilo activation of IKACh would display relaxation characteristics opposite to that of ACh. Indeed, we show that membrane hyperpolarization induces a time-dependent decrease in IKACh recorded from atrial myocytes in the presence of Pilo. We propose a novel molecular mechanism to describe the enigmatic relaxation gating process: IKACh relaxation represents a voltage-dependent change in agonist affinity as a consequence of a voltage-dependent conformational change in the muscarinic receptor.
Methods
Ethical approval
All animal studies were performed in accordance with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996) and after securing approval by the University of Utah and University of Colima Institutional Animal Care and Use Committees. All animal studies conform to the principles of UK regulations, as described in Drummond (2009). The experiments presented in this paper involved the isolation of left atrial myocytes from adult cats. The experimental protocol, including the use of sodium pentobarbitone as the anaesthetic agent, was approved by the Institutional Animal Care and Use Committee of the University of Colima. Animals were killed by excision of the heart en bloc while under anaesthesia.
Cell isolation and preparation
Isolated left atrial myocytes from adult cats (>2 kg) were prepared using an enzymatic perfusion method (Navarro-Polanco & Sanchez-Chapula, 1997) in accordance with the Public Health Service Policy on the Humane Care and Use of Laboratory Animals. The animals were anaesthetized with sodium pentobarbitone (35 mg kg−1, i.p.) 30 min after having received heparin (1000 u kg−l, i.p.). The hearts were excised and mounted via the aorta to a Langendorff perfusion system. Hearts were initially perfused with Tyrode solution for 5 min, and then with nominally zero calcium solution. After 5 min of perfusion, the perfusate was switched to a zero calcium solution plus collagenase (0.33 mg ml−1; Type I, Sigma-Aldrich, St Louis, MO, USA) and protease (0.033 mg ml−1; Type XIV, Sigma-Aldrich) for 30 min. The enzymes were washed out by perfusion with low-chloride, low-sodium, high-potassium solution, the Kraft–Brühe (KB) medium, for 5 min. The left atrium was dissected out, cut into small pieces and placed in a beaker with KB medium. Isolated cells were obtained by mechanical agitation with a flamed Pasteur pipette and stored in KB medium at 4°C for later electrophysiological experiments.
Solutions
The Tyrode solution contained (mm): 118 NaCl, 24 NaHCO3, 0.42 NaH2PO4, 5.4 KCl, 1.8 CaCl2, 1.05 MgCl2, 11 glucose and 20 taurine; the solution was equilibrated with 95% O2 and 5% CO2 (pH 7.4). Nominally Ca2+-free Tyrode solution was prepared by omitting CaCl2 from the Tyrode solution. The KB medium had the following composition (mm): 80 potassium glutamate, 40 KCl, 20 taurine, 10 KH2PO4, 5 MgSO4, 10 glucose, 10 Hepes, 0.5 creatine, 10 succinic acid and 0.2 EGTA; pH was adjusted to 7.4 with KOH. The solution was bubbled with 100% O2.
Patch pipettes were filled with (in mm): 80 potassium aspartate, 10 KH2PO4, 1.0 MgSO4, 20 KCl, 5 Hepes, 5 K4-BAPTA, 0.2 GTP-Na and 3 ATP-Na2 (pH 7.25 with KOH). The standard bath solution contained (in mm): 136 NaCl, 4 KCl, 1.0 MgCl2, 10 Hepes-Na+, 0.5 CaCl2, 2.0 CoCl2 and 11 glucose (pH 7.35 with NaOH). ACh and Pilo were dissolved directly in the external solution at the desired concentrations and prepared fresh daily.
IKACh recording in isolated myocytes
Whole-cell voltage-clamp experiments were performed at a temperature of 36 ± 0.5°C and using an Axopatch-200B amplifier (Molecular Devices, Sunnyvale, CA, USA). Data acquisition and command potentials were controlled by pCLAMP 10.0 software (Molecular Devices). Patch pipettes with a resistance of 1.5–3 MΩ were made from borosilicate capillary glass (WPI, Sarasota, FL, USA). The capacitance and series resistance with the cell membrane were compensated to provide the fastest possible capacitive transient, without ringing (oscillations). Currents were filtered with a four-pole Bessel filter at 2 kHz, digitized at 5 kHz. An agar–KCl bridge was used to ground the bath. To ensure a good intracellular dialysis, all current recordings were acquired at least 10 min after establishing the whole-cell configuration. Recordings were carried out in the presence of 3 μm E-4031 and 50 μm chromanol 293B to block rapid delayed rectifier (IKr) and slow delayed rectifier (IKs), respectively. External cobalt (2 mm) and internal BAPTA (5 mm) were included to block L-type calcium and calcium-activated currents, respectively. In addition, the IKATP blocker glibenclamide was included in the external solution for experiments performed in Figs 1 and 6. The ACh- and Pilo-induced currents traces were obtained by digitally subtracting currents recorded under control conditions from those recorded in the presence of agonist. Sequential concentration–response curves were performed as previously described in the preceding manuscript.
Figure 1. IKACh relaxation gating and opposite voltage-dependent IKACh modulation by ACh and Pilo.
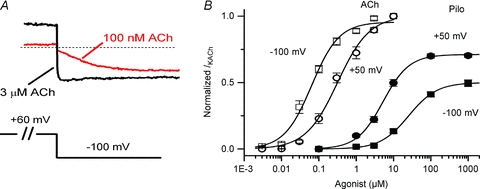
A, examples of IKACh elicited by low and high ACh concentrations highlight the properties of relaxation gating. IKACh relaxation refers to a slow increase in current magnitude elicited by membrane hyperpolarization following a depolarizing pre-pulse (100 nm ACh, red trace). This time-dependent change in current is absent at saturating ACh concentrations (3 μm ACh, black trace). B, concentration–response curves for ACh (open symbols) and Pilo (filled symbols) activation of IKACh at Vh+50 mV (circles) and −100 mV (squares). Data were normalized to current elicited by a saturating concentration of ACh (10 μm) and plotted as a function of ligand concentration. The lines represent data fits to a Hill equation. The EC50 and Hill coefficient for ACh were 64 ± 10 nm and 1.1 ± 0.1 (Vh−100 mV) and 317 ± 49 nm and 1.0 ± 0.1 (Vh+50 mV). The EC50 and Hill coefficient for Pilo were 25.5 ± 2.6 μm and 1.1 ± 0.1 (Vh−100 mV) and 5.2 ± 0.7 μm and 1.2 ± 0.1 (Vh+50 mV). n = 10–14 cells.
