

Abstract
Glycogen storage disease type Ib is caused by deficiencies in the glucose-6-phosphate transporter (G6PT), a phosphate (Pi)-linked antiporter capable of homologous (Pi:Pi) and heterologous (G6P:Pi) exchanges similar to the bacterial hexose-6-phosphate transporter, UhpT. Protease protection and glycosylation scanning assays have suggested that G6PT is anchored to the endoplasmic reticulum by 10 transmembrane domains. However, recent homology modeling proposed that G6PT may contain 12 helices and that amino acids essential for the functions of UhpT also play important roles in G6PT. Site-directed mutagenesis and in vitro expression assays demonstrated that only one of the four residues critical for UhpT activity is essential in G6PT. Furthermore, glycosylation scanning and protease sensitivity assays showed that the 10-domain model of G6PT is more probable than the 12-domain UhpT-like model.
Keywords: glycogen storage disease type I, protein glycosylation, antiporter, glycosylation scanning, G6P transport, phosphate transport, endoplasmic reticulum
Introduction
Glycogen storage disease type Ib (GSD-Ib, MIM232220) is an autosomal recessive disorder caused by a deficiency in the endoplasmic reticulum (ER)-bound glucose 6-phosphate transporter (G6PT or SLC37A4) [1, 2]. The primary function of G6PT is to translocate glucose-6-phosphate (G6P) from the cytoplasm into the lumen of the ER for hydrolysis to glucose and inorganic phosphate (Pi) either by the liver/kidney/intestine-restricted glucose-6-phosphatase-α (G6Pase-α) [1, 2] or the ubiquitously expressed G6Pase-β [3, 4]. The concerted action of G6PT and G6Pase-α is required to maintain glucose homeostasis between meals and a deficiency of either protein results in a phenotype of disturbed glucose homeostasis [1, 2]. The concerted action of G6PT and G6Pase-β is vital for normal neutrophil functions and a deficiency of either protein results in a phenotype of neutropenia and myeloid dysfunctions [5, 6]. Therefore, understanding of the structure-function requirements of G6PT will provide valuable insight into the functional coupling between G6PT and both G6Pases.
The topology of the G6PT protein has been determined through protease protection and glycosylation scanning experiments [7]. Protease protection analysis has shown that G6PT contains an even number of transmembrane helices with both N- and C-termini facing the cytoplasm [7]. Hydropathy profile analysis of the G6PT amino acid sequence predicts either 10 [8] or 12 [9] putative transmembrane helices, with glycosylation scanning analysis supporting the 10-domain model [7]. By sequence homology, G6PT belongs to the organophosphate:Pi antiporter family of the major facilitator superfamily [10] that includes the glycerol-3-phosphate transporter, GlpT [11] and the bacterial hexose-6-phosphate transporter, UhpT [12]. Indeed, the G6PT cDNA was originally isolated based on a comparison of the sequence of a Lactobacillus lactis UhpT with liver expressed sequence tags [9]. Both G6PT and UhpT are Pi-linked antiporter capable of both homologous (Pi:Pi) and heterologous (G6P:Pi) exchanges [13,14]. Structure-function studies of UhpT have identified amino acid residues absolutely required for transport activities [15,16]. These include R46 and R275, proposed to form the substrate-binding site in UhpT [15], and D388 and K391, proposed to involve in intra-helical salt bridge formation [16]. The corresponding residues in G6PT are R28, K240, H366, and V369 [9].
Recently, the structure of GlpT from E. coli was determined to 3.3 Å resolution using X-ray crystallography [17,18] which revealed that GlpT contains 12 transmembrane helices. Because G6PT and UhpT shares significant sequence homology with GlpT, it was proposed that G6PT and UhpT may adopt the same 12-helical topology [19]. A three-dimensional structural model of G6PT consisting of 12 helices was subsequently built by homology modeling with GlpT [19]. The model predicts that amino acids involved in substrate-binding in G6PT are R28 and K240, like R46 and R275 in UhpT [19]. We have previously shown that the R28C and R28H mutations identified in the G6PT gene of GSD-Ib patients abolish microsomal G6P uptake activity [20]. We now show that the G6PT K240C and K240R mutations retain significant G6P and Pi transport activity. The individual G6PT H366D and V369K mutations reduce G6PT transport activities. The H366D/V369K double G6PT mutant that produces a potential site for intra-helical salt bridge formation like D388 and K391 in UhpT [16] retains a similar activity to the G6PT V369K mutant. Taken together, these results indicate that the structural requirements of G6PT and UhpT are different. Furthermore, we present evidence showing that the 10-domain model of G6PT is more probable than the 12 domain UhpT-like model.
