

Abstract
Orotidine 5′-monophosphate decarboxylase (OMPDC) catalyzes the exchange for deuterium from solvent D2O of the C-6 proton of 1-(β-D-erythrofuranosyl)-5-fluorouracil (FEU), a phosphodianion truncated product analog. The deuterium exchange reaction of FEU is accelerated 1.8 × 104-fold by 1 M phosphite dianion (HPO32−). This corresponds to a 5.8 kcal/mol stabilization of the vinyl carbanion-like transition state, which is similar to the 7.8 kcal/mol stabilization of the transition state for OMPDC-catalyzed decarboxylation of a truncated substrate analog by bound HPO32−. These results show that the intrinsic binding energy of phosphite dianion is used in stabilization of the vinyl carbanion-like transition state common to the decarboxylation and deuterium exchange reactions.
Orotidine 5′-monophosphate decarboxylase (OMPDC) employs no metal ions or other cofactors but yet effects an enormous 1017-fold acceleration of the decarboxylation of orotidine 5′-monophosphate (OMP, Scheme 1A) to give uridine 5′-monophosphate (UMP).1–3 Several mechanisms utilizing different types of covalent or Brønsted acid catalysis by catalytic side chains have been proposed as pathways to avoid formation of the unstable UMP carbanion intermediate of a direct decarboxylation reaction (Scheme 1A). However, it is now known that OMPDC effectively catalyzes the exchange of the C-6 proton of UMP (Scheme 1B, X = H)4 and of 5-fluorouridine 5′-monophosphate (FUMP, Scheme 1B, X = F)5 for deuterium from solvent D2O. The kinetic data for the enzymatic deuterium exchange reaction of UMP show that OMPDC stabilizes a bound UMP carbanion intermediate relative to UMP by at least 14 kcal/mol, compared to the proton transfer reaction in water.4 This provides compelling evidence that the decarboxylation reaction also proceeds through the same enzyme-stabilized UMP carbanion intermediate (Scheme 1B).
Scheme 1.

The interactions between OMPDC and the phosphodianion group of OMP provide a large 12 kcal/mol stabilization of the transition state for enzyme-catalyzed decarboxylation.6 A part of this total 12 kcal/mol of transition state binding energy is utilized in stabilization of the Michaelis complex. However, the binding of exogenous phosphite dianion (HPO32−) to OMPDC results in an 8 × 104-fold increase in kcat/Km for enzyme-catalyzed decarboxylation of the truncated substrate 1-(β-D-erythrofuranosyl)orotic acid (EO) that lacks a 5′-phosphodianion moiety (Scheme 2A). This shows that the phosphodianion binding interactions do not simply anchor OMP to OMPDC, but rather that they are also utilized to activate OMPDC towards catalysis of the decarboxylation reaction.6 Phosphite dianion provides similar activation of the enzyme-catalyzed reactions of truncated substrates for the proton transfer reaction catalyzed by triosephosphate isomerase,7 and the hydride transfer reaction catalyzed by glycerol 3-phosphate dehydrogenase.8 This transmission of binding energy from a nonreactive binding determinant to a distant reaction center is a special property of enzymatic catalysis.9,10 It has not yet been mimicked in the de novo design of protein catalysts, in part because the mechanisms for this utilization of binding energy are not fully understood.
Scheme 2.

The binding interactions between OMPDC and HPO32− may be utilized to introduce destabilizing electrostatic stress between carboxylate side chains at OMPDC and the 6-CO2− group of bound OMP at the ground-state Michaelis complex, which is relieved at a product-like unstressed transition state for the decarboxylation reaction.11,12 However, such ground state effects cannot explain the 8 × 104-fold difference in kcat/Km for the OMPDC-catalyzed reaction of EO in the absence and presence of bound phosphite dianion,6 because this is a comparison of the activation barriers for conversion of unstressed free OMPDC to transition states in which any HPO32−-induced stress has necessarily been relieved.10
We now consider this question: Is the HPO32− binding energy utilized in the stabilization of a UMP-carbanion intermediate, or in the stabilization of some other feature of the transition state for decarboxylation of OMP? The removal of the 6-CO2− group from the pyrimidine ring of EO gives the truncated product analog 1-(β-D-erythrofuranosyl)uracil (EU). Now, the observation that the OMPDC-catalyzed deuterium exchange reaction of EU is also strongly activated by HPO32− would provide compelling evidence that the dianion binding interactions are utilized in stabilization of an enzyme-bound vinyl carbanion intermediate common to both the enzyme-catalyzed decarboxylation and deuterium exchange reactions (Scheme 1). Conversely, the failure to observe strong phosphite activation of the deuterium exchange reaction of EU would suggest that the specificity in the binding of HPO32− to the transition state also involves interactions between OMPDC and the 6-CO2− group of the pyrimidine ring of OMP.