Figure 6. Atropine and tertiapin Q inhibit Pilo-activated currents in atrial myocytes.
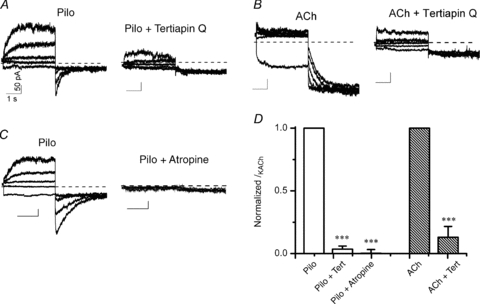
Macroscopic current recordings from an isolated atrial myocyte activated by 3 μm Pilo (A) and 30 nm ACh (B) before (left) and during perfusion with 300 nm tertiapin Q (right). Currents were elicited by 3.5 s pre-pulses between −100 and +60 mV in steps of 40 mV, followed by step hyperpolarization to −120 mV. Vh was −40 mV. C, 3 μm Pilo-activated current before (left) and after perfusion with 100 nm atropine (right). The x–y scale bars indicate 1 s and 50 pA, respectively, for all current traces in A–C. D, fraction of IKACh inhibited by tertiapin Q or atropine. Current at the end of the 3.5 s voltage to +60 mV (Pilo) and −100 mV (ACh) after tertiapin Q or atropine treatment was normalized to pre-treatment value to obtain the relative amount of IKACh inhibited by tertiapin Q or atropine. n = 4 cells each group.
Drugs
Tertiapin Q and chromanol 293B were obtained from Tocris Bioscience (Ellisville, MO, USA). All other reagents were from Sigma-Aldrich. In experiments using pertussis toxin (PTX), half of the left atrial cells were stored in KB medium at 36°C, and the other half in KB medium plus PTX at a final concentration of 5 μg ml−1 for 2–6 h. Before recording, PTX-treated myocytes were washed for 20–30 min in the recording chamber. Additionally, PTX (5 μg ml−1) was added to the internal solution.
Modelling of IKACh
Two Markov models of IKACh were developed to reconstruct experimental data in the presence of 100 nm ACh and 3 μm Pilo. The models are based on a more complete model of receptor systems (Weiss et al. 1996). A diagram of the 4-state model is shown in Fig. 7. Supplementary Table SI lists the states of the ACh and Pilo models. Rate coefficients were either dependent on transmembrane voltage Vm or concentration of the ligand [L]. Voltage-dependent forward rates α and backward rates β were defined as:
where α0 and β0 are the rates at 0 mV, zα and zβ the charges, T temperature, F Faraday constant, and R gas constant. Ligand concentration [L]-dependent forward rates γ and backward rates δ were defined as:
with the parameter γL and rate constant δL. IKACh was defined based on (Zhang et al. 2002):
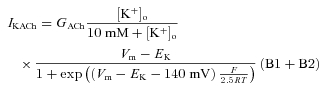 |
where GACh is the conductance, [K+]o the extracellular K+ concentration, EK the reversal voltage, and B1 and B2 the states. Parameters of the models are compiled in Supplemental Table S2.
Figure 7. Diagram of 4-state Markov model to reconstruct IKACh.
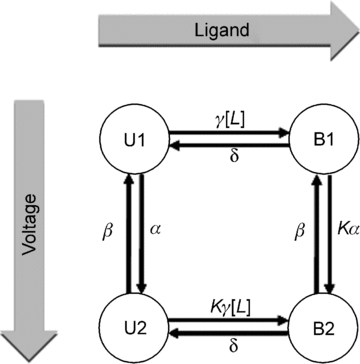
Rate coefficients were dependent on membrane voltage or ligand concentration. Stochastic optimization methods were used to generate models of IKACh in the presence of ACh and Pilo as described in Methods.
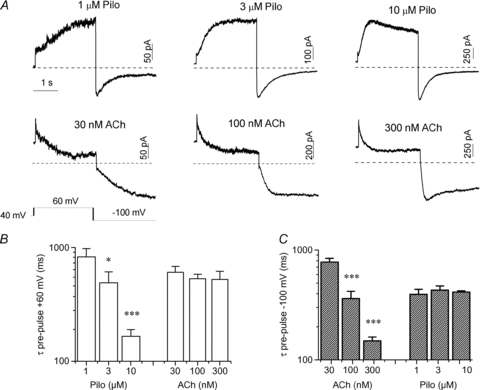
Parameters of ACh and Pilo models were determined by computational methods described by us previously (Abbruzzese et al. 2010). In short, an iterative stochastic approach for numerical fitting of feature vectors extracted from experimental and model data was applied. The feature vectors comprised parameters from mono-exponential fitting of IKACh activation and deactivation (Supplemental Table S3). The fit error E was defined as:
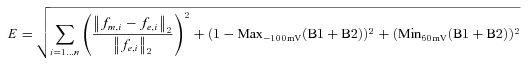 |
where n is the number of features, fe features from experiments, fm features from the model,  the Euclidean norm, Max-100mV the maximum of states at voltage of −100 mV, and Min60V the minimum of states at voltage of 60 mV. Supplemental Table S3 presents the fit errors of the ACh and Pilo models using the stochastic approach. The rate constant parameters for the ACh and Pilo models with the smallest fit error E are listed in Supplemental Table S4.
the Euclidean norm, Max-100mV the maximum of states at voltage of −100 mV, and Min60V the minimum of states at voltage of 60 mV. Supplemental Table S3 presents the fit errors of the ACh and Pilo models using the stochastic approach. The rate constant parameters for the ACh and Pilo models with the smallest fit error E are listed in Supplemental Table S4.