Materials and methods
Construction of G6PT mutants
The template for G6PT mutant construction by PCR was nucleotides 1 to 1286 of the human SLC37A4 cDNA in the pAdlox shuttle vector [9,20], which contains the entire coding region, with the translation initiation codon, ATG, at nucleotides 1–3. The two outside PCR primers are nucleotides 1 to 20 (sense) and 1270 to 1290 (antisense). The sense and antisense mutant primers are 20 nucleotides in length with the codon to be mutated in the middle. The nucleotide changes in the mutant constructs include: K240C (nucleotides 841 to 843, GGC to TGC); K240R (nucleotides 841 to 843, GGC to CGC); H366D (nucleotides 1096 to 1098, CAC to GAC); V369K (nucleotides 1105 to 1107, GTG to AAG). The G6PT-5Flag and G6PT-3Flag constructs have been described [7]. The eight-amino-acid Flag marker peptide, DYKDDDDK was also used to tag the N- and C-termini of G6PT-T53N and G6PT-S55N mutants using the respective mutant construct [7] as a template. The 5′-primer for N-terminal-Flag G6PT constructs contained an ATG initiation codon followed by the 24 bp Flag coding sequence (5′-GACTACAAGGACGACGATGACAAG-3′) and nucleotides 1 to 20 of human G6PT; the 3′-primer contained nucleotides 1270 to 1290 of human G6PT. The 5′-primer for C-terminal-Flag G6PT constructs contained nucleotides 1 to 20 of human G6PT; the 3′-primer at nucleotides 1267 to 1287 containing the last coding nucleotides of human G6PT, followed by the 24 bp Flag coding sequence and a termination codon. After PCR, the amplified fragment was ligated into the pAdlox vector. The nucleotide sequence in all constructs was verified by DNA sequencing.
Recombinant Adenoviruses containing mutant G6PT were generated by the Cre-lox recombination system [21] as described [20]. The recombinant virus was plaque purified and amplified to produce viral stocks with titers of approximately 1 to 3 × 1010 plaque forming unit (pfu) per ml.
Gene expression in COS-1 cells and microsomal G6P uptake assays
Recombinant adenovirus carrying wild-type G6PT (Ad-G6PT) and G6Pase-α (Ad-G6Pase-α) have been described [20,22]. COS-1 cells were grown at 37 °C in HEPES-buffered Dulbecco’s modified minimal essential medium supplemented with 4% fetal bovine serum. Cells in 150-cm2 flasks were infected with Ad-G6PT or an Ad-G6PT mutant, or co-infected with Ad-G6Pase-α and a wild-type or a mutant Ad-G6PT. The multiplicity of infection for Ad-G6PT or Ad-G6PT mutant was 50 pfu/cell and for Ad-G6Pase-α, 25 pfu/cell. Mock infected COS-1 cells were used as controls. After incubation at 37 °C for 24 h, the infected cultures were used to isolate microsomes for G6P uptake, proteoliposome reconstitution, and Western-blot analysis.
Microsomal G6P uptake measurements were performed essentially as described previously [20]. Briefly, microsomes (40 μg) were incubated for 3 min in a reaction mixture (100 μl) containing 50 mM sodium cacodylate buffer, pH 6.5, 250 mM sucrose, and 0.2 mM [U-14C]G6P (50 μCi/μmol, American Radiolabeled Chemicals, St Louis, MO). The reaction was stopped by filtering immediately through a nitrocellulose membrane (0.45 μm, Millipore Co., Billerica, MA) and the washed and dried filters were counted in a liquid scintillation counter. The 3 min incubation time was chosen based on the results of the kinetic studies G6P uptake in microsomes co-expressing G6PT and G6Pase-α [20]. In microsomes expressing both G6Pase-α and G6PT, the [U-14C]G6P taken up by the microsomes is hydrolyzed by G6Pase-αto [U-14C]glucose and Pi and the radioactive molecule(s) accumulated inside the microsomes is primarily [U-14C]glucose [23].
Solubilization and reconstitution of membrane proteins
Microsomal membrane protein solubilization and proteoliposome reconstitution were performed as previously described [13,14]. Briefly, membrane proteins were solubilized on ice by mixing 2 mg of microsomes in 1 ml of a solution containing 20 mM Tris-HCl, pH 7.5, 20% glycerol, 1.25% (w/v) n-octyl-β-D-glucopyranoside (Calbiochem, San Diego, CA), 2 mM DTT, protease inhibitors (1% aprotinin, 1 mM AEBSF, 2 μg/ml pepstatin A, and 2 μg/ml leupeptin, all from Roche Diagnostics, Indianapolis, IN, and 0.4% (w/v) lipid mixture in 2 mM β-mercaptoethanol that contains the E coli polar lipid extract, L-α phosphotidylcholine, L-α phosphotidylserine, and cholesterol (60:17.5:10:12.5 w/w), all from Avanti Polar lipids, Inc., Alabaster, AL.