The OMPDC-catalyzed deuterium exchange reaction of EU is calculated to be too slow to detect at room temperature. We therefore examined the OMPDC-catalyzed deuterium exchange reaction of the truncated substrate 1-(β-D-erythrofuranosyl)-5-fluorouracil (FEU, Scheme 2B), where the electron-withdrawing 5-F provides a large stabilization of the carbanion-like transition state.5 The preparation of FEU is described in Supporting Information.
The exchange for deuterium of the C-6 proton of h-FEU to give d-FEU catalyzed by OMPDC from S. cerevisiae (C155S mutant) in D2O was monitored by 19F NMR spectroscopy at 470 MHz.5,13 Figure 1 shows 19F NMR spectra obtained during the OMPDC-catalyzed (360 μM) deuterium exchange reaction of FEU (4.2 mM) in the presence of 4.7 mM HPO32− in D2O buffered by 50 mM glycylglycine at pD 8.1, 25 °C and I = 0.14 (NaCl). Deuterium exchange results in a decrease in the area of the doublet at 46.87 ppm due to h-FEU and the appearance of an upfield shifted singlet at 46.58 ppm due to d-FEU. Observed first-order rate constants for OMPDC-catalyzed deuterium exchange were determined as the slopes of linear plots (not shown) of reaction progress against time over the first 5–10% reaction (eq 1), where AH and AD are the integrated areas of the signals for h-FEU and d-FEU, respectively (Figure 1). Observed second-order rate constants (kcat/Km)obsd for the OMPDC-catalyzed deuterium exchange reaction, determined in the absence and presence of phosphite dianion, were calculated according to eq 2. Details of the kinetic protocols and the experimentally-determined rate constants are given in the Supporting Information.
Figure 1.

19F NMR spectra obtained during the OMPDC-catalyzed deuterium exchange reaction of FEU (4.2 mM) in the presence of 4.7 mM phosphite dianion in D2O at pD 8.1 and 25 °C.
| (1) |
| (2) |
A value of (kcat/Km)S = ko/fND = 9.89 × 10−5 M−1 s−1 (Table 1) was determined as the second-order rate constant for the OMPDC-catalyzed deuterium exchange reaction of FEU in the absence of HPO32− at pD 8.1 (Scheme 3), where ko = 5.44 × 10−5 M−1 s−1 is the observed second-order rate constant for enzyme-catalyzed deuterium exchange in the absence of phosphite, and fND = 0.55 is the fraction of FEU present in the reactive neutral N3-D form.14 The value of fND was calculated from pKa = 8.19 for ionization of the N3-D of the pyrimidine ring of FEU in D2O at 25 °C and I = 0.10 (NaCl), determined by spectrophotometeric titration at 269 nm.
Table 1.