Data analysis
Data are reported as mean ± SEM (n, number of cells). pCLAMP 10.0 software was used to perform non-linear least-squares kinetic analyses of time-dependent currents based on the simplex algorithm. Where appropriate, Student's paired t test or ANOVA followed by Bonferroni's test were used for evaluating statistical difference. Significance was determined for P < 0.05 (*), P < 0.01 (**) and P < 0.001 (***).
Results
Opposite voltage-dependent activation of atrial IKACh by ACh and Pilo
In the preceding manuscript, we demonstrated that the potency of the muscarinic agonists ACh and Pilo varied with membrane potential in an opposite manner, as determined by IKACh in single, isolated feline atrial myocytes. Experiments in the preceding manuscript were performed in external K+ concentration ([K+]o) of 20 mm. The experimental protocol was repeated in a physiological [K+]o of 4 mm (see Methods). The relationship between membrane potential and potency was still apparent, with Pilo demonstrating increased potency at depolarized potentials and ACh demonstrating increased potency at negative potentials. However, for both agonists the concentration–response curve was shifted slightly rightward along the concentration axis for the lower [K+]o (Fig. 1B). The EC50 for ACh was 64 ± 10 nm and 317 ± 49 nm at −100 and +50 mV, respectively. The EC50 for Pilo was 25.5 ± 2.6 μm and 5.2 ± 0.4 μm at −100 and +50 mV, respectively.
IKACh activation and relaxation induced by ACh
Figure 2 depicts the classical properties of IKACh relaxation. Following a 3.5 s pre-pulse to variable potentials, a test pulse to −120 mV elicited an instantaneous and time-dependent current that varied with voltage and agonist concentration. For example, following a pre-pulse to +60 mV in the presence of 100 nm ACh, a voltage step to −120 mV elicited a small instantaneous current that represented rapid unblock of open IKACh channels during the pre-pulse, followed by a time-dependent increase in current that represented the relaxation gating process (Fig. 2A, left panel red trace). By contrast, a hyperpolarizing pre-pulse to −100 mV followed by a voltage step to −120 mV elicited solely an instantaneous current (Fig. 2A, left panel blue trace). The time- and voltage-dependent relaxation property was absent at high concentrations of ACh, such as 3 μm (Fig. 2A). The relationship between the pre-pulse potential and the fraction of open IKACh channels (I/Imax) elicited by various ACh concentrations is plotted in Fig. 2B. This graph highlights the voltage- and agonist concentration-dependence of IKACh relaxation. For sub-saturating ACh concentrations, stronger depolarizing voltage steps activated fewer KACh channels than hyperpolarizing pre-pulses. However, saturating ACh concentrations activated all available KACh channels, independent of pre-pulse voltage.
Figure 2. IKACh relaxation elicited by ACh in feline atrial myocytes.
A, macroscopic currents were induced by 100 nm (left) and 3 μm ACh (right) obtained with a double-pulse protocol (inset above). Currents at −120 mV were recorded after 3.5 s pre-pulses between −100 and +60 mV in steps of 40 mV. Current magnitude at the arrow is a measure of the number of KACh channels activated during the pre-pulse. Zero current level is indicated by the horizontal dashed line. B, relationship between the pre-pulse voltage and I/Imax ratio for currents induced by three ACh concentrations (n = 4–5). Current magnitude measured at arrow was normalized to the maximum current elicited by pre-pulse to −100 mV. For sub-saturating ACh concentrations, the fraction of KACh channels activated during the pre-pulse decreased with stronger depolarizations.
In the preceding manuscript and Fig. 1B, we determined that the half-maximal effective concentration (EC50) for feline IKACh activation by ACh was 4-fold less when measured at a holding potential (Vh) of −100 mV compared to +50 mV. For example, 30 nm ACh activated ∼30% of available IKACh channels at a Vh of −100 mV, compared to ∼5% at +50 mV. For saturating ACh concentrations (such as 3 μm), the magnitude of the response was the same at positive and negative membrane potentials. We propose that the time-dependent increase in IKACh (i.e. relaxation) is a consequence of a voltage-dependent change in receptor affinity for ACh (a lower affinity conformation during a depolarizing pre-pulse transitioning to a higher affinity conformation at the hyperpolarized potential).
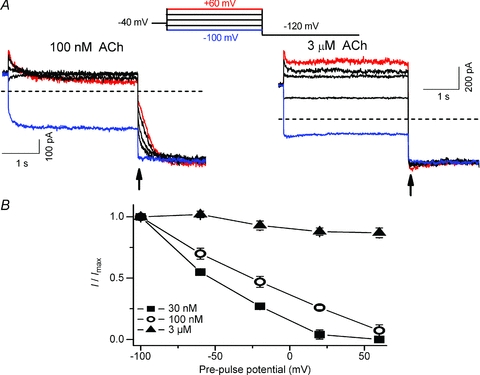
IKACh activation and relaxation induced by Pilo
In the preceding manuscript and Fig. 1B, we showed that IKACh activation by Pilo was also voltage dependent, but in a manner opposite to that of ACh. For example, 3 μm Pilo activated ∼5% of available IKACh channels at a Vh of −100 mV, compared to ∼25% at +50 mV. If IKACh relaxation gating was due to a voltage-dependent change in the agonist affinity of M2R, then we would predict that Pilo would induce relaxation gating in a manner opposite to ACh. Indeed, unlike ACh, Pilo induced a time-dependent increase in current during depolarizing pre-pulse voltage steps and almost no current during a hyperpolarizing pre-pulse voltage step (Fig. 3A). Following pre-pulse voltage steps to variable potentials, a test pulse to −120 mV also elicited a rapid current whose magnitude varied with pre-pulse potential, but the dependence on pre-pulse potential was opposite to that of ACh. For example, the magnitude of the rapid component increased as a function of pre-pulse potential. The relationship between the pre-pulse potential and the fraction of open KACh channels elicited by Pilo is illustrated in Fig. 3B. Unlike ACh, the fraction of open KACh channels elicited by Pilo at negative pre-pulse potentials was small and increased with stronger depolarizing pre-pulse potentials. Similar to ACh, this relationship was agonist concentration dependent. Finally, the behaviour of the time-dependent component of Pilo-induced currents at −120 mV was opposite to those of ACh. Pilo-induced currents at −120 mV decreased slowly (Fig. 3A) while ACh-induced currents slowly increased (Fig. 2A). These observations are consistent with the idea that IKACh relaxation gating properties induced by Pilo are opposite to those of ACh.