To reconstitute proteoliposomes, the detergent solubilized microsomal membrane extracts (500 to 700 μg) were mixed with the sonicated lipid mixture at a ratio of 1:10 protein:lipid (w/w). Proteoliposomes were then formed by placing the reaction inside a dialysis cassette from PIERCE (Rockford, IL) and dialyzing extensively overnight against a phosphate buffer (50 mM KH2PO4, pH 7.0, 1mM DTT, and protease inhibitors) or a MOPS/K buffer (20 mM MOPS, pH 7.5 adjusted with KOH; 75 mM K2SO4; 2.5 mM MgSO4, 1 mM DTT, and protease inhibitors) at 4 °C. The resulting Pi-loaded or MOPS-loaded proteoliposomes were pelleted, resuspended in MOPS/K buffer, and used immediately in transport assays. The MOPS-loaded proteoliposomes were used as negative controls. In addition, proteoliposomes prepared from detergent solubilized microsomal membrane extracts of mock-infected cells and liposomes lacking microsomal proteins were prepared in parallel and also used as negative controls. The protein content in microsomes, detergent extract and proteoliposomes was quantified by the Amido black B protein estimation method as described [24].
Transport assays
Transport assays were performed at room temperature using reaction mixtures containing 20 mM MOPS/K buffer, 25 μg/ml of 50 mM Pi,-loaded G6PT-proteoliposomes or MOPS-loaded proteoliposomes, 0.1 mM [U-14C]G6P or 0.5 mM 32Pi. The 32Pi was prepared by boiling carrier-free 32Pi (MP Biochemical, Inc., Irvine, CA) in 1 ml of 1 N HCl for 3 h and diluting with an equal volume of 2 M K2HPO4 as described [25]. After incubation at room temperature for 9 min, the reaction mixture was filtered immediately through presoaked 0.22 μm nitrocellulose filters (Millipore), and the washed and dried filters were counted in a liquid scintillation counter.
Western-blot analysis
For Western-blot analysis, proteins were resolved by electrophoresis through a 10% polyacrylamide-SDS gel and trans-blotted onto polyvinylidene fluoride membranes (Invitrogen, Carlsbad, CA). The membranes were incubated overnight with a rabbit anti-G6PT antibody raised against amino acids 25 to 79 of human G6PT [20] or a monoclonal antibody against the Flag epitope (Sigma-Aldrich, St Louis, MO). The membranes were then incubated with a horseradish peroxidase-conjugated second antibody and the immunocomplex visualized using the SuperSignal Dura West Pico Chemiluminescent substrate from PIERCE (Rockford, IL).
Tryptic digestion of microsomes expressing G6PT wild-type and glycosylation mutants
One mg of intact microsomes expressing G6PT wild-type, T53N, or S55N mutant were digested with 500 μg of trypsin in a buffer containing 20 mM Tris-HCl, pH 7.5 and 150 mM NaCl at room temperature for 10 min. The reaction was quenched with a 10-fold excess of soybean trypsin inhibitor (Sigma-Aldrich, Saint Louis, MO) and 11 mM AEBSF and the resulting digests analyzed by Western blot analysis. The intactness of microsomes was evaluated by latency values which were assessed by mannose-6-P phosphohydrolysis in intact (I) versus detergent-disrupted (D) microsomes, defined as (1-I/D) × 100 [26]. Microsomes having a latency value of 95% or greater (intact microsomes) were used in this study.
Statistical analysis
The unpaired t test was performed using the GraphPad Prism Program, version 4 (GraphPad Software, San Diego, CA). Values were considered statistically significant at p < 0.05.
Results
G6PT and UhpT differ in structural requirements
The homology modeling predicts that amino acid residues essential for UhpT functions might play important roles in G6PT [19]. In UhpT, mutagenesis and functional studies have demonstrated that R46 and R275, located in the substrate-translocation pore, are essential residues implicated in substrate binding [15]. Indeed, substituting R46 or R275 with either a cysteine or a lysine inactivated UhpT, and Western-blot analysis showed that both R46K and R275K mutations destabilized UhpT [15]. In addition, amino acids D388 and K391 in UhpT are proposed to be involved in intrahelical salt bridge formation [16]. While G6P transport was abolished after individual replacement of D388 (D388C) or K391 (K391C) in UhpT, G6P transport was retained when uncharged residues replaced both residues as in the D388C/K391C double UhpT mutant [16].