Kinetic parameters for the OMPDC-catalyzed decarboxylation and deuterium exchange reactions of the whole substrates OMP and FUMP and of the truncated substrate pieces EO and FEU and the calculated intrinsic phosphodianion and phosphite binding energies.a
| Reaction | Whole Substrate | (kcat/Km)SPib M−1s−1 | Substrate Piece | (kcat/Km)Sc M−1s−1 | ΔGROPid | (kcat/Km)E·HPi/Kde M−2 s−1 | ΔGHPif | |
|---|---|---|---|---|---|---|---|---|
|
|
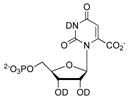
|
(5.50 ± 0.55) × 106 |
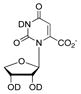
|
0.030 ±0.003 | −11.2 kcal/mol | (1.73 ± 0.35) × 104 | −7.8 kcal/mol | |
|
|
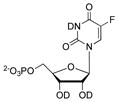
|
620 ± 62 g |
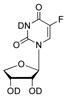
|
(9.89 ± 0.99) × 10−5 h | −9.2 kcal/mol
|
1.76 ± 0.35 h | −5.8 kcal/mol
|
|
| ΔΔGROPi i | ΔΔGHPi j | |||||||
| 2.0 kcal/mol | 2.0 kcal/mol |
In D2O buffered by either 20 or 50 mM glycylglycine at pD 8.1, 25 °C and I = 0.14 (NaCl). Quoted errors are the range of uncertainty based on multiple determinations.
Second-order rate constant for OMPDC-catalyzed reaction of the whole substrate.
Second-order rate constant for OMPDC-catalyzed reactions of EO or FEU in the absence of phosphite dianion.
The intrinsic phosphodianion binding energy, calculated from the ratio (kcat/Km)SPi/(kcat/Km)S.
Third-order rate constant for the phosphite-activated OMPDC-catalyzed reactions of the substrate pieces EO or FEU (Scheme 3).
The intrinsic phosphite binding energy, calculated from the ratio [(kcat/Km)E·HPi/Kd]/(kcat/Km)S.
Calculated from (kcat/Km)obsd = 400 M−1 s−1 at pD 8.1 and pKa = 8.37 for ionization of N3-D of FUMP. The ionized substrate is assumed to be unreactive towards OMPDC-catalyzed deuterium exchange.
Calculated from ko = 5.44 × 10−5 M−1 s−1 at pD 8.1 and pKa = 8.19 for ionization of N3-D of FEU (see text).
The difference in the phosphodianion binding energies for the decarboxylation and deuterium exchange reactions of the whole substrate.
The difference in the phosphite binding energies for the decarboxylation and deuterium exchange reactions of the truncated substrate pieces.
Scheme 3.

Figure 2 shows the dependence of (kcat/Km)obsd/ko for the deuterium exchange reaction of FEU on the concentration of phosphite dianion in D2O buffered by 50 mM glycylglycine at pD 8.1, 25 °C and I = 0.14 (NaCl), where (kcat/Km)obsd is the observed second-order rate constant for the exchange reaction (eq 3, derived for Scheme 3). The data were fit to eq 4, with (kcat/Km)S = 9.89 × 10−5 M−1 s−1, to give (kcat/Km)E·HPi/Kd = 1.76 M−2 s−1 as the third-order rate constant for OMPDC-catalyzed deuterium exchange into FEU activated by phosphite dianion (Table 1). In this nomenclature E·HPi denotes an enzyme that is saturated with phosphite dianion. Table 1 also reports the kinetic parameters for the OMPDC-catalyzed decarboxylation of the whole substrate OMP and of the truncated substrate EO in D2O buffered by 20 mM glycylglycine at pD 8.1, 25 °C and I = 0.14 (NaCl) that were determined using published procedures.6
Figure 2.

The dependence of (kcat/Km)obsd/ko for the OMPDC-catalyzed deuterium exchange reaction of FEU on the concentration of phosphite dianion in D2O at pD 8.1, 25 °C and I = 0.14 (NaCl).
| (3) |
| (4) |
A value of (kcat/Km)SPi = 620 M−1 s−1 for reaction of the whole substrate was determined for the OMPDC-catalyzed deuterium exchange reaction of FUMP in D2O under the conditions used for the exchange reaction of FEU (Table 1).