Figure 3. Opposite IKACh relaxation gating induced by Pilo.
A, macroscopic current recordings induced by 1 μm (left) and 3 μm Pilo (right) obtained with the same voltage protocol described in Fig. 2. B, relationship between the pre-pulse voltage and I/Imax ratio for currents induced by three Pilo concentrations (n = 4–6). Current magnitude measured at arrow was normalized to the maximum current elicited by pre-pulse to +60 mV. Opposite to ACh, the fraction of KACh channels activated by Pilo during the pre-pulse increased with stronger depolarizations. Note that Pilo-induced currents at −120 mV decayed slowly, while ACh-induced currents at −120 mV increased.
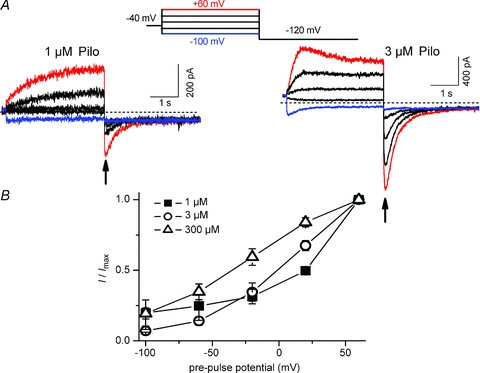
The kinetics of IKACh relaxation induced by ACh and Pilo
The classical kinetic properties of IKACh relaxation are illustrated in Fig. 4A (bottom panel). A depolarizing pre-pulse to +60 mV elicited a decaying current whose magnitude was ACh-concentration dependent (Fig. 4A, note scale bars). However, the kinetics of current decay were independent of ACh concentration (Fig. 4B). By contrast, both the current magnitude and the kinetics of current increase elicited by a hyperpolarizing test potential of −100 mV were ACh-concentration dependent (Fig. 4A and C). The kinetic behaviour of IKACh elicited by Pilo was opposite to that by ACh. For example, the time constants for current elicited by the depolarizing pre-pulse decreased with increasing concentrations of Pilo (Fig. 4B). The kinetics of current decay elicited by the hyperpolarizing test pulse were independent of Pilo concentration (Fig. 4C). Thus, IKACh relaxation induced by Pilo behaves in a manner opposite to that induced by ACh. Membrane depolarization opens KACh channels in the presence of Pilo, while closing channels in the presence of ACh.
Figure 4. Agonist concentration dependence of ACh and Pilo IKACh relaxation.
A, representative currents elicited by various concentrations of Pilo (top) and ACh (bottom) with a depolarizing pre-pulse to +60 mV from a holding potential of −40 mV, followed by a hyperpolarized test pulse to −100 mV (voltage protocol shown in inset). The horizontal time bar indicating 1 s duration applies to all traces in the panel. B, relationship of agonist concentration and τ of current decay (with ACh) or current increase (with Pilo) evoked by the pre-pulse to +60 mV (n = 5–8). C, relationship of agonist concentration and time constants (τ) of current increase (with ACh) or current decay (with Pilo) produced by the test pulse to −100 mV (n = 5–12).
Kinetics of activation and deactivation of KACh channels by ACh and Pilo
As the voltage range for IKACh activation by ACh and Pilo was opposite, we used opposite voltage protocols to assess IKACh activation and deactivation by these agonists (Fig. 5). The kinetics of IKACh activation by 100 nm ACh were determined by a depolarizing pre-pulse to +60 mV to close KACh channels, followed by hyperpolarizing pulses to open channels. The time constants of ACh-induced IKACh activation were voltage dependent and varied from 288 ± 68 ms at −100 mV to 642 ± 95 ms at +20 mV (Fig. 5C). Deactivation of ACh-induced IKACh was assessed by a hyperpolarized pre-pulse to −100 mV to open KACh channels, followed by depolarizing pulses to close channels. Unlike activation, the kinetics of ACh-induced IKACh deactivation were not voltage dependent (Fig. 5D).
Figure 5. Kinetics of IKACh activation by ACh and Pilo are voltage dependent, but vary in an opposite manner with voltage.
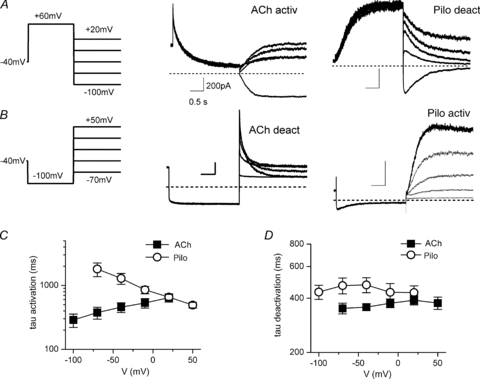
A and B, currents evoked by 100 nm ACh (middle) and 3 μm Pilo (right) using the protocol shown (left). Protocol A: depolarizing pre-pulse to +60 mV, followed by hyperpolarizing pulses from −100 mV to +20 mV in +30 mV steps were used to measure the time constants of ACh-IKACh activation and Pilo-IKACh deactivation. Protocol B, hyperpolarizing pre-pulse to −100 mV, followed by depolarizing pulses from −70 mV to +50 mV in +30 mV steps were used to measure ACh-IKACh deactivation and Pilo-IKACh activation. The x–y scale bars indicate 0.5 s and 200 pA, respectively, for all traces displayed in A and B. C, relationship between membrane voltage and time constants (τ) of ACh- and Pilo-IKACh activation (n = 5–12). D, relationship between membrane voltage and time constants (τ) of ACh- and Pilo-IKACh deactivation (n = 5–12).