The corresponding residues in G6PT are R28, K240, H366, and V369 (Fig. 1). The homology modeling predicts that R28 and K240 in G6PT are also essential residues forming part of the substrate-binding site [19]. We have previously shown that R28H and R28C mutations identified in the G6PT gene of GSD-Ib patients abolish microsomal G6P uptake activity [20], demonstrating that R28 is an essential amino acid in G6PT. To delineate further the structural requirements of G6PT and its relationship to UhpT, we constructed Ad-G6PT constructs carrying p.K240R, p.K240C, p.H366D, and p.V369K mutations as well as an Ad-G6PT-H366D/V369K double mutant capable of salt bridge formation. Unlike R275 in UhpT, K240R and K240C G6PT mutants retained 49–54% and 16–18% wild-type activity, respectively (Table 1). Likewise, the H366D and V369K G6PT mutants retained 19–20% and 8–14% of wild-type activity and the H366D/V369K mutant retained similar activity as the V369K mutant (Table 1). Western-blot analysis showed that K240R, K240C, and V369K mutants supported the synthesis of wild-type levels of G6PT proteins and H366D and H366D/V369K mutants supported the synthesis of increased levels of G6PT proteins (Fig. 2A). Taken together, our results show that the structural requirements of G6PT and UhpT are different.
Fig. 1.
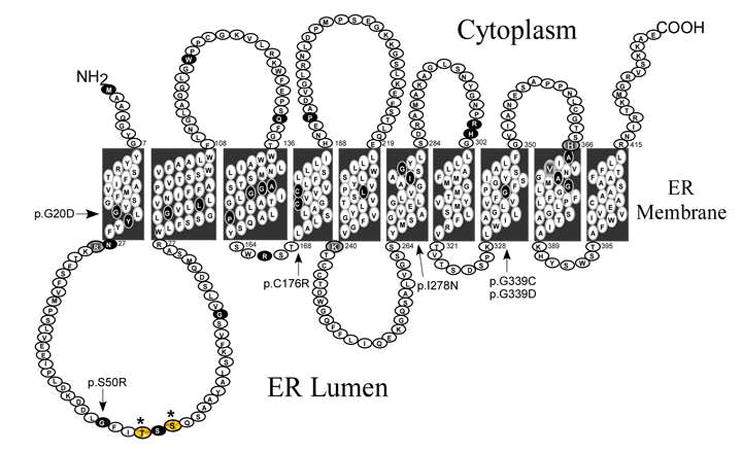
The 10-transmembrane helical structure of human G6PT. The locations of missense mutations identified in the SLC37A4 gene of GSD-Ib patients are shown in black. The mutations, p.G20D in helix 1, p.S50R in luminal loop 1, p.C176R in helix 4, p.I278N in helix 6, and p.G339C/p.G339D in helix 8 that destabilize the G6PT [20] are indicated by arrows. The Asn-linked glycosylation sites introduced at amino acids T53 and S55 to generate G6PT T53N and S55N mutants, respectively, are indicated by asterisks. R28, K240, H366, and V369 are indicated by shaded larger circles. Amino acids 50 to 71 in luminal loop 1 of the 10-domain model are predicted to constitute helix 2 in the 12-helical model of G6PT [19].
Table 1.
Structure-function analysis of human G6PT
| Ad-G6PT | Microsomal G6P Uptake Activity | Proteoliposomal G6P Uptake Activity | Proteoliposomal Pi Uptake Activity |
|---|---|---|---|
| Mock | 0.085 ± 0.003 | 0.31 ± 0.04 | 9.3 ± 0.3 |
| Wild-type | 0.332 ± 0.002 (100) | 1.52 ± 0.01 (100) | 112.7 ± 0.4 (100) |
| K240C | 0.125 ± 0.006 (16.2) | 0.50 ± 0.02 (15.7) | 28.2 ± 0.5 18.3) |
| K240R | 0.219 ± 0.008 (54.3) | 0.90 ± 0.01 (48.8) | 63.2 ± 1.2 (52.1) |
| H366D | 0.131 ± 0.006 (18.6) | 0.55 ± 0.03 (19.8) | 29.7 ± 0.8 (19.7) |
| V369K | 0.104 ± 0.003 (7.7) | 0.48 ± 0.03 (14) | 19.1 ± 0.3 (9.5) |
| H366D/V369K | 0.105 ± 0.004 (8.1) |
Microsomal G6P uptake activity (nmol/mg microsomal protein/3 min) was analyzed in microsomes isolated from COS-1 cells co-infected with Ad-G6Pase-α and Ad-G6PT or an Ad-G6PT mutant. Proteoliposomal G6P or Pi uptake activity (nmol/mg proteoliposomal protein/9 min) was analyzed in proteoliposomes reconstituted from detergent solubilized COS-1 microsomal membrane extracts expressing a wild-type or a mutant Ad-G6PT and were loaded with 50 mM Pi as described under Materials and methods. Results shown are from 3 independent experiments, each point determined in triplicate. Values represent mean ± SEM. Numbers in parentheses represent the percentage of wild-type transport activity.
