

Abstract
A concise approach to the tetrahydroxanthone natural products has been developed employing vinylogous addition of siloxyfurans to benzopyryliums and a late stage Dieckmann cyclization. Using this methodology, chiral, racemic forms of the natural products blennolides B and C have been synthesized in a maximum of four steps from a 5-hydroxychromone substrate. The regio- and diastereoselectivity of vinylogous additions was probed using computational studies which suggest involvement of Diels-Alder-like transition states.
Tetrahydroxanthones are a class of mycotoxins1 bearing both monomeric and dimeric frameworks. The recently isolated tetrahydroxanthones blennolides A (1) and B (2) (Figure 1)2 are monomer units of the antitumor agents secalonic acids B (3) and D (4),3 the latter which exhibits antibacterial, cytostatic, and anti-HIV properties.4 Blennolide C (5), the methyl isomer of 1, and the antifungal agent parnafungin A (6)5 also possess the characteristic dihydro-2H-xanthenone framework found in many tetrahydroxanthones. Related, isomeric natural products including paecilin B (7) (stereochemistry unassigned) containing the isomeric chromone lactone moiety have also been reported.6 Recently, Bräse and Nicolaou have reported elegant approaches to blennolide C (5) and the related natural product diversonol employing biomimetic construction of the tetrahydroxanthone core.7 Herein, we describe a concise approach to racemic blennolides and related tetrahydroxanthones employing a “retrobiomimetic” process8 involving vinylogous addition of siloxyfurans to benzopyryliums.
Figure 1.

Tetrahydroxanthones and related natural products.
Biosynthetically, the blennolides appear to be derived from a sequence involving oxidation of benzophenone ester 8, oxa-Michael addition, and reduction to dihydro-2H-xanthenone 9 (Figure 2, (a)).9 The chromone lactone structure 10 found in paecilin B (7)6 appears to be derived from hydrolysis/lactonization of the tetrahydroxanthone framework.6b,c We envisioned that precursor 11 may be obtained by vinylogous addition of siloxyfurans10 to activated benzopyrylium salts 12.11 Conjugate reduction of butenolide 11 should afford chromone lactone 10. The last step in the sequence entails a “retrobiomimetic” transformation8 in which tetrahydroxanthones 9 may be produced by Dieckmann cyclization7k of chromone lactones 10.
Figure 2.

Biosynthetic pathway vs. “retrobiomimetic” synthesis.
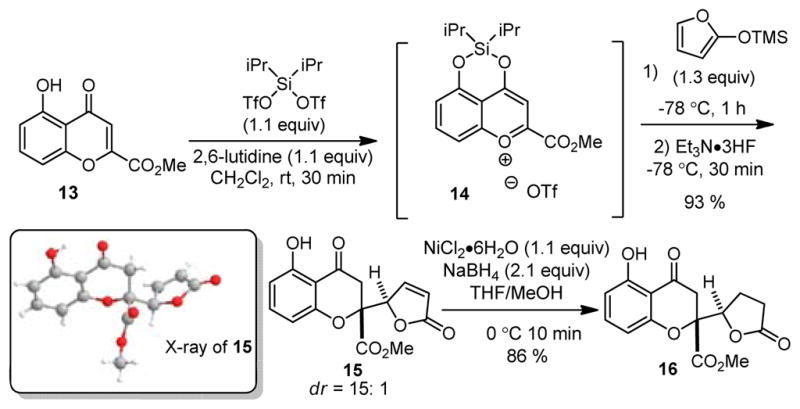
We initiated our study by treating the readily available 5-hydroxychromone 1312,13 with a number of Lewis acids in an effort to promote vinylogous addition of 2-trimethylsilyloxy furan (Scheme 1). Unfortunately, in our initial experiments we did not observe substantial adduct formation. In light of the high reactivity of 4-siloxy-1-benzopyrylium salts towards carbon nucleophiles,11a,b we focused our efforts on silyl triflate activation of chromone 13. In particular, we reasoned that dialkylsilyl ditriflate reagents, generally used to protect diols as silylenes,14 may directly afford activated siloxybenzopyrylium species. In the event, treatment of 13 with diisopropyl silyl ditriflate in the presence of 2,6-lutidine led to formation of benzopyrylium 14.13 Treatment of 14 with 2-trimethylsiloxy furan at −78 °C cleanly led to formation of chromone butenolide 15 (dr = 15: 1) after desilylation with Et3N·3HF. Crystallization of 15 facilitated X-ray crystal structure analysis of the major diastereomer.13 Finally, conjugate reduction of butenolide 15 with nickel boride15 provided chromone lactone 16.