The kinetics of IKACh activation by 3 μm Pilo were determined by a hyperpolarizing pre-pulse to −100 mV to close KACh channels, followed by depolarizing pulses to open channels. While the time constants of Pilo-induced IKACh activation were also voltage dependent, the voltage dependence was inverted compared to ACh (Fig. 5C). That is, activation was slower at hyperpolarized potentials and accelerated with depolarization: 1806 ± 437 ms at −70 mV and 492 ± 50 ms at +50 mV. Similar to ACh, the kinetics of Pilo-induced IKACh deactivation were independent of membrane voltage (Fig. 5D).
The data above suggest that membrane depolarization opens KACh channels in the presence of Pilo, while hyperpolarization opens channels in the presence of ACh. In order to further clarify the nature of the current activated by Pilo and ACh, we measured the instantaneous current at various potentials following a depolarizing pre-pulse to +60 mV (Pilo) or hyperpolarizing pulse to −100 mV (ACh) in order to measure a ‘fully activated’I–V (Supplemental Fig. S1). The current activated by 3 μm Pilo and 100 nm ACh reversed near −80 mV (as expected for a potassium current) and inwardly rectified (as expected for IKACh). The inwardly rectifying relationship between current and voltage was similar for Pilo- and ACh-activated current.
Pilo activates an atropine-sensitive, tertiapin-sensitive channel via a pertussis toxin-sensitive G-protein-coupled pathway
Next, we explored the possibility that Pilo might activate a current distinct from IKACh and thereby give the appearance of opposite relaxation gating. For this purpose, we characterized the effect of the muscarinic antagonist atropine and the cardiac KACh channel blocker tertiapin Q (Drici et al. 2000; Kitamura et al. 2000) on Pilo-activated current in atrial myocytes. Currents were recorded using the double-pulse protocol previously described (Fig. 2) in myocytes before and after perfusion with 100 nm atropine or 300 nm tertiapin Q. As shown in Fig. 6, tertiapin inhibited both Pilo- and ACh-activated currents and completely abolished the relaxation process. Similarly, atropine completely inhibited the Pilo-activated current. Pertussis toxin (PTX) irreversibly inhibits the activity of the Gαi/o family of G-proteins. PTX reduced Pilo-activated current at both Vh−100 mV and +50 mV to ∼25% of current activated in non-PTX-treated myocytes (Supplemental Fig. S2; n = 6 cells in each group). Thus, Pilo activates a tertiapin-sensitive channel via a Gαi/o-protein-coupled pathway, consistent with IKACh.
A Markov model of IKACh
Two Markov models were developed to reconstruct the effects of ACh and Pilo on IKACh. Model parameters were determined from measurements in the presence of 100 nm ACh and 3 μm Pilo (Fig. 5). The models do not differ in topology, but rather, the parameters differ for ACh- and Pilo-activation. The models are comprised of four states coupled with rate coefficients dependent on Vm and ligand concentration. U1 represents the receptor state in the absence of ligand and at a hyperpolarized membrane potential; U2, the absence of ligand at a depolarized potential; B1, the presence of ligand at a hyperpolarized membrane potential; and B2, the presence of ligand at a depolarized membrane potential (Fig. 7). The model design satisfies the condition of microscopic reversibility. Features and fit errors associated with parameterization of the model are listed in Supplemental Tables S1–S4. Simulated IKACh elicited by step depolarizations in the presence of 100 nm ACh and 3 μm Pilo are presented in Fig. 8, using the pulse protocols from Figs 2A and 3A. The simulations recapitulate the experimental features of opposite relaxation gating induced by ACh and Pilo. Simulated currents elicited by a step depolarization to +60 mV followed by voltage steps to variable potentials in the presence of 100 nm ACh and 3 μm Pilo are presented in Fig. 9A and B (based on experimental data from Fig. 5A). Simulated currents elicited by a voltage step to −100 mV followed by voltage steps to variable potentials in the presence of 100 nm ACh and 3 μm Pilo are presented in Fig. 9C and D (based on experimental data from Fig. 5B). A comparison of features extracted from the measured and simulated ACh and Pilo IKACh is shown in Supplemental Figs S3 and S4. While the model is simplistic in its design, it faithfully recapitulates the features of activation and deactivation of IKACh elicited by ACh and Pilo.
Figure 8. Simulated IKACh.
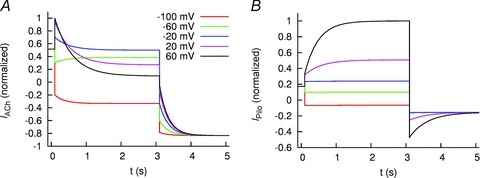
Simulated IKACh elicited by 100 nm ACh (A) and 3 μm Pilo (B). Cells were stepped to voltages between −100 and +60 mV and then returned to −120 mV as in Figs 2 and 3.
Figure 9. Simulated IKACh evoked by 100 nm ACh (left) and 3 μm Pilo (right) using a voltage-clamp protocol as in Fig. 5.
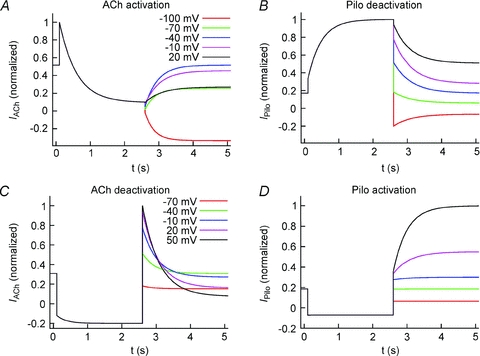
A, currents elicited by step depolarization to +60 mV, followed by voltage steps between −100 to 20 mV to measure ACh activation and Pilo deactivation. B, currents elicited by step hyperpolarization to −100 mV, followed by voltage steps between −100 to 20 mV to measure ACh deactivation and Pilo activation.