Fig. 2.
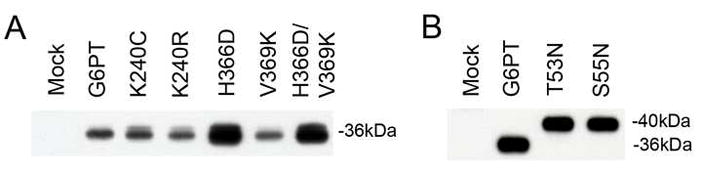
Western-blot analysis of G6PT synthesis in COS-1 cells. COS-1 cells were co-infected with Ad-G6Pase-α and a wild-type or a mutant Ad-G6PT construct as described under Materials and methods. Mock-infected cultures were used as controls. Each lane contained 20 μg of microsomal proteins. The G6PT proteins were visualized with an anti-human G6PT luminal loop 1 antibody [20]. Similar results were obtained using COS-1 microsomes infected only with a wild-type or a mutant Ad-G6PT construct.
Human G6PT contains10-transmembrane helices
Protease protection and glycosylation scanning studies suggest that G6PT is anchored in the ER by 10 domains [7]. The homology modeling, however, proposed that G6PT contains 12 helices [19]. The main difference between the two topological models of G6PT is that residues 50 to 71, which constitute helix 2 in the 12-domain model, are situated in a 51-residue luminal loop 1 in the 10-domain model (Fig. 1). The 10-domain model was supported by studies showing that an introduced glycosylation site at either residues 53–55 (N53SS) in G6PT-T53N or at residues 55–57 (N55QS) in G6PT-S55N (Fig. 1) was utilized [7]. To support the 12-domain model, Almqvist et al [19] suggested that the G6PT-T53N and G6PT-S55N mutants were probably misfolded in the ER membrane.
Western-blot analysis showed that G6PT T53N and S55N mutants supported the synthesis of wild-type levels of G6PT proteins (Fig. 2B), indicating that these mutations do not destabilize G6PT. Protease sensitivity assays were then used to examine whether the T53N and S55N G6PT mutants are similarly folded in the ER membrane as the wild-type transporter. To do this we generated additional T53N and S55N constructs carrying either an N-terminal Flag-tag (G6PT-T53N-5Flag or G6PT-S55N-5Flag) or a C-terminal Flag-tag (G6PT-T53N-3Flag or G6PT-S55N-3Flag). Intact microsomes expressing either wild-type or mutant G6PT were subjected to limited trypsin digestion. The resulting digests were then analyzed by Western-blot using an antibody against amino acids 25 to 79 in G6PT [20], encompassing the entire luminal loop 1 in the 10-domain model (Fig. 1) or a monoclonal antibody against the Flag-tag. Using the anti-G6PT loop 1 antibody, G6PT wild-type and S55N (or T53N) mutant migrated as 36-kDa and 40-kDa polypeptides, respectively and after trypsin digestion migrated as 16-kDa and 23-kDa polypeptides, respectively (Fig. 3A). We have previously shown that the G6PT S55N and T53N mutants directed the synthesis of a 36-kDa polypeptide in the presence of glycosylation inhibitor tunicamysin [7]. Consistent with this, Western blot analysis using the anti-G6PT loop 1 antibody showed that the G6PT-S55N mutant synthesized in the presence of tunicamycin migrated as a 36-kDa polypeptide and after trypsin digestion, a 16-kDa polypeptide (Fig. 3B) similar to wild-type G6PT.
Fig. 3.
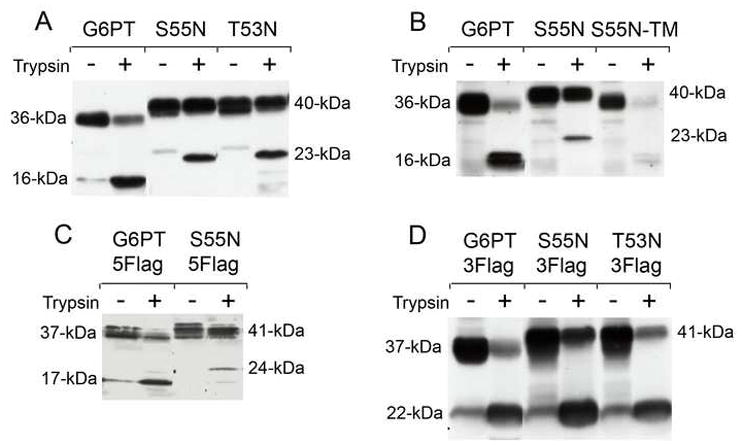
Protease sensitivity of G6PT wild-type and glycosylation mutants. Intact microsomes having a latency value of 95% or greater were isolated from COS-1 cells infected with Ad-G6PT, Ad-G6PT-S55N, Ad-G6PT-T53N, Ad-G6PT-5Flag, Ad-G6PT-S55N-5Flag, Ad-G6PT-3Flag Ad-G6PT-T53N-3Flag or Ad-G6PT-S55N-3Flag. Intact microsomes were also isolated from COS-1 cells infected with Ad-G6PT or Ad-G6PT-S55N after incubated at 37 °C for 48 h in the presence of tunicamycin (1 μg/ml). The microsomal preparations were thensubjected to limited trypsin digestion as described under Materials and methods and the resulting digests analyzed by Western-blot. (A) (B) The G6PT proteins visualized with an anti-G6PT luminal loop 1 antibody [20]; and (C) (D) The G6PT proteins visualized with an anti-Flag monoclonal antibody.