Scheme 1.
Model Studies
We next evaluated the effect of time and temperature for vinylogous additions (Table 1). Interestingly, increased reaction temperature led to reduced diastereoselectivities in additions to 14 leading to a preference for diastereomer 17 at higher temperature (cf. entries 1 and 2). Conducting the reaction at 0 °C for 0.5 h (entry 3) led to an inseparable 3: 1 mixture of 15: 17 which supports epimerization at higher temperatures and longer reaction times (vide infra).16 Similar results were obtained for vinylogous additions to chromone 18 leading to adducts 19 and 20 (entries 5 and 6). Addition of 4-methyl-2-trimethylsiloxyfuran to 14 (entry 7) led to reduced diastereoselectivities (2: 1) in comparison to 2-trimethylsiloxyfuran (dr = 15: 1, cf. Scheme 1). Based on our experimental data, we propose the generalized mechanism shown in Scheme 2. Initial vinylogous addition of 2-trimethylsiloxyfuran to 14 at −78 °C leads to the kinetic adduct 23 which may lose TMSOTf to afford silylene 24, a precursor to chromone 15. At higher temperature, thermodynamic equilibration of 23 to 25 may occur by butenolide enolization16b through silylated intermediate 26. The equilibration process was confirmed by 1H NMR studies.13 Computational studies indicate that adduct 27 is approximately 1 kcal/mol more stable than diastereomer 24.13
Table 1.
Evaluation of Time and Temperature
 | ||||||
|---|---|---|---|---|---|---|
| entry | R1 | R2 | temp (°C) | time (h) | yield (%)a | ratiob |
| 1 | H | H | −30 | 3 | 95 | 3: 1 (15: 17) |
| 2 | H | H | 0 | 3 | 90 | 1: 2 (15: 17) |
| 3 | H | H | 0 | 0.5 | 91 | 3: 1 (15: 17) |
| 4 | H | H | 40 | 3 | 64 | 1: 2 (15: 17) |
| 5 | Me | H | −78 | 1 | 96 | 20: 1(19: 20) |
| 6 | Me | H | 0 | 3 | 97 | 1: 2 (19: 20) |
| 7 | H | Me | −78 | 1 | 89 | 2: 1 (21: 22) |
| 8 | H | Me | 0 | 3 | 85 | 1: 2 (21: 22) |
Yield determined based on crude H-NMR analysis using 1,3,5-trimethoxybenzene as internal standard.
Ratio determined based on crude 1H-NMR analysis.
Scheme 2.

Proposed Mechanism for Vinylogous Addition
In order to understand the observed regio- and diastereoselectivity, we employed DFT methods to model the reaction of benzopyrylium 14 with 2-trimethylsiloxy furan.13 FMO analyses showed that C2 of 14 and C5 of the siloxyfuran should be the most reactive sites (Figure 3). Thirteen candidate transition state structures were generated by conformational variation about the nascent C2-C5 bond and were optimized at the B3LYP/6-31G(d) level of theory.13 Similar transition states have been proposed for vinylogous Mukaiyama aldol reactions.17 The lowest energy Re-Si (or Si-Re) structure (TS-A in Figure 4) which leads to the observed major product bears striking resemblance to an asynchronous endo [4+2] transition state; other TS candidates of like stereochemistry were greater than 4.3 kcal/mol higher in energy. The most favorable Re-Re (or Si-Si) TS structure (TS-B) is 2.68 kcal/mol above TS-A, consistent with the stereochemistry observed for the minor, kinetic product. With 4-methyl-2-trimethylsiloxy furan (cf. Table 1, entry 7), TS structures similar to TS-A would be disfavored by steric factors, thus explaining the loss of diastereoselectivity.
Figure 3.