Discussion
IKACh plays a crucial role in regulating heart rate variability and vulnerability to atrial arrhythmias (Wickman et al. 1998; Kovoor et al. 2001). Relaxation gating of IKACh was first described by Noma and Trautwein as a time-dependent change in current magnitude following a depolarizing or hyperpolarizing voltage step (Noma & Trautwein, 1978). In essence, relaxation gating refers to the time course of opening or closing KACh channels. Over the past 30 years, this gating phenomenon has been extensively studied. A critical role for RGS proteins in the relaxation process was revealed by the observation that co-expression of RGS4 proteins was required (together with M2R, Kir3.1 and Kir3.4) to reconstitute relaxation gating behaviour in a heterologous expression system (Inanobe et al. 2001). RGS proteins accelerate the rate of GTP hydrolysis at the level of the G-protein and thereby terminate the G-protein-mediated signal. Mice lacking the cardiac RGS4 protein display enhanced responses to muscarinic agonists (Cifelli et al. 2008). Changes in subsarcolemmal Ca2+ throughout the cardiac cycle are proposed to regulate RGS activity to decrease the opening of KACh channels at depolarized membrane potentials, while increasing opening at hyperpolarized potentials (Ishii et al. 2001). In this study, we propose a novel explanation for IKACh relaxation gating based upon the recent observation that GPCRs are capable of sensing transmembrane voltage.
GPCRs are classically activated by external stimuli including photons, hormones, neurotransmitters, odorants and chemokines. The intrinsic voltage sensitivity of GPCRs provides an additional mechanism to fine-tune cellular signalling in excitable tissue. For muscarinic receptors, the potency of ACh varies in a voltage- and receptor-specific manner. Depolarization increases the potency of ACh for activation of M1R, while decreasing that for M2R. Moreover, direct radio-labelled binding experiments confirmed that depolarization reduces the ACh affinity of M2R (Ben-Chaim et al. 2003). In the preceding manuscript, we demonstrated that the relationship between potency and voltage is also agonist dependent. ACh and Pilo manifested opposite voltage-dependent IKACh modulation and exerted opposite effects on the magnitude of M2R gating charge displacement. In addition, we showed that depolarization increased a calculated measure of Pilo affinity for M2R. Based on these observations, we propose that voltage alters the conformation of the M2R agonist binding pocket, with the depolarized conformation favouring binding of Pilo and the hyperpolarized conformation favouring ACh binding.
Based on the opposite voltage-dependent changes in agonist affinity, we predicted that Pilo activation of IKACh would display relaxation gating behaviour opposite to that of ACh. Indeed, our results supported this prediction. For sub-saturating agonist concentrations, membrane depolarization opened KACh channels in the presence of Pilo, while closing channels in the presence of ACh. Moreover, the kinetics of IKACh induced by Pilo were opposite to those for ACh. That is, depolarization increased the rate of Pilo-induced IKACh activation, while decreasing the rate of ACh- induced IKACh activation. These observations are difficult to reconcile with the hypothesis that IKACh relaxation is due to changes in RGS activity as a consequence of voltage-dependent changes in intracellular Ca2+ (Ishii et al. 2001). If true, both agonists should behave in a similar manner with a depolarization-evoked elevation in Ca2+. Certainly, RGS proteins are indisputably crucial for G−protein cycling. However, our data are more consistent with the idea that voltage sensitivity is a property of the receptor itself, rather than a phenomenon intrinsic to RGS activity. Indeed, the M2R is capable of sensing transmembrane voltage and agonist affinity varies with voltage.
We considered the possibility that Pilo might activate a current distinct from IKACh and thereby give the appearance of opposite relaxation gating. For example, Pilo was reported to activate a ‘delayed rectifier’ K+ current in guinea-pig atrium that was coupled to M3R, IKM3 (Wang et al. 1999). Our main evidence to identify the Pilo-activated current as IKACh was the observation that the current was blocked by tertiapin Q and PTX. While tertiapin Q is a potent blocker of inward rectifier K+ channels Kir3.1/3.4 (Drici et al. 2000; Kitamura et al. 2000), it is known to block other K+ channels, including a Ca2+-activated large-conductance K+ channel (BK channel) and the inward rectifiers, Kir1.1 and Kir3.2 (Jin & Lu, 1998; Kanjhan et al. 2005). Nevertheless, we consider it unlikely that these other K+ conductances are involved in the Pilo-activated current in atrial myocytes. First, our recording conditions included a calcium-channel blocker (2 mm Co2+) and an intracellular Ca2+ quelant (5 mm BAPTA) which would inhibit calcium activation of BK channels. Second, Kir1.1 and Kir3.2 are not expressed in heart (Hibino et al. 2010). While the slow time course of Pilo-induced IKACh activation (see Fig. 3) is reminiscent of a delayed rectifier current, our experimental and in silico data support the notion that indeed the current elicited by Pilo is IKACh. The fully activated current–voltage relationship of Pilo-activated current inwardly rectifies in a manner similar to ACh-activated current. The time course of ACh-induced IKACh is also slow, but does not give the appearance of a delayed rectifier because the affinity for ACh decreases with membrane depolarization, while that for Pilo increases.
New Markov models of IKACh activated by ACh or Pilo were formulated based on a simplified four-state model that incorporated ligand-specific voltage-dependent parameters. The models faithfully reproduced the general features of opposite IKACh relaxation gating induced by ACh and Pilo. The Pilo model also recapitulated the slowly activating currents elicited by membrane depolarization in the presence of Pilo that resemble a ‘delayed rectifier’ K+ current (Figs 3 and 8B). Thus, incorporation of ligand-specific voltage-dependent parameters reproduces the opposite relaxation gating behaviour of IKACh induced by ACh and Pilo.