Using the anti-Flag antibody, wild-type G6PT-5Flag and mutant S55N-5Flag migrated as 37-kDa and 41-kDa polypeptides, respectively and as 17-kDa and 24-kDa polypeptides, respectively, after tryspin digestion (Fig. 3C). While wild-type G6PT-3Flag and mutant S55N-3Flag (or T53N-3Flag) migrated as 37-kDa and 41-kDa polypeptides, respectively in the absence of trypsin digestion, all migrated as a 22-kDa polypeptide after trypsin digestion (Fig. 2D), indicating that the C-terminal tryptic peptides of wild-type and mutant G6PT are identical.
Discussion
G6PT [9,14], GlpT [11], and UhpT [12] are members the organophosphate:Pi antiporter family of the major facilitator superfamily [10]. G6PT was shown to contain 10 transmembrane helices by protease protection and glycosylation scanning analysis [7]. However, recent homology modeling [19] based on the crystal structural of GlpT [17,18] predicts that G6PT contains 12 helices. Homology modeling [19] also proposes that amino acids essential for the activity of UhpT may play vital role in G6PT. In this study, we conducted structure-function studies of G6PT to determine if the predictions on which residues are critical for the activity of UhpT are pertinent to the structure of G6PT. We also re-examined the topology of G6PT. We show that structural requirements of G6PT and UhpT differ and that G6PT wild-type and glycosylation mutants exhibit similar sensitivity towards limited trypsin digestion. Taken together, the results support the 10 domain model of G6PT.
Both G6PT and UhpT are Pi-linked antiporters [13,14]. Mutagenesis studies have shown that in UhpT, R46, R275, D388 and K391 are essential residues [15,16]. R46 and R275 are proposed to be involved in substrate binding [15], and D388 and K391 in intrahelical salt bridge formation [16]. The corresponding residues in G6PT are R28, K240, H366, and V369. Homology modeling predicted that R28 and K240 in G6PT are 10.1 Å apart that could form part of the substrate-binding site [19]. We show that with the exception of R28 which is an essential residue in G6PT, K240, D388 and K391 are not, indicating that the structural requirements of G6PT and UhpT differed.
The main difference between the two topological models of G6PT is that residues 50 to 71, which constitute helix 2 in the 12-domain model [19], are situated in a 51-residue luminal loop 1 in the 10-domain model (Fig. 1). The glycosylation scanning study showed that the two G6PT mutants, T53N and S55N that generate potential glycosylation sites at N53SS and N55QS, respectively were utilized, which could only occur if the glycosylation sites were in the 51-residue luminal loop 1 in the 10-domain model [7] but not in helix-2 in the 12-domain model [19]. To explain the inconsistence to the 12-domain model G6PT, Almqvist et al [19] suggested that the two G6PT glycosylation mutants are probably misfolded in the ER membrane. Studies have shown that glycosylation of membrane proteins is critical for their membrane targeting and folding [27,28], and incorrect glycosylation can cause misfolding and preferential degradation of the misfolded proteins [29,30]. If the T53N and S55N mutants were misfolded in the ER membrane then they should exhibit differential stability and protease sensitivity as compared to the wild-type transporter. Our results show the contrary. First, the T53N and S55N mutants supported the synthesis of wild-type levels of G6PT protein in COS-1 cells, indicating that the mutations failed to destabilize mutant G6PT proteins. Secondly, limited trypsin digestion of wild-type G6PT produced an N-terminal polypeptide of 16-kDa and a C-terminal polypeptide of 22-kDa and limited trypsin digestion of G6PT T53N or S55N mutants produced an N-terminal polypeptide of 23-kDa and a C-terminal polypeptide of 22-kDa. However, in the presence of tunicamycin, limited trypsin digestion of G6PT S55N produced an N-terminal polypeptide of 16-kDa, indicating that the apparent difference in mobility between wild-type and mutant G6PT resulted from the added oligosaccharide side chains in the glycosylation mutants. Therefore, the introduced glycosylation site at either N53SS or N55QS in G6PT is utilized and the secondary structure of the glycosylated G6PT proteins is indistinguishable from the wild-type G6PT. Taken together, our data demonstrate that the glycosylation of G6PT does not cause G6PT to be misfolded in the ER membrane, again confirming the 10-transmembrane domain model of G6PT.