The data in Table 1 show that 1 M HPO32− is calculated to result in a 1.8 × 104-fold increase (kcat/Km)obsd for the OMPDC-catalyzed deuterium exchange reaction of FEU. This corresponds to a 5.8 kcal/mol stabilization of the vinyl carbanion-like transition state (Figure 3). By comparison, HPO32− shows a 2.0 kcal/mol larger (7.8 kcal/mol) affinity for the transition state for decarboxylation of the truncated substrate EO (Table 1).6 If OMPDC acts similarly in catalysis of the deuterium exchange reactions of EU and FEU, and the only role of the 5-F is to provide electrostatic stabilization of a vinyl carbanion intermediate, then the binding of HPO32− should likewise result in a 2.0 kcal/mol larger stabilization of the transition state for decarboxylation of EO compared with the deuterium exchange reaction of EU (Table 1).
Figure 3.

Partial free energy profiles for the unactivated and phosphite-activated OMPDC-catalyzed deuterium exchange reactions of the truncated substrate h-FEU in D2O. The slowest step for the enzyme-catalyzed reaction is deprotonation of substrate by the alkyl amino side chain of Lys-93 to form the enzyme bound carbanion. The carbanion-like transition state is stabilized by 5.8 kcal/mol by the binding of phosphite dianion. In this figure, the barrier to formation of the enzyme-bound carbanion also includes rotation of the CH2-ND2H+ bond at Lys-93 that is required to exchange the positions of the substrate derived –H and the solvent derived –D prior to hydron transfer to form d-FEU.4,13,15
Similarly, there is a 2.0 kcal/mol difference in the intrinsic phosphodianion binding energies determined for the OMPDC-catalyzed decarboxylation reaction of OMP (11.2 kcal/mol) and the deuterium exchange reaction of FUMP (9.2 kcal/mol, Table 1). In other words, ca. 80% of the intrinsic phosphodianion binding energy in the decarboxylation reaction is utilized in stabilization of the carbanion-like transition state common to both the decarboxylation and the deuterium exchange reactions. These results provide strong evidence that the interactions between bound HPO32− and the flexible phosphate gripper loop of OMPDC are directed towards effecting thermodynamic stabilization of a vinyl carbanion intermediate.4
We are unsure of the explanation for the 2.0 kcal/mol larger dianion binding energy for the transition state of the decarboxylation compared to the deuterium exchange reactions. The D70N mutation at OMPDC from M. thermautotrophicus results in a 200-fold decrease in kcat for decarboxylation of OMP, but only a 2-fold decrease in the rate of the deuterium exchange reaction of FUMP.5 This provides evidence that interactions between the carboxylate groups of D70 and enzyme-bound OMP promote decarboxylation of OMP, but not deuterium exchange of FUMP. However, the results from Table 1 are not relevant to the question of whether phosphite- driven loop closure induces destabilizing electrostatic interactions between carboxylate groups, which are relieved at the transition state for decarboxylation of OMP. This is because the rate constants reported in Table 1 provide a measure of the relative barriers to conversion of the free enzyme and reactants in the ground state to the respective transition states (see Figure 3). Destabilizing interactions at the intermediate Michaelis complexes cannot lead to a rate-enhancing reduction in the barrier for formation of these transition states from free enzyme and substrates.10 We note that one difference between the decarboxylation and deuterium exchange reactions is the presence of neutral CO2 at the product complex for the decarboxylation reaction. We suggest that the absence of CO2 from the transition state and product complex for the deuterium exchange reaction may lead to small changes in the position of other catalytic side chains that result in the observed 2.0 kcal/mol smaller phosphite activation of the deuterium exchange reaction of EU, compared with the decarboxylation of EO. This is consistent with the notion that a precise orientation of these side chains relative to reactant is required to observe the full catalytic power of OMPDC.
In conclusion, the substrate phosphodianion and the substrate piece phosphite dianion interact strongly with catalytic side chains at the flexible gripper loop of OMPDC.16 These interactions drive loop closure over the ligand and this leads to a large stabilization of carbanion-like transition states for both the decarboxylation and deuterium exchange reactions. The results may be rationalized by the model shown in Scheme 4, where OMPDC in the open form (EO) is inactive and the rare unliganded closed enzyme (EC) and the HPO32−-liganded enzyme (EC·HPO32−) exhibit essentially equal high reactivities towards both carbon deprotonation and decarboxylation of the truncated substrates FEU and EO, respectively, so that kcat/Km = (kcat/Km)′. The intrinsic binding energy of HPO32− is utilized to drive the unfavorable conformational change from EC to give EC.17,18 This model suggests that an important remaining challenge in understanding the mechanism of action of OMPDC is to provide a physical explanation for the proposed large effect of loop closure on the stability of the vinyl carbanion intermediate that is common to the OMPDC-catalyzed decarboxylation and deuterium exchange reactions.