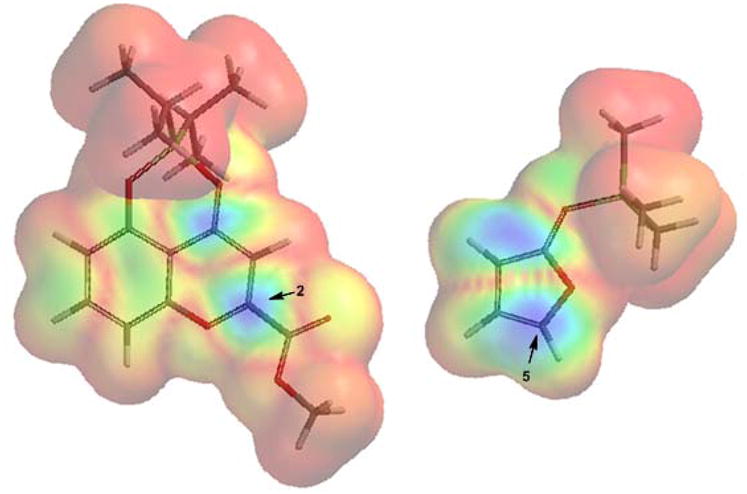
Composite surfaces of the B3LYP/6-31G(d) LUMO of 14 and HOMO of 2-trimethylsiloxyfuran, mapped onto an electron density isosurface using Spartan ‘08. Reactive sites are shown in blue.
Figure 4.
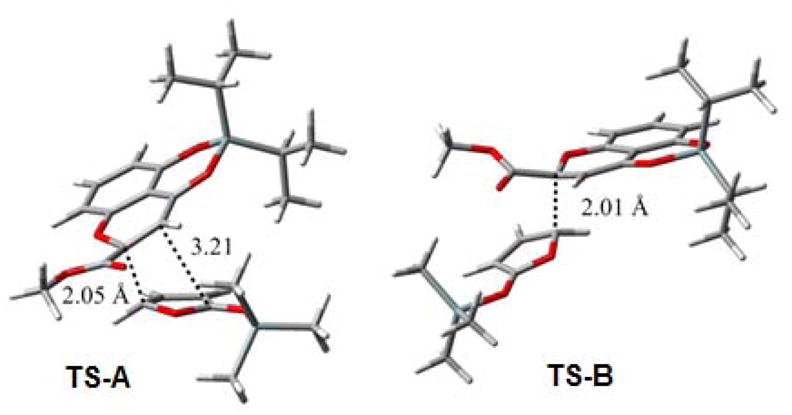
Lowest energy transition state structures for Re-Si (TS-A) and Re-Re (TS-B) addition in the reaction of 14 and 2-trimethylsiloxyfuran.
Having achieved the synthesis of chromone lactone structures, we next turned our attention to Dieckmann-type cyclizations (Scheme 3).7k,18 Treatment of 16 with NaOMe in MeOH led exclusively to the ring-opened hydroxy ester 28. Gratifyingly, we found that treatment of 16 with NaOMe in THF19 led to observable precipitation to a presumed dianion intermediate and formation of dihydro-2H-xanthenones 29 and 30 after workup. After evaluating several bases, NaH was found to be superior to NaOMe to afford 29/30 in 67 % yield (dr = 20: 1 by 1H NMR). In order to evaluate the cyclization on the diastereomer of 16, we subjected a mixture of butenolides 17 and 15 (2: 1) to conjugate reduction (NiB2) which afforded an inseparable mixture of 31 and 16 in a 2: 1 ratio. Subsequent Dieckmann cyclization (NaH/THF) afforded 30 and 29 in a 1: 2 ratio. These studies support a mechanism for equilibration to favor the syn hydroxy configuration in 29 as shown in Scheme 5.5,6b,c,20 Enolate 32 derived from 31 may condense with the lactone to form tetrahedral intermediate 33. After ring-opening, the resulting dianion 34 (a precursor to 30) may equilibrate by retro-Michael addition to 35 which may be followed by oxa-Michael addition7a,b to provide diastereomer 36 and thence 29 after workup.
Scheme 3.

Dieckmann Cyclization
Scheme 5.

Proposed Mechanism for Cyclization/Isomerization
After completion of the model studies, we synthesized both (±)-epi-blennolide C (Scheme 6) and (±)-blennolide C (Scheme 7) from butenolides 19 and 20. Conjugate reduction of 19 and 20 (20: 1) using NiB2 led to chromone lactones 37 (dr = 20: 1). Dieckmann cyclization of 37 using NaH/THF led to production of epi-blennolide C 39 (72 % isolated yield). Similar transformations were used to obtain blennolide C (5) from a 2: 1 mixture of 20: 19 via lactone 38. The spectroscopic properties of synthetic 5 and epi-blennolide C 39 were in complete agreement with previously published data.2,7b,c
Scheme 6.