In summary, we demonstrate that the structural requirements of the two antiporters, G6PT and UhpT, are different and that G6PT is anchored in the ER by 10 transmembrane domains in contrast to the 12 domains used by UhpT.
Acknowledgments
This research was supported in part by the Intramural Research Programs of the NICHD, NIH.
The abbreviations used are
- GSD-Ib
glycogen storage disease type Ib
- G6PT
glucose-6-phosphate transporter
- ER
endoplasmic reticulum
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Chou JY, Matern D, Mansfield BC, Chen YT. Type I glycogen storage diseases: disorders of the glucose-6-phosphatase complex. Curr Mol Med. 2002;2:121–143. doi: 10.2174/1566524024605798. [DOI] [PubMed] [Google Scholar]
- 2.Chou JY, Mansfield BC. Glucose-6-phosphate transporter: the key to glycogen storage disease type Ib. In: Broer S, Wagner CA, editors. Membrane Transporter Diseases. Kluwe Academic/Plenum Publishers; New York: 2003. pp. 191–205. [Google Scholar]
- 3.Shieh JJ, Pan CJ, Mansfield BC, Chou JY. Glucose-6-phosphate hydrolase, widely expressed outside the liver, can explain age-dependent resolution of hypoglycemia in glycogen storage disease type Ia. J Biol Chem. 2003;278:47098–47103. doi: 10.1074/jbc.M309472200. [DOI] [PubMed] [Google Scholar]
- 4.Ghosh A, Shieh JJ, Pan CJ, Chou JY. Histidine-167 is the phosphate acceptor in glucose-6-phosphatase-β forming a phosphohistidine-enzyme intermediate during catalysis. J Biol Chem. 2004;279:12479–12483. doi: 10.1074/jbc.M313271200. [DOI] [PubMed] [Google Scholar]
- 5.Cheung YY, Kim SY, Yiu WH, Pan CJ, Jun HS, Ruef RA, Lee EJ, Westphal H, Mansfield BC, Chou JY. Impaired neutrophil activity and increased susceptibility to bacterial infection in mice lacking glucose-6-phosphatase-β. J Clin Invest. 2007;117:784–793. doi: 10.1172/JCI30443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim SY, Jun HS, Mead PA, Mansfield BC, Chou JY. Neutrophil stress and apoptosis underlie myeloid dysfunction in glycogen storage disease type Ib. Blood. 2008;111:5704–5711. doi: 10.1182/blood-2007-12-129114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pan CJ, Lin B, Chou JY. Transmembrane topology of human Glucose-6-phosphate transporter. J Biol Chem. 1999;274:13865–13869. doi: 10.1074/jbc.274.20.13865. [DOI] [PubMed] [Google Scholar]
- 8.Hoffman K, Stoffel W. TMbase – a database of membrane spanning protein segments. Biol Chem Hoppe-Seyler. 1993;347:166–170. [Google Scholar]
- 9.Gerin I, Veiga-da-Cunha M, Achouri Y, Collet J-F, Van Schaftingen E. Sequence of a putative glucose-6-phosphate translocase, mutated in glycogen storage disease type 1b. FEBS Lett. 1997;419:235–238. doi: 10.1016/s0014-5793(97)01463-4. [DOI] [PubMed] [Google Scholar]
- 10.Pao SS, Paulsen IT, Saier MH., Jr Major facilitator superfamily. Microbiol Mol Biol Rev. 1998;62:1–34. doi: 10.1128/mmbr.62.1.1-34.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goldrick D, Yu GQ, Jiang SQ, Hong JS. Nucleotide sequence and transcription start point of the phosphoglycerate transporter gene of Salmonella typhimurium. J Bacteriol. 1988;179:3421–3426. doi: 10.1128/jb.170.8.3421-3426.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maloney PC, Ambudkar SV, Anatharam V, Sonna LA, Varadhachary A. Anion-exchange mechanisms in bacteria. Microbiol Rev. 1990;54:1–17. doi: 10.1128/mr.54.1.1-17.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ambudkar SV, Larson TJ, Maloney PC. Reconstitution of sugar phosphate transport systems of Escherichia coli. J Biol Chem. 1986;261:9083–9086. [PubMed] [Google Scholar]
- 14.Chen SY, Pan CJ, Krishnamachary N, Mansfield BC, Ambudkar SV, Chou JY. The Glucose-6-phosphate transporter is a phosphate-linked antiporter deficient in glycogen storage disease type Ib and Ic. FASEB J. 2008;22:2206–2213. doi: 10.1096/fj.07-104851. [DOI] [PubMed] [Google Scholar]