Scheme 4.

Supplementary Material
Acknowledgments
We acknowledge the National Institutes of Health (Grant GM39754 to J.P.R. and Grant GM65155 to J.A.G.) for generous support of this work.
Footnotes
Supporting Information Available: Experimental procedures for the synthesis of FEU and the kinetic protocols and pD titrations, two Tables of values of kobsd and (kcat/Km)obsd for the reactions of FEU and EO, and a graph similar to that in Figure 2 showing the phosphite activation of the OMPDC-catalyzed decarboxylation of EO in D2O. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Callahan BP, Miller BG. Bioorg Chem. 2007;35:465–469. doi: 10.1016/j.bioorg.2007.07.004. [DOI] [PubMed] [Google Scholar]
- 2.Miller BG, Wolfenden R. Ann Rev Biochem. 2002;71:847–885. doi: 10.1146/annurev.biochem.71.110601.135446. [DOI] [PubMed] [Google Scholar]
- 3.Radzicka A, Wolfenden R. Science. 1995;267:90–93. doi: 10.1126/science.7809611. [DOI] [PubMed] [Google Scholar]
- 4.Amyes TL, Wood BM, Chan K, Gerlt JA, Richard JP. J Am Chem Soc. 2008;130:1574–1575. doi: 10.1021/ja710384t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chan KK, Wood BM, Fedorov AA, Fedorov EV, Imker HJ, Amyes TL, Richard JP, Almo SC, Gerlt JA. Biochemistry. 2009;48:5518–5531. doi: 10.1021/bi900623r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Amyes TL, Richard JP, Tait JJ. J Am Chem Soc. 2005;127:15708–15709. doi: 10.1021/ja055493s. [DOI] [PubMed] [Google Scholar]
- 7.Amyes TL, Richard JP. Biochemistry. 2007;46:5841–5854. doi: 10.1021/bi700409b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tsang WY, Amyes TL, Richard JP. Biochemistry. 2008;47:4575–4582. doi: 10.1021/bi8001743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morrow JR, Amyes TL, Richard JP. Acc Chem Res. 2008;41:539–548. doi: 10.1021/ar7002013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jencks WP. Adv Enzymol Relat Areas Mol Biol. 1975;43:219–410. doi: 10.1002/9780470122884.ch4. [DOI] [PubMed] [Google Scholar]
- 11.Gao J, Byun KL, Kluger R. Top Curr Chem. 2004;238:113–136. [Google Scholar]
- 12.Gao J. Curr Op Struct Biol. 2003;13:184–192. doi: 10.1016/s0959-440x(03)00041-1. [DOI] [PubMed] [Google Scholar]
- 13.Toth K, Amyes TL, Wood BM, Chan K, Gerlt JA, Richard JP. J Am Chem Soc. 2010;132:7018–7024. doi: 10.1021/ja102408k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ionization of the substrate at N3 is presumed to result in a large decrease in binding affinity and render the substrate ureactive towards enzyme-catalyzed deuterium exchange.
- 15.Toth K, Amyes TL, Wood BM, Chan K, Gerlt JA, Richard JP. J Am Chem Soc. 2007;129:12946–12947. doi: 10.1021/ja076222f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miller BG, Hassell AM, Wolfenden R, Milburn MV, Short SA. Proc Natl Acad Sci U S A. 2000;97:2011–2016. doi: 10.1073/pnas.030409797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Go MK, Amyes TL, Richard JP. Biochemistry. 2009;48:5769–5778. doi: 10.1021/bi900636c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Malabanan MM, Amyes TL, Richard JP. Curr Opin Struct Biol. 2010;20:702–710. doi: 10.1016/j.sbi.2010.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.