Synthesis of (±)-epi-Blennolide C
Scheme 7.

Synthesis of (±)-Blennolide C
The natural product blennolide B (2)2 has syn, anti stereochemistry of ester, hydroxyl, and methyl groups on the dihydro-2H-xanthenone core. Based on our model studies and the hypothesis that butenolide reduction should occur anti to the 5-substituent,15a we initiated our synthesis from a 2: 1 mixture of butenolides 21 and 22 (Table 1, Entry 7). In this case, nickel boride chemoselectively reduced the butenolide to afford a mixture of four chromone lactone diastereomers. Interestingly, when Rh/Al2O3 was used for conjugate reduction,21 we obtained 40 as a single diastereomer as well as the separable, overreduced hydroxyl chromone lactones 41 and 42. Alcohols 41 and 42 could be reoxidized to lactones 40 and 44, respectively, using the Bobbitt reagent 43 (50 wt% on SiO2).22 The stereochemistry of chromone lactones 40 and 44 were confirmed by X-ray crystal structure analyses (Figure 5). NMR data for chromone lactones 40 and 44 were not in agreement with data reported for paecilin B6 (Figure 1) indicating that 7 is a diastereomer of both 40 and 44. Treatment of 40 with NaH in THF afforded (±)-blennolide B (2) whose spectroscopic properties were identical to reported data.2,13 Interestingly, cyclization of chromone lactone 44 (NaH) led to the isolation of (±)-blennolide B (73 %) with negligible amounts of diastereomer 45 observed in the crude 1H NMR spectrum. This result further supports the isomerization process shown in Scheme 5 which in this case likely occurs due to unfavorable repulsion between the ester and methyl groups in diastereomer 45.13
Figure 5.
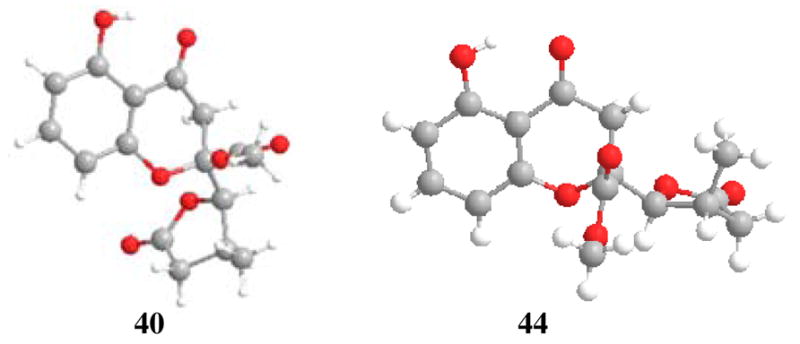
X-ray crystal structures of compounds 40 and 44.
In conclusion, we have developed a concise and “retrobiomimetic” approach to tetrahydroxanthones employing vinylogous addition of siloxyfurans to benzopyryliums as a key step. The regio- and diastereoselectivity of vinylogous additions was probed using computational studies which suggest involvement of Diels-Alder-like transition state. Using this methodology, the natural products (±)-blennolides B and C were synthesized in a maximum of 4 steps from readily available 5-hydroxychromones. Further studies, including development of an asymmetric variant of the vinylogous addition, are currently under investigation and will be reported in due course.
Supplementary Material
Scheme 4.

Equilibration of Dieckmann products
Scheme 8.

Synthesis of (±)-Blennolide B
Acknowledgments
We thank the National Institutes of Health (GM-073855, J.A.P., Jr.) and the National Science Foundation (CHE-0910826, R.P.J.) for research support and Drs. Jeff Bacon (Boston University) and Emil Lobkovsky (Cornell University) for X-ray crystal structure analyses. We also thank the NSF (CHE-0443618) for the high resolution mass spectrometer used in this work.