- 15.Fann MC, Davies AH, Varadhachary A, Kuroda T, Sevier C, Tsuchiya T, Maloney PC. Identification of two essential arginine residues in UhpT, the sugar phosphate antiporter of Escherichia coli. Membrane Biol. 1998;164:187–195. doi: 10.1007/s002329900404. [DOI] [PubMed] [Google Scholar]
- 16.Hall JA, Fann MCPC. Maloney, altered substrate selectivity in a mutant of an intrahelical salt bridge in UhpT, the sugar phosphate carrier of Escherichia coli. J Biol Chem. 1999;274:6148–6153. doi: 10.1074/jbc.274.10.6148. [DOI] [PubMed] [Google Scholar]
- 17.Lemieux MJ, Song J, Kim MJ, Huang Y, Villa A, Auer M, Li XD, Wang DN. Three-dimensional crystallization of the Escherichia coli glycerol-3-phosphate transporter: a member of the major facilitator superfamily. Protein Sci. 2003;12:2748–2756. doi: 10.1110/ps.03276603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang Y, Lemieux MJ, Song J, Auer M, Wang DN. Structure and mechanism of the glycerol-3-phosphate transporter from Escherichia coli. Science. 2003;301:616–620. doi: 10.1126/science.1087619. [DOI] [PubMed] [Google Scholar]
- 19.Almqvist J, Huang Y, Hovmöller S, Wang DN. Homology modeling of the human microsomal glucose 6-phosphate transporter explains the mutations that cause the glycogen storage disease type Ib. Biochemistry. 2004;43:9289–9297. doi: 10.1021/bi049334h. [DOI] [PubMed] [Google Scholar]
- 20.Chen LY, Pan CJ, Shieh JJ, Chou JY. Structure-function analysis of the glucose-6-phosphate transporter deficient in glycogen storage disease type Ib. Hum Mol Genet. 2002;11:3199–3207. doi: 10.1093/hmg/11.25.3199. [DOI] [PubMed] [Google Scholar]
- 21.Hardy S, Kitamura M, Harris-Stansil T, Dai Y, Phipps ML. Construction of adenovirus vectors through Cre-lox recombination. J Virol. 1997;71:1842–1849. doi: 10.1128/jvi.71.3.1842-1849.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ghosh A, Shieh JJ, Pan CJ, Sun MS, Chou JY. The catalytic center of glucose-6-phosphatase: His176 is the nucleophile forming the phosphohistidine-enzyme intermediate during catalysis. J Biol Chem. 2002;277:32837–32842. doi: 10.1074/jbc.M201853200. [DOI] [PubMed] [Google Scholar]
- 23.Berteloot A, Vidal H, van de Werve G. Evidence for a membrane exchangeable glucose pool in the functioning of rat liver glucose-6-phosphatase. J Biol Chem. 1991;266:5497–5507. [PubMed] [Google Scholar]
- 24.Schaffner W, Weissmann C. A rapid, sensitive, and specific method for the determination of protein in dilute solution. Anal Biochem. 1973;56:502–514. doi: 10.1016/0003-2697(73)90217-0. [DOI] [PubMed] [Google Scholar]
- 25.Ambudkar SV, Zlotnick GW, Rosen BP. Calcium efflux from Escherichia coli. Evidence for two systems. J Biol Chem. 1984;259:6142–6146. [PubMed] [Google Scholar]
- 26.Nordlie RC, Sukalski KA. Multifunctional glucose-6-phosphatase: a critical review. In: Martonosi AN, editor. The Enzymes of Biological Membranes. 2. Plenum Press; New York: 1985. pp. 349–398. [Google Scholar]
- 27.Parodi AJ. Protein glucosylation and its role in protein folding. Annu Rev Biochem. 2000;69:69–93. doi: 10.1146/annurev.biochem.69.1.69. [DOI] [PubMed] [Google Scholar]
- 28.Goder V, Spiess M. Topogenesis of membrane proteins: determinants and dynamics. FEBS Lett. 2001;504:87–93. doi: 10.1016/s0014-5793(01)02712-0. [DOI] [PubMed] [Google Scholar]
- 29.Molinari M. N-glycan structure dictates extension of protein folding or onset of disposal. Nat Chem Biol. 2007;3:313–320. doi: 10.1038/nchembio880. [DOI] [PubMed] [Google Scholar]
- 30.Caramelo JJ, Parodi AJ. How sugars convey information on protein conformation in the endoplasmic reticulum. Semin Cell Dev Biol. 2007;18:732–742. doi: 10.1016/j.semcdb.2007.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]