Limitations
IKACh desensitization reduces the response of muscarinic agonists in a manner dependent upon agonist concentration and duration of agonist exposure. Fast IKACh desensitization occurs over a time-scale of seconds (∼20 s) and is rapidly reversible with washout. Slow desensitization requires both higher agonist concentrations and more prolonged agonist exposure (minutes to hours) and does not reverse rapidly (Boyett & Roberts, 1987; Bunemann et al. 1996). IKACh relaxation gating occurred with sub-saturating agonist concentrations (≤300 nm ACh and ≤10 μm Pilo) that do not induce desensitization. Moreover, desensitization would not be expected to induce the opposite voltage-dependent kinetic behaviour of Pilo- and ACh-activated current. However, exposure to maximal ACh concentrations (3 μm) probably induced some degree of desensitization. While we propose that the lack of IKACh relaxation gating induced by maximal ACh concentrations simply represents the opening of all available channels independent of voltage, we cannot completely exclude a possible role for desensitization.
Our experiments were performed using whole-cell patch-clamp recordings and thus examine the properties of IKACh relaxation on a macroscopic level. Earlier work demonstrates that single KACh channels gate in a modal fashion and that agonist activation involves shifts in gating behaviour between various gating modes (Okabe et al. 1991; Ivanova-Nikolova & Breitwieser, 1997; Ivanova-Nikolova et al. 1998; Yakubovich et al. 2000). Whether Pilo and membrane potential differentially modulate KACh gating modes will be defined in future experiments.
Taken together, our results suggest a novel molecular mechanism to describe the enigmatic relaxation gating of IKACh. We propose that IKACh relaxation represents a voltage-dependent change in agonist affinity as a consequence of a voltage-dependent conformational change in the muscarinic receptor. Moreover, this voltage sensitivity is also agonist specific. As such, depolarization induces a conformational change in the M2R agonist binding pocket that decreases the affinity for ACh while increasing that for Pilo. Thus, IKACh relaxation gating is mediated by the intrinsic voltage sensitivity of the muscarinic receptor.
Acknowledgments
This work was supported by Consejo Nacional de Ciencia y Tecnología, México (CONACyT-054577 to R.A.N.-P). We recognize the support of the Nora Eccles Treadwell Foundation. The authors wish to thank Miguel Angel Flores-Virgen for technical assistance.
Glossary
Abbreviations
- ACh
acetylcholine
- GPCR
G-protein-coupled receptor
- IKACh
acetylcholine-activated K+ current
- M2R
muscarinic receptor type-2
- Pilo
pilocarpine
- RGS
regulators of G-protein signalling
Author contributions
All research studies were done at the Centro Universitario de Investigaciones Biomédicas. E.G.M.-G., J.A.S.-C., M.T.-F. and R.A.N.-P. participated in the conception and design of the studies. E.G.M.-G., F.B.S., J.A.R.-P and R.A.N.-P contributed to the experiment performance. E.G.M.-G., M.T.-F. and R.A.N.-P performed data analysis and were involved in the interpretation of data. E.G.M.-G., M.T.-F. and R.A.N.-P had equal contributions in writing the paper with critical input from all authors. All authors approved the final version.
Supplementary material
Suppl. Fig. 1
Suppl. Fig. 2
Suppl. Fig. 3
Suppl. Fig. 4
Suppl. Table 1
Suppl. Table 2
Suppl. Table 3
Suppl. Table 4
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer-reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors
References
- Abbruzzese J, Sachse FB, Tristani-Firouzi M, Sanguinetti MC. Modification of hERG1 channel gating by Cd2+ J Gen Physiol. 2010;136:203–224. doi: 10.1085/jgp.201010450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Chaim Y, Chanda B, Dascal N, Bezanilla F, Parnas I, Parnas H. Movement of ‘gating charge’ is coupled to ligand binding in a G-protein-coupled receptor. Nature. 2006;444:106–109. doi: 10.1038/nature05259. [DOI] [PubMed] [Google Scholar]
- Ben-Chaim Y, Tour O, Dascal N, Parnas I, Parnas H. The M2 muscarinic G-protein-coupled receptor is voltage-sensitive. J Biol Chemi. 2003;278:22482–22491. doi: 10.1074/jbc.M301146200. [DOI] [PubMed] [Google Scholar]
- Boyett MR, Kodama I, Honjo H, Arai A, Suzuki R, Toyama J. Ionic basis of the chronotropic effect of acetylcholine on the rabbit sinoatrial node. Cardiovasc Res. 1995;29:867–878. [PubMed] [Google Scholar]
- Boyett MR, Roberts A. The fade of the response to acetylcholine at the rabbit isolated sino-atrial node. J Physiol. 1987;393:171–194. doi: 10.1113/jphysiol.1987.sp016818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitwieser GE, Szabo G. Uncoupling of cardiac muscarinic andβ-adrenergic receptors from ion channels by a guanine nucleotide analogue. Nature. 1985;317:538–540. doi: 10.1038/317538a0. [DOI] [PubMed] [Google Scholar]
- Bunemann M, Brandts B, Pott L. Downregulation of muscarinic M2 receptors linked to K+ current in cultured guinea-pig atrial myocytes. J Physiol. 1996;494:351–362. doi: 10.1113/jphysiol.1996.sp021497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cifelli C, Rose RA, Zhang H, Voigtlaender-Bolz J, Bolz SS, Backx PH, Heximer SP. RGS4 regulates parasympathetic signaling and heart rate control in the sinoatrial node. Circ Res. 2008;103:527–535. doi: 10.1161/CIRCRESAHA.108.180984. [DOI] [PubMed] [Google Scholar]