Footnotes
Supporting Information Available: Experimental procedures, mechanistic studies, calculation studies, complete ref. 5a, and characterization data for all new compounds described herein, CIF files for compounds 15, 40, and 44. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Bräse S, Encinas A, Keck J, Nising CF. Chem Rev. 2009;109:3903. doi: 10.1021/cr050001f. [DOI] [PubMed] [Google Scholar]
- 2.Zhang W, Krohn K, Ullah Z, Flörke U, Pescitelli G, Di Bari L, Antus S, Kurtán T, Rheinheimer J, Draeger S, Schulz B. Chem Eur J. 2008;14:4913. doi: 10.1002/chem.200800035. [DOI] [PubMed] [Google Scholar]
- 3.(a) Franck B, Gottschalk EM, Ohnsorge U, Baumann G. Angew Chem Int Ed Engl. 1964;3:441. [Google Scholar]; (b) Franck B, Baumann G, Ohnsorge U. Tetrahedron Lett. 1965;6:2031. doi: 10.1016/s0040-4039(00)90148-5. [DOI] [PubMed] [Google Scholar]; (c) Franck B, Gottschalk EM, Ohnsorge U, Hüper F. Chem Ber. 1966;99:3842. [Google Scholar]; (d) Steyn PS. Tetrahedron. 1970;26:51. doi: 10.1016/0040-4020(70)85006-2. [DOI] [PubMed] [Google Scholar]
- 4.(a) Stoll A, Renz J, Brack A. Helv Chim Acta. 1952;35:2022. [Google Scholar]; (b) Kurobane I, Iwahashi S, Fukuda A. Drugs Exp Clin Res. 1987;13:339. [PubMed] [Google Scholar]; (c) McPhee F, Caldera PS, Bemis GW, McDonagh AF, Kuntz ID, Craik CS. Biochem J. 1996;320:681. doi: 10.1042/bj3200681. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Liao G, Zhou J, Wang H, Mao Z, Xiao W, Wang H, She Z, Zhu Y. Oncol Rep. 2010;23:387. [PubMed] [Google Scholar]
- 5.Parish CA, et al. J Am Chem Soc. 2008;130:7060. doi: 10.1021/ja711209p.For synthetic studies, see: Zhou Q, Snider BB. Org Lett. 2009;11:2936. doi: 10.1021/ol901054r.Zhou Q, Snider BB. J Org Chem. 2010;75:8224. doi: 10.1021/jo101826p.
- 6.Guo Z, She Z, Shao C, Wen L, Liu F, Zheng Z, Lin Y. Magn Reson Chem. 2007;45:777. doi: 10.1002/mrc.2035.Xanthonquinodins: Tabata N, Tomada H, Matsuzaki K, Ōmura S. J Am Chem Soc. 1993;115:8558.Tabata N, Tomoda H, Iwai Y, Ōmura S. J Antibiotics. 1996;49:267. doi: 10.7164/antibiotics.49.267.Chaetomanone: Kanokmedhakul S, Kanokmedhakul K, Phonkerd N, Soytong K, Kongsaeree P, Suksamrarn A. Planta Med. 2002;68:834. doi: 10.1055/s-2002-34415.Noduliprevenone: Pontius A, Krick A, Kehraus S, Foegen SE, Müller M, Klimo K, Gerhäuser C, König GM. Chem Eur J. 2008;14:9860. doi: 10.1002/chem.200801574.
- 7.Nising CF, Ohnemüller UK, Bräse S. Angew Chem Int Ed. 2006;45:307. doi: 10.1002/anie.200502913.Nicolaou KC, Li A. Angew Chem Int Ed. 2008;47:6579. doi: 10.1002/anie.200802632.Gérard EMC, Bräse S. Chem Eur J. 2008;14:8086. doi: 10.1002/chem.200801507.Additional synthetic studies: Franck B, Stöckigt J, Zeidler U, Franckowiak G. Chem Ber. 1973;106:1198.Gabbutt CD, Hepworth JD, Urquhart MWJ, Vazquez de Miguel LM. J Chem Soc Perkin Trans. 1997;1:1819.Letcher RM, Yue TY, Chiu KF, Kelkar AS, Cheung KK. J Chem Soc Perkin Trans. 1998;1:3267.Lesch B, Bräse S. Angew Chem Int Ed. 2004;43:115. doi: 10.1002/anie.200352154.Ohnemüller UK, Nising CF, Nieger M, Bräse S. Eur J Org Chem. 2006:1535.Nising CF, Friedrich A, Bräse S. Synlett. 2007:2987.Ohnemüller UK, Nising CF, Encinas A, Bräse S. Synthesis. 2007:2175.Tietze LF, Spiegl DA, Stecker F, Major J, Raith C, Große C. Chem Eur J. 2008;14:8956. doi: 10.1002/chem.200800967.Tatsuta K, Yoshihara S, Hattori N, Yoshida S, Hosokawa S. J Antibiotics. 2009;62:469. doi: 10.1038/ja.2009.52.Volz N, Bröhmer MC, Nieger M, Bräse S. Synlett 2009, 550 and further reference within.