- Corey S, Krapivinsky G, Krapivinsky L, Clapham DE. Number and stoichiometry of subunits in the native atrial G-protein-gated K+ channel, IKACh. J Biol Chem. 1998;273:5271–5278. doi: 10.1074/jbc.273.9.5271. [DOI] [PubMed] [Google Scholar]
- DiFrancesco D, Ducouret P, Robinson RB. Muscarinic modulation of cardiac rate at low acetylcholine concentrations. Science. 1989;243:669–671. doi: 10.1126/science.2916119. [DOI] [PubMed] [Google Scholar]
- Drici MD, Diochot S, Terrenoire C, Romey G, Lazdunski M. The bee venom peptide tertiapin underlines the role of IKACh in acetylcholine-induced atrioventricular blocks. Br J Pharmacol. 2000;131:569–577. doi: 10.1038/sj.bjp.0703611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond GB. Reporting ethical matters in The Journal of Physiology: standards and advice. J Physiol. 2009;587:713–719. doi: 10.1113/jphysiol.2008.167387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita S, Inanobe A, Chachin M, Aizawa Y, Kurachi Y. A regulator of G protein signalling (RGS) protein confers agonist-dependent relaxation gating to a G protein-gated K+ channel. J Physiol. 2000;526:341–347. doi: 10.1111/j.1469-7793.2000.00341.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibino H, Inanobe A, Furutani K, Murakami S, Findlay I, Kurachi Y. Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol Rev. 2010;90:291–366. doi: 10.1152/physrev.00021.2009. [DOI] [PubMed] [Google Scholar]
- Inanobe A, Fujita S, Makino Y, Matsushita K, Ishii M, Chachin M, Kurachi Y. Interaction between the RGS domain of RGS4 with G proteinα subunits mediates the voltage-dependent relaxation of the G protein-gated potassium channel. J Physiol. 2001;535:133–143. doi: 10.1111/j.1469-7793.2001.t01-1-00133.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii M, Inanobe A, Fujita S, Makino Y, Hosoya Y, Kurachi Y. Ca2+ elevation evoked by membrane depolarization regulates G protein cycle via RGS proteins in the heart. Circ Res. 2001;89:1045–1050. doi: 10.1161/hh2301.100815. [DOI] [PubMed] [Google Scholar]
- Ivanova-Nikolova TT, Breitwieser GE. Effector contributions to Gβγ-mediated signaling as revealed by muscarinic potassium channel gating. J Gen Physiol. 1997;109:245–253. doi: 10.1085/jgp.109.2.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanova-Nikolova TT, Nikolov EN, Hansen C, Robishaw JD. Muscarinic K+ channel in the heart. Modal regulation by G proteinβγ subunits. J Gen Physiol. 1998;112:199–210. doi: 10.1085/jgp.112.2.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin W, Lu Z. A novel high-affinity inhibitor for inward-rectifier K+ channels. Biochemistry. 1998;37:13291–13299. doi: 10.1021/bi981178p. [DOI] [PubMed] [Google Scholar]
- Kanjhan R, Coulson EJ, Adams DJ, Bellingham MC. Tertiapin-Q blocks recombinant and native large conductance K+ channels in a use-dependent manner. J Pharmacol Exp Ther. 2005;314:1353–1361. doi: 10.1124/jpet.105.085928. [DOI] [PubMed] [Google Scholar]
- Kitamura H, Yokoyama M, Akita H, Matsushita K, Kurachi Y, Yamada M. Tertiapin potently and selectively blocks muscarinic K+ channels in rabbit cardiac myocytes. J Pharmacol Exp Ther. 2000;293:196–205. [PubMed] [Google Scholar]
- Kovoor P, Wickman K, Maguire CT, Pu W, Gehrmann J, Berul CI, Clapham DE. Evaluation of the role of IKACh in atrial fibrillation using a mouse knockout model. J Am Coll Cardiol. 2001;37:2136–2143. doi: 10.1016/s0735-1097(01)01304-3. [DOI] [PubMed] [Google Scholar]
- Navarro-Polanco RA, Sanchez-Chapula JA. 4-Aminopyridine activates potassium currents by activation of a muscarinic receptor in feline atrial myocytes. J Physiol. 1997;498:663–678. doi: 10.1113/jphysiol.1997.sp021891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noma A, Trautwein W. Relaxation of the ACh-induced potassium current in the rabbit sinoatrial node cell. Pflugers Arch. 1978;377:193–200. doi: 10.1007/BF00584272. [DOI] [PubMed] [Google Scholar]
- Okabe K, Yatani A, Brown AM. The nature and origin of spontaneous noise in G protein-gated ion channels. J Gen Physiol. 1991;97:1279–1293. doi: 10.1085/jgp.97.6.1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaffinger PJ, Martin JM, Hunter DD, Nathanson NM, Hille B. GTP-binding proteins couple cardiac muscarinic receptors to a K channel. Nature. 1985;317:536–538. doi: 10.1038/317536a0. [DOI] [PubMed] [Google Scholar]
- Wang H, Shi H, Lu Y, Yang B, Wang Z. Pilocarpine modulates the cellular electrical properties of mammalian hearts by activating a cardiac M3 receptor and a K+ current. Br J Pharmacol. 1999;126:1725–1734. doi: 10.1038/sj.bjp.0702486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss JM, Morgan PH, Lutz MW, Kenakin TP. The cubic ternary complex receptor–occupancy model I. Model description. J Theor Biol. 1996;178:151–167. doi: 10.1006/jtbi.1996.0139. [DOI] [PubMed] [Google Scholar]
- Wickman K, Nemec J, Gendler SJ, Clapham DE. Abnormal heart rate regulation in GIRK4 knockout mice. Neuron. 1998;20:103–114. doi: 10.1016/s0896-6273(00)80438-9. [DOI] [PubMed] [Google Scholar]
- Yakubovich D, Pastushenko V, Bitler A, Dessauer CW, Dascal N. Slow modal gating of single G protein-activated K+ channels expressed in Xenopus oocytes. J Physiol. 2000;524:737–755. doi: 10.1111/j.1469-7793.2000.00737.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Holden AV, Noble D, Boyett MR. Analysis of the chronotropic effect of acetylcholine on sinoatrial node cells. J Cardiovasc Electrophysiol. 2002;13:465–474. doi: 10.1046/j.1540-8167.2002.00465.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.