- 8.For “retrobiomimetic” synthesis, see: Andriamialisoa RZ, Langlois N, Langlois Y. J Org Chem. 1985;50:961. doi: 10.1021/jo01336a006.Ebner T, Rebell J, Fischer P, Meese CO. J Lab Comp Radiopharm. 1989;27:485.Martynow JG, Kirst HA. J Org Chem. 1994;59:1548.
- 9.(a) Kurobane I, Vining LC, McInnes AG, Walter JA, Wright JLC. Tetrahedron Lett. 1978;19:1379. [Google Scholar]; (b) Kurobane I, Vining LC, McInnes AG. J Antibiotics. 1979;32:1256. doi: 10.7164/antibiotics.32.1256. [DOI] [PubMed] [Google Scholar]
- 10.For recent review on vinylogous additions of siloxyfurans, see: Casiraghi G, Zanardi F, Appendino G, Rassu G. Chem Rev. 2000;100:1929. doi: 10.1021/cr990247i.Casiraghi G, Zanardi F, Battistini L, Rassu G. Synlett. 2009:1525.
- 11.For addition of carbon nucleophiles to 4-siloxy-1-benzopyrylium salts, see: Ohkata K, Ishimaru K, Lee YG, Akiba K. Chem Lett. 1990:1725.Lee YG, Ishimaru K, Iwasaki H, Ohkata K, Akib K. J Org Chem. 1991;56:2058.For vinylogous addition of siloxyfurans to isoquinolinium salts, see: Hermange P, Tran Huu Dau ME, Retaileau P, Dodd RH. Org Lett. 2009;11:4044. doi: 10.1021/ol901452d.
- 12.Wu L, Lal J, Simon KA, Burton EA, Luk YY. J Am Chem Soc. 2009;131:7430. doi: 10.1021/ja9015149. [DOI] [PubMed] [Google Scholar]
- 13.See Supporting Information for complete experimental and computational details.
- 14.Corey EJ, Hopkins PB. Tetrahedron Lett. 1982;23:4871. [Google Scholar]
- 15.Bekish AV, Prokhorevich KN, Kulinkovich OG. Eur J Org Chem. 2006:5069.For a review on the chemistry of nickel boride, see: Khurana JM, Gogia A. Org Prep Proc Int. 1997;29:1.
- 16.For temperature-dependent siloxyfuran additions, see: Wieland LC, Vieira EM, Snapper ML, Hoveyda AH. J Am Chem Soc. 2009;131:570. doi: 10.1021/ja8062315.For isomerization of butenolides, see: Lee H, Kim KW, Park J, Kim H, Kim S, Kim D, Hu X, Yang W, Hong J. Angew Chem Int Ed. 2008;47:4200. doi: 10.1002/anie.200705663.
- 17.López CS, Álvarez R, Vaz B, Niet Faza O, De Lera ÁR. J Org Chem. 2005;70:3654. doi: 10.1021/jo0501339. [DOI] [PubMed] [Google Scholar]
- 18.For a review on the Dieckmann reaction, see: Davis BR, Garratt PJ. In: Comprehensive Organic Synthesis. Trost BM, Fleming I, editors. Vol. 2. Pergamon: Oxford; 1991. p. 806.
- 19.Gerard B, Jones G, II, Porco JA., Jr J Am Chem Soc. 2004;126:13620. doi: 10.1021/ja044798o. [DOI] [PubMed] [Google Scholar]
- 20.Burobane I, Vining LC, McInnes AG. Ger Offen. 1980;41:DE 80-3002761. [Google Scholar]
- 21.For hydrogenation of butenolides using Rh/Al2O3, see: Matsubara J, Nakao K, Hamada Y, Shioiri T. Tetrahedron Lett. 1992;33:4187.Gurjar MK, Cherian J, Ramana CV. Org Lett. 2004;6:317. doi: 10.1021/ol035908j.
- 22.Bobbitt JM. J Org Chem. 1998;63:9367